
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of ACP–CCS–PVA composite membrane for a potential application in guided bone regeneration†
Qiaolin Du‡
a,
Jian Sun‡a,
Yanyan Zhoub,
Yadong Yuc,
Weijing Kongb,
Chaoqun Chena,
Yifeng Zhoua,
Ke Zhaoa,
Changyu Shao *b and
Xinhua Gu*a
*b and
Xinhua Gu*a
aDepartment of Stomatology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310003, China. E-mail: guxh@zju.edu.cn
bStomatology Hospital, School of Stomatology, Clinical Research Center for Oral Diseases of Zhejiang Province, Key Laboratory of Oral Biomedical Research of Zhejiang Province, Cancer Center of Zhejiang University, Zhejiang University School of Medicine, Hangzhou 310006, China
cDepartment of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, China
First published on 1st September 2023
Abstract
The barrier membranes of guided bone regeneration (GBR) have been widely used in clinical medicine to repair bone defects. However, the unmatched mechanical strength, unsuitable degradation rates, and insufficient regeneration potential limit the application of the current barrier membranes. Here, amorphous calcium phosphate–carboxylated chitosan–polyvinyl alcohol (ACP–CCS–PVA) composite membranes are fabricated by freeze–thaw cycles, in which the ATP-stabilized ACP nanoparticles are uniformly distributed throughout the membranes. The mechanical performance and osteogenic properties are significantly improved by the ACP incorporated into the CCS–PVA system, but excess ACP would suppress cell proliferation and osteogenic differentiation. Our work highlights the pivotal role of ACP in GBR and provides insight into the need for biomaterial fabrication to balance mechanical strength and mineral content.
1. Introduction
The barrier membrane of guided bone regeneration (GBR) forms a biological barrier between soft tissue and the bone defect area to prevent faster migrating fibroblasts and epithelial cells from entering the bone defect area, therefore creating a favorable environment for bone tissue growth.1 They have been widely used for periodontal treatment and oral implants. Previous studies suggested that an ideal GBR membrane should have barrier function, good biocompatibility, biodegradability, and well-matched mechanical properties.2 However, the current GBR-based technology for bone regeneration, which employs barrier membranes, requires properties to boost osteoblast proliferation and differentiation as well as promote bone deposition.Natural bone is an organic-inorganic composite mainly consisting of calcium phosphate (CaP) minerals and collagen fibrils.3 Currently, the CaP minerals (e.g., tricalcium phosphate (β-TCP), hydroxyapatite (HAP), amorphous calcium phosphate (ACP)) and phosphate and borate bioactive glass are widely used as superior bone graft substitutes and as the inorganic component of biomimetic barrier membranes, which have gained attention due to their excellent osteoconductive and osteoinductive properties.4–10 However, it is challenging to transform these crystalline CaP minerals into the low-crystalline apatite needed for bone formation since they are stable under physiological conditions,11 and these bioactive glass tends to lead to inhomogeneities and agglomerate formation within composites.12
A growing body of evidence shows that the amorphous precursor, i.e., ACP, is a vital transition phase and bears a fundamental role in bone formation. Amorphous calcium phosphate exhibits better osteoconductivity in vivo than apatite and higher biodegradability than tricalcium phosphate.13–15 In addition, ACP, as nano-sized inorganic structural units, can be uniformly dispersed between polymers, improving the strength of polymers.16–18 Because the ACP is unstabilized and readily nucleation in an aqueous solution, a stabilizer is required in preparing the stabilized ACP. Polyacrylic acid (PAA), poly aspartic acid (pAsp), and other polyelectrolytes, for example, are used to stabilize the ACP through cation binding,19 but these polymers may have potential toxicity for cells.20 The biomolecule adenine nucleoside triphosphate (ATP) is biosafety. It can significantly stabilize the ACP, but the effect of the formed composite on the cell behavior, such as adhesion, proliferation, and differentiation, is poorly understood.
Although collagen membranes are widely used in clinical applications due to their high biocompatibility, their mechanical strength is lower, and are susceptible to enzymatic degradation in vivo.21 To some extent, the mechanical properties of these collagen membranes can be improved by calcium phosphate mineralisation or complexation with other biomaterials (e.g. nanohydroxyapatite, silicate ceramic), but this also results in increased brittleness and reduced toughness of the collagen membranes, which is not conducive to clinical manipulation.22,23 As a result, other natural polymers such as chitosan, hyaluronic acid, silk fibroin, and synthetic polymers such as polylactic acid, polycaprolactone, and polyvinyl alcohol are also commonly used in biological materials.24–26 In particular, the N-acetylglucosamine moiety of chitosan is similar to the glycosaminoglycan in bone extracellular matrix (ECM), which can attract growth factors and proteins to induce new bone regeneration.27 In addition, the positive charge of chitosan can improve cell adhesion and diffusion to alter cell behavior.28 Compared to chitosan, its derivative carboxylation chitosan (CCS) has improved antioxidant properties, higher water retention, excellent biodegradability, biocompatibility, and antibacterial activity. However, the mechanical strength of pure CCS is too weak to maintain the osteogenic space. Another polymer, PVA, with high tensile strength, flexibility, elongation at break, and slow degradation properties, gives PVA-based biomaterials the ability to absorb more of the mechanical strain of the muscle/skeleton.29,30 However, these polymers lack osteoinductivity and cannot provide osteoblasts with an environment conducive to their proliferation and development.
In the present study, a composite membrane consisting of ATP-stabilized ACP, CCS, and PVA was prepared in which the amorphous particles were uniformly distributed throughout the membrane. Incorporating ACP could dramatically improve the mechanical properties of the composite membrane and enhance osteoblast proliferation and differentiation. However, the excess ACP would lead to suppression, indicating that the ACP should be kept within an optimal concentration range first and then the mechanical properties. These findings provide a clue for the manufacture of biomaterials for GBR and further deepen our understanding of ACP and how it affects bone regeneration.
2. Materials and methods
2.1 Preparation of highly stable ACP nanoparticles
CaCl2·2H2O (Aladdin, Shanghai, China) and ATP (BBI, Shanghai, China) were dissolved in deionized water at room temperature to prepare solution A, and the pH was adjusted to 9.0 with 1 M NaOH (Aladdin, Shanghai, China). Solution B was prepared by dissolving NaH2PO4·2H2O (Aladdin, Shanghai, China) in deionized water. Solution B was added to solution A under magnetic stirring, and the pH was maintained at 9.0 and reacted until solution B dropped off. The final solution concentration was 25 mM CaCl2, 25 mM NaH2PO4, and 5 mM ATP. The precipitate was centrifuged, washed 3 times with deionized water and 100% ethanol, and dried in a freeze dryer (FreeZone®, Labconco, USA) for 24 hours to obtain ACP nanoparticles.2.2 Fabrication of ACP–CCS–PVA composite membranes
The composite membrane used in this study was prepared using a freeze–thaw cycle method. CCS (Aladdin, Shanghai, China, 3 wt%) was completely dissolved in deionized water at room temperature. PVA (Aladdin, Shanghai, China, 6 wt%) was completely dissolved in deionized water at 95 °C and then cooled to room temperature. Mixing the two solutions in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, adding a certain mass of ACP nanoparticles, and fully vibrating to prepare composite membranes with 0 wt%, 10 wt%, and 20 wt% ACP named 0 wt% ACP–CCS–PVA, 10 wt% ACP–CCS–PVA, and 20 wt% ACP–CCS–PVA, respectively. Pour the homogeneous mixture into the mold. Then, the sample was sealed and frozen at −20 °C for 8 h and thawed at room temperature for 3 h. The freeze–thaw cycle process was repeated five times. Eventually, the frozen samples were placed in a freeze dryer (FreeZone®, Labconco, USA) and freeze-dried at −60 °C for 20 h before characterization.
:1, adding a certain mass of ACP nanoparticles, and fully vibrating to prepare composite membranes with 0 wt%, 10 wt%, and 20 wt% ACP named 0 wt% ACP–CCS–PVA, 10 wt% ACP–CCS–PVA, and 20 wt% ACP–CCS–PVA, respectively. Pour the homogeneous mixture into the mold. Then, the sample was sealed and frozen at −20 °C for 8 h and thawed at room temperature for 3 h. The freeze–thaw cycle process was repeated five times. Eventually, the frozen samples were placed in a freeze dryer (FreeZone®, Labconco, USA) and freeze-dried at −60 °C for 20 h before characterization.
2.3 Characterization
The samples were sputter-coated (E-1010, Hitachi, Japan) with AuePd and morphologically characterized by SEM (Ultra-55, Carl Zeiss, Germany), and performed the element analysis by using energy-dispersive spectrometry (EDS).
2.4 In vitro analysis
2.5 Statistical analysis
Data were expressed as mean ± standard deviation. The statistical significance of the results was evaluated by one-way ANOVA analysis. P < 0.05 was considered statistically significant.3. Results
3.1 Characterization of ACP nanoparticles and composite membranes
Stabilized ACP nanoparticles were prepared in an aqueous solution using ATP as a stabilizer. The ACP nanoparticles were uniformly dispersed in ethanol and observed by TEM, having a spherical structure and diameters of less than 20 nm (Fig. 1). Their amorphous nature was confirmed by selected area electron diffraction (SAED, Fig. 1). In addition, the ACP nanoparticles were further examined by SEM, whose contour is discernible, and subjected to elemental analysis by EDS, which detected the carbon derived from the ATP, confirming that the ATP interacts with ACP (Fig. 2a and b). The CCS–PVA and ACP–CCS–PVA composite membranes were fabricated through the freeze-drying process, and the internal structure was examined by SEM (Fig. 2c–f), where the hierarchical porous structure of mesoporous and macropore can be seen and also confirmed by N2 adsorption–desorption isotherms (Fig. S1†). In addition, the calcium and phosphorus elements were evenly mixed with the CCS and PVA, as shown by the EDS results, rather than aggregating together to form larger ACP particles. | ||
| Fig. 1 TEM and SAED patterns of the ATP-stabilized ACP nanoparticles. | ||
 | ||
| Fig. 2 (a) SEM of the ACP nanoparticles. (b) The EDS spectra show the element composition of ACP nanoparticles. (c) SEM of the CCS–PVA composite membrane. (d) The EDS spectra show the element composition of the CCS–PVA composite membrane. (e) SEM of the ACP–CCS–PVA composite membrane. (f) The EDS spectra show the element composition of the ACP–CCS–PVA composite membrane. | ||
The ACP nanoparticles and the composite membranes of CCS–PVA and ACP–CCS–PVA were examined using FTIR (Fig. 3a). Two characteristic peaks at 567 cm−1 and 1049 cm−1 appeared in ACP nanoparticles, attributed to the asymmetric bending vibration of P–O (ν4) and the asymmetric stretching vibration of P–O (ν1), respectively. The ν4 peak of P–O blue shifted to 594 cm−1 implying that the ACP was successfully incorporated in the ACP–CCS–PVA composite membrane and confirmed that the ACP may interact with the CCS and PVA via a cation binding to carboxyl groups. However, the ν1 peak of P–O was covered because the peak 1080 cm−1 arose from the C–O tensile vibration peak of PVA is too strong. In addition, a strong absorption peak arises at 1651 cm−1 in the ACP samples (Fig. S2†), attributed to the stretching vibration mode of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group of the ATP,31 indicating that the ATP interacts with ACP. Since the strong interaction, the ATP-stabilized ACP nanoparticles could be stabilized without phase change within 27 days when immersed in PBS solution at pH 7.4 (Fig. S3†). Furthermore, these materials were further examined by XRD, where the broad peak at around two thetas of 30° suggested the amorphous nature of the mineral particles, and at around two thetas of 20° suggested that CCS–PVA was in the amorphous state (Fig. 3b).
C group of the ATP,31 indicating that the ATP interacts with ACP. Since the strong interaction, the ATP-stabilized ACP nanoparticles could be stabilized without phase change within 27 days when immersed in PBS solution at pH 7.4 (Fig. S3†). Furthermore, these materials were further examined by XRD, where the broad peak at around two thetas of 30° suggested the amorphous nature of the mineral particles, and at around two thetas of 20° suggested that CCS–PVA was in the amorphous state (Fig. 3b).
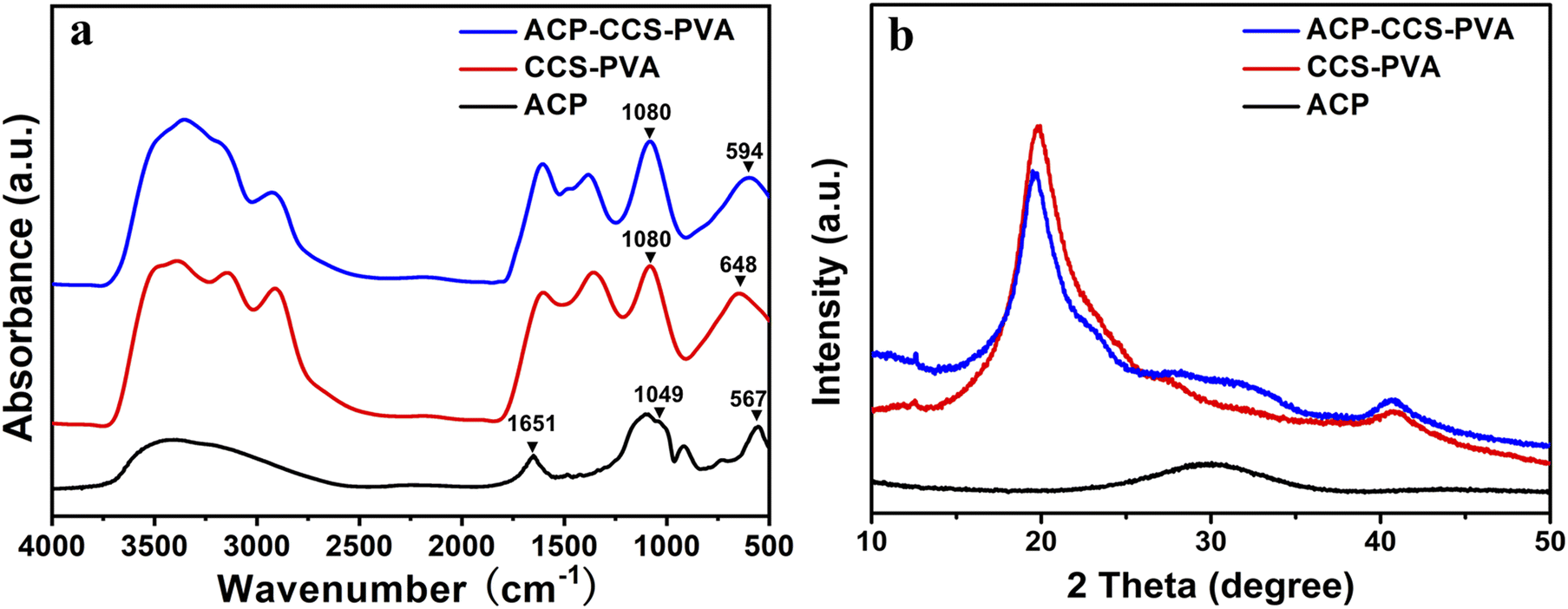 | ||
| Fig. 3 (a) FTIR spectra and (b) XRD patterns of the ACP nanoparticles, the CCS–PVA composite membranes, and the ACP–CCS–PVA composite membranes. | ||
3.2 Features of the composite membrane
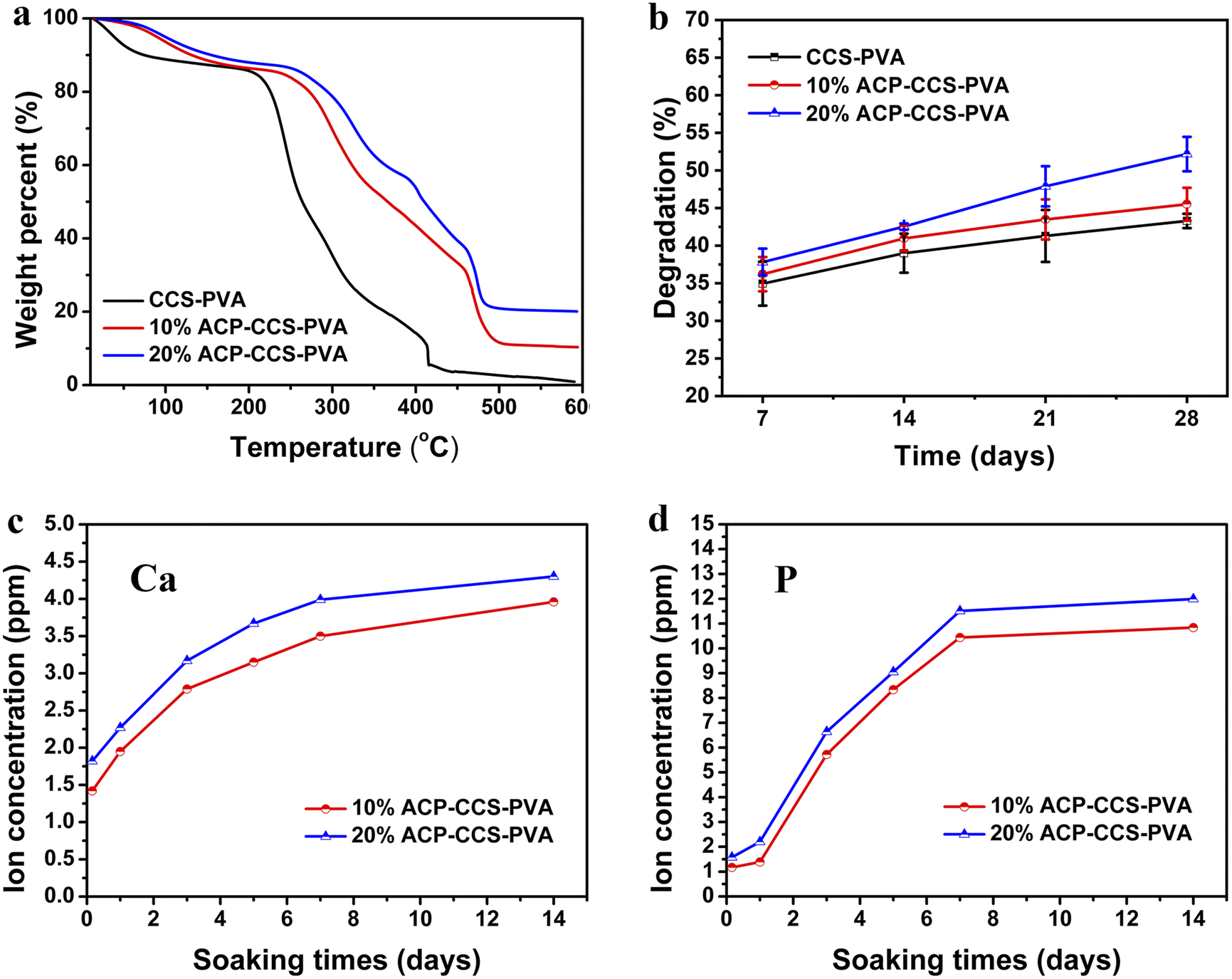 | ||
| Fig. 4 (a) TGA curves, (b) degradation, (c) calcium ion, and (d) phosphate ion release curves of composite membranes. | ||
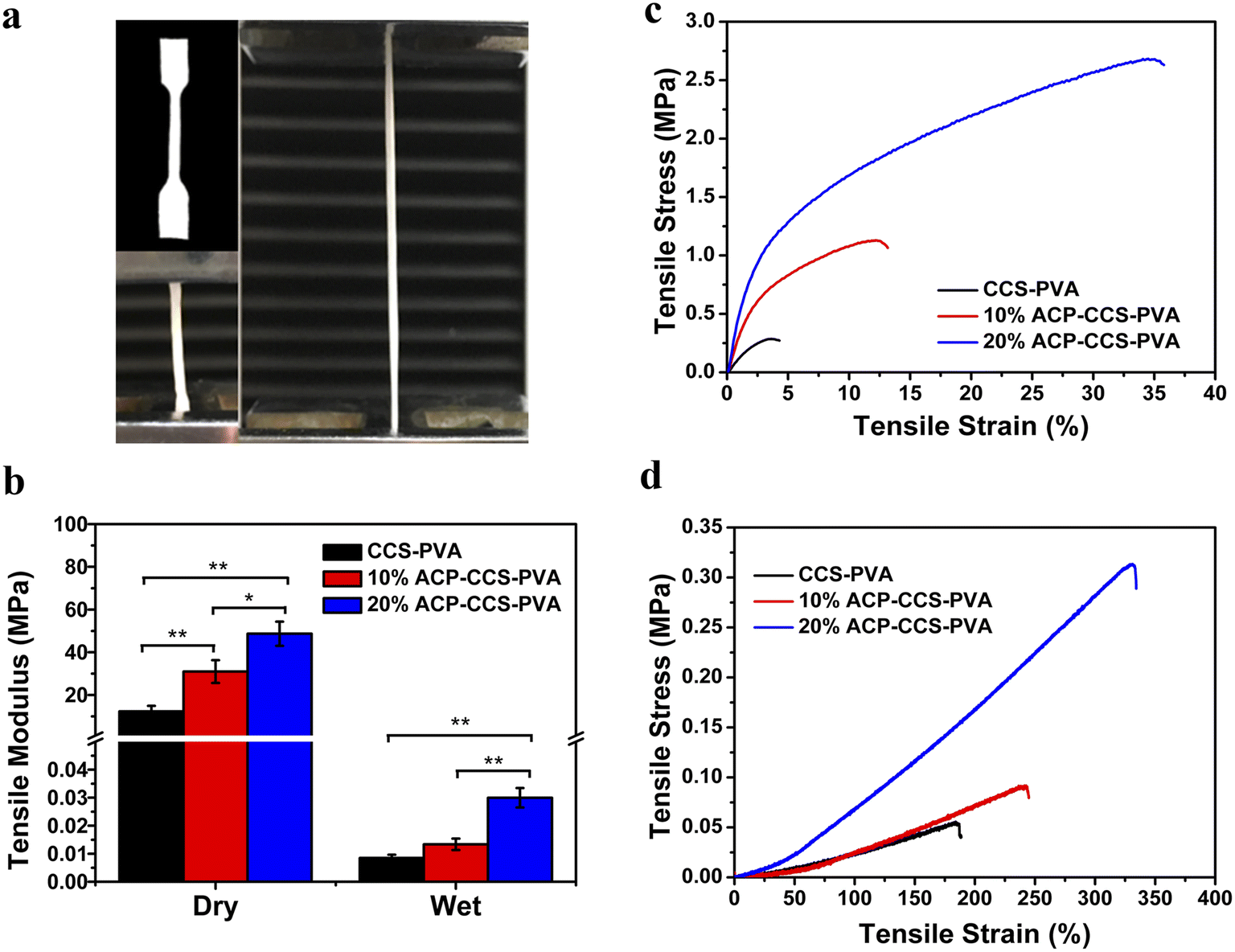 | ||
| Fig. 5 (a) Mechanical properties testing device diagram. (b) Composite membranes' tensile modulus and stress–strain curves were obtained in (c) dry and (d) wet conditions (*P < 0.05, **P < 0.01, n = 3). | ||
3.3 Biological behavior of composite membranes
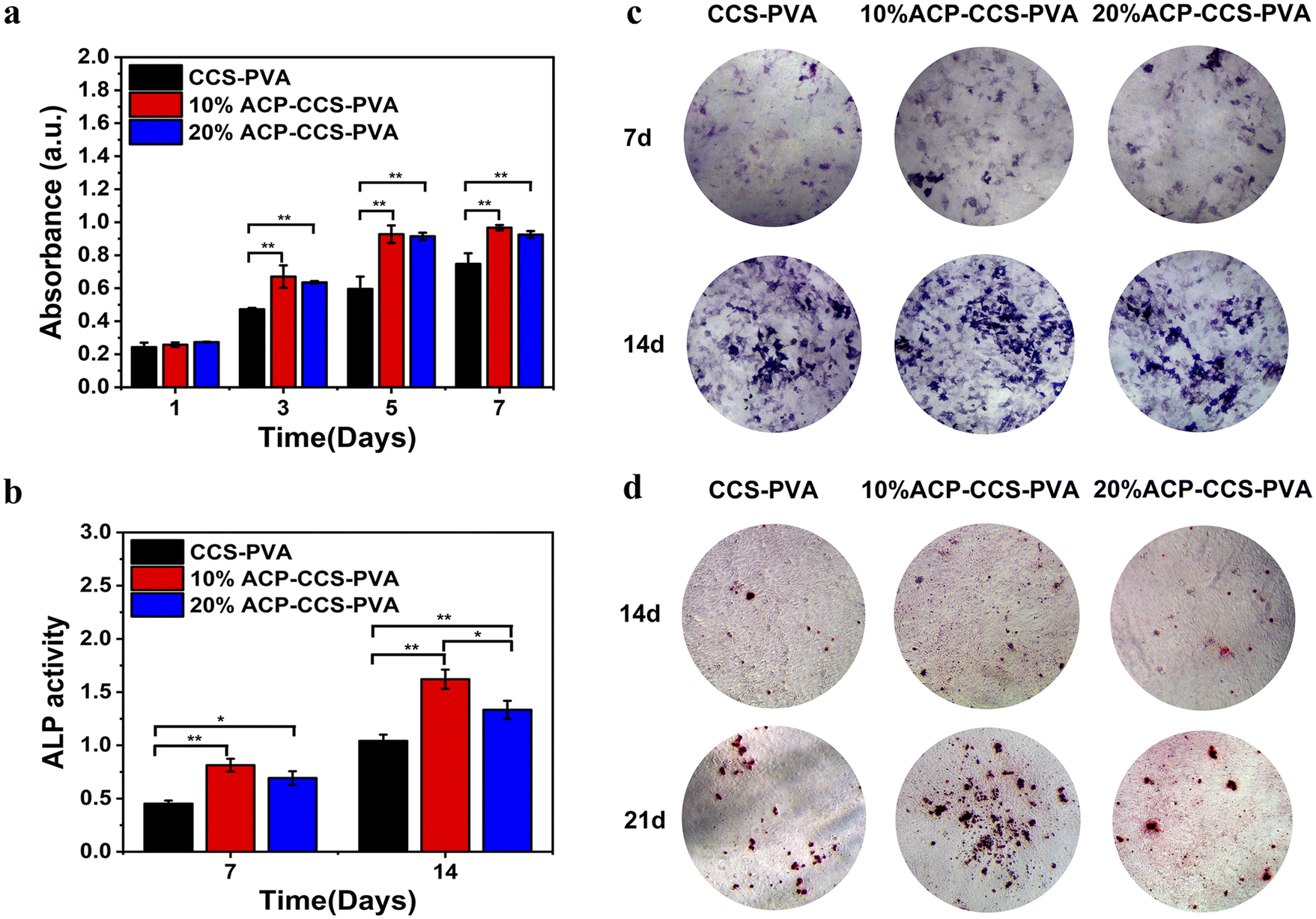 | ||
| Fig. 6 (a) CCK-8 assay results of CCS–PVA, 10 wt%, and 20 wt% ACP–CCS–PVA composite membranes. The ALP activity (b) and staining (c) were on the 7th and 14th days. (d) The alizarin red staining was on the 14th and 21st days (*P < 0.05, **P < 0.01, n = 3). | ||
Furthermore, alizarin red staining was conducted on the 14th and 21st days to further assess composite membranes' capacity to promote osteogenesis (Fig. 6d). The 10 wt% ACP–CCS–PVA can effectively stimulate the mineralization nodules formation compared with the CCS–PVA and 20 wt% ACP–CCS–PVA.
The transcription levels of osteogenic genes on days 14 were presented in Fig. 7, and those of osteogenic genes in the 10 wt% ACP–CCS–PVA group were significantly increased compared with the CCS–PVA and 20 wt% ACP–CCS–PVA groups. These results demonstrate osteogenic differentiation of cells, whereas excess ACP could suppress cell differentiation.
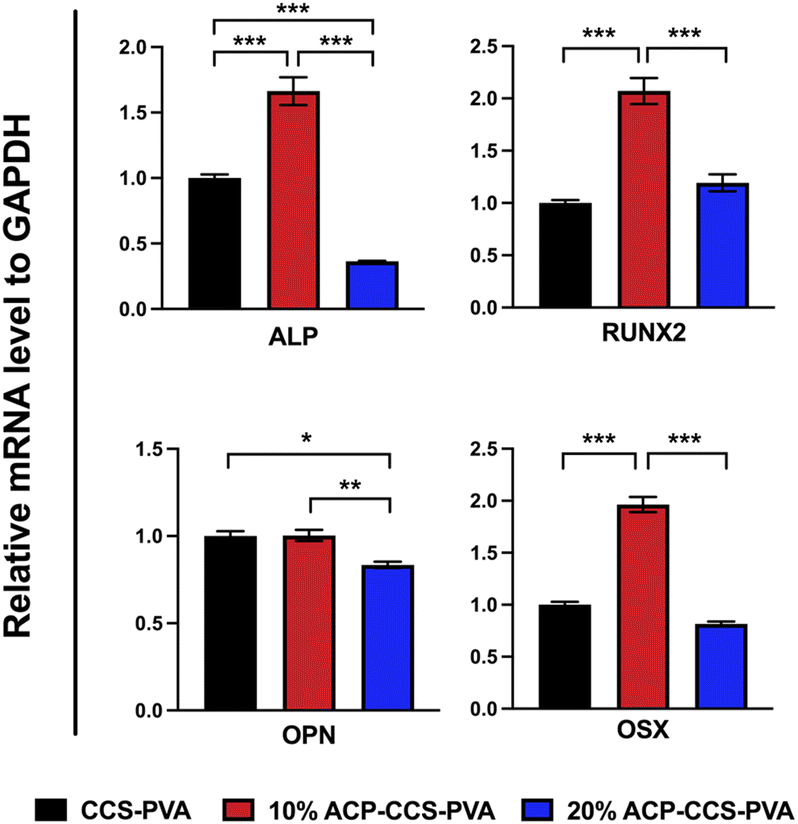 | ||
| Fig. 7 The mRNA expression of osteogenesis-related genes was analyzed after incubated for 14 days (*P < 0.05, **P < 0.01, ***P < 0.001, n = 3). | ||
4. Discussion
Numerous studies have demonstrated that using GBR membranes can improve bone formation,33–35 but rather than focusing on the cellular and molecular mechanisms of GBR, researchers have mainly studied the histological assessment of bone formation under the membrane cover. The mechanism is currently thought to be that GBR membranes act as a passive barrier to soft tissue invasion while ignoring their fundamental role in promoting bone regeneration.36 Some studies suggest that the GBR membranes stimulate the local environment in the bone defect area, which is beneficial for the recruitment and differentiation of various cell types, including osteoblasts and osteoclasts.37 These findings accelerate understanding of the mechanism by which GBR membranes promote bone regeneration and the manufacture and application of new biomaterials.In the present study, we fabricated an organic–inorganic composite membrane, which could serve as a barrier membrane for the potential application of GBR. Pure ACP is unstabilized in an aqueous solution and quickly transforms into thermodynamically more stable HAP crystals.38 Therefore, a biomolecule, ATP, was used here to stabilize ACP for a long time (in our case, we could stabilize it for 27 days; see Fig. S3†). Then we could directly investigate how ACP affects the cell behavior rather than crystallizing calcium phosphate. The organic components are CCS and PVA, which could form a strong network structure by forming hydrogen bonds between the hydroxyl groups of PVA and the carboxyl groups of CCS.39 When ACP is incorporated into the CCS and PVA system, there is an interaction between the mineral and organic matter through the binding of calcium to carboxyl groups, which enhances the mechanical performance of ACP–CCS–PVA compared to the pure CCS–PVA membrane. Due to the biological material's open porosity and excellent hydrophilicity, the ACP–CCS–PVA composite membrane produced a rigid and elastic hydrogel state, particularly in wet conditions similar to body fluids. These properties are consistent with a suitable biomaterial that could provide a well-matched mechanical microenvironment of the extracellular matrix, which is critical in regulating cell adhesion, migration, proliferation, and differentiation.40 In our case, the ACP–CCS–PVA composite membrane's higher elastic modulus might activate the cells' mechanical signaling pathway and effectively promote the osteogenic differentiation of cells. Although the swelling curves reflected that the incorporation of ACP made the ACP–CCS–PVA composite membranes denser in structure (Fig. S5†), they still have good pore structure, and the good pores can promote the diffusion of liquid, oxygen, and bioactive substances.41
The evaluation of the osteogenesis of the composite membrane shows that the ACP–CCS–PVA membranes are more robust than the CCS–PVA, and 10 wt% ACP incorporation is optimal, rather than the ACP amount higher and better. The slow, continuous release of calcium and phosphate ions from ATP-stabilized ACP nanoparticles not only selectively binds serum proteins and integrin receptors but also attracts osteoblasts, improving the microenvironment for bone formation.42,43 In addition, ACP may act as a calcium and phosphate reservoir to promote the mineral deposition in collagen fibrils, a significant component of bone. These factors suggest that calcium phosphate minerals may improve bone formation to some extent through chemical and biological pathways. Nevertheless, we discovered that the amount of ACP added to the composite membrane was not proportionate to the enhancement of osteogenic differentiation of MC3T3-E1 cells by ALP activity assay, alizarin red staining assay (Fig. 6b–d) and expression of osteogenic genes (Fig. 7). Because excess ACP is thought to cause excessive ion release, and local high ion concentrations interfere with related signaling pathways in osteoblasts, thereby affecting bone metabolism.44 For example, ACP NPs/macrophage-modulated environments attenuated the osteogenic capacity of BMSCs,45 sustained calcium ions in bone-forming solutions facilitated osteogenic differentiation of the cells, and ACP in its non-crystalline state was susceptible to transformation into HAP, which induced a decrease in calcium concentration leading to downregulation of the expression of osteogenesis-related proteins.46 Therefore, the ATP-stabilised ACP in this study can continuously release calcium and phosphate ions, which is considered to be a factor for increasing ALP activity, but whether the high concentration of ACP is a potential inflammatory factor in the process of cellular osteogenic differentiation also needs to be further investigated.
5. Conclusions
In conclusion, we found that ACP–CCS–PVA composite membranes have improved mechanical properties and promoted osteogenic differentiation of cells compared to CCS–PVA composite membranes, demonstrating their great potential in GBR. Although a higher ACP content has a superior mechanical strength, the excess ACP would negatively influence cell proliferation and osteogenic differentiation. Further research is required to investigate in vivo effect of the ACP–CCS–PVA composite membranes and their molecular mechanism for guiding bone tissue regeneration. These findings highlight the significant role of calcium phosphate in bone regeneration and provide a strategy for biomaterial preparation to balance the mechanical properties and mineral content.Author contributions
Qiaolin Du and Jian Sun: methodology, investigation, writing – original draft, writing – review & editing. Yanyan Zhou: investigation, formal analysis. Yadong Yu: validation. Weijing Kong: project administration. Chaoqun Chen: funding acquisition. Yifeng Zhou and Ke Zhao: software. Changyu Shao: conceptualization, supervision, writing – review & editing. Xinhua Gu: conceptualization, supervision, funding acquisition, methodology, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (grant no. 2023KY092) and the Research and Development Project of Stomatology Hospital Zhejiang University School of Medicine (grant no. RD2022JCYL07).References
- J. I. Sasaki, G. L. Abe, A. Li, P. Thongthai, R. Tsuboi, T. Kohno and S. Imazato, Biomater. Invest. Dent., 2021, 8, 54–63 CAS.
- J. Caballe-Serrano, Y. Abdeslam-Mohamed, A. Munar-Frau, M. Fujioka-Kobayashi, F. Hernandez-Alfaro and R. Miron, Arch. Oral Biol., 2019, 100, 57–68 CrossRef CAS PubMed.
- S. Jin, J. Li, J. Wang, J. Jiang, Y. Zuo, Y. Li and F. Yang, Int. J. Nanomed., 2018, 13, 4591–4605 CrossRef CAS.
- A. R. Padalhin, N. Thuy Ba Linh, Y. Ki Min and B. T. Lee, J. Biomater. Sci., Polym. Ed., 2014, 25, 487–503 CrossRef CAS.
- Y. Tu, C. Chen, Y. Li, Y. Hou, M. Huang and L. Zhang, Bio-Med. Mater. Eng., 2017, 28, 223–233 CAS.
- F. Han, P. Zhang, Y. Sun, C. Lin, P. Zhao and J. Chen, Int. J. Nanomed., 2015, 10, 7333–7343 CAS.
- N. Eliaz and N. Metoki, Materials, 2017, 10, 334 CrossRef PubMed.
- D. Ege, Q. Nawaz, A. M. Beltran and A. R. Boccaccini, ACS Biomater. Sci. Eng., 2022, 8, 5273–5283 CrossRef CAS PubMed.
- D. Ege, K. Zheng and A. R. Boccaccini, ACS Appl. Bio Mater., 2022, 5, 3608–3622 CrossRef CAS PubMed.
- W. C. Lepry and S. N. Nazhat, Adv. NanoBiomed Res., 2021, 1, 2000055 CrossRef CAS.
- M. Schweikle, S. H. Bjornoy, A. T. J. van Helvoort, H. J. Haugen, P. Sikorski and H. Tiainen, Acta Biomater., 2019, 90, 132–145 CrossRef CAS.
- S. Gorodzha, T. E. Douglas, S. K. Samal, R. Detsch, K. Cholewa-Kowalska, K. Braeckmans, A. R. Boccaccini, A. G. Skirtach, V. Weinhardt, T. Baumbach, M. A. Surmeneva and R. A. Surmenev, J. Biomed. Mater. Res., Part A, 2016, 104, 1194–1201 CrossRef CAS PubMed.
- G. Balasundaram, M. Sato and T. J. Webster, Biomaterials, 2006, 27, 2798–2805 CrossRef CAS PubMed.
- D. Tadic, F. Peters and M. Epple, Biomaterials, 2002, 23, 2553–2559 CrossRef CAS PubMed.
- B. M. Whited, D. Skrtic, B. J. Love and A. S. Goldstein, J. Biomed. Mater. Res., Part A, 2006, 76, 596–604 CrossRef.
- G. F. Smaisim, K. J. Mohammed, S. K. Hadrawi, H. Koten and E. Kianfar, Bionanoscience, 2023, 13, 819–839 CrossRef.
- Z. S. Abbas, M. M. Kadhim, A. M. Rheima, A. D. J. Al-bayati, Z. T. Abed, F. M. D. Al-Jaafari, A. S. Jaber, S. K. Hachim, Z. H. Mahmoud, F. K. Ali, H. Koten and E. Kianfar, Bionanoscience, 2023, 13, 983–1011 CrossRef.
- A. Taloni, M. Vodret, G. Costantini and S. Zapperi, Nat. Rev. Mater., 2018, 3, 211–224 CrossRef.
- S. Yao, X. Lin, Y. Xu, Y. Chen, P. Qiu, C. Shao, B. Jin, Z. Mu, N. Sommerdijk and R. Tang, Adv. Sci., 2019, 6, 1900683 CrossRef CAS PubMed.
- J. S. Martinez, T. C. Keller 3rd and J. B. Schlenoff, Biomacromolecules, 2011, 12, 4063–4070 CrossRef CAS.
- Y. Ren, L. Fan, S. Alkildani, L. Liu, S. Emmert, S. Najman, D. Rimashevskiy, R. Schnettler, O. Jung, X. Xiong and M. Barbeck, Int. J. Mol. Sci., 2022, 23, 14987 CrossRef CAS.
- C. Chu, J. Deng, X. Sun, Y. Qu and Y. Man, Tissue Eng., Part B, 2017, 23, 421–435 CrossRef CAS PubMed.
- M. Ahmadipour, H. Mohammadi, A. L. Pang, M. Arjmand, T. A. Otitoju, P. U. Okoye and B. Rajitha, Int. J. Polym. Mater. Polym. Biomater., 2022, 71, 180–195 CrossRef CAS.
- S. Chen, H. Tian, J. Mao, F. Ma, M. Zhang, F. Chen and P. Yang, Int. J. Biol. Macromol., 2023, 226, 410–422 CrossRef CAS PubMed.
- D. T. Wu, J. G. Munguia-Lopez, Y. W. Cho, X. Ma, V. Song, Z. Zhu and S. D. Tran, Molecules, 2021, 26, 7043 CrossRef CAS PubMed.
- O. Kamaci, N. Yucel, H. Koten, E. Bulus and G. S. Bulus, J. Polytech., 2023, 1, 1 Search PubMed.
- J. M. Unagolla, T. E. Alahmadi and A. C. Jayasuriya, Carbohydr. Polym., 2018, 199, 426–436 CrossRef CAS PubMed.
- K. Hozumi and M. Nomizu, Int. J. Mol. Sci., 2018, 19, 2713 CrossRef PubMed.
- D. Coelho, A. Sampaio, C. J. S. M. Silva, H. P. Felgueiras, M. T. P. Amorim and A. Zille, ACS Appl. Mater. Interfaces, 2017, 9, 33107–33118 CrossRef CAS.
- M. J. Pei, D. Zhu, J. F. Yang, K. D. Yang, H. J. Yang, S. J. Gu, W. Q. Li, W. L. Xu, P. Xiao and Y. S. Zhou, Eur. Polym. J., 2023, 182, 111737 CrossRef CAS.
- C. Qi, Y. J. Zhu, B. Q. Lu, X. Y. Zhao, J. Zhao, F. Chen and J. Wu, Small, 2014, 10, 2047–2056 CrossRef CAS.
- A. Ressler, M. Antunovic, L. Teruel-Biosca, G. G. Ferrer, S. Babic, I. Urlic, M. Ivankovic and H. Ivankovic, Carbohydr. Polym., 2022, 277, 118883 CrossRef CAS PubMed.
- G. I. Benic, D. S. Thoma, F. Munoz, I. Sanz Martin, R. E. Jung and C. H. Hammerle, Clin. Oral Implants Res., 2016, 27, 567–576 CrossRef PubMed.
- S. Pajoumshariati, H. Shirali, S. K. Yavari, S. N. Sheikholeslami, G. Lotfi, F. Mashhadi Abbas and A. Abbaspourrad, Sci. Rep., 2018, 8, 7513 CrossRef.
- M. Lian, B. Sun, Z. Qiao, K. Zhao, X. Zhou, Q. Zhang, D. Zou, C. He and X. Zhang, Colloids Surf., B, 2019, 176, 219–229 CrossRef CAS PubMed.
- I. Elgali, O. Omar, C. Dahlin and P. Thomsen, Eur. J. Oral Sci., 2017, 125, 315–337 CrossRef.
- O. Omar, I. Elgali, C. Dahlin and P. Thomsen, J. Clin. Periodontol., 2019, 46, 103–123 CrossRef PubMed.
- S. V. Dorozhkin, Biomater. Sci., 2021, 9, 7748–7798 RSC.
- L. Nie, Y. Deng, P. Li, R. Hou, A. Shavandi and S. Yang, ACS Omega, 2020, 5, 10948–10957 CrossRef CAS PubMed.
- N. Alcorta-Sevillano, I. Macias, A. Infante and C. I. Rodriguez, Cells, 2020, 9, 2630 CrossRef CAS PubMed.
- P. Xia and Y. Luo, J. Biomed. Mater. Res., Part B, 2022, 110, 1206–1214 CrossRef CAS PubMed.
- W. He, M. Andersson, P. P. de Souza, C. A. de Souza Costa, E. M. Munoz, H. O. Schwartz-Filho, M. Hayashi, A. Hemdal, A. Fredel, A. Wennerberg and R. Jimbo, Biomed. Mater., 2013, 8, 035007 CrossRef CAS.
- X. Niu, S. Chen, F. Tian, L. Wang, Q. Feng and Y. Fan, Mater. Sci. Eng., C, 2017, 70, 1120–1124 CrossRef CAS PubMed.
- P. Berube, Y. Yang, D. L. Carnes, R. E. Stover, E. J. Boland and J. L. Ong, J. Periodontol., 2005, 76, 1697–1709 CrossRef CAS PubMed.
- L. Chen, P. Qiao, H. Liu and L. Shao, Inflammation, 2021, 44, 278–296 CrossRef CAS PubMed.
- O. Groninger, S. Hess, D. Mohn, E. Schneider, W. Stark, S. Marsmann, P. Wolint, M. Calcagni, P. Cinelli and J. Buschmann, Int. J. Mol. Sci., 2020, 21, 2627 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04498j |
| ‡ Qiaolin Du and Jian Sun contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |