
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled electrochemical surface exfoliation of graphite pencil electrodes for high-performance supercapacitors†
Ayman A. AbdelHamid,
Abdelaziz Elgamouz and
Abdel-Nasser Kawde *
*
Pure and Applied Chemistry Group, Department of Chemistry, College of Sciences, University of Sharjah, P.O. Box 27272, Sharjah, United Arab Emirates. E-mail: akawde@sharjah.ac.ae
First published on 14th July 2023
Abstract
A controlled surface exfoliation method for graphite pencil electrodes using an environmentally friendly, low cost and scalable electrochemical process is reported. A simple direct current power supply in a neutral medium is used for inducing graphene formation on the electrode surface in a controlled manner. The electrochemical properties of the surface exfoliated electrode are characterized, displaying a >300× increase in the electrochemical surface area and >50× decrease in the electrode resistance after exfoliation. The surface graphene layer is characterized using electron microscopy, Raman, infrared, X-ray photoelectron, and energy dispersive X-ray spectroscopies and X-ray diffractometry showing a fully exfoliated surface, formation of surface defects and mild surface graphene oxidation while maintaining an intact graphitic crystal structure. The surface exfoliated electrode is tested as a supercapacitor demonstrating more than 2 orders of magnitude improvement over non-exfoliated electrode in both 3-electrode and 2-electrode setups and achieving a high areal capacitance of ∼54 mF cm−2. The benign nature, low cost, scalability of our controlled surface exfoliation methodology, and its significant impact on the electrochemical properties of the electrode make it very promising for further investigation in various applications such as energy storage and conversion, sensors, and catalysis.
Introduction
Energy storage has been in the spotlight of global affairs due to the rapid drive for electrification of the transport market and boosting renewable energy output. Both directions aim to reduce urban pollution and decrease reliance on fossil fuels, which are detrimental to the environment, a major cause of global warming and are not sustainable in the long term.1,2 A supercapacitor (SC) is an energy storage system characterized by high power capability, fast charging, and very long cycle life,3 acting as a bridge between conventional capacitors and batteries.2,4,5 It is a vital component of the energy storage landscape, offering unique advantages and supplementing other energy storage systems. SCs have been used in various applications, especially those that require high-rate capability and long cycle life, such as acceleration and regenerative breaking in electric vehicles, backup power supply, electronic devices, healthcare, power tools, energy management, and solar energy harvesting.1,6–8 Interestingly, they have been used lately in electric buses, demonstrating the huge potential market of this technology.1,8 The main type of SCs is electric double-layer SCs (EDLCs), which store charge electrostatically on the electrode surface via Helmholz double-layer formation with no faradaic reactions involved. EDLCs require high electric conductivity and large surface area, two parameters that are well fulfilled by carbon.4,9 Carbon is a very versatile material with a wide variety of allotropes, forms, surface areas, and textures. Moreover, it is of low cost, has high mechanical stability, and is environmentally friendly.2,10 Graphite pencil electrode (GPE) is an inexpensive carbon-based electrode that is readily available and easy to process, modify and use in different applications. Its conductive graphitic nature allows for high electric conductivity, and its easy surface modification allows for the preparation of highly performing SC electrodes. Several studies have reported very promising SC performance using GPEs, applying different materials to enhance capacitance, such as polymers and/or inorganic nanoparticles.4,11,12Graphene is one of the most promising SC carbon materials. The two-dimensional morphology of graphene, together with the sp2 nature of its carbon structure, impart superior electrical, mechanical, chemical, and surface properties. The sp2-bonded carbon allows for a very high electrical conductivity of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1 and a highly mechanically strong structure with Young's modulus of 1 TPa. The layered morphology allows for high flexibility and a very large specific surface area of ∼2630 m2 g−1.8,13–15 The exceptional electrical, mechanical, and surface properties of graphene make it an excellent candidate for EDLCs, whose sole requirements are high surface and electrical conductivity. A single graphene sheet is estimated to store ∼21 μF cm−2, the highest attainable limit for any carbon material, with an equivalent theoretical specific capacitance of ∼550 F g−1.15 The introduction of graphene to the surface of GPE could enhance its energy storage performance significantly. The modified electrode would retain the advantages of GPE as availability, low cost, and easy processing while using the superior properties of graphene on the surface. Although graphene deposition on the surface of GPE has been reported earlier,16,17 such an approach requires separate synthesis of graphene oxide or graphene, followed by their deposition on the surface of GPE. This complicates the electrode preparation process and increases its cost and processing time. A more practical and efficient method would be partially exfoliating the GPE, forming surface graphene sheets. This would facilitate the electrode preparation and reduce its cost, critical parameters in the energy storage field. Direct surface exfoliation would also produce a more robust and stable electrode as the graphene is generated from the body of the GPE itself. Thus, it would be structurally better connected to the graphite core. A few reports have investigated this strategy for energy storage applications, mainly by Şahin's group, who could electrochemically graphenize the surface of GPE using cyclic voltammetry in concentrated acids, whereby the surface of GPE was exfoliated via acid-mediated oxidization during the anodic sweep forming graphene oxide that was then reduced to graphene during the cathodic sweep.14 Moreover, they could introduce dopants to the exfoliated graphene by tuning the acid composition, such as introducing N,10,18 S,18 and P19 dopants, using nitric, sulfuric, and phosphoric acids, respectively. Although this strategy achieved one-step GPE surface exfoliation with impressive SC performance, it has some limitations. The reliance on highly concentrated acids for the oxidative exfoliation of graphite is a critical challenge for large-scale applications due to the hazardous and corrosive nature of such acids, which would stifle and complicate industrial-scale applications. The second critical limitation is using cyclic voltammetry as the exfoliation technique which is only suitable for small-scale preparation in a 3-electrode cell and cannot be implemented on a large scale.
000 cm2 V−1 s−1 and a highly mechanically strong structure with Young's modulus of 1 TPa. The layered morphology allows for high flexibility and a very large specific surface area of ∼2630 m2 g−1.8,13–15 The exceptional electrical, mechanical, and surface properties of graphene make it an excellent candidate for EDLCs, whose sole requirements are high surface and electrical conductivity. A single graphene sheet is estimated to store ∼21 μF cm−2, the highest attainable limit for any carbon material, with an equivalent theoretical specific capacitance of ∼550 F g−1.15 The introduction of graphene to the surface of GPE could enhance its energy storage performance significantly. The modified electrode would retain the advantages of GPE as availability, low cost, and easy processing while using the superior properties of graphene on the surface. Although graphene deposition on the surface of GPE has been reported earlier,16,17 such an approach requires separate synthesis of graphene oxide or graphene, followed by their deposition on the surface of GPE. This complicates the electrode preparation process and increases its cost and processing time. A more practical and efficient method would be partially exfoliating the GPE, forming surface graphene sheets. This would facilitate the electrode preparation and reduce its cost, critical parameters in the energy storage field. Direct surface exfoliation would also produce a more robust and stable electrode as the graphene is generated from the body of the GPE itself. Thus, it would be structurally better connected to the graphite core. A few reports have investigated this strategy for energy storage applications, mainly by Şahin's group, who could electrochemically graphenize the surface of GPE using cyclic voltammetry in concentrated acids, whereby the surface of GPE was exfoliated via acid-mediated oxidization during the anodic sweep forming graphene oxide that was then reduced to graphene during the cathodic sweep.14 Moreover, they could introduce dopants to the exfoliated graphene by tuning the acid composition, such as introducing N,10,18 S,18 and P19 dopants, using nitric, sulfuric, and phosphoric acids, respectively. Although this strategy achieved one-step GPE surface exfoliation with impressive SC performance, it has some limitations. The reliance on highly concentrated acids for the oxidative exfoliation of graphite is a critical challenge for large-scale applications due to the hazardous and corrosive nature of such acids, which would stifle and complicate industrial-scale applications. The second critical limitation is using cyclic voltammetry as the exfoliation technique which is only suitable for small-scale preparation in a 3-electrode cell and cannot be implemented on a large scale.
An ideal electrochemical GPE surface exfoliation process should use a low-cost, environmentally friendly electrolyte and a simple electrochemical technique that can be implemented industrially. (NH4)2SO4 is a green and mild electrolyte that is very effective for the electrochemical exfoliation of graphite using a simple direct current (DC) power supply and has been used for the electrochemical synthesis of graphene.20,21 The OH− generated by water oxidation at the cathode at a high voltage bias conducted a nucleophilic attack on the graphite anode oxidizing the edges and grain boundaries of the graphite layers, allowing SO42− and water intercalation, followed by SO2 an O2 gas generation that exfoliated the graphene sheets.20 In this work, we modified this facile electrochemical setup for controlled GPE surface exfoliation instead of complete electrode exfoliation and applied the electrochemically surface-exfoliated GPE (SEGPE) as a high-performance SC electrode. The controlled GPE surface exfoliation could be achieved by tuning the bias voltage and exfoliation time. The GPE surface exfoliation process was optimized, achieving a >300× increase in the electrode's electrochemical surface area, together with a >50× decrease in the electrode's resistance. The exfoliated surface was studied using electron microscopy; Raman, infrared, X-ray photoelectron, and energy dispersive X-ray spectroscopies; and X-ray diffractometry, showing complete surface coverage by graphene sheets, surface defect generation, minor surface oxygenation, and stability of the graphitic crystalline structure, which are highly conducive properties for electrochemical applications. The optimal SEGPE was used as an SC electrode showing >2 orders of magnitude higher energy storage capacity, as compared to pristine GPE, both in half and full symmetric cells, demonstrating the high impact of our surface exfoliation technique for GPE applications.
The GPE surface exfoliation methodology reported herein bypassed the concentrated acids and complex techniques used in previous work, providing a path for large-scale implementation. We have also demonstrated the efficiency of our approach and its significant impact on the energy storage performance of GPE. These excellent results open the door for further applications of our surface exfoliation strategy for energy storage, sensing, and catalysis.
Experimental
Materials and chemicals
Pentel Hi-polymer HB Pencil leads (diameter = 0.5 mm) were used as the graphite pencil electrodes. (NH4)2SO4, H2SO4, and K3[Fe(CN)6] were purchased from Sigma-Aldrich. K4[Fe(CN)6]·3H2O and KCl were purchased from Wardle Chemicals and Eurolab, respectively. Ultrapure water was generated by Milli-Q Elix Essential® 5 system.Surface exfoliation of GPE
A power supply (IRWiN POWERBASE V8) was used in the DC mode as the power source. The positive and negative terminals were connected to the GPE and Pt foil as anode and cathode, respectively. The two electrodes were immersed into a 0.1 M (NH4)2SO4 solution at a distance of 2 cm and the GPE was maintained at a depth of 1 cm. Different voltages and exfoliation times were used to optimize the surface exfoliation process. The SEGPEs were washed well using ultrapure water after the exfoliation process, and the excess water was drained before further electrochemical testing. The SEGPEs were dried in an oven at 60 °C till completely dry before physicochemical characterization.Physicochemical characterization
Scanning electron microscopy (SEM) was conducted using Tescan Vega3 fitted with an Oxford energy dispersive X-ray spectroscopy (EDX) analyzer. Fourier transform infrared (FTIR) and Raman spectroscopies were carried out using Bruker Tensor II and Renishaw inVia system, respectively. X-ray photoelectron spectroscopy (XPS) was carried out by Nexsa G2 Surface Analysis System (Thermo Scientific) with monochromatic Al Kα X-ray (1486.6 eV) and an ultra-high vacuum of ∼10−9 mbar. Powder X-ray diffractometry (XRD) was performed using Bruker D8 Advance with a Cu source (λ = 0.15406 nm) at a voltage of 40 kV, current of 40 mA, using a step of 0.02° and time per step of 0.15 s.Electrochemical testing
All electrochemical experiments were conducted using a CHI660E electrochemical workstation. Electrochemical characterization of SEGPEs was conducted in a three-electrode cell comprising GPE/SEGPE, Pt wire, and Ag/AgCl (1 M KCl) as working, counter, and reference electrodes, respectively, in an equimolar (5 mM) solution of K3[Fe(CN)6] and K4[Fe(CN)6]·3H2O and 0.1 M KCl as the supporting electrolyte. Cyclic voltammetry (CV) was conducted at a voltage range of −0.4 V to 0.8 V using different scan rates. Supercapacitor testing was done in two and three-electrode setups in 1 M H2SO4 at a voltage range of 0–1 V using CV, and Galvanostatic charge–discharge (GCD) at different scan rates and current densities, respectively. Electrochemical impedance spectroscopy (EIS) was employed at a frequency range of 106 to 0.05 Hz and an amplitude of 5 mV. The three-electrode setup comprised GPE and SEGPE as working electrodes, Pt wire, and Ag/AgCl (1 M KCl) as counter and reference electrodes, respectively. The two-electrode setup had a symmetric configuration comprising two identical SEGPEs.Results and discussion
The surface exfoliation process used in our work is based on an electrochemical system that has been developed for the electrochemical exfoliation of graphene from graphite sources. In such a system, a high voltage bias is applied by a DC power source in a 2-electrode setup, with the graphite source used as the anode. The OH− generated by water electrolysis attacks the highly polarized graphite source, opening up the graphite structure for intercalation by the SO42− anions together with H2O molecules, undergoing reduction and oxidation and producing SO2 and O2 gases, respectively. In addition, CO gas is produced by carbon oxidation.20,21 This gas generation is observed during the exfoliation process and is mainly responsible for tearing up the graphite structure resulting in graphene exfoliation. Our study modified this exfoliation process for surface-confined exfoliation of GPE rather than complete electrode exfoliation. In our modified methodology, graphite exfoliation is only confined to the surface of the electrode, leading to a surface-exfoliated electrode that is covered by graphene sheets (Fig. 1a). This approach is much more facile, less costly, and more environmentally and industrially friendly, as compared to previously reported methods for GPE surface graphenization that synthesized graphene oxide and graphene separately followed by their deposition on GPE16,17 or used highly concentrated acids to conduct oxidative acid-mediated exfoliation.10,14,19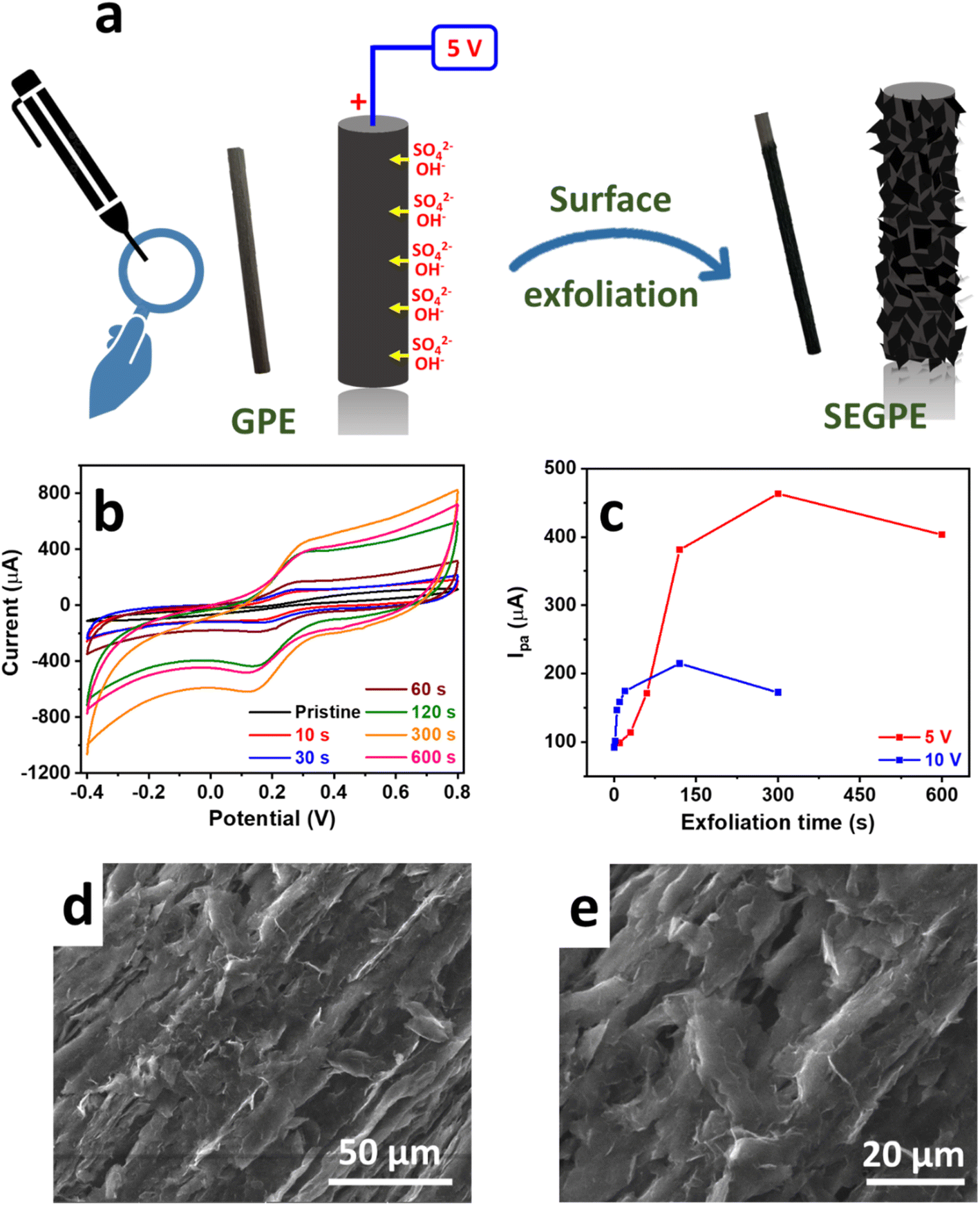 | ||
| Fig. 1 (a) Scheme showing graphite electrochemical surface exfoliation process. (b) CV profiles of SEGPEs exfoliated at 5 V for different durations. (c) Comparison between CV anodic peak currents of SEGPEs exfoliated at 5 V and 10 V. CV in (b) and (c) was conducted in 5 mM Fe(CN)63−/4−/0.1 M KCl at a scan rate of 100 mV s−1. (d and e) SEM images of SEGPE exfoliated at 5 V for 300 s at different magnifications. | ||
The exfoliation process was confined to the GPE surface by optimizing the exfoliation DC voltage and duration. To evaluate our surface exfoliation process, we used a Fe(CN)63−/4− redox couple which is a standard electrochemical highly reversible one-electron transfer system used to study the electrode surface.2 CV was conducted in a solution of 5 mM Fe(CN)63−/4− in 0.1 M KCl at a voltage range of −0.4 V to 0.8 V and a scan rate of 100 mV for preliminary evaluation. Since our objective was only surface exfoliation, we started with a low voltage bias of 2.5 V; however, only minimal change of the CV profile of the Fe(CN)63−/4− redox couple was observed, as compared to the pristine GPE, even at a relatively long exfoliation duration of 300 s, where 2 broad redox peaks with high polarization were observed, indicating inefficient charge transfer process (Fig. S1a, ESI†). Thus, 2.5 V was deemed not high enough to induce sufficient surface exfoliation. Upon the application of a voltage bias of 5 V, the exfoliation process proceeded effectively, as was observed visually by the bubbling and graphene exfoliation at the GPE surface. After only 10 s, the Fe(CN)63−/4− CV profile changed significantly with the sharp redox peaks observed with much lower polarization (ΔE = 0.1 V), as compared to ΔE of 0.65 V in the case of pristine GPE (Fig. 1b). More importantly, the current and area under the curve increased significantly, reflecting enhanced redox kinetics on the electrode surface, which could be attributed to the higher surface area, exposed surface graphene sheets, and also the surface functional groups generated during exfoliation, all of which provided significantly more electrochemically active sites for the redox reactions to take place. Such a phenomenon was augmented as the exfoliation time increased, where the peak current increased with the exfoliation duration with more than one order of magnitude increase over 300 s, as compared to pristine GPE. This was explained by the highly exfoliated electrode surface with graphene sheets covering the entire surface. The current response decreased at a long exfoliation time of 600 s indicating electrode consumption. At an exfoliation voltage of 10 V, the reaction proceeded much faster as noted by the sharper redox peaks and higher area under the curve, as compared to 5 V, till ∼90 s exfoliation time (Fig. 1c and S1b, ESI†). At longer exfoliation time than 90 s, the current response decreased significantly, as compared to 5 V. This could be explained by the very rapid exfoliation process at 10 V that supplied ∼5× power as compared to 5 V, thus the rate of change of electrode surface was much faster and the electrode was consumed very rapidly, thus complicating the controlled surface exfoliation process. Therefore, we concluded that 5 V would be the optimal voltage bias to use in our surface exfoliation strategy because it was high enough to induce very effective exfoliation but not too high to cause uncontrolled exfoliation.
The surface of pristine GPE appeared rather smooth under SEM (Fig. S2f, ESI†). After surface exfoliation for 1 s at 5 V, its roughness started to increase (Fig. S2g, ESI†), and at 10 s, exfoliated sheets could be observed (Fig. S2h, ESI†). The exfoliated graphene sheets covered the entire surface at 300 s (Fig. 1d, e and S2i, ESI†) and appeared much deeper into the core of the GPE at 600 s (Fig. S2j, ESI†). The morphology change agreed with the color and texture changes that could be observed visually, where the GPE color changed from light grey (Fig. S2a, ESI†) to dark grey and black at 1 s (Fig. S2b, ESI†) and 10 s (Fig. S2c, ESI†), respectively. At 300 s, the texture appeared rather rough, and surface etching was apparent (Fig. S2d, ESI†), and it became deeper at 600 s (Fig. S2e, ESI†). The color, texture, and morphological changes agreed with the results obtained electrochemically, where the surface exfoliation occurred gradually and seemed complete at 300 s, which matched the conclusion drawn earlier in the Fe(CN)63−/4− system.
GPE surface exfoliation was studied more extensively at a voltage bias of 5 V to further understand and optimize the process. SEGPEs exfoliated at 5 V were characterized via the Fe(CN)63−/4− to evaluate their electrochemical surface area and resistance change, as compared to the pristine GPE. CV was conducted at an increasing scan rate from 5 mV to 200 mV (Fig. S3, ESI†). As expected, the redox current increased linearly with the square root of the scan rate with a coefficient of determination (R2) as high as 0.997 (Fig. 2a), indicating a diffusion-controlled redox reaction. The electrochemical surface area was calculated using the Randles–Sevcik equation (eqn (1)).22,23
| IPa = 2.69 × 105 × n3/2 × D1/2 × v1/2 × C × A | (1) |
| Ip ∝ vα | (2) |
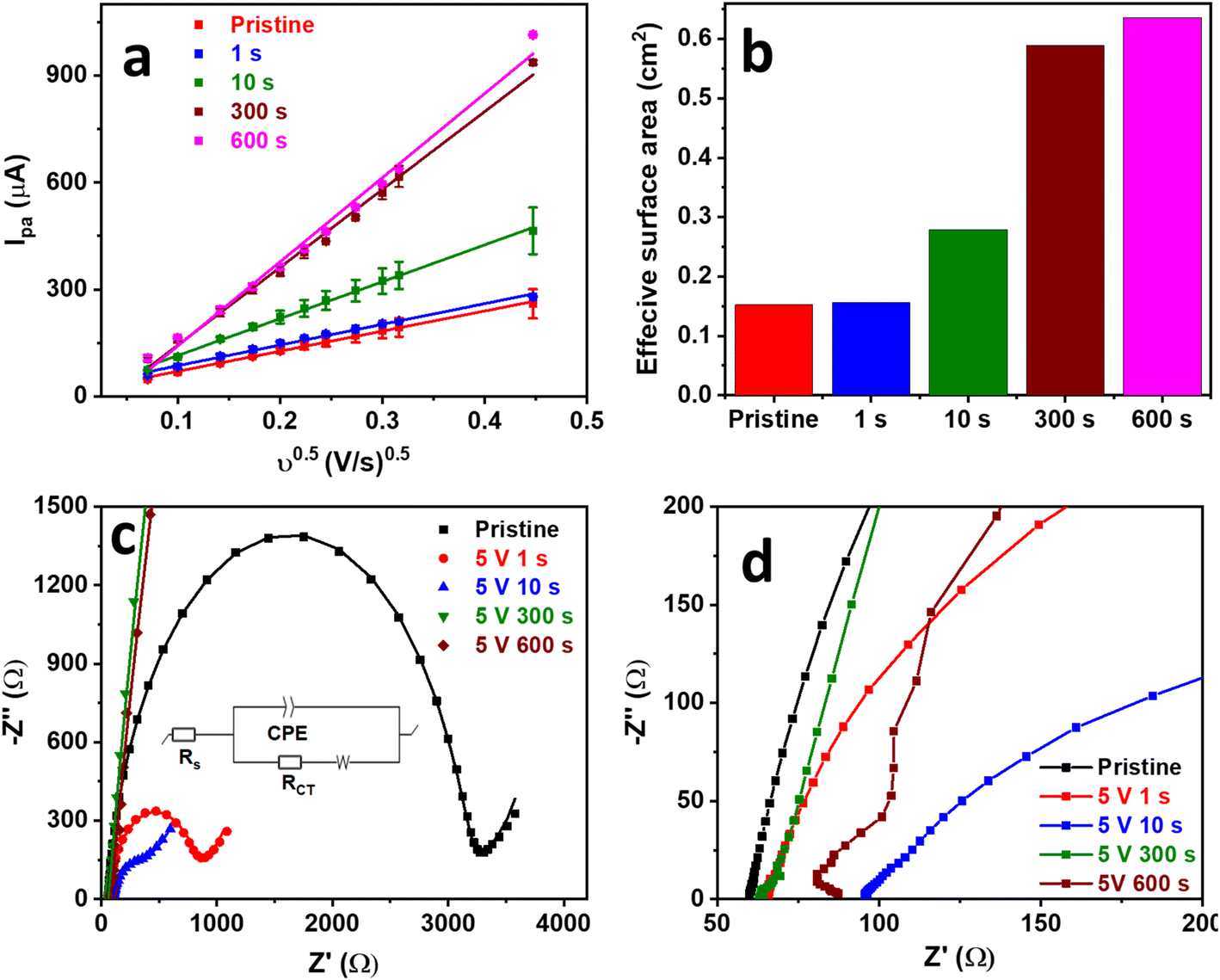 | ||
| Fig. 2 (a) Ipa vs. ν0.5 plots, (b) effective surface area, (c) full-range (points: raw data, lines: fitting, inset: equivalent circuit used in fitting), and (d) partial-range Nyquist plots of pristine GPE and SEGPEs (5 V) tested in 5 mM Fe(CN)63−/4−/0.1 M KCl. | ||
As expected, the logIp vs. logv plot of pristine GPE and SEGPE showed linear relationships with slopes of 0.45 and 0.59, respectively (Fig. S4, ESI†). The fractal dimension (Df), which is a measure of surface roughness, could be calculated according to eqn (3).4 Df has a value of 2 for flat surfaces. A Df value of less than 2 indicates inactive areas in the electrode surface, while values higher than 2 indicate high electrode surface roughness, 3D structure, and abundant microscopic domains. Pristine GPE had a Df value of 1.9, indicating a flat surface with low surface activity. SEGPE displayed a higher Df of 2.2 due to surface exfoliation that created a rough 3D surface.
| Df = 2α + 1 | (3) |
The SEGPEs were studied using EIS in the Fe(CN)63−/4− couple system to investigate the effect of surface exfoliation on their solution (Rs) and charge transfer (RCT) resistances (Fig. 2c and d and Table 1).4,12 Rs of the surface exfoliated electrodes did not show much change as compared to pristine GPE, reflecting no significant change in electrode conductivity. However, the surface exfoliation process had a major impact on RCT, where RCT decreased from ∼3126 Ω for pristine GPE to 758.0 Ω and 312.1 Ω for SEGPE exfoliated for 1 s and 10 s, respectively. RCT completely disappeared at 300 s and 600 s exfoliation, indicating the significant increase in the rate of charge transfer after surface exfoliation, mainly attributed to the large increase in the electrochemical surface area and the generation of electrochemically active sites due to exfoliation-induced surface functionalization. The >50-fold decrease in total resistance after surface exfoliation for 300 s showed the significant impact of our exfoliation methodology on the electrode's charge transfer kinetics.
| Exfoliation time (s) | Rs (Ω) | RCT (Ω) | RT (Ω) |
|---|---|---|---|
| 0 | 59.9 | 3126 | 3186 |
| 1.00 | 64.7 | 758.0 | 822.7 |
| 10.0 | 98.6 | 312.1 | 410.7 |
| 300 | 63.0 | 0 | 63.00 |
| 600 | 83.3 | 0 | 83.30 |
To get an insight into the capacitive contribution of the huge increase of the redox current upon exfoliation. CV was conducted in 0.1 M KCl without the Fe(CN)63−/4− couple.24 The capacitive component of the redox reaction increased significantly after surface exfoliation, accounting for ∼72% of the total charge, compared to only ∼2% in the case of pristine GPE (Fig. S5, ESI†). This could be attributed to the >300% increase in the electrochemical surface area upon surface exfoliation; in addition to the creation of surface functional groups, both phenomena could contribute to physical charge storage. This indicated the high potential of our surface exfoliation methodology for energy storage applications.
To understand the physical changes taking place during surface exfoliation, the composition, defects, and surface chemistry were characterized. EDX was used to study the composition of the SEGPE (Fig. 3a), especially the C/O ratio, which is a very good indicator of the oxygenated functional groups formed on the SEGPE surface. C/O ratio of pristine GPE was ∼25% for pristine GPE and only decreased to ∼23% for SEGPE exfoliated at 1 s; however, it underwent a major reduction to ∼4% at 10 s and remained almost constant thereafter (Fig. S6a, ESI†). In other words, the oxygen content on the electrode's surface increased from 3.9 at% for pristine GPE to ∼20 at% for SEGPE exfoliated at 10 s and 300 s, respectively. These results clearly showed the induction of surface oxygenated functional groups as a result of the exfoliation process and could have resulted mainly from the nucleophilic attack by the OH− that induced the initial graphite oxidation and led to opening up its structure for later anionic intercalation and exfoliation. EDX spectrum and mapping images of SEGPE (5 V, 300 s) are shown in Fig. S6c and d, ESI,† displaying C and O as the major elements with atomic percentages of 79.2% and 19.6%, respectively. Minor amounts of Si and S were also detected, the former due to the inherent clay constituent in GPE, and the latter due to residual SO42− groups from the exfoliation process. The Raman spectrum of graphite showed 3 main peaks at 1355.6 cm−1, 1581.7 cm−1, and 2724.6 cm−1 (Fig. 3b), corresponding to D, G, and 2D bands, originating from A1g symmetry mode due to sp3 carbon and defects, doubly degenerate phonon E2g symmetry mode at the Brillouin zone center due to in-plane sp2 vibration, and second order zone boundary phonons in the graphene structure, respectively.25,26 SEGPE exhibited major changes in all 3 bands. The D band intensity increased significantly mainly due to the introduction of defects, sp3 carbon, and oxygenated functional groups during exfoliation (Fig. 3b). The ID/IG ratio is a well-known defect indicator,25,26 it has been shown to increase >3× from 0.36 till ∼1.2 after 10 s exfoliation, in agreement with previous reports on graphene,27,28 with minimal changes upon extending the exfoliation time (Fig. S6b, ESI†) with corresponding sp2 carbon domains (La) of 12.1 nm for pristine GPE, and 3.7 nm, 4.1 nm, and 3.3 nm for SEGPE at 10 s, 300 s, and 600 s, respectively, as calculated using Kinghts empirical equation (eqn (4)),29 which indicated major disruption of sp2 carbon during exfoliation.
| La = 4.35 × (ID/IG)−1 | (4) |
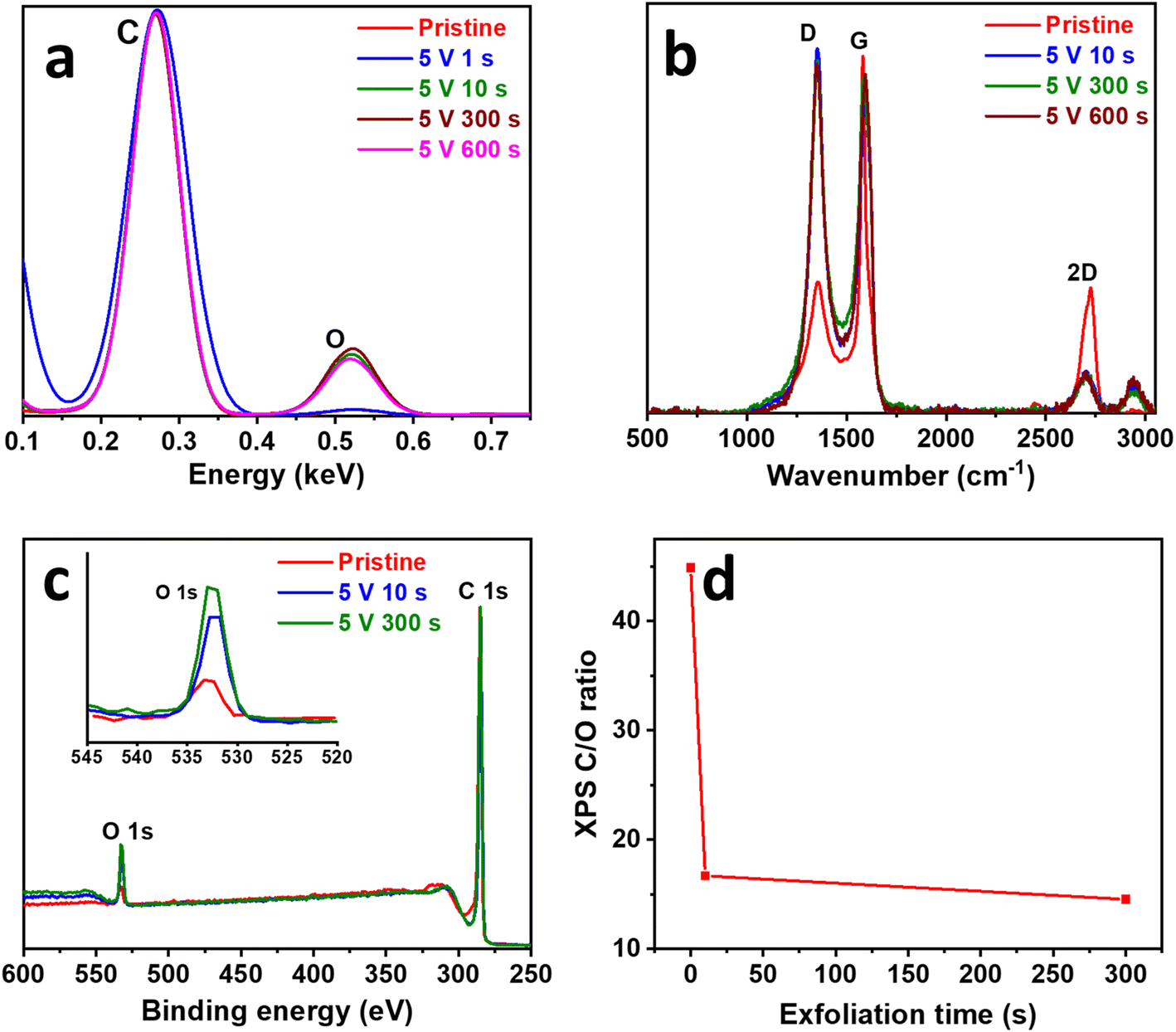 | ||
| Fig. 3 (a) EDX, (b) Raman, and (c) XPS survey spectra of pristine GPE and SEGPEs (5 V). (d) XPS-derived C/O ratio vs. exfoliation time plot. Inset in (c): magnified O 1s region. | ||
This observation agreed well with the C/O ratio displayed earlier and showed the correlation between the generated surface defects, disruption of large sp2 carbon domains, and oxygenated functional group formation. Another change in the Raman spectra after exfoliation was that of the G band that showed peak broadening and blue shift, two phenomena that have been reported earlier and attributed to structural disorder, stress, and isolated double bonds30,31 all of which could be associated with the reactions taking place during the exfoliation process. Finally, the 2D band intensity was reduced, which could be attributed to structural defects and disorders, and agreed with earlier reports.26,31 This analysis demonstrated the effect of surface exfoliation on the structure of graphite indicating disorder, defects, sp3 carbon, and oxygenated functional group formation. The decoration of SEGPE with oxygenated functional groups was analyzed using FTIR (Fig. S6e, ESI†). Pristine GPE only showed a peak at ∼1616 cm−1 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.19,32 At 10 s exfoliation, an extra small peak appeared at ∼1235 cm−1, which could be assigned to epoxy C–O bond,32,33 and was more pronounced at 300 s exfoliation, which also showed 2 additional peaks at ∼1446 cm−1 and ∼954 cm−1 that could be assigned to CH234 and epoxy or peroxide33 functional groups, respectively. The effect of the introduced functional groups on the structure of graphite has been examined by XRD (Fig. S6f, ESI†) by comparing the most intense (002) plane for pristine GPE and SEGPE at 10 s and 300 s. SEGPE (002) plane did not show any change in peak position or broadening, which indicated an intact graphitic crystalline structure with no change in interlayer spacing and no detectable disorder.35–37 This could be explained by the confinement of the surface exfoliation to the GPE surface and thus no significant structural disorder was inflicted on the electrode and the core remained intact.
C bond.19,32 At 10 s exfoliation, an extra small peak appeared at ∼1235 cm−1, which could be assigned to epoxy C–O bond,32,33 and was more pronounced at 300 s exfoliation, which also showed 2 additional peaks at ∼1446 cm−1 and ∼954 cm−1 that could be assigned to CH234 and epoxy or peroxide33 functional groups, respectively. The effect of the introduced functional groups on the structure of graphite has been examined by XRD (Fig. S6f, ESI†) by comparing the most intense (002) plane for pristine GPE and SEGPE at 10 s and 300 s. SEGPE (002) plane did not show any change in peak position or broadening, which indicated an intact graphitic crystalline structure with no change in interlayer spacing and no detectable disorder.35–37 This could be explained by the confinement of the surface exfoliation to the GPE surface and thus no significant structural disorder was inflicted on the electrode and the core remained intact.
An in-depth compositional analysis of the electrodes' surface was done using XPS. Survey spectra showed 2 main peaks at ∼285 eV and ∼533 eV, corresponding to C 1s and O 1s, respectively (Fig. 3c).38,39 The oxygen content increased as exfoliation proceeded with the C/O ratio decreasing from 44.9 for pristine GPE to 16.7 and 14.6 for SEGPE exfoliated at 10 s and 300 s (Fig. 3d), corresponding to an oxygen at% of 2.2, 5.7 and 6.4, respectively. C 1s core level spectra were analyzed to study the development of oxygenated functional groups over exfoliation time (Fig. S7a–c, ESI†). The spectra could be deconvoluted into 4 different components at 284.8 eV, 285.9 eV, 286.8 eV, and 288.8 eV, corresponding to CC, C–O, CO, and O–CO groups, respectively.39 It was observed that the ratio of the oxygenated carbon components, especially C–O, increased over exfoliation time from 0.32 for pristine GPE to 0.34 and 0.37 for SEGPE at 10 s and 300 s, respectively (Fig. S7d, ESI†). This analysis confirmed the formation of oxygenated groups on the exfoliated surface and agreed with the previous EDX, Raman, and FTIR results.
The gradual surface exfoliation process shown by SEM, which displayed the typical layered graphene structure with the fingerprint wavy and wrinkled morphology,40 together with EDX and XPS analyses that showed carbon to be the major element, and Raman analysis that demonstrated a significant enhancement of the D band upon exfoliation and a 3× increase in ID/IG ratio to ∼1.2, typical of graphene,27,28 confirm electrode surface graphenization during our controlled surface exfoliation process. The ∼84% and ∼68% decrease in C/O ratio as calculated using EDX and XPS, respectively, upon surface exfoliation, in addition to the significant increase in oxygenated carbon species as detected by FTIR and XPS confirm the formation of oxygenated groups during the surface exfoliation process. The absence of change to the whole electrode's graphitic crystal structure could be explained by the controlled surface electrode treatment, which confined exfoliation and oxygenated group formation only to the surface layer while maintaining the graphitic highly conductive GPE core in the pristine condition to act as a directly attached electrode current collector.
The mechanism of our controlled surface exfoliation strategy is based on the electrochemical graphite exfoliation method for graphene synthesis;20,21 however, the exfoliation voltage and time were controlled to confine the exfoliation process only to the surface. After applying the voltage bias between the graphite anode and Pt cathode, water was oxidized on the Pt surface, producing nucleophilic hydroxyl ions that attacked the positively-charged graphite, expanding its interlayer spaces. Such expansion allowed for the intercalation of electrolyte sulfate ions and water molecules into the graphite layers. The evolution of SO2 and O2 gases upon reduction and oxidation of SO42− and H2O resulted in graphite exfoliation. We have demonstrated that by fine-tuning the exfoliation voltage and time, the exfoliation process could be confined to the surface, thus changing the method outcome from graphene synthesis to controlled surface graphenization. We carefully optimized the exfoliation voltage and duration. We found that an exfoliation voltage of 5 V and duration of 300 s resulted in complete surface graphenization, together with the highest electrochemical activity, lowest resistance, and excellent capacitive properties.
The pristine GPE and SEGPE were tested for their supercapacitor performance initially using a 3-electrode setup in 1 M H2SO4. The open circuit potential (OCP) of the SEGPE was ∼1.1 V, ∼2.5× higher than that of pristine GPE (Fig. S8a, ESI†), mainly due to its higher surface area and functionalized surface. EIS is a very informative technique that sheds light on the conductivity and capacitive behavior of the electrodes. Nyquist plots showed the low resistance of both pristine and SEGPE (Fig. 4a), with negligible charge transfer resistance as shown by the non-existent semi-circles.41 The intersection with the real impedance axis was taken as the total electrode resistance (RT), including both solution and charge transfer resistances.4,12 SEGPE had a slightly lower RT of 18.12 Ω, as compared to 20.23 Ω shown by the pristine GPE. The Bode plots showed a higher phase angle of SEGPE at lower frequencies, as compared to pristine GPE (Fig. S8b, ESI†), indicating better capacitive behavior.12,42 However, the phase angle decreased faster in SEGPE with increasing frequency, which also showed pseudocapacitive behavior.42 This could be explained by the surface functional groups and defects introduced to the surface of the electrode during exfoliation that could have contributed to the pseudocapacitive charge storage,43,44 and could also be attributed to the clay content in pencil leads,45 which is mainly composed of silicates46 that exhibit pseudocapacitive behavior,47 and whose surface exposure could have been increased upon surface exfoliation.
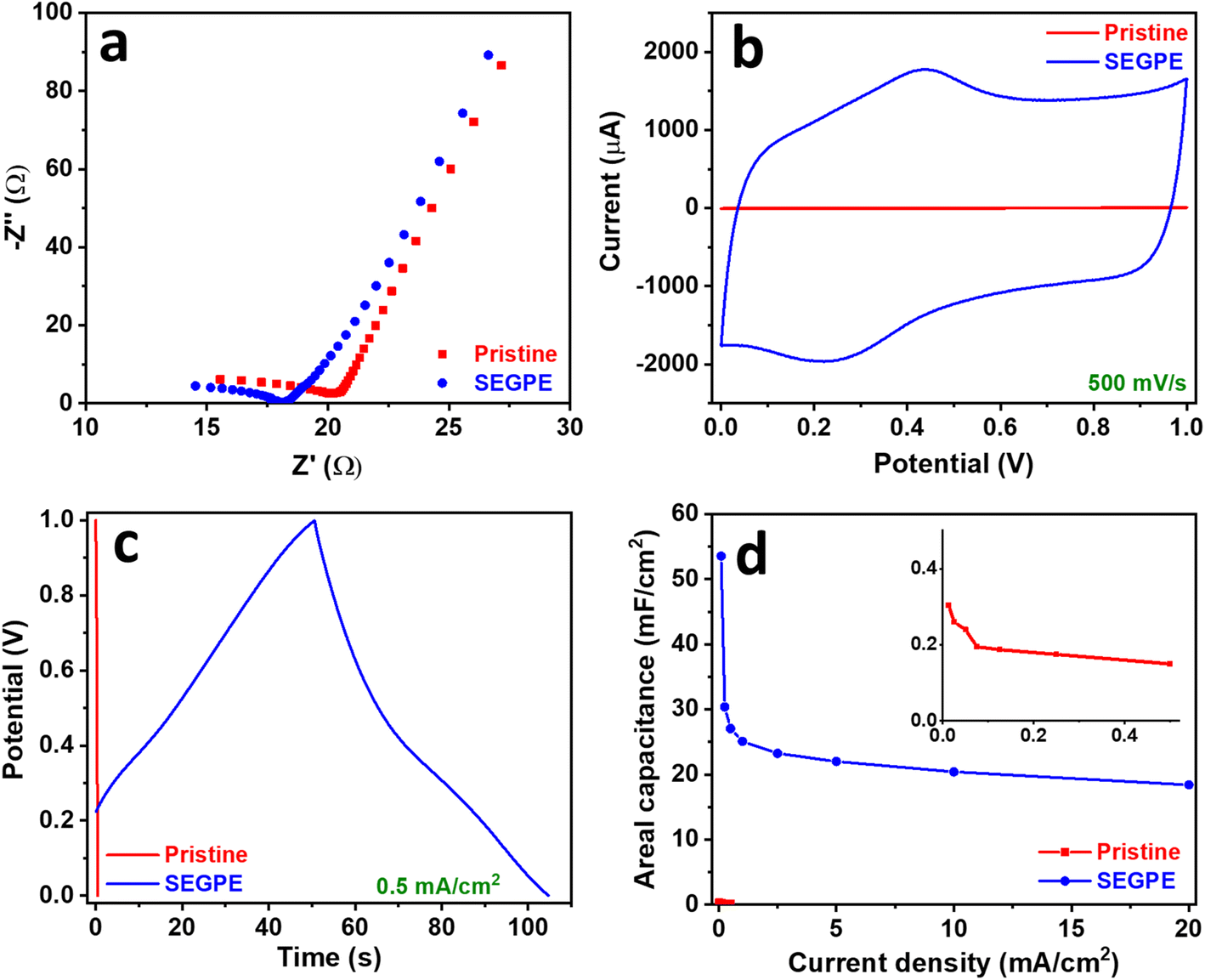 | ||
| Fig. 4 (a) Nyquist, (b) CV, (c) GCD, and (d) areal capacitance vs. current density plots of pristine GPE and SEGPE (5 V, 300 s) in a 3 electrode cell. Inset in (d): magnified plot of pristine GPE. | ||
CV was used to evaluate the electrode capacitance at a voltage range of 0–1 V. A typical rectangular-shaped CV was observed for pristine GPE (Fig. S9a, ESI†), which indicated an EDLC-type supercapacitor.11 Only slight bumps could be seen at 0.2–0.4 V indicating a minor faradaic contribution, which could be attributed to the clay content present in the pencil leads, where the HB grade used in this study contains 26% clay.45 The rectangular CV shape did not change at different rates till 500 mV s−1, indicating good rate capability. However, a low current response of <10 μA was observed even at a high scan rate of 500 mV s−1, indicating the limited energy storage capacity of pristine GPE. After surface exfoliation, the rectangular CV shape was maintained at all scan rates (Fig. S9b, ESI†), however, the faradaic contribution was more pronounced, which could be explained by the pseudocapacitive contribution of the oxygenated groups and the increased exposure of the pseudocapacitive clay content. The current response significantly increased as compared to pristine GPE, reaching ∼2000 μA at 500 mV s−1 (Fig. 4b), showing more than 2 orders of magnitude improvement in capacitive energy storage over pristine GPE. This major charge storage increase is due to the complete coverage of the electrode surface by graphene sheets as demonstrated earlier, which enhanced the electrochemical surface area and thus increased the capacitive charge storage. Another reason could be the formation of surface oxygenated groups and the increased exposure of the GPE clay content during surface exfoliation, causing a significant pseudocapacitive contribution. The electrode charge storage capability was evaluated in more depth by GCD, and the areal capacitance was calculated using the discharge curve according to eqn (5).10
| C = (i × Δt)/(ΔV × A) | (5) |
The ultimate energy storage parameters are energy density and power density. The first considers the electrode capacitance and working voltage, and the second the rate capability. Thus, the Ragone plot of energy density vs. power density is the most important display of the electrode energy storage performance. Energy and power densities were calculated according to eqn (6) and (7).18,48
| E = (C × ΔV2)/(2 × 3.6) | (6) |
| P = (E × 3.6)/Δt | (7) |
| Electrode | Electrolyte | Configuration | Areal capacitance (mF cm−2) | Current density (mA cm−2) | Ref. |
|---|---|---|---|---|---|
| a CNT: carbon nanotubes. NCS: nickel cobalt sulfide. PVA: polyvinyl alcohol. | |||||
| Oxidized GPE (6H) | — | 3 electrode | 48.0 | 0.300 | 2 |
| GPE (HB) | — | 3 electrode | 15.0 | 0.200 | 2 |
| GPE (1H) | 1 M Na2SO4 | 3 electrode | 15.6 | 2.00 | 11 |
| P-doped treated GPE (HB) | 1 M H2SO4 | 3 electrode | 49.7 | 0.500 | 19 |
| S, N co-doped treated GPE | 1 M H2SO4 | 3 electrode | 71.5 | 10.0 | 18 |
| Graphene coated Si | 0.5 M Na2SO4 | 3 electrode | 8.16 | 5.00 mV s−1 (scan rate) | 51 |
| Paper@CNT | 3 M KOH | 3 electrode | 15.3 | 0.100 | 52 |
| Paper@CNT@NCS | 3 M KOH | 3 electrode | 38.3 | 0.100 | 52 |
| Paper@CNT@NCS | 3 M KOH | 2 electrode | <2.00 | 0.400 | 52 |
| Laser-induced graphene | 1 M H2SO4 | 2 electrode | 9.00 | 0.0200 | 53 |
| Laser-induced graphene | LiCl/PVA | 2 electrode | 3.90 | 0.250 | 54 |
| Graphite | H3PO4/PVA | 2 electrode | 10.4 | 0.100 | 55 |
| This work | 1 M H2SO4 | 3 electrode | 53.6 | 0.100 | This work |
| 3 electrode | 30.4 | 0.250 | |||
| 3 electrode | 23.3 | 2.50 | |||
| 2 electrode | 4.70 | 0.125 | |||
| 2 electrode | 4.31 | 0.469 | |||
| 2 electrode | 3.75 | 3.13 | |||
The 2-electrode setup is more representative of the supercapacitor performance and is closer to the practical device than the 3-electrode system, which tends to overestimate capacitance.4 Therefore, we assembled a symmetric 2-electrode cell using identical SEGPEs.4 Areal capacitance (C in mF cm−2) in symmetric cells was calculated according to eqn (8) (volumetric capacitance in F cm−3 was also calculated using eqn (8), using a volumetric current density in A cm−3 instead of an areal current density in mA cm−2),48 while the energy and power densities were calculated according to eqn (6) and (7),48 as explained earlier.
| C = (2 × i × Δt)/(ΔV × A) | (8) |
EIS of pristine GPE and SEGPE symmetric cells showed similar behavior as that in the 2-electrode setup. SEGPE symmetric cell has an RT of 7.9 Ω, which was significantly lower than that of pristine GPE (36.9 Ω) (Fig. 5a), indicating better charge transfer capability of the surface exfoliated electrode.56 Bode plots also showed a typical capacitive performance at low frequencies, with the SEGPE displaying a higher phase angle than pristine GPE, indicating better capacitive behavior (Fig. S8c, ESI†). CV and GCD of both pristine GPE and SEGPE (Fig. 5b, c and 6a, b) showed typical capacitor rectangular and triangular shapes, respectively. Pristine GPE symmetric cell could not be tested at CV scan rates higher than 50 mV s−1, beyond which very high signal noise was observed, probably due to its limited charge transfer that could not tolerate high current currents. At 50 mV s−1, only a small current of <0.3 μA was detected (Fig. S12a, ESI†). SEGPE symmetric cell could be tested up to 500 mV s−1 with the current response ∼40 μA and ∼250 μA at 40 mV s−1 and 500 mV s−1, respectively, 200× better than pristine GPE (Fig. 5b and S12b, ESI†).
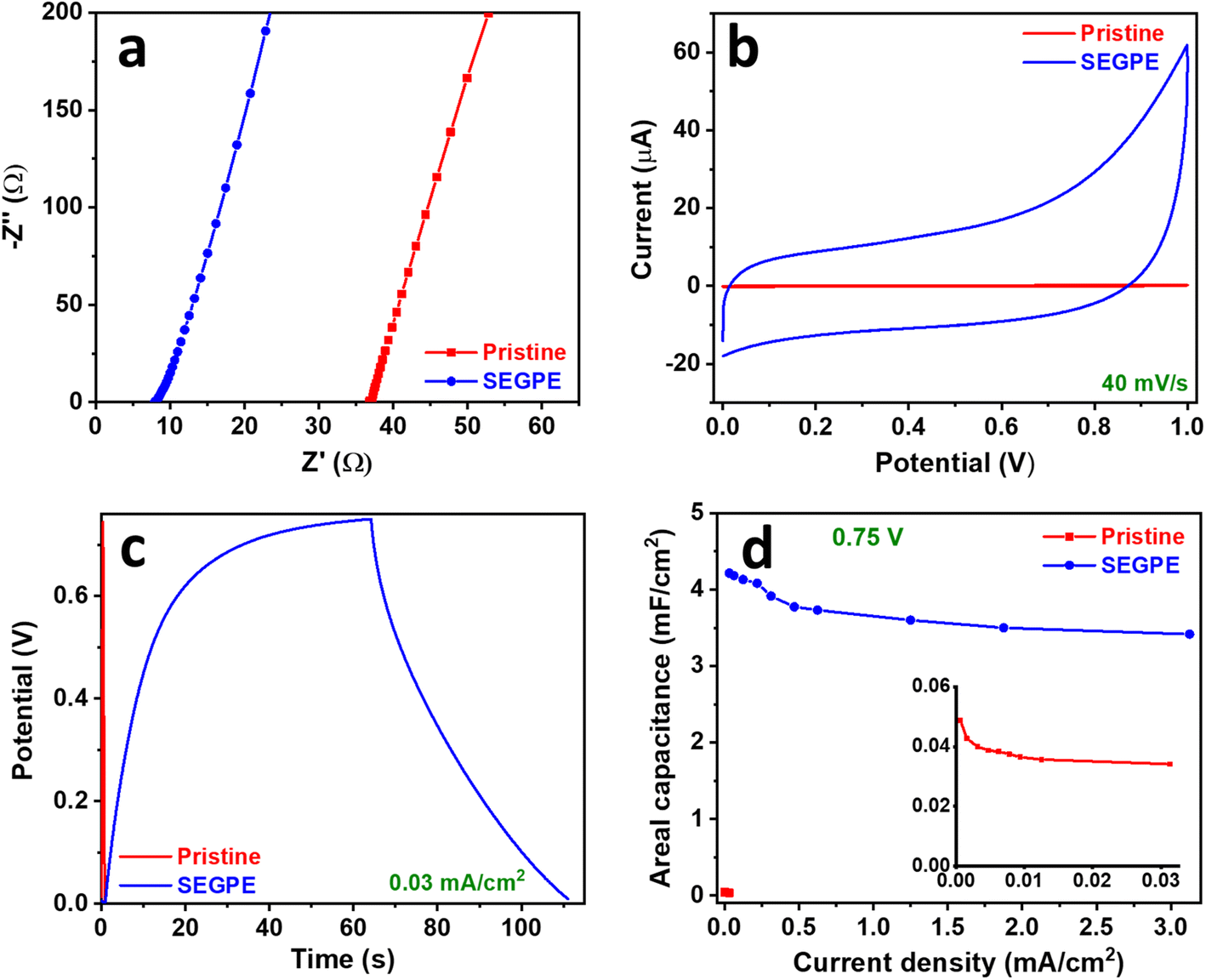 | ||
| Fig. 5 (a) Nyquist, (b) CV, (c) GCD, and (d) areal capacitance vs. current density plots of pristine GPE and SEGPE (5 V, 300 s) in a symmetric cell. Inset in (d): magnified plot of pristine GPE. | ||
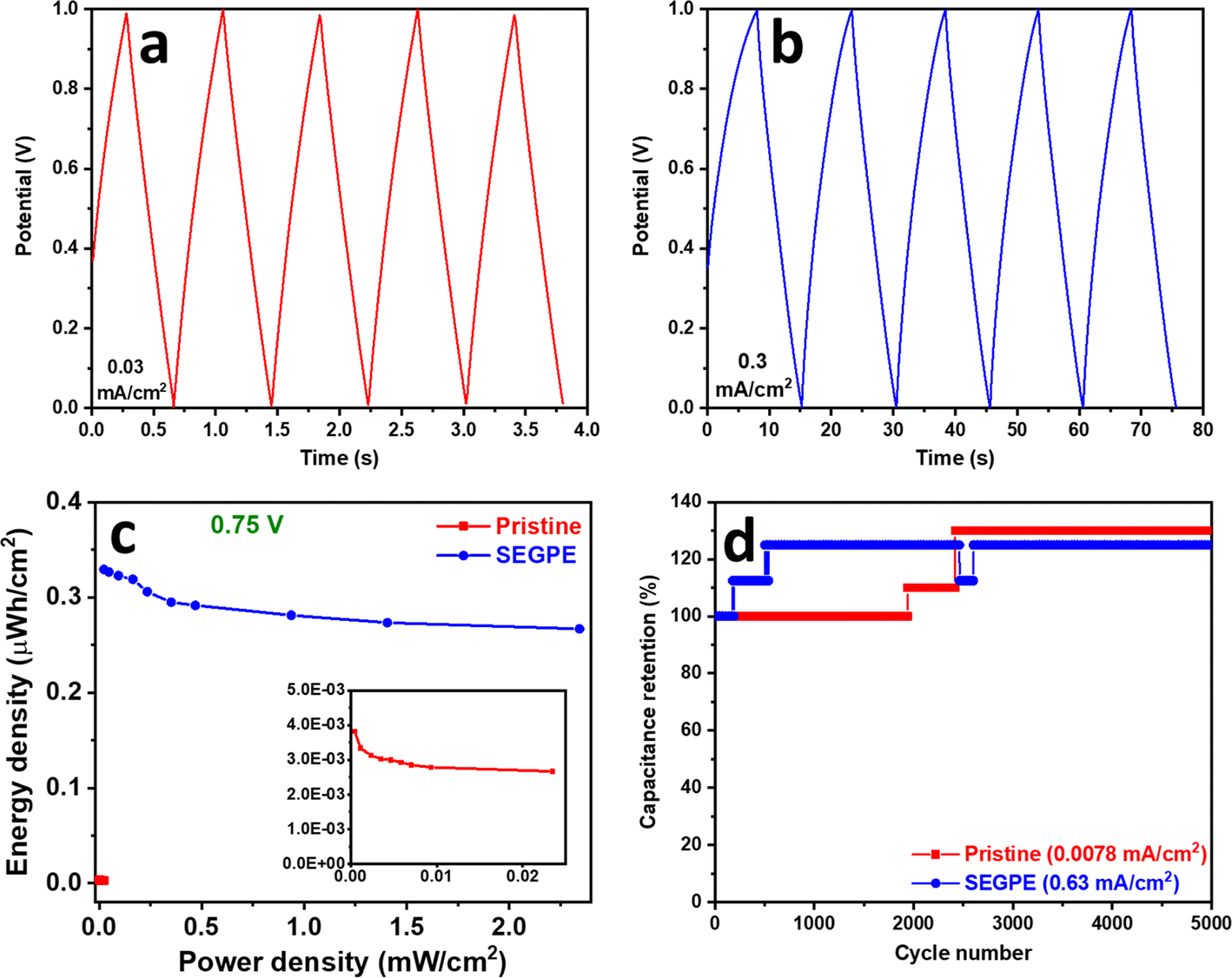 | ||
| Fig. 6 (a and b) GCD profiles, (c) areal Ragone plot, and (d) stability profiles of (a) pristine GPE, (b) SEGPE (5 V, 300 s), and (c and d) pristine GPE and SEGPE (5 V, 300 s) in a symmetric cell setup. Inset in (c): magnified pristine GPE plot. | ||
GCD of the symmetric cell was conducted in the voltage range of 0–1 V. Pristine GPE symmetric cell showed GCD discharge times of 43.1 s and 0.4 s (Fig. S12c, ESI†), corresponding to areal capacitances of 0.05 mF cm−2 and 0.02 mF cm−2 (Fig. S12e, ESI†), at current densities of 6 × 10−4 mA cm−2 and 0.03 mA cm−2, and volumetric capacitances of 0.004 F cm−3 and 0.002 F cm−3 at current densities of 5 × 10−5 A cm−3 and 0.003 A cm−3, respectively (Fig. S11c, ESI†). On the other hand, SEGPE symmetric cell showed GCD discharge times of 76.1 s and 0.6 s (Fig. S12d, ESI†), corresponding to areal capacitances of 4.8 mF cm−2 and 3.8 mF cm−2 (Fig. S12e, ESI†), at current densities of 0.03 and 3.13 mA cm−2, and volumetric capacitances of 0.4 F cm−3 and 0.3 F cm−3 at current densities of 0.003 A cm−3 and 0.3 A cm−3, respectively (Fig. S11c, ESI†). SEGPE symmetric cell showed >200× higher areal capacitance, as compared to pristine GPE symmetric cell at the same current density of 0.03 mA cm−2, which was consistent with the CV results and also the results of 3-electrode setup. Interestingly, SEGPE symmetric cell also showed very good rate capability with ∼80% capacitance retention over 2 orders of magnitude increase in current density from 0.03 mA cm−2 to 3.13 mA cm−2. It achieved an energy density of 0.7 μW h cm−2 and 0.5 μW h cm−2 at power densities of 0.03 mW cm−2 and 3.13 mW cm−2 (Fig. S12f, ESI†), corresponding to volumetric energy densities of 0.05 mW h cm−3 and 0.04 mW h cm−3 at power densities of 0.003 W cm−3 and 0.3 W cm−3, respectively (Fig. S11d, ESI†). Both pristine and SEGPE symmetric cells showed excellent stability over 5000 cycles (Fig. 6d), with both devices showing a capacitance increase over cycling, indicating induced activation, as explained earlier.
It was observed that SEGPE symmetric cell showed long charge curves at low current densities of 0.031–0.13 mA cm−2 (Fig. S12d†), while the GCD plots were balanced at higher current densities (Fig. 6b), indicating an overcharge process taking place at low current densities. Therefore, we conducted the GCD testing of symmetric cells at a lower voltage range of 0–0.75 V (Fig. S13a and b†). At a current density of 0.031 mA cm−2 (0.0026 A cm−3), corresponding to a power density of 0.023 mW cm−2 (0.0019 W cm−3), GPE and SEGPE showed discharge times of 0.41 s and 51 s (Fig. 5c), capacitances of 0.0342 mF cm−2 (0.0028 F cm−3) and 4.22 mF cm−2 (0.344 F cm−3) (Fig. 5d and S11e, ESI†), and energy densities of 0.0027 μW h cm−2 (2.18 × 10−4 mW h cm−3) and 0.329 μW h cm−2 (0.027 mW h cm−3), respectively (Fig. 6c and S11f, ESI†), with SEGPE showing >2 orders of magnitude higher performance, as compared to GPE. Moreover, SEGPE could maintain an excellent performance up to a current density of 3.13 mA cm−2 (0.255 A cm−3), corresponding to a power density of 2.34 mW cm−2 (0.191 W cm−3), achieving a capacitance of 3.42 mF cm−2 (0.279 F cm−3) (Fig. 5d and S11e, ESI†) and an energy density of 0.267 μW h cm−2 (0.0218 mW h cm−3) (Fig. 6c and S11f, ESI†). The differences between the supercapacitor performance of pristine GPE and SEGPE are highlighted in Table 3.
| Parameter | Pristine | SEGPE |
|---|---|---|
| 3-electrode cell | ||
| RT (Ω) | 20.2 | 18.1 |
| Discharge time (s) at 0.500 mA cm−2 | 0.300 | 54.1 |
| Areal capacitance (mF cm−2) at 0.500 mA cm−2 | 0.150 | 27.1 |
| Volumetric capacitance (F cm−3) at 0.0408 A cm−3 | 0.0122 | 2.21 |
| Areal energy density (μW h cm−2) at 0.250 mW cm−2 | 0.0208 | 3.76 |
| Volumetric energy density (mW h cm−3) at 0.0204 W cm−3 | 0.00170 | 0.307 |
| OCP (V) | 0.438 | 1.11 |
|
||
| Symmetric cell (1 V) | ||
| RT (Ω) | 36.9 | 7.88 |
| Discharge time (s) at 0.0313 mA cm−2 | 0.380 | 76.1 |
| Areal capacitance (mF cm−2) at 0.0313 mA cm−2 | 0.0238 | 4.80 |
| Volumetric capacitance (F cm−3) at 0.00255 A cm−3 | 0.00194 | 0.388 |
| Energy density (μW h cm−2) at 0.0313 mW cm−2 | 0.00330 | 0.661 |
| Volumetric energy density (mW h cm−3) at 0.00255 W cm−3 | 2.69 × 10−4 | 0.0539 |
|
||
| Symmetric cell (0.75 V) | ||
| Discharge time (s) at 0.0313 mA cm−2 | 0.41 | 50.6 |
| Areal capacitance (mF cm−2) at 0.0313 mA cm−2 | 0.0342 | 4.22 |
| Volumetric capacitance (F cm−3) at 0.00255 A cm−3 | 0.00279 | 0.344 |
| Energy density (μW h cm−2) at 0.0313 mW cm−2 | 0.00267 | 0.329 |
| Volumetric energy density (mW h cm−3) at 0.00255 W cm−3 | 2.18 × 10−4 | 0.0269 |
Conclusion
We have developed a new strategy for controlled surface exfoliation of GPE and demonstrated a significant impact of the surface exfoliated electrode in energy storage. The surface exfoliation parameters of voltage and time were optimized and the electrochemical properties of SEGPE were fully characterized, demonstrating a >300× increase in the electrochemical surface area and >50× decrease in the electrode's total resistance. This major improvement was mainly attributed to the complete electrode surface coverage by graphene sheets and the surface defects and functional groups induced by the surface exfoliation process. The optimal SEGPE was tested as a supercapacitor electrode in both 3-electrode and 2-electrode symmetric setups, demonstrating ∼200-fold higher areal capacitance as compared to pristine GPE in both configurations. This significant improvement showed the high impact of our controlled surface exfoliation strategy for supercapacitor applications. The environmentally friendly, low cost and scalable process used in surface exfoliation in our work, together with the significant improvement in the electrochemical properties of the surface exfoliated electrode, also showed the high potential of our approach for various other applications, including energy storage and conversion, sensors, and catalysis.Author contributions
Ayman AbdelHamid (A. A. A.): conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – initial draft preparation, visualization; Abdelaziz Elgamouz (A. E.), Abdel-Nasser Kawde (A. K.): conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing – original draft preparation, writing review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is funded by the Research Institute of Science and Engineering (RISE), University of Sharjah, Sharjah, United Arab Emirates, Seed Research Project No. (22021440119), V.C.R.G./R. 447/2022 and Collaborative Research Project No. (22021440122), V.C.R.G./R. 447/2022.References
- P. Manasa, S. Sambasivam and F. Ran, J. Energy Storage, 2022, 54, 105290 CrossRef.
- N. Vishnu, A. Gopalakrishnan and S. Badhulika, Electrochim. Acta, 2018, 269, 274–281 CrossRef CAS.
- G. Xiong, P. He, Z. Lyu, T. Chen, B. Huang, L. Chen and T. S. Fisher, Nat. Commun., 2018, 9, 790 CrossRef PubMed.
- S. Mondal, N. Aravindan and M. V. Sangaranarayanan, Electrochim. Acta, 2019, 324, 134875 CrossRef CAS.
- A. A. AbdelHamid, X. Yang, J. Yang, X. Chen and J. Y. Ying, Nano Energy, 2016, 26, 425–437 CrossRef CAS.
- W. Du, Z. Zhu, Y. Wang, J. Liu, W. Yang, X. Qian and H. Pang, RSC Adv., 2014, 4, 6998 RSC.
- S. Banerjee, B. De, P. Sinha, J. Cherusseri and K. K. Kar, in Handbook of Nanocomposite Supercapacitor Materials I: Characteristics, ed., K. K. Kar, Springer Nature Switzerland AG, 2020 Search PubMed.
- M. Horn, B. Gupta, J. MacLeod, J. Liu and N. Motta, Curr. Opin. Green Sustainable Chem., 2019, 17, 42–48 CrossRef.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dinca, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- M. B. Arvas, M. Gencten and Y. Sahin, Ionics, 2021, 27, 2241–2256 CrossRef CAS.
- R. Sha and S. Badhulika, Nanotechnology, 2019, 30, 035402 CrossRef CAS.
- E. Karaca, D. Gökcen, N. Ö. Pekmez and K. Pekmez, Int. J. Energy Res., 2019, 44, 158–170 CrossRef.
- S. W. Bokhari, A. H. Siddique, P. C. Sherrell, X. Yue, K. M. Karumbaiah, S. Wei, A. V. Ellis and W. Gao, Energy Rep., 2020, 6, 2768–2784 CrossRef.
- H. Gürsu, M. Gençten and Y. Şahin, Electrochim. Acta, 2017, 243, 239–249 CrossRef.
- M. F. El-Kady, Y. Shao and R. B. Kaner, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS.
- N. Baig, A. N. Kawde, A. Elgamouz, M. Morsy, A. M. Abdelfattah and R. Othaman, RSC Adv., 2022, 12, 2057–2067 RSC.
- A. H. Oghli and A. Soleymanpour, Biochem. Eng. J., 2021, 167, 107920 CrossRef CAS.
- M. B. Arvas, H. Gürsu, M. Gencten and Y. Sahin, ChemistrySelect, 2022, 7, e202200360 CrossRef CAS.
- M. B. Arvas, H. Gürsu, M. Gencten and Y. Sahin, J. Energy Storage, 2022, 55, 105766 CrossRef.
- K. Parvez, Z. S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Mullen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed.
- K. Chen, D. Xue and S. Komarneni, J. Colloid Interface Sci., 2017, 487, 156–161 CrossRef CAS PubMed.
- M. Ibrahim, H. Ibrahim, N. B. Almandil, M. A. Sayed and A. N. Kawde, Anal. Methods, 2020, 12, 2846–2857 RSC.
- A. Ganguly and K. Y. Hwa, Mater. Today Chem., 2022, 24, 100862 CrossRef CAS.
- S. Ben-Amor, E. Vanhove, F. Sékli Belaïdi, S. Charlot, D. Colin, M. Rigoulet, A. Devin, N. Sojic, J. Launay, P. Temple-Boyer and S. Arbault, Electrochim. Acta, 2014, 126, 171–178 CrossRef CAS.
- F. Yin, S. Wu, Y. Wang, L. Wu, P. Yuan and X. Wang, J. Solid State Chem., 2016, 237, 57–63 CrossRef CAS.
- B. Prakoso, Y. Ma, R. Stephanie, N. H. Hawari, V. Suendo, H. Judawisastra, Y. Zong, Z. Liu and A. Sumboja, RSC Adv., 2020, 10, 10322–10328 RSC.
- B. Li, X. Jin, J. Lin and Z. Chen, J. Cleaner Prod., 2018, 189, 128–134 CrossRef CAS.
- A. S. AlShammari, M. M. Halim, F. K. Yam and N. H. M. Kaus, Mater. Sci. Semicond. Process., 2020, 116, 105140 CrossRef CAS.
- S. W. Park, B. Jang, H. Kim, J. Lee, J. Y. Park, S. O. Kang and Y. H. Choa, Front. Chem., 2021, 9, 699231 CrossRef CAS PubMed.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 2, 032183 CrossRef.
- D. Mhamane, W. Ramadan, M. Fawzy, A. Rana, M. Dubey, C. Rode, B. Lefez, B. Hannoyer and S. Ogale, Green Chem., 2011, 13, 1990 RSC.
- M. B. Arvas, H. Gürsu, M. Gencten and Y. Sahin, J. Energy Storage, 2021, 35, 102328 CrossRef.
- D. He, Z. Peng, W. Gong, Y. Luo, P. Zhao and L. Kong, RSC Adv., 2015, 5, 11966–11972 RSC.
- X. Colom, F. Carrillo, F. Nogués and P. Garriga, Polym. Degrad. Stab., 2003, 80, 543–549 CrossRef CAS.
- J. Lin, Y. Huang, S. Wang and G. Chen, Ind. Eng. Chem. Res., 2017, 56, 9341–9346 CrossRef CAS.
- B. Zhao, L. Jiang, X. Zeng, K. Zhang, M. M. F. Yuen, J.-B. Xu, X.-Z. Fu, R. Sun and C.-P. Wong, J. Mater. Chem. A, 2016, 4, 14595–14604 RSC.
- S. Hou, J. Li, X. Huang, X. Wang, L. Ma, W. Shen, F. Kang and Z.-H. Huang, Appl. Sci., 2017, 7, 852 CrossRef.
- S. K. Bikkarolla, P. Cumpson, P. Joseph and P. Papakonstantinou, Faraday Discuss., 2014, 173, 415–428 RSC.
- F. T. Johra, J.-W. Lee and W.-G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- B. Vasić, A. Zurutuza and R. Gajić, Carbon, 2016, 102, 304–310 CrossRef.
- A.-K. Hjelm and G. Lindbergh, Electrochim. Acta, 2002, 47, 1747–1759 CrossRef CAS.
- J. S. Ko, M. B. Sassin, D. R. Rolison and J. W. Long, Electrochim. Acta, 2018, 275, 225–235 CrossRef CAS.
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- S. Zhang, Y. Yang, R. Xiao, M. Yu, Y. Zhang, X. Sun, L. Lu, X. Wu and Y. Chen, Appl. Clay Sci., 2021, 200, 105821 CrossRef CAS.
- M. C. Sousa and J. W. Buchanan, Comput. Graph. Forum, 2000, 19, 27–49 CrossRef.
- M. He, Z. Wang, M. J. Moldowan and K. Peters, Org. Geochem., 2022, 163, 104331 CrossRef CAS.
- M. Wang, H. Wang, J. Wang and J. Zhang, J. Electroanal. Chem., 2022, 905, 115960 CrossRef CAS.
- H. Zhou, G. Han, Y. Xiao, Y. Chang and H.-J. Zhai, J. Power Sources, 2014, 263, 259–267 CrossRef CAS.
- H. Gul, A. A. Shah and S. Bilal, Polymers, 2019, 11, 1678 CrossRef CAS PubMed.
- S. Chandra Sekhar, G. Nagaraju, B. Ramulu, S. K. Hussain, D. Narsimulu and J. S. Yu, Nano Res., 2019, 12, 2597–2608 CrossRef CAS.
- T. H. Wu, C. T. Chang, C. C. Wang, S. Parwaiz, C. C. Lai, Y. Z. Chen, S. Y. Lu and Y. L. Chueh, Nanoscale Res. Lett., 2018, 13, 242 CrossRef PubMed.
- K. Yu, W. M. Tang and J. Y. Dai, Flexible Solid-state Supercapacitors Using Paper-based Electrodes for Energy Storage, presented at 2018 IEEE International Conference on Electron Devices and Solid State Circuits (EDSSC), 6–8 June 2018, 2018 Search PubMed.
- Z. Peng, J. Lin, R. Ye, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 3414–3419 CrossRef CAS.
- B. Xie, Y. Wang, W. Lai, W. Lin, Z. Lin, Z. Zhang, P. Zou, Y. Xu, S. Zhou, C. Yang, F. Kang and C.-P. Wong, Nano Energy, 2016, 26, 276–285 CrossRef CAS.
- S. Zhu, Y. Li, H. Zhu, J. Ni and Y. Li, Small, 2019, 15, 1804037 CrossRef.
- P. Xu, Q. Gao, L. Ma, Z. Li, H. Zhang, H. Xiao, X. Liang, T. Zhang, X. Tian and C. Liu, Carbon, 2019, 149, 452–461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03952h |
| This journal is © The Royal Society of Chemistry 2023 |