
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl sulfonate anion stabilized aromatic triangular cation [Pd3]+: syntheses, structures and properties †
Miaomiao Wang,
Zhixin He,
Meng Chen and
Yanlan Wang *
*
Department of Chemistry and Chemical Engineering, Liaocheng University, 252059 Liaocheng, China. E-mail: wangyanlan@lcu.edu.cn
First published on 10th October 2023
Abstract
A series of sulfonate anions paired aromatic triangular palladium clusters 3–7, abbreviated as [Pd3]+[ArSO3]−, were synthesized using a simple “one pot” method, and gave excellent isolated yields (90–95%). Their structures and properties have been fully characterized and further investigated by fluorescence, single crystal X-ray Diffraction (XRD) and X-ray Photoelectron Spectroscopy (XPS). In varying organic solvents, they presented apparently stronger absorption and emission in MeOH, driven by the combined interactions of hydrogen bonds and polarity. The crystallographic data demonstrated that the methyl orange ion stabilized complex 7 possessed a D3h symmetric metallic core which was still coplanar and almost equilateral, jointly influenced by the giant hindrance and milder donating effect from the sulfonate. The binding energies for Pdn+ 3d5/2 and Pdn+ 3d3/2 measured by XPS presented at 336.55 and 342.00 eV, respectively. These data were much lower than that of a usual Pd2+ 3d and significantly higher than that of a Pd0 species, further proving the unified palladium valence state (+4/3) in the tri-palladium core and its aromaticity featured by the cyclic electron delocalization.
Introduction
Among multi-palladium composed complexes, aromatic triangular tri-palladium complexes [LPd]3+ (L = ligand)1–5 and their heteroaromatics6,7 are excellent species as the metallic analogs of cyclopropene positive ion [C3H3]+. The unique all-metal aromaticity and excellent stability endow such three-membered palladium cores with excellent molecular stability and Lewis alkalinity, which has brought them much attention in theoretical chemistry,8–11 coordination chemistry,12 catalytic reactions13–22 and nanomaterials.23Compared with the usual negative ions like SbF6−, PF6−, BF4− and CF3COO−, from the perspective of electronic factors of ion pairs for organometallic complexes, aryl sulfonate anions (ArSO3−) were recognized as much weaker σ-donors and usually present milder coordination abilities with transition metals,24–26 which could potentially bring distinctive structural and characteristic properties for aromatic tri-palladium frameworks. As the foundation of this work, the Zwitterionic analogue reflected high stability, catalytic activity and stereoselectivity in the selective reduction of alkynes.27 While, the full crystal structures and properties were still undiscovered perhaps owing to the reduced symmetry. In this work, we designed and synthesized a series of C3-symmetry aromatic triangular cations 3–7 (Scheme 1) stabilized by substituted aryl sulfonate anions and revealed their extraordinary structures and properties.
 | ||
| Scheme 1 Syntheses of substituted aryl sulfonates stabilized tri-palladium complexes 3–7. | ||
Results and discussion
Aryl silver sulfonates 2
Even the aromatic sulfonates sodium salts are more commercially available, their corresponding silver sulfonates analogues are experimentally proved to be much more effective under identical conditions. Besides the commercial silver 4-methylbenzenesulfonate (2e), we synthesized the other employed substituted silver benzenesulfonates (2a–d) through two pathways (see Scheme S1†) according to literature,28–31 The employed silver salts were prepared either by mixing corresponding sulfonic acids with Ag2CO3 under dark (for 2a, 2b) or reacting their sodium sulfonates with AgNO3 (for 2c, 2d), gave the isolated yields in the range of 74–99%.Tri-palladium complexes 3–7
The aryl sulfonate anion stabilized new aromatic triangular tri-palladium complexes 3–7 were synthesized using a facile “one pot” method (Scheme 1). Simply mixed [Pd(dba)2] (1.0 equiv.), tri-4-fluorophenylphosphate (1.0 equiv.), and 4-chlorophenyldisulfide (0.5 equiv.) in chloroform and kept stirring for two hours under N2. Hitherto, the original purple solution became deep red and the trimetallic cationic core was proposed to be formed by then, while, adding the prepared silver benzenesulfonates (2a–e) (0.033 equiv.) was critical for the ions exchanges which were crucial for the subsequent facile purification and high isolated yields (90–95%). For comparisons, we prepared five analogues tri-palladium complexes, including 3 stabilized by 4-methylbenzenesulfonate, 4 stabilized by 4-hydroxybenzenesulfonate, 5 stabilized by 4-chlorobenzenesulfonate, 6 stabilized by 4-vinylbenzenesulfonate and 7 stabilized by (E)-(((4-((4-(dimethylamino)phenyl)diazenyl)phenyl)sulfonyl)oxy), abbreviated as methyl orange anion. These air-stable deep red solids were obtained by filtration and repeated precipitations using CHCl3/hexane. The purified products 3–7 were fully characterized by 1H, 13C, 31P, 19F NMR, ESI-MS, UV-visible, X-ray Photoelectron Spectroscopy (XPS) and single crystal X-ray Diffractions (XRD).High resolution mass spectroscopies (HRMS)
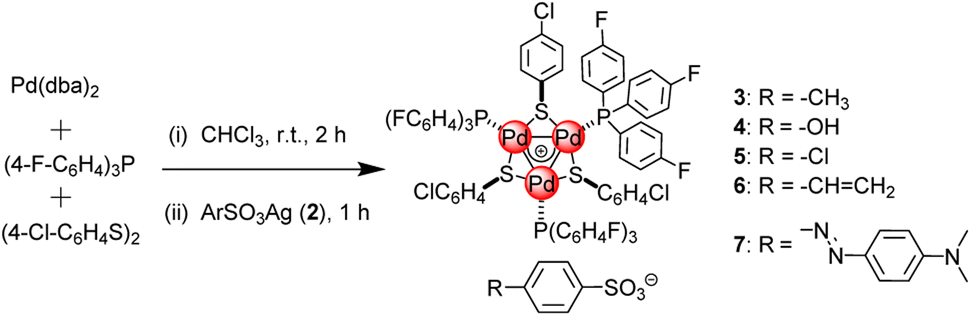
The calculated high resolution mass for the tri-palladium cation in complexes 3–7 was 1698.7048, for example, the founded mass for 3 was 1698.7062 by ESI-MS, which was perfectly consistent with the calculated value (Fig. 1a). As the cationic fragment was identical and the found mass for this part were extremely close for all these complexes. In addition, the counter anions 4-methylbenzenesulfonate in 3 (Fig. 1b), 4-hydroxybenzenesulfonate in 4 (Fig. 1c), 4-chlorobenzenesulfonate in 5 (Fig. 1d), 4-vinylbenzenesulfonate in 6 (Fig. 1e) and methyl orange anion in 7 (Fig. 1f) were found at 171.0169, 172.9959, 190.9623, 183.0165 and 304.0802, respectively, which were also fully consistent with calculated values. The above perfect cationic and anionic modes for complexes 3–7 by ESI-MS definitely proved the correct combination and structural integrity.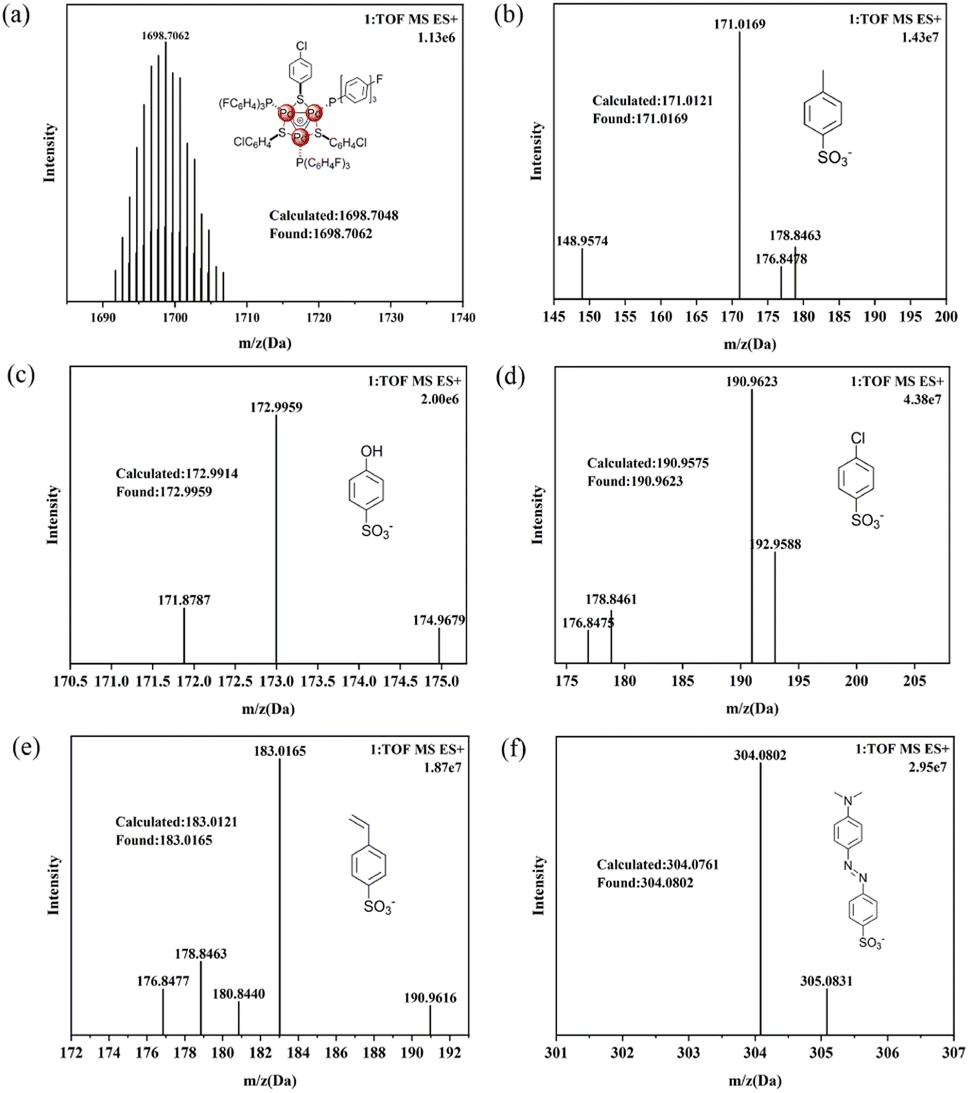 | ||
| Fig. 1 (a) The ESI-MS for cations of complexes 3; (b) anion for complex 3; (c) anion for complex 4; (d) anion for complex 5; (e) anion for complex 6; (f) anion for complex 7. | ||
Single crystal X-ray diffraction
The deep-red single crystal suitable for X-ray diffraction for the most representative complex 7 was obtained by vapor diffusion method. From the plane defined by the three Pd atoms and the plane defined by the aromatic ring in the counter ion (Fig. 2), the structure and orientation for the triangular tri-palladium cation and the methyl orange anion could be undoubtedly confirmed. Noteworthy, the Pd–Pd–Pd bond angles for 7 are 61.11(2)°, 59.24(2)° and 59.65(2)° which were slightly deviated from the examples in the literature (59.5–60.5°).5,6 This deviation might be caused by the first introduced anionic benzenesulfonate which probably contributed larger spatial site resistance and milder coordination modes. Even though, the palladium triangles were almost equilateral. The three Pd–Pd bond lengths are 2.8458(10) Å, 2.8577(9) Å and 2.8996(10) Å, respectively, which were much below the sum of van der Waals radii (3.26 Å) for metal–metal bonding and confirmed the Pd–Pd bonding modes.32 Compared to the tri-palladium analogue stabilized by SbF6−,6 the cationic part for complex 7 did not exactly coincide with C3 symmetry and possessed a D3h symmetric core. Some of the benzene rings on the coordinated S/P ligands in complex 7 tilted to the opposite direction to the negative methyl orange ion mainly due to low energy interactions are preferred when the cluster contains bulky aromatic moieties (Fig. S6†). Combined with the NMR and ESI-MS values, these crystal data further proved that our synthetic strategy allowed for the successful construction and isolation for these aryl sulfonate anion paired aromatic triangular tri-palladium cations.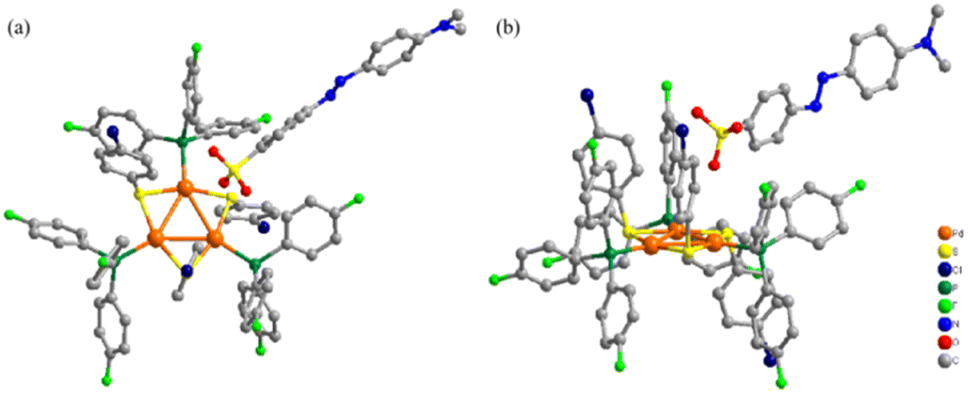 | ||
| Fig. 2 X-ray diffraction for complex 7 with 30% probability ellipsoids. Hydrogen atoms have been omitted for the sake of clarity. (a) Plane defined by the three Pd atoms, (b) plane defined by the aromatic ring in the counterion. | ||
Absorption and emission
In order to understand the luminescent properties for the aryl sulfonate anion stabilized new aromatic triangular tri-palladium complexes 3–7, we investigated their absorptions and fluorescence emissions. Under UV-visible light irradiation, all these complexes showed two distinct absorption peaks at around 245 nm and 400 nm in CH2Cl2 solutions, which could be ascribed to the occurrence of π–π* and n–π* transitions of phosphine and sulfur ligands (Fig. 3a).33,34 For example, 4-methylbenzenesulfonate paired complex 3 presented the maximum absorption at the wavelength of 399 nm. While, the presence of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group and the enlarged conjugation area in the methyl orange anion stabilized complex 7 caused red shift (to 404 nm), and the absorption intensity became much stronger than complex 3 (increased about 30%). So, the substituted aryl sulfonate anions possessing different types of chromophores and auxochromes indeed brought variations in the maximum absorption wavelength and absorption intensities under identical conditions.35 For comparison, we further measured the absorption for complexes (3–7) in MeOH (Fig. S8†), their absorption intensity presented obvious solvent dependence. Interestingly, complex 7 also presented red shift compared with complex 3 when MeOH was used as the solvent.
N group and the enlarged conjugation area in the methyl orange anion stabilized complex 7 caused red shift (to 404 nm), and the absorption intensity became much stronger than complex 3 (increased about 30%). So, the substituted aryl sulfonate anions possessing different types of chromophores and auxochromes indeed brought variations in the maximum absorption wavelength and absorption intensities under identical conditions.35 For comparison, we further measured the absorption for complexes (3–7) in MeOH (Fig. S8†), their absorption intensity presented obvious solvent dependence. Interestingly, complex 7 also presented red shift compared with complex 3 when MeOH was used as the solvent.
 | ||
| Fig. 3 (a) UV-visible spectra for complexes 3–7 in CH2Cl2 (1 × 10−4 M); (b) fluorescence emissions for complexes 3–7 in CH3OH (1 × 10−4 M); (c) PLQY for complexes 3–7 (5.00%, 10.33%, 8.68%, 4.66%, 12.55%, respectively) in CH3OH at room temperature (1 × 10−4 M); (d) fluorescence emissions for complexes 3–7 in THF (1 × 10−4 M); (e) PLQY for complexes 3–7 (2.12%, 2.30%, 3.27%, 1.13%, 1.12%, respectively) in THF at room temperature (1 × 10−4 M); (f) fluorescence emissions for complex 7 (1 × 10−4 M) in different organic solvents (CH3OH, CH3COCH3, CHCl3, CH3CN, THF); (g) the CIE coordinates for complex 7 (1 × 10−4 M) from THF to CH3OH; (h) from left to right: color of methyl orange-sodium (1d), methyl orange-silver (2d), complex 3, complex 7 in DMF. | ||
For better understanding their luminescence properties, we further examined the fluorescence emission for all complexes 3–7 in polar/protic CH3OH (Fig. 3b), or less polar/nonprotic THF (Fig. 3d). The fluorescence quantum yields (PLQY) for complexes 3–7 in CH3OH were 5.00%, 10.33%, 8.68%, 4.66%, and 12.55%, respectively (Fig. 3c). Among these analogues, the methyl orange anion stabilized complex 7 exhibited the highest PLQY in the polar CH3OH. Interestingly, the PLQY for all these complexes were dropped sharply in less polar THF and complex 7 presented the lowest value (Fig. 3e), which was roughly consistent with their emission intensities. For better comparison, we also measured the PLQY for these clusters in (CH3COCH3, CHCl3 and CH3CN) and similar moderate values were obtained (Fig. S10†). It was worth noting that all these complexes exhibited higher PLQY in polar CH3OH or CH3CN than less polar solvents (CH3COCH3, CHCl3 and THF). This result could be attributed by the complex strength of solvent polarity and the possible hydrogen bonding interactions between the negative ions and protic CH3OH.
Besides the PLQY, the emission intensity of complex 7 also varied significantly when the solvents polarity changes with the excitation wavelength set at 250 nm (Fig. 3f). This result could be attributable to that the polarity of solvents could significantly impact the energies of π–π* and n–π* transitions, therefore, the fluorescence intensity of substances may also change as the polarity of the solvent varies.36 Basically, the excited states possessed greater polarity and was more beneficial from increasing solvent polarity.38–40 Generally, complex 7 conformed to the rule of fluorescence intensity enhancement with increasing solvent polarity and presented apparently stronger emission in CH3OH. Meanwhile, the fluorescence wavelengths also presented red shift when the polarity of organic solvents were increased. This clear difference was also probably caused by the interactions of hydrogen bonds with hydroxyl in CH3OH.37 Overall, the CIE coordinates of complex 7 changed from blue to teal with the increase of solvent polarity (Fig. 3g). Besides 7, the luminescence properties for complexes 3–6 were also examined in different solvents, and obvious varying for emission Intensities and wavelengths were observed, probably caused by the introduction of different groups on the anionic aryl sulfonates (see ESI, Fig. S5†).
In addition, the color comparisons for methyl orange-sodium, methyl orange-silver (2d), complex 3 or 7 in DMF were presented (Fig. 3h). Different with the commercial sodium analogue, the synthesized silver salt 2d showed a deeper red color as solid but decreased solubility in water or DMF, resulting in yellow color in solutions. Comparing complexes 3 and 7, we found that the methyl orange anion introduced complex 7 showed more pronounced red color, which was probably caused by the color enhancement effect from the dye-based anionic fragment.41
X-ray photoelectron spectroscopy (XPS)
In order to know the bonding forms and valence states for these aryl sulfonate anions stabilized new aromatic triangular tri-palladium complexes 3–7, we further examined them by XPS. Taking complex 7 as an example, the observed binding energies for their involved elements: Pd, C, O, S, Cl, P, N, and F presented to be completely consistent with atoms in analogous structures in literature49 and definitely manifested their existence by comparing with established values (Fig. 4a). In addition, the high-resolution XPS spectrum for C 1s in complex 7 presented five characteristic peaks at 284.00, 284.35, 284.60, 286.75, and 290.90 eV (Fig. 4b), which were ascribed to the sp2 carbon, sp3 carbon, C–P, C–Cl, and C–F, respectively.42,43 Among them, the presence of bonding energies for sp3 hybridized carbons which belonged to the two methyl groups on the methyl orange anion illustrated the intimate paring between the sulfonate anion and the tri-palladium cation.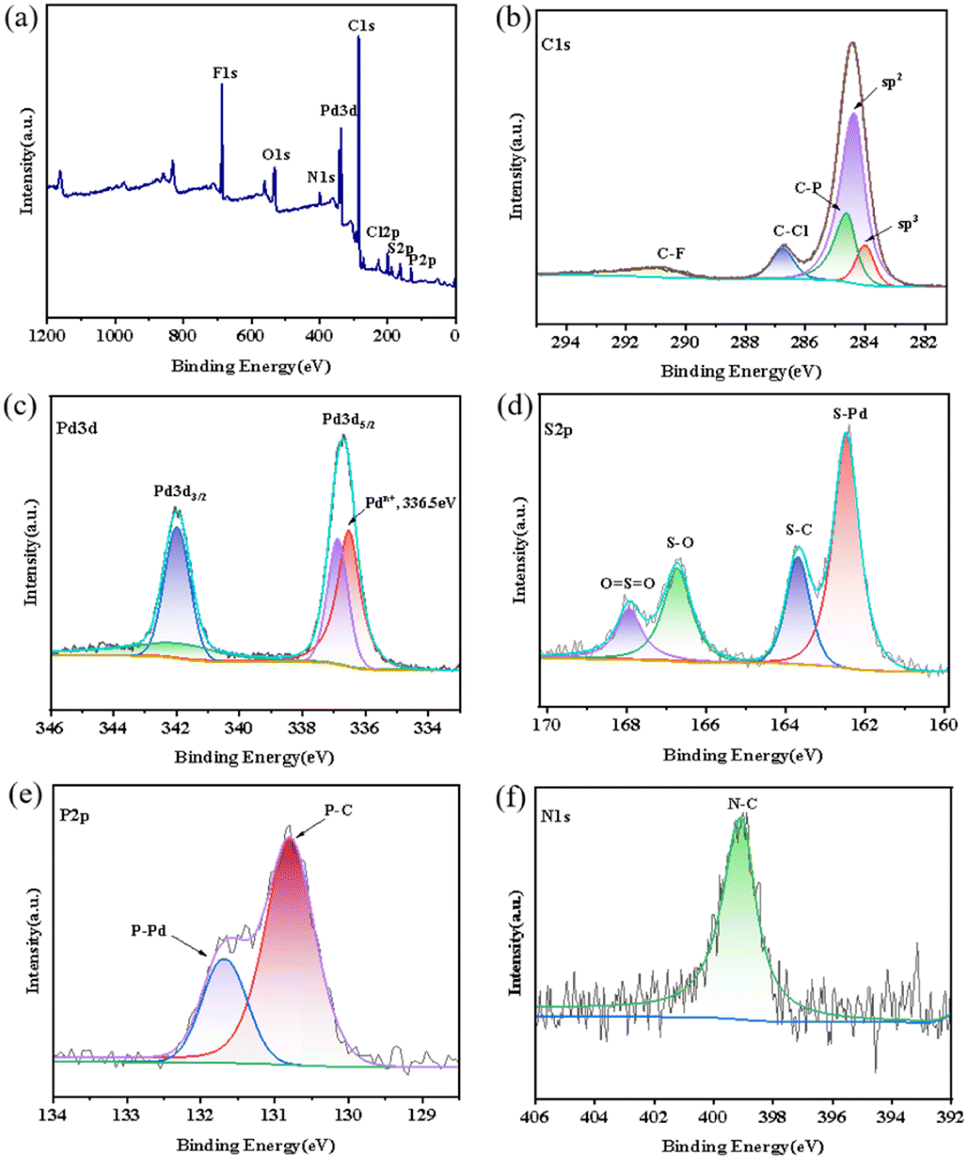 | ||
| Fig. 4 (a) XPS survey spectrum for complex 7, high-resolution for (b) C 1s, (c) Pd 3d, (d) S 2p, (e) N 1s, (f) P 2p spectra in complex 7. | ||
Theoretically, the Pd2+, Pdn+ (0 < n< 2) and Pd0 species have relatively fixed and different binding energies according to literature.44 As expected, the XPS for Pd 3d in complex 7 showed two peaks at 336.55 and 342.00 eV, which were certainly assigned to Pdn+ 3d5/2 and Pdn+ 3d3/2, respectively (Fig. 4c). The found Pd 3d5/2 bond energies for cyclic tri-palladium core [Pd3]+ was much lower than Pd2+ 3d5/2 (about 337.60 eV) and significantly higher than that of Pd0 3d5/2 in species like nanoparticles (about 335.00 eV).45,46 Similarly, the Pd 3d3/2 bond energies for metallic core [Pd3]+ was perfectly appeared between the Pd2+ 3d3/2 (about 342.91 eV) and Pd0 3d3/2 (about 341.75 eV).47,48 So, our recorded XPS data certainly proved the unified palladium valence state (+4/3) in these tri-palladium cation [Pd3]+ and their aromaticity characterized by definite cyclic electron delocalization.49
In addition, the high-resolution XPS for S2p in complex 7 presented four peaks at 162.50, 163.70, 166.70, and 167.95 eV, responding to the S–Pd, S–C, S–O, and OSO, respectively (Fig. 4d). As expected, the XPS spectrum for N 1s showed only one peak at 399.10 eV (Fig. 4e), representing the unique N–C bonding in the anionic fragment. Clearly, the XPS spectrum for P2p showed two peaks at 130.8 and 131.70 eV, assignable to the P–C and P–Pd bonding modes, respectively (Fig. 4f).50 Overall, the above measured electron binding energies by XPS are perfectly in line with the high resolution mass and single crystal X-ray diffraction results, definitely revealed the chemical compositions, bonding forms, and valence states for elements in these aryl sulfonates stabilized aromatic triangular tri-palladium complexes.
Conclusions
In this work, we synthesized a series of new aromatic triangular tri-palladium complexes 3–7 stabilized by substituted aryl sulfonate anions, and investigated their structures and properties. Besides the methyl orange anion, several functional groups (methyl, hydroxy, chloro, vinyl) were successfully introduced to the benzenesulfonates. Firstly, the cationic and anionic modes for these complexes by ESI-MS were fully consistent with calculated values and confirmed the structural integrity. Secondly, the single crystal X-ray diffraction confirmed that complex 7 possessed a D3h symmetric metallic core and became less C3 symmetry compared to its hexafluoroantimonate analogue due to its increased steric effect and weaker σ-donating effect from the negative methyl orange ion. While, the three palladium atoms were still coplanar and the palladium triangles were almost equilateral, especially, the short Pd–Pd bond lengths were supportive for metal–metal bonding. Thirdly, the absorption and fluorescence properties for these complexes were examined in different organic solvents, and presented apparently stronger absorption and emissions in CH3OH, caused by the interactions of hydrogen bonds and solvent polarity. Finally, the measured XPS for Pd 3d in complex 7 showed two peaks at 336.55 and 342.00 eV, which were assigned to Pdn+ 3d5/2 and Pdn+ 3d3/2, respectively. This recorded XPS data certainly proved the unified palladium valence state (+4/3) in these ArSO3− paired aromatic tri-palladium complexes.Overall, these experimental results are consistent with the formulation and cognition that the metallic core [Pd3]+ in these complexes are aromatic framework. The milder coordination effect between the aryl sulfonates anions and the tri-palladium cations endows their paired complexes with good stability and stronger Lewis-alkalinity. Therefore, the introduction of aryl sulfonates to these new triangular aromatic palladium complexes could potentially realize their practical applications in the fields of coordination chemistry, catalytic reactions, and functionalization for macromolecular materials.
Author contributions
MW, ZH performed the synthetic chemistry and measured the NMR, HRMS, and luminescence data. MW collected and solved the XRD data. MW and MC collected and analysed the XPS data. YW and MW conceived the idea. All authors wrote/edited the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank for National Natural Science Foundation of China (Grant No. 21901097); Natural Science Foundation of Shandong province (Grant No. ZR2023MB075); Science and Technology Foundation of Liaocheng University (Grant No. 318051728); Introduction and Cultivation Project for Young Innovative Talents in Shandong Provincial Colleges and Universities (Innovation Team of Functional Organometallic Materials Presided by Prof. Yanlan Wang).Notes and references
- A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
- N. V. Tkachenko, I. A. Popov, M. Kulichenko, N. Fedik, Z.-M. Sun, A. Muñoz-Castro and A. I. Boldyrev, Eur. J. Inorg. Chem., 2021, 41, 4239–4250 CrossRef.
- F. Bigi, D. Cauzzi, N. Della Ca, M. Malacria, R. Maggi, E. Motti, Y. Wang and G. Maestri, ACS Org. Inorg. Au, 2022, 2, 373–385 CrossRef CAS PubMed.
- S. Blanchard, L. Fensterbank, G. Gontard, E. Lacôte, G. Maestri and M. Malacria, Angew. Chem., Int. Ed., 2014, 126, 2018–2022 CrossRef.
- Y. Wang, P.-A. Deyris, T. Caneque, F. Blanchard, Y. Li, F. Bigi, R. Maggi, S. Blanchard, G. Maestri and M. Malacria, Chem. - Eur. J., 2015, 21, 12271–12274 CrossRef CAS PubMed.
- M. Kulichenko, N. Fedik, A. Monfredini, A. Muñoz-Castro, D. Balestri, A. I. Boldyrev and G. Maestri, Chem. Sci., 2021, 12, 477–486 RSC.
- F. Bigi, G. Cera, R. Maggi, Y. Wang, M. Malacria and G. Maestri, J. Phys. Chem. A, 2021, 125, 10035–10043 CrossRef CAS PubMed.
- I. A. Popov, A. A. Starikova, D. V. Steglenko and A. I. Boldyrev, Chem. - Eur. J., 2018, 24, 292–305 CrossRef CAS PubMed.
- X. R. You and H. J. Zhai, Chin. J. Chem., 2019, 37, 126–130 CrossRef CAS.
- P. von R. Schleyer and R. B. King, Chem.–Eur. J., 2007, 13, 978–984 CrossRef PubMed.
- Y. Wang, A. Monfredini, P.-A. Deyris, F. Blanchard, E. Derat, G. Maestri and M. Malacria, Chem. Sci., 2017, 8, 7394–7402 RSC.
- P.-A. Deyris, T. Caneque, Y. Wang, P. Retailleau, F. Bigi, R. Maggi, G. Maestri and M. Malacria, ChemCatChem, 2015, 7, 3266–3269 CrossRef CAS.
- A. Monfredini, V. Santacroce, L. Marchiò, R. Maggi, F. Bigi, G. Maestri and M. Malacria, ACS Sustain. Chem. Eng., 2017, 5, 8205–8212 CrossRef CAS.
- M. Lanzi, T. Cañeque, L. Marchiò, R. Maggi, F. Bigi, M. Malacria and G. Maestri, ACS Catal., 2018, 8, 144–147 CrossRef CAS.
- X. Li, J. Li, X. Wang, L. Wu, Y. Wang, G. Maestri, M. Malacria and X. Liu, Dalton Trans., 2021, 50, 11834–11842 RSC.
- F. Fu, J. Xiang, H. Cheng, L. Cheng, H. Chong, S. Wang and Y. Li, ACS Catal., 2017, 7, 1860–1867 CrossRef CAS.
- C. J. Diehl, T. Scattolin, U. Englert and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 211–215 CrossRef CAS PubMed.
- C. Cecchini, M. Lanzi, G. Cera, M. Malacria and G. Maestri, Synthesis, 2019, 51, 1216–1224 CrossRef CAS.
- A. Serafino, N. Camedda, M. Lanzi, N. Della Ca, G. Cera and G. Maestri, J. Org. Chem., 2021, 86, 15433–15452 CrossRef CAS PubMed.
- X. Wang, L. Sun, M. Wang, G. Maestri, M. Malacria, X. Liu, Y. Wang and L. Wu, Eur. J. Org Chem., 2022, 10, e20220 Search PubMed.
- N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682–9693 CrossRef CAS PubMed.
- J. Ren, P. C. Lan, M. Chen, W. Zhang and S. Ma, Organometallics, 2019, 38, 3460–3465 CrossRef CAS.
- S. S. Deshmukh, S. R. Gaikwad, N. R. Mote, M. Manod, R. G. Gonnade and S. H. Chikkali, ACS Omega, 2019, 4, 9502–9511 CrossRef CAS PubMed.
- M. Zabransky, I. Císarova and P. Štěpnička, Organometallics, 2018, 37, 1615–1626 CrossRef CAS.
- N. Li, J. Wang, X. Zhang, R. Qiu, X. Wang, J. Chen, S. F. Yin and X. Xu, Dalton Trans., 2014, 43, 11696–11708 RSC.
- A. Monfredini, V. Santacroce, P.-A. Deyris, R. Maggi, F. Bigi, G. Maestri and M. Malacria, Dalton Trans., 2016, 45, 15786–15790 RSC.
- A. Fedorchuk, Y. Slyvka, V. Kinzhybalo, T. Lis and M. Myskiv, Inorg. Chim. Acta, 2019, 484, 79–86 CrossRef CAS.
- H. Liu, H. Wu, J. Ma, S. Song, J. Yang, Y. Liu and Z. Su, Inorg. Chem., 2007, 46, 7299–7311 CrossRef CAS PubMed.
- R. Şuteu, A. Toma, M. Mehring and A. Silvestru, J. Organomet. Chem., 2020, 920, 121343 CrossRef.
- A. Toma, C. Raţ, O. Pavel, C. Hardacre, T. Rüffer, H. Lang, M. Mehring, A. Silvestru and V. Pârvulescub, Catal. Sci. Technol., 2017, 7, 5343–5353 RSC.
- L. M. Mirica and J. R. Khusnutdinova, Coord. Chem. Rev., 2013, 257, 299–314 CrossRef CAS.
- J. Li, X. Li, L. Sun, X. Wang, L. Yuan, L. Wu, X. Liu and Y. Wang, Eur. J. Inorg. Chem., 2021, 40, 4230–4237 CrossRef.
- J. R. Shakirova, E. V. Grachova, V. V. Gurzhiy, I. O. Koshevoy, A. S. Melnikov, O. V. Sizova, S. P. Tunik and A. Laguna, Dalton Trans., 2012, 41, 2941–2949 RSC.
- G. J. Zhao, B. H. Northrop, K. L. Han and P. J. Stang, J. Phys. Chem. A, 2010, 114, 9007–9013 CrossRef CAS PubMed.
- M. Liu, H. Hao, G. Liao, C. Li, K. Liu, N. Wang, Q. Niu and X. Yin, Adv. Opt. Mater., 2023, 2202918 CrossRef CAS.
- M. Fan, Y. Tang, C. Li, B. Chen, T. Wang, P. Zhou and X. Cui, J. Phys. Chem. B, 2023, 127, 3187–3196 CrossRef CAS PubMed.
- G. Singh, Pawan, S. Sharma, Mohit, P. Satija, Diksha, Priyanka, Y. Thakur and A. Kaur, Inorg. Chem., 2022, 61, 12043–12061 CrossRef CAS PubMed.
- A. Barak, N. Dhiman, F. Sturm, F. Rauch, Y. Lakshmanna, K. Findlay, A. Beeby, T. Marder and S. Umapathy, J. Phys. Chem. C, 2023, 127, 5855–5865 CrossRef CAS.
- R. Hamze, J. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. Djurovich, G. Bertrand and M. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- H. Danjo, K. Asai, T. Tanaka, D. Ono, M. Kawahata and S. Iwatsuki, Chem. Commun., 2022, 58, 2196–2199 RSC.
- Y. Liu, Y. Zhou, J. Li, Q. Wang, Q. Qin, W. Zhang, H. Asakura, N. Yan and J. Wang, Appl. Catal., B, 2017, 209, 679–688 CrossRef CAS.
- Y. Liu, C. Zou, K. Wang, Z. Bian, S. Jiang, Y. Zhou and J. Wang, Mol. Catal., 2021, 505, 111487 CrossRef CAS.
- K. Wang, W. Cui, Z. Bian, Y. Liu, S. Jiang, Y. Zhou and J. Wang, Appl. Catal., B, 2021, 281, 119425 CrossRef CAS.
- Y. Lou, J. Ma, W. Hu, Q. Dai, L. Wang, W. Zhan, Y. Guo, X. Cao, Y. Guo, P. Hu and G. Lu, ACS Catal., 2016, 6, 8127–8139 CrossRef CAS.
- Y. Zhang, Q. Sun, G. Guo, Y. Cheng, X. Zhang, H. Ji and X. He, Chem. Eng. J., 2023, 451, 138867 CrossRef CAS.
- W. Shi, G. Xu, X. Han, Y. Wang, Z. Liu, S. Xue, N. Sun, X. Shi, Y. Yu and H. He, J. Environ. Sci., 2023, 126, 333–347 CrossRef CAS PubMed.
- A. Ibrahim, U. B. Memon, S. P. Duttagupta, R. K. Singh Raman and A. Sarkar, J. Electron. Mater., 2023, 52, 446–462 CrossRef CAS.
- Y. Yun, H. Sheng, J. Yu, L. Bao, Y. Du, F. Xu, H. Yu, P. Li and M. Zhu, Adv. Synth. Catal., 2018, 360, 4731–4743 CrossRef CAS.
- Z. Jiang, S. Guo and T. Fang, ACS Appl. Energy Mater., 2019, 2, 7233–7243 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2270588. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04460b |
| This journal is © The Royal Society of Chemistry 2023 |