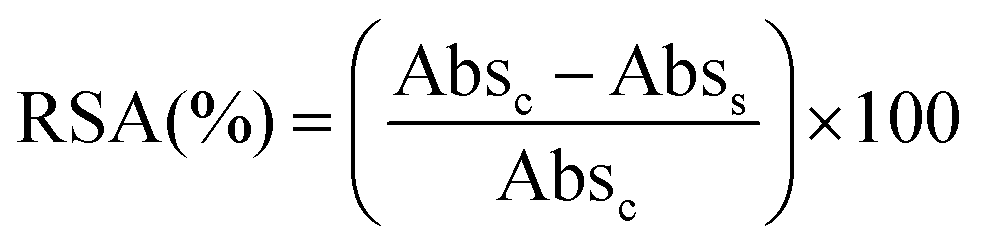DOI:
10.1039/D3RA04527G
(Paper)
RSC Adv., 2023,
13, 29477-29488
Mitochondria-targeting EGCG derivatives protect H9c2 cardiomyocytes from H2O2-induced apoptosis: design, synthesis and biological evaluation†
Received
7th July 2023
, Accepted 2nd October 2023
First published on 9th October 2023
Abstract
Pathologies related to cardiovascular diseases mostly emerge as a result of oxidative stress buildup in cardiomyocytes. The heavy load of mitochondrial oxidative phosphorylation in cardiac tissues corresponds to a surge in oxidative stress leading to mitochondrial dysfunction and cellular apoptosis. Thus, scavenging the reactive oxygen species (ROS) linked to mitochondria can significantly improve cardio-protection. Epigallocatechin-3-gallate (EGCG), the major polyphenol found in green tea has been extensively studied for its profound health-beneficial activities. Herein, we designed and synthesized a series of mitochondrial-targeting EGCG derivatives, namely MitoEGCGn (n = 4, 6, 8) by incorporating triphenylphosphonium ion onto it using different linkers. MitoEGCGn were found to be non-toxic to H9c2 rat cardiomyocyte cells even at higher doses in comparison to its parent molecule EGCG. Interestingly, MitoEGCG4 and MitoEGCG6 protected the H9c2 cardiomyocyte cells from the oxidative damage induced by H2O2 whereas EGCG was found to be toxic and ineffective in protecting the cells from H2O2 damage. MitoEGCG4 and MitoEGCG6 also protected the cells from the H2O2-induced disruption of mitochondrial membrane potential as well as activation of apoptosis as revealed by pro-caspase 3 expression profile, DNA fragmentation assay, and AO/EtBr staining. Taken together, our study shows that the mitochondria targeting EGCG derivatives were able to effectively combat the H2O2-induced oxidative stress in H9c2 cardiomyocytes. They eventually augmented the mitochondrial health of cardiomyocytes by maintaining the mitochondrial function and attenuating apoptosis. Overall, MitoEGCG4 and MitoEGCG6 could provision a cardioprotective role to H9c2 cardiomyocytes at the time of oxidative insults related to mitochondrial dysfunction-associated injuries.
1. Introduction
Cardiovascular diseases are counted among the leading reasons for a very large number of human deaths worldwide.1 The World Health Organization (WHO) has predicted that the mortality rate due to cardiovascular related diseases might elevate to 25 million by 2030.2,3 The pathological progression of cardiac damage is mostly linked to the elevated degree of oxidative stress in cells, which can be brought on by a number of factors including fat accumulation, a sedentary lifestyle, consuming food high in saturated fat and sodium ion, etc. These damages are multifaceted as oxidative stress results in the accumulation of free radicals which harms several biomolecules like DNA, lipids and proteins.4,5 It also plays a central role in activating a redox-regulated signaling cascade which can culminate into mitochondrial-associated cell death.6,7 Mitochondria are considered as the powerhouse of the cell. The production of ATP in the mitochondria through oxidative phosphorylation intrinsically produces ROS and this makes the mitochondrion as the primary source for intracellular ROS. Due to the higher aerobic metabolism in heart tissues, mitochondria are highly susceptible to oxidative damage leading to mitochondrial dysfunction which can ultimately initiate intrinsic apoptotic pathways.8,9 Hence, the development of therapeutic strategies to overcome the mitochondrial dysfunction-associated injuries to heart tissues caused by ROS can prove helpful in installing a check in the progression of cardiovascular diseases.
Green tea has gained global attention in recent times due to its remarkable antioxidant, anti-carcinogenic, cardioprotective, neuroprotective and anti-inflammatory properties.10–13 (−)-Epigallocatechin-3-gallate (EGCG) is the most abundant polyphenol in green tea accounting for about 50–75% of the catechins and is responsible for the majority of health benefits attributed to green tea. It has also been classified as ‘Generally Recognized as Safe (GRAS)’ for its health beneficial effects by the United States Food and Drug Administration (FDA) and European Food Safety Authority (EFSA). EGCG is well studied for its antioxidant property which is due to the presence of the eight phenolic hydrogens. Its structure allows electron delocalization, conferring it with radical scavenging activity. It is known to react with ROS such as superoxide radical (O2˙−), singlet oxygen (1O2), hydroxyl radical (OH˙), peroxyl radical (ROO˙), nitric oxide (NO), etc.14 However, the therapeutic effect of EGCG is hampered by its poor systemic absorption after oral administration, poor gastrointestinal tract stability and pharmacokinetics, low bioavailability, first-pass metabolism, constrained membrane permeability across the intestine, and low accumulation in the related tissues.15 In our recent report, we conducted a chemoproteomics-based protein profiling of EGCG in HeLa cells. The post-proteomics analysis provided insightful information on the molecular mechanism of action behind the plethoric biological activities exhibited by EGCG.16 Previously, we have successfully demonstrated that an alkyl substitution at the 4′′-position on the D-ring hydroxyl group can be explored to generate EGCG analogues with increased bioavailability and thus enhanced biological activity.17,18 The characteristic resonance stabilization of the phenolate at the 4′′-position provides scope for regioselective alkylation at this position.
Herein, we strategically designed mitochondria-targeting EGCG analogues (MitoEGCGn) by conjugating EGCG with a triphenylphosphonium cation (TPP) through hydrophobic alkyl linkers. The linker serves a dual purpose: (i) an optimum distance between EGCG and TPP to avoid any potential steric hindrance and (ii) the required balance between the hydrophobicity and hydrophilicity to cross the mitochondrial membrane. TPP is a widely used lipophilic cation for mitochondrial targeting due to its positive charge which is delocalized throughout the aromatic rings making it membrane permeant.19,20 Due to the large mitochondrial membrane potential (∼120–180 mV, negative inside), MitoEGCGn can readily accumulate in the mitochondria and can exert its antioxidant activity to safeguard mitochondrial health from severe oxidative damages. Herein, we have used the H9c2 rat cardiomyocytes with H2O2-induced oxidative stress as a cardiac injury model for current investigation.21,22 Even at high concentrations, MitoEGCGn (n = 4, 6 and 8) was found to be substantially less toxic than EGCG. The structural modifications on EGCG did not compromise on the biological activities exhibited by their mitochondrial analogues as shown by in vitro radical scavenging activity analysis. MitoEGCG4 and MitoEGCG6 both were able to reduce H2O2-induced cytotoxicity in rat cardiomyocytes in comparison to EGCG. The inhibition of the mitochondrial outer membrane potential (Ψm) reduction upon MitoEGCGn treatment demonstrates that those agents protected the cells from H2O2-induced oxidative mitochondrial damage brought on by ROS formation. Thus, our study provides a strategic model for developing mitochondria-targeting natural polyphenols as a cardioprotective agent during cardiovascular pathologies.
2. Materials and methods
2.1. Chemicals
EGCG was purchased from Biosynth Carbosynth, USA. Anhydrous NaOAc and triphenylphosphine were purchased from NICE Chemicals, India and Spectrochem, India, respectively. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), anhydrous Na2SO4, and NaHCO3 were purchased from Sisco Research Laboratories (SRL), India. All solvents used were of reagent grades and used as purchased (NICE Chemicals, India) without any further purification. Amphotericin, tris buffer, sodium dodecyl sulphate (SDS), tween-20, Triton X 100, glycerol, glycine, Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), antibiotics comprised of Penicillin and Streptomycin were purchased from HiMedia, India. H2O2, TMRE and DCFHDA dyes were purchased from Sigma-Aldrich, India. Anti-pro-caspase 3 and anti-rabbit IgG HRP-linked antibodies were procured from Cell Signaling Technology, USA.
2.2. Synthesis of MitoEGCGn
MitoEGCGn derivatives were synthesized in three steps as described below.
2.2.1 Step 1: Synthesis of 1,n-dibromoalkane (where n = 4, 6, and 8). To a mixture of alkane-1,n-diol (11.47 mmol, 1 equiv.) in water (20 mL), hydrobromic acid (HBr) (172.14 mmol, 15 equiv.) was added. The mixture was stirred under reflux for 48 h. After the completion of the reaction, the dibromoalkane compounds were extracted with chloroform (CHCl3) (20 mL × 3). The CHCl3 layer was then neutralized by washing with saturated sodium bicarbonate (NaHCO3) solution (20 mL × 2). The organic layer was dried by passing over anhydrous sodium sulphate (Na2SO4). The pale-yellow liquid dibromoalkane compounds were obtained by the evaporation of the solvent under reduced pressure. The obtained product was directly used for the next step without any purification.
2.2.2 Step 2: Synthesis of EGCG-CnBr (where n = 4, 6, and 8). To a mixture of EGCG (0.45 mmol, 1 equiv.) in dry dimethylformamide (DMF) (2 mL), anhydrous sodium acetate (NaOAc) (1.31 mmol, 3 equiv.) and 1,n-dibromoalkane (2.25 mmol, 5 equiv.) was added one by one. The mixture was stirred for 2 h at 85 °C. The progress of the reaction was monitored by thin-layer chromatography (TLC). The reaction was quenched by the addition of H2O (5 mL) and the desired compound was extracted with ethyl acetate (EA) (5 mL × 3). The EA layer was washed well with H2O (5 mL × 3) to remove the DMF. The organic layer was dried by passing over anhydrous sodium sulphate (Na2SO4) and the crude compounds were obtained by the evaporation of the solvent under reduced pressure. The desired compounds, (EGCG-CnBr) were purified by silica gel column chromatography (silica: 230–400 mesh size and solvent: 20% acetone–DCM).
2.2.3 Step 3: Synthesis of MitoEGCGn (where n = 4, 6, and 8). EGCG-CnBr (0.16 mmol, 1 equiv.) obtained from the previous step was dissolved in acetonitrile (50 mL) and the mixture was then treated with triphenylphosphine (TPP) (0.81 mmol, 5 equiv.). The reaction was allowed to stir for 36 h at 100 °C. The progress of the reaction was monitored by thin-layer chromatography (TLC). At the end of the reaction, the crude compounds were obtained by the evaporation of the solvent under reduced pressure. The desired compounds, MitoEGCGn were purified by silica gel column chromatography (silica: 230–400 mesh size and solvent: 20% methanol–EA) as pale brown solids.
2.3. Instruments used for the characterization of MitoEGCGn compounds
Shimadzu LC-MS-8045 instrument is used for the LC-MS (liquid chromatography-mass spectrometry) characterization of the MitoEGCGn compounds by utilizing the Sprite TARGA C18 column with dimensions 40 × 2.1 mm, 5 μm and monitored at 210 and 254 nm in positive mode of ionization for mass detection. 1H NMR and 13C NMR spectra were recorded in Bruker Ascend 400 NMR spectrometer.
2.4. Characterization of MitoEGCGn
2.4.1 MitoEGCG4. The compound was obtained with 59% yield and 99.2% purity. MS (ESI): m/z calculated for C44H40O11P+ = 775.23, observed = 775.20. 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.89–7.73 (15H, m), 6.84 (2H, s), 6.43 (2H, s), 5.94 (1H, d, J = 2.24 Hz), 5.84 (1H, d, J = 2.24 Hz), 5.38 (1H, m), 4.97 (1H, m), 3.97 (2H, t, J = 5.88 Hz), 2.95 (1H, dd, J = 17.14 and 4.38 Hz), 2.66 (1H, d, J = 16.52 Hz), 1.82 (2H, m), 1.74 (2H, m), 1.29 (2H, m). 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 164.95, 156.57, 156.50, 155.57, 150.68, 145.66, 138.51, 134.93, 133.51, 132.36, 130.22, 128.38, 124.25, 118.95, 118.10, 108.70, 105.45, 97.26, 95.55, 94.38, 76.34, 70.48, 68.47, 30.73, 28.39, 22.42, 13.93, 10.84.
2.4.2 MitoEGCG6. The compound was obtained with 56% yield and 99.4% purity. MS (ESI): m/z calculated for C46H44O11P+ = 803.26, observed = 803.35. 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.92–7.70 (15H, m), 6.81 (2H, s), 6.40 (2H, s), 5.93 (1H, d, J = 2.24 Hz), 5.82 (1H, d, J = 2.32 Hz), 5.37 (1H, m), 4.95 (1H, m), 3.87 (2H, t, J = 6.12 Hz), 2.93 (1H, dd, J = 16.68 and 4.14 Hz), 2.65 (1H, d, J = 16.68 Hz), 1.51 (6H, m), 1.37 (2H, m), 1.21 (2H, m). 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 164.99, 156.58, 156.51, 155.59, 150.70, 145.71, 138.86, 134.90, 133.53, 132.42, 130.21, 128.49, 124.15, 119.01, 118.16, 108.69, 105.46, 97.28, 95.55, 94.37, 76.38, 71.59, 68.47, 48.63, 29.05, 24.44, 21.69, 20.40, 19.90.
2.4.3 MitoEGCG8. The compound was obtained with 52% yield and 98.6% purity. MS (ESI): m/z calculated for C48H48O11P+ = 831.29, observed = 831.25. 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.90–7.74 (15H, m), 6.84 (2H, s), 6.42 (2H, s), 5.94 (1H, d, J = 2.24 Hz), 5.83 (1H, d, J = 2.24 Hz), 5.35 (1H, m), 4.95 (1H, m), 3.87 (2H, t, J = 6.72 Hz), 2.93 (1H, dd, J = 17.14 and 4.50 Hz), 2.66 (1H, d, J = 17.12 Hz), 1.60 (6H, m), 1.27 (8H, m). 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 165.03, 156.57, 156.49, 155.58, 150.75, 145.71, 138.91, 134.90, 133.54, 132.40, 130.19, 128.69, 124.11, 119.02, 118.17, 108.65, 105.47, 97.24, 95.54, 94.36, 71.73, 68.53, 67.44, 48.62, 38.10, 29.82, 29.36, 27.99, 25.12, 22.42, 13.92, 10.83.
2.5. Cell culture and treatments
The H9c2 cell line was bought from the National Centre for Cell Science (NCCS) Pune, India. They were grown in Dulbecco's Modified Eagle Medium (DMEM, provided with 10% fetal bovine serum (FBS), 1% pen-strep, and 0.2% amphotericin) at 37 °C in a humid environment with 5% CO2. A mother stock with a concentration of 100 mM of EGCG and MitoEGCGn was prepared and stored at −20 °C until further usage. All treatments were done while maintaining the final DMSO concentration below 0.02%. DMSO treatment was considered as the control in every experiment. Cells were pretreated with different concentrations of EGCG, MitoEGCGn (where n = 4, 6, and 8) for 24 h and were thoroughly washed with 1X PBS twice post incubation. After washing, they were replenished with fresh medium containing 400 μM of H2O2 followed by incubation for another 20 min.
2.6. Measurement of in vitro radical scavenging activity
The total radical scavenging capacity of EGCG and MitoEGCGn compounds was measured using the DPPH assay protocol as reported earlier by our group.16 Briefly, 0.2 mM stock solution of diphenyl picryl radical (DPPH) was prepared in 95% methanol at room temperature in dark. To obtain a final concentration of 0.1–200 μM for the EGCG or MitoEGCGn compounds (n = 4, 6, or 8), 150 μL of 0.2 mM DPPH solution was added to 50 μL, 4X DMSO stocks of the respective compounds. DMSO and ascorbic acid was taken as a negative and positive control respectively. The samples were incubated in a 96-well plate for 1 h at room temperature and in the dark. Using a microplate reader (BioTek, Epoch 2), the absorbance was measured at 517 nm after the allotted incubation time. The following formula was used to calculate the percentage of radical scavenging activity (RSA), where Abss and Absc stand for the sample's and the control's respective absorbances. Using GraphPad Prism software and non-linear regression analysis, IC50 values were calculated.
2.7. Analysis of cell viability using MTT assay
The cell viability profile of rat cardiomyocyte cells, H9c2 after treatment with newly synthesized MitoEGCGn compounds, EGCG and in the experiments associated with H2O2 treatment were measured by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In short, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well were seeded into the 96-well plates and treated according to the procedure mentioned in Section 2.5. Following the proper treatment and allotted incubation time, 10 μL of MTT solution (5 mg mL−1) was added to each well and incubated for an additional 2 h. After carefully removing the medium, 100 μL DMSO was added to each well to dissolve the hydrophobic formazan crystals formed. Using a microplate reader (BioTek, Epoch 2), the absorbance of the resultant blue solution was assessed at 590 nm and 620 nm (reference reading). IC50 values were determined using GraphPad Prism software and non-linear regression analysis.
000 cells per well were seeded into the 96-well plates and treated according to the procedure mentioned in Section 2.5. Following the proper treatment and allotted incubation time, 10 μL of MTT solution (5 mg mL−1) was added to each well and incubated for an additional 2 h. After carefully removing the medium, 100 μL DMSO was added to each well to dissolve the hydrophobic formazan crystals formed. Using a microplate reader (BioTek, Epoch 2), the absorbance of the resultant blue solution was assessed at 590 nm and 620 nm (reference reading). IC50 values were determined using GraphPad Prism software and non-linear regression analysis.
2.8. Measurement of intracellular ROS using DCFH-DA staining
The intracellular ROS was accessed using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescence assay. Briefly, H9c2 cells were seeded in 35 mm dishes at a density of 0.6 × 106 cells per dish and were allowed to adhere to the substratum overnight. Cells were incubated with EGCG and MitoEGCGn at 50 μM for 24 h in triplicates. After the stipulated time, one set of MitoEGCGn and EGCG treated cells was subjected to treatment with 400 μM H2O2 for 20 min and then incubated with 10 μM DCFH-DA reagent for 30 min. The media was removed after 30 min, cells were washed in 1X PBS and later supplemented with fresh media. The fluorescence was imaged using a fluorescence microscope (Olympus IX83) using ex/em wavelength of 485/530 nm.
2.9. Measurement of mitochondrial membrane potential
The change in mitochondrial membrane potential was measured fluorescently using TMRE reagent. Briefly, H9c2 cells were seeded in 35 mm dishes at a density of 0.3 × 106 cells per dish and were allowed to adhere to the substratum overnight. Cells were incubated with MitoEGCGn at various concentrations ranging from 0 to 100 μM for 24 h in duplicates. After the stipulated time, one set of MitoEGCGn treated cells was subjected to treatment with 400 μM H2O2 for 20 min and then incubated with TMRE 100 nM for 15 min. The media was removed after 30 min, cells were washed in 1X PBS and later supplemented with fresh media. The fluorescence images were acquired using a fluorescence microscope with ex/em wavelength of 544/570 nm.
2.10. Protein expression of pro-caspase 3 using western blotting
H9c2 cells were grown in 35 mm culture dishes and were subjected to pretreatment with 50 μM MitoEGCG4, MitoEGCG6 and EGCG for 24 h. After the pretreatment cells were washed twice with 1X PBS of pH 7.4 and replenished with fresh medium containing 400 μM H2O2 for 4 h. Following the treatment, cells were lysed in a lysis buffer containing 1% Triton X-100, 1% protease inhibitor cocktail, 100 mM DTT, 1 M Tris pH 7.4, 2 M NaCl, 0.1 M EDTA, and 10% glycerol. Bicinchoninic acid assay was used for estimating protein concentration. An equal amount of protein (40 μg) was separated on a 15% SDS-PAGE gel followed by transferring them to a PVDF membrane (Amersham) via western blotting. Afterwards, the membrane was blocked in 5% non-fat milk made with 1X PBST. The membrane was then incubated with the anti-pro-caspase 3 primary antibody at 4 °C overnight, after several washes in 1X PBST (3 times, 5 min each). It was again incubated further with HRP-conjugated anti-rabbit IgG secondary antibodies in 1X PBST for 2 h at room temperature, followed by the previously mentioned washing procedure. The same membrane was blotted against HRP conjugated anti β-actin antibody for loading control. Standard chemiluminescent substrate (ECL reagent, BioRad) was used for developing the membrane in both the cases. Densitometric intensities of bands of pro-caspase 3 were quantified using ImageJ software (NIH, USA) and normalized with respect to their corresponding loading controls (β-actin). The statistical analysis and graph were obtained using GraphPad Prism Software.
2.11. Statistical analysis
For the statistical analysis, GraphPad Prism 7 (GraphPad Software Inc., San Diego, CA, USA) was utilized. All the data are represented as mean ± standard error of the mean (SEM) from a minimum of three independent tests. A non-linear regression analysis was used to determine the IC50 values. One-way analysis of variance (ANOVA) with Dunnett's multiple comparison tests was used to analyze the statistical differences. The statistical significance was represented by the following symbols, ‘ns’ represents non-significance and is given for p > 0.05, * for p < 0.05, ** for p < 0.01, *** for p < 0.001, and **** for p < 0.0001.
3. Results and discussion
3.1. Synthesis and characterization of MitoEGCGn
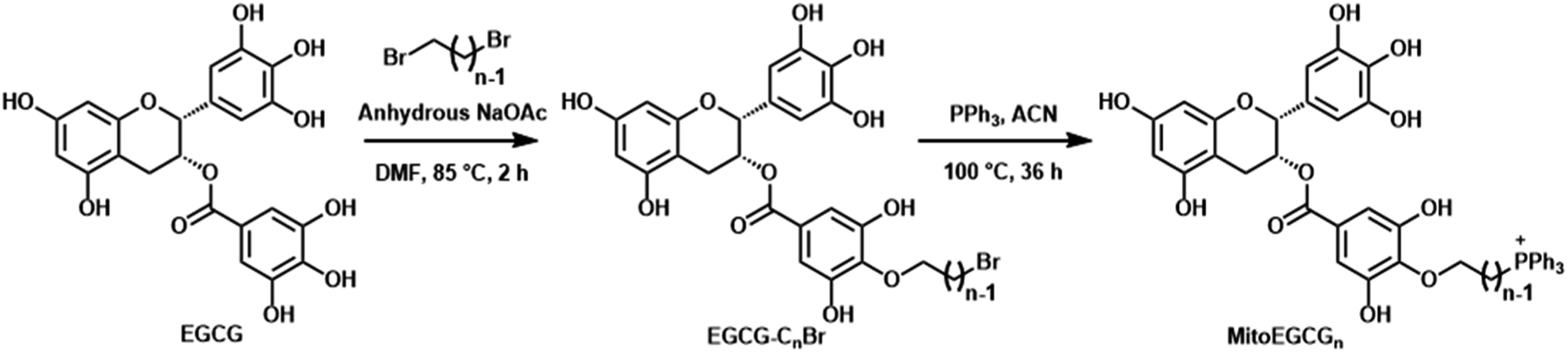
MitoEGCGn compounds were synthesized through a three-step process as shown in Scheme 1 starting from the dibromination of 1,n-alkanediols, where n = 4, 6, and 8. The resulting dibromoalkanes were reacted with EGCG to introduce the alkane linker to the 4′′ position of EGCG via a nucleophilic substitution reaction in the presence of a weak base, NaOAc. The TPP was then introduced by the substitution of bromine in EGCG-CnBr, where n = 4, 6, and 8. The MitoEGCGn, (n = 4, 6, and 8) compounds were obtained with an overall 50–60% yield and >98.5% purity. They were characterized using LC-MS, HPLC and NMR (ESI Fig. S1–S12†).
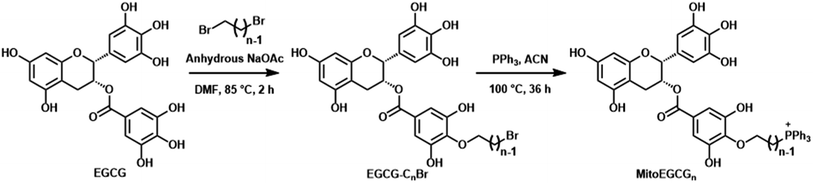 |
| | Scheme 1 Synthetic route for MitoEGCGn where n = 4, 6, and 8. | |
The presence of multiple phenolic hydroxyl groups in EGCG makes it a good antioxidant substance by virtue of single-electron transfer mechanism. However, it is reported that EGCG tends to get destabilized and undergo rapid degradation and auto-oxidation in alkaline environments and physiological conditions.15 Hence, several attempts have been made to stabilize the core pharmacophore of EGCG via small chemical modifications. Among the eight hydroxyl groups, one that is present in the 4′′ position is the most acidic due to the presence of electron-withdrawing ester group at its para position and the characteristic resonance stabilization of the phenoxide ions. Previous studies from our group as well as other researchers have suggested that the substitution at 4′′-OH does not alter the biological activities of EGCG. The introduction of alkyl groups of different chain lengths was found to increase the stability of EGCG.16,17,23,24 The alkyl chain will also contribute to the increase in the lipophilicity of the compound and thus, the cellular uptake. Hence, we introduced the TPP moiety at the 4′′ position of EGCG via an alkane chain linker anticipating its favorable cellular uptake and mitochondrial targeting. Of note, TPP is a well-established moiety to target compounds into the mitochondria.
3.2. Antioxidant property of MitoEGCGn compounds
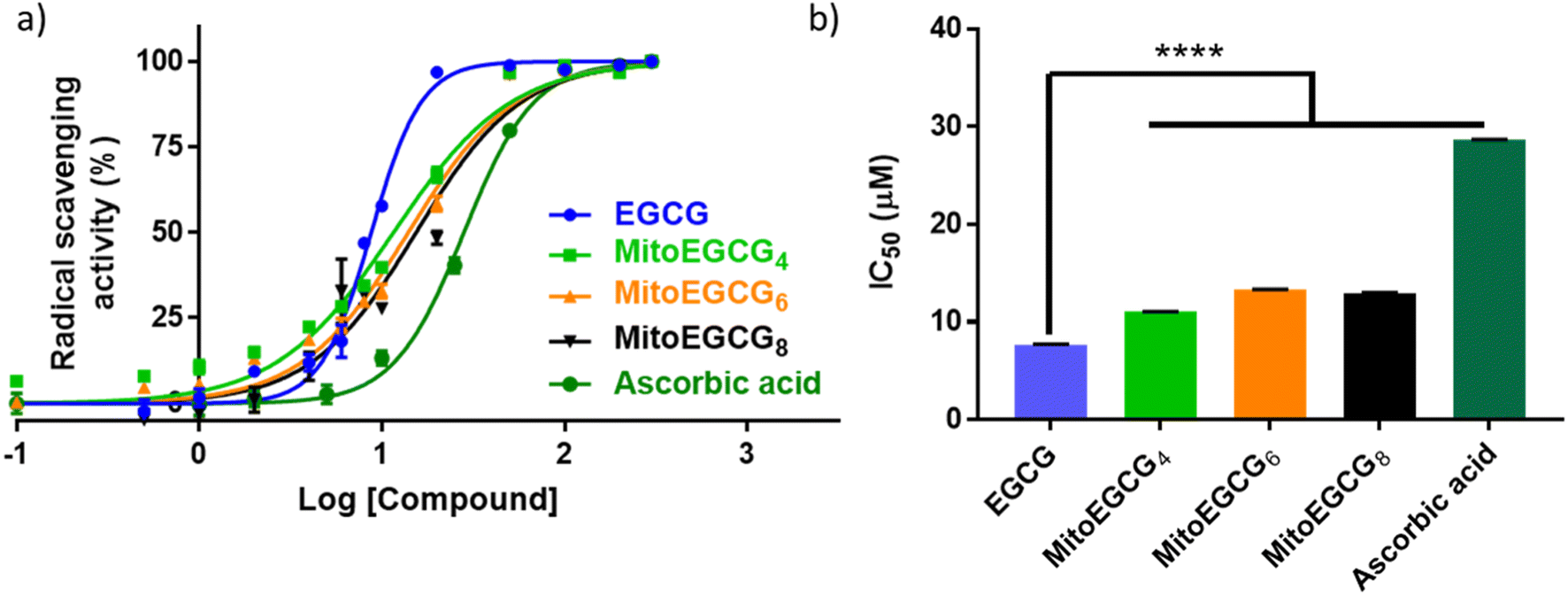
EGCG is widely known as an antioxidant agent as it can effectively scavenge the ROS. In order to evaluate whether the structural modification performed at 4′′ position of EGCG to introduce TPP has any influence on the native antioxidant activity of EGCG, we performed a radical scavenging activity analysis using in vitro DPPH assay. In DPPH assay, the antioxidant activity of EGCG and MitoEGCGn compounds were measured as the capacity to reduce the stable DPPH radical (violet) into a colorless reduced DPPH molecule. Here, ascorbic acid was taken as the positive control or reference compound which is known to be an effective antioxidant compound. The antioxidant properties of MitoEGCGn (IC50 of MitoEGCG4 = 11.04 ± 0.02 μM, MitoEGCG6 = 13.32 ± 0.02 μM and MitoEGCG8 = 12.98 ± 0.04 μM) were not compromised upon the addition of TPP linked alkyl chains to EGCG (IC50 of EGCG being 7.69 ± 0.02 μM) (Fig. 1a and b). In fact, they remain better antioxidant agents than well-known antioxidant, ascorbic acid (IC50 = 28.67 ± 0.01 μM). Mitochondria are considered as the primary source of ROS generation mainly due to the transport of electrons through the various complexes of electron transport chain for the sake of ATP production in the cells.25,26 Thus, targeting EGCG directly to mitochondria using TPP ion conjugation is expected to exert potent reduction in the damage induced by the ROS.
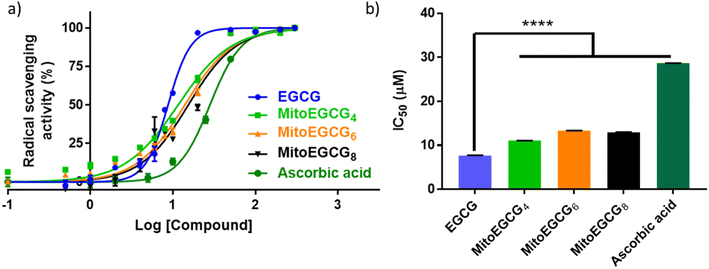 |
| | Fig. 1 (a) In vitro antioxidant profiles of EGCG, MitoEGCG4, MitoEGCG6, MitoEGCG8 and ascorbic acid as derived from DPPH assay, (b) comparison and evaluation of IC50 values of EGCG, MitoEGCG4, MitoEGCG6, MitoEGCG8 and ascorbic acid obtained from DPPH assay. Data are expressed as mean ± standard error of the mean (SEM) and **** represents p < 0.0001. | |
3.3. MitoEGCGn derivatives do not show toxicity to H9c2 cells
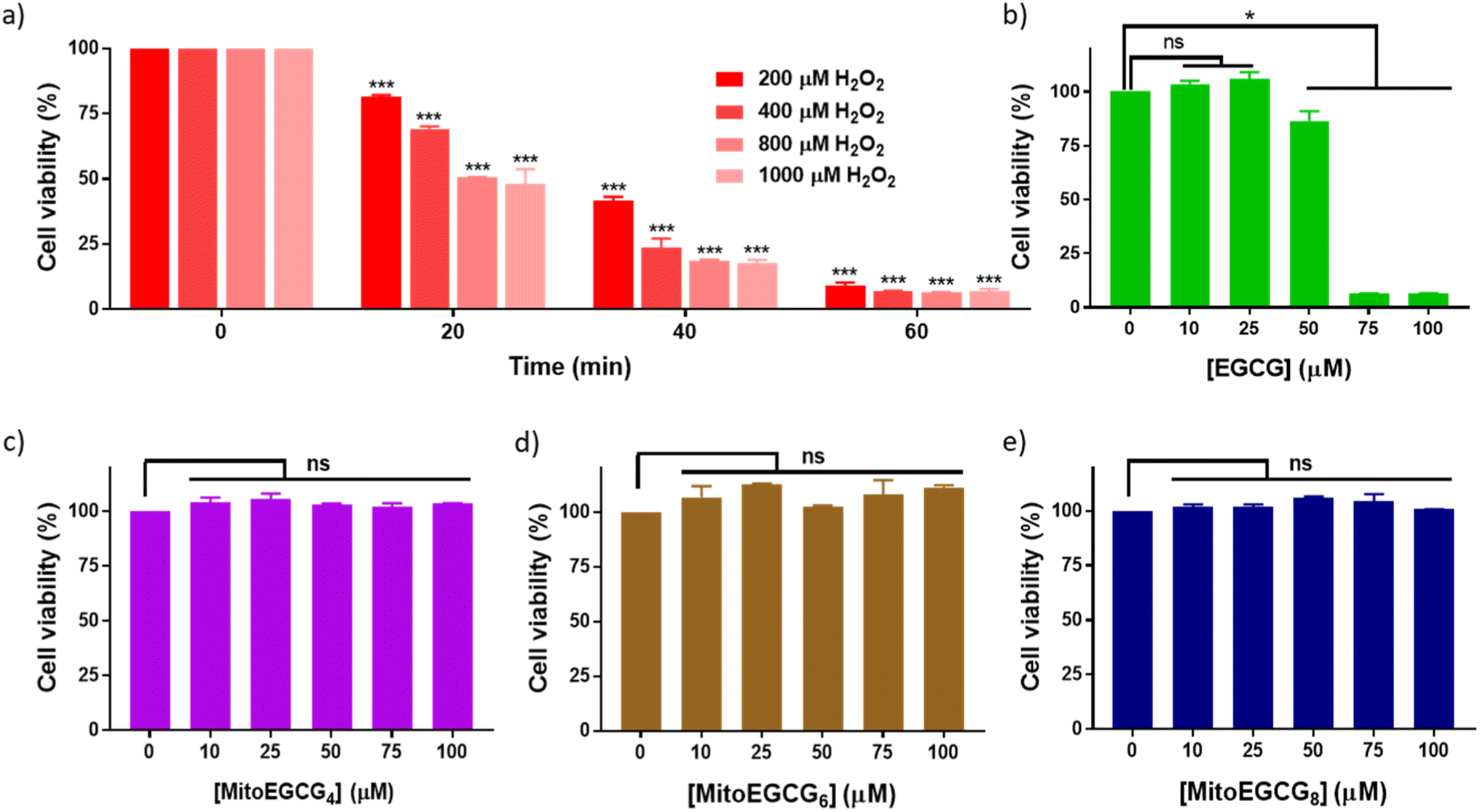
Progression of cardiovascular disease-related conditions are commonly associated with the accumulation of oxidative stress.27,28 The H9c2 cell lines are widely used for studying cardiac function and disease conditions because they have many characteristics similar to cardiac myocytes, the cells responsible for contraction in the heart. We used rat cardiomyocytes (H9c2 cell line) treated with H2O2 to create a cellular model for cardiac tissues in response to oxidative stress conditions.22,29 In order to select the effective concentration of H2O2 in inducing cellular damage to H9c2 cells, cell viability was assessed in the presence of different concentrations of H2O2 (0–1000 μM) with different duration of treatments (Fig. 2a). Treatment with 400 μM H2O2 for 20 min induced 30.81% loss in cell viability of H9c2 cells which further increased with increase in time of incubation. The effect of MitoEGCGn compounds on the cell viability of H9c2 cells was also analyzed using the MTT assay and was compared with that of the parent compound, EGCG. We observed that, where EGCG was non-toxic to cells only up to 50 μM (Fig. 2b), MitoEGCG4 (Fig. 2c), MitoEGCG6 (Fig. 2d), and MitoEGCG8 (Fig. 2e) were not toxic to cells even at higher concentrations of 100 μM. This observation suggested that MitoEGCGn compounds could predominantly behave like an antioxidant at higher concentrations where the parent molecule (EGCG) exerts pro-oxidant activity thus, killing the cells. Hence, it was imperative to analyze the action of MitoEGCGn compounds in the presence of H2O2 treatment, a potential stress condition leading to further cellular damage.
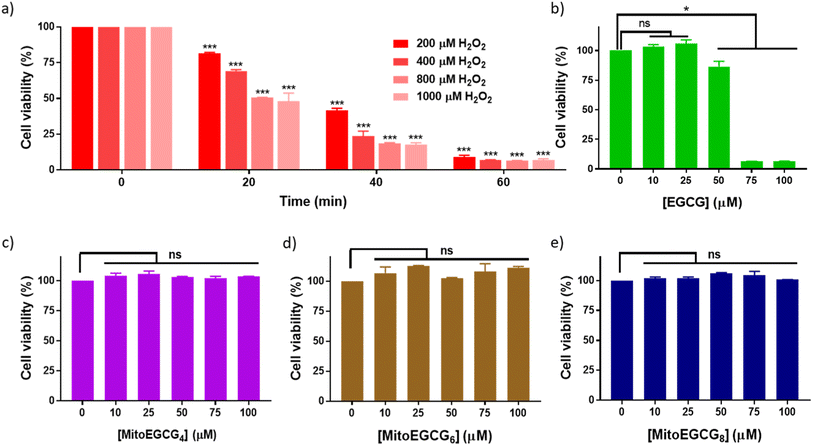 |
| | Fig. 2 Cell viability of H9c2 cells treated with (a) different concentrations of H2O2 (0–1000 μM) at different time points (0–60 min), (b) EGCG for 24 h, (c) MitoEGCG4 for 24 h, (d) MitoEGCG6 for 24 h and (e) MitoEGCG8 for 24 h. All the experiments have been conducted in triplicates. ***p < 0.001 compared with respective treatment for 0 min, ns denotes p > 0.05 (non-significance) and *p < 0.05 as compared to DMSO control. | |
3.4. MitoEGCGn alleviated the damage induced by H2O2 on H9c2 cells
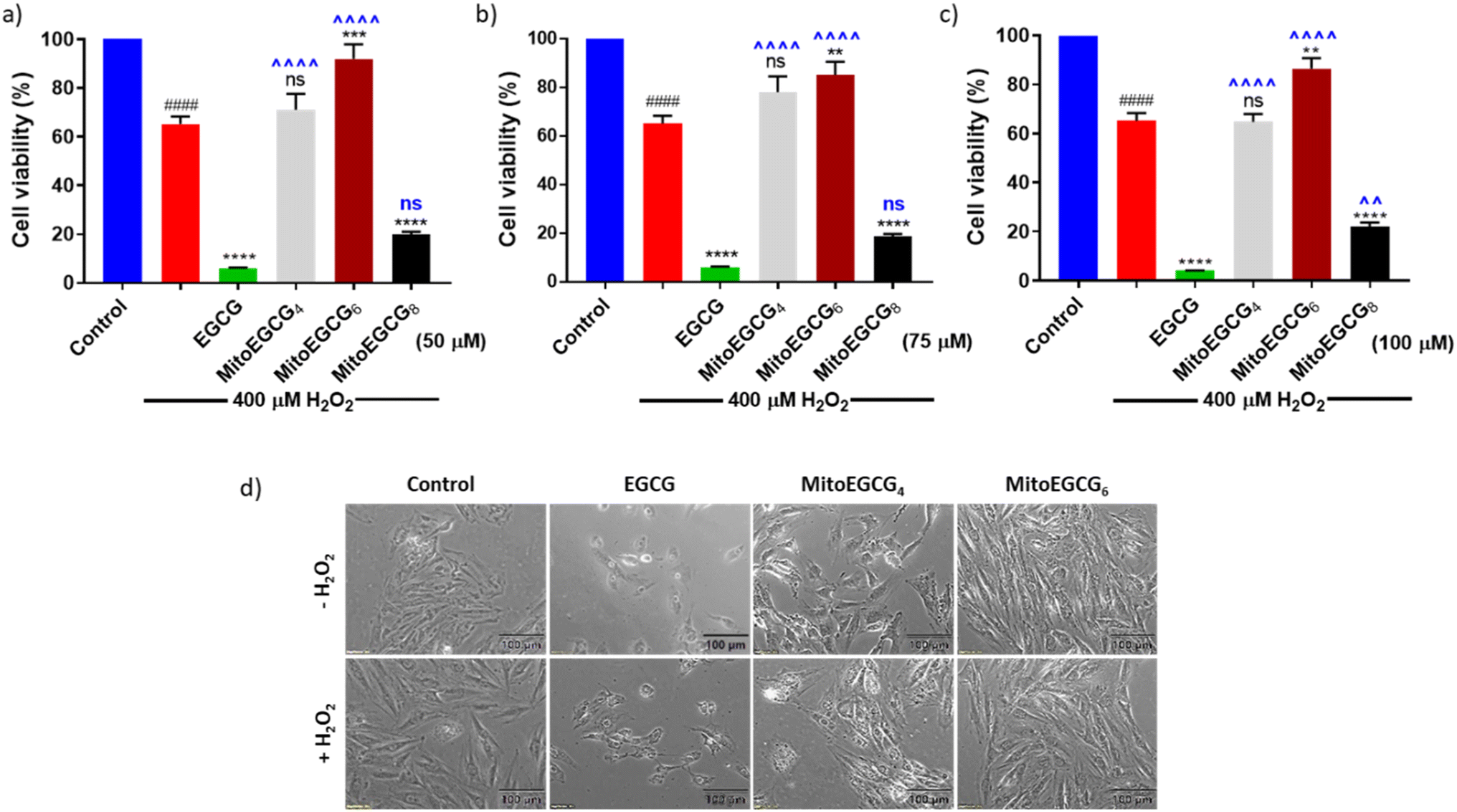
In order to evaluate the protective effect of MitoEGCGn compounds against H2O2-induced oxidative stress, we pretreated H9c2 cells with different concentrations of EGCG, MitoEGCG4, MitoEGCG6 and MitoEGCG8 separately for 24 h and later subjected to oxidative stress by treating them further with 400 μM H2O2 for 20 min. The cell viability analysis demonstrated that MitoEGCG4, and MitoEGCG6 both protected the H9c2 cell lines from the oxidative damage induced by H2O2 from the concentration of 50 μM whereas EGCG failed to exhibit its antioxidant activity (Fig. 3a and b). MitoEGCG8 was however unable to show similar results as exhibited by its lower alkyl chain counterparts (Fig. 3a–c) This observation could be attributed to the increased hydrophobicity of MitoEGCG8 which restricts its entry inside the mitochondrial matrix.30 The inverted bright field image of cells in Fig. 3d and ESI Fig. S10 and S11† clearly show that both MitoEGCG4 and MitoEGCG6 protected H9c2 cells from the damage induced by H2O2 in comparison to EGCG. Importantly, EGCG alone was found to be toxic. Our findings substantiate the potential of mitochondrial targeting of EGCG to protect cells from severe oxidative stress. The antioxidative property of EGCG is widely utilized therapy for healing and cell survival in case of several life-threatening pathological diseases including cardiovascular diseases.31,32 However, depending on the concentration used and the prevalent redox environment, EGCG is known to exert prooxidant activity in addition to its antioxidant action.33 Interestingly, MitoEGCG4 and MitoEGCG6 were able to exhibit their antioxidant activity even at higher doses where EGCG mostly start behaving as a prooxidant. These observations strongly suggest that MitoEGCG4 and MitoEGCG6 targeted EGCG into the mitochondria and were involved in quenching the ROS produced inside the mitochondria. Further experiments to understand the mechanistic underlying were performed with MitoEGCG4 and MitoEGCG6.
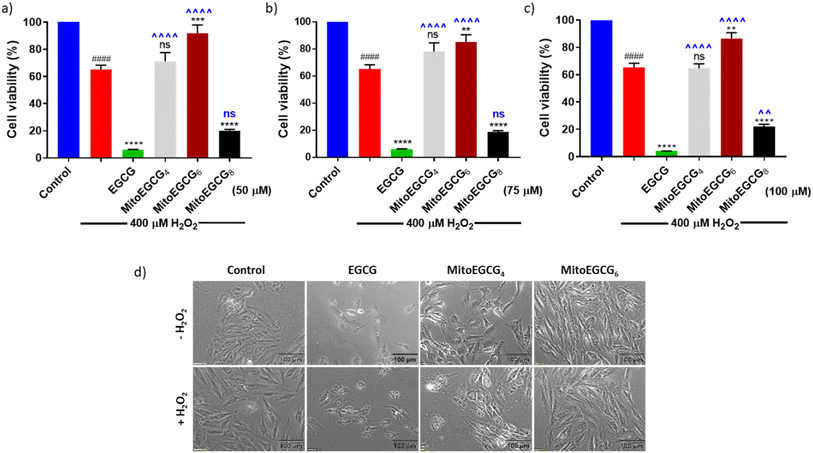 |
| | Fig. 3 Bar diagram showing cell viability of H9c2 cells treated with (a) 50 μM, (b) 75 μM, (c) 100 μM of EGCG, MitoEGCG4, MitoEGCG6 and MitoEGCG8 in the presence of 400 μM H2O2 for 20 min. All experiments were conducted in triplicates and statistical significance was denoted as follows: ####p < 0.0001 as compared to control. ns represents non-significance and **p < 0.01, ***p < 0.001 and ****p < 0.0001 as compared to H2O2 treated samples. ns represents non-significance ^^p < 0.01, and ^^^^p < 0.0001 as compared to EGCG treated samples (one-way ANOVA with Dunnett's multiple-comparison post-test) and (d) Inverted microscopic images of H9c2 cells pretreated with 50 μM of EGCG, MitoEGCG4, and MitoEGCG6 and then kept in media with and without H2O2 (200 μM). Magnification 20×. Cell viability was assessed using an MTT assay. | |
3.5. MitoEGCGn scavenged the ROS generated by the action of H2O2 treatment
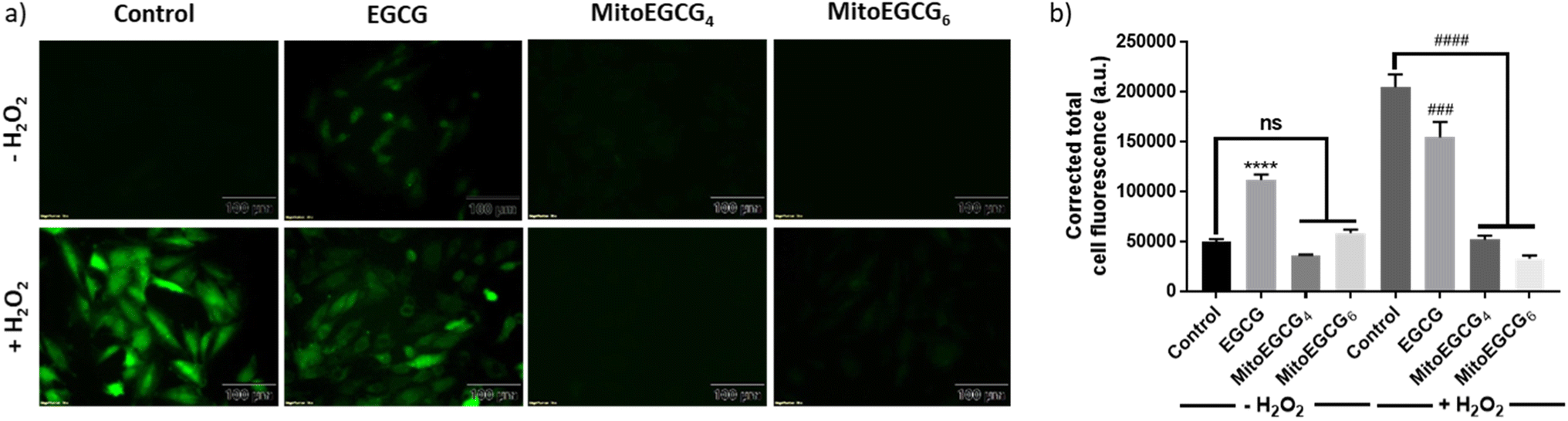
H2O2 treatment to cells is a widely used procedure to induce ROS generation which ultimately promotes cellular injury. In order to determine the mechanism of action behind the activity of MitoEGCGn compounds in effectively reducing H2O2-induced cell death, intracellular ROS generation was determined using 2′,7′-dichlorodihydro fluorescein diacetate (DCFH-DA) staining analysis. DCFH-DA is a cell-permeable non-fluorescent dye which in the presence of esterases inside the cell gets hydrolyzed upon cellular absorption to produce 2′,7′-dichlorodihydrofluorescein (DCFH), which is then oxidized by ROS to produce a green fluorescent 2′,7′-dichlorofluorescein (DCF) molecule. As the intracellular ROS generation is directly proportional to the intensity of the fluorescence, this reagent can be used to determine the amount of ROS produced inside the cell in the presence of different antioxidants. From the DCFH-DA staining results it is evident that MitoEGCG4 and MitoEGCG6 effectively protected the H9c2 cells by scavenging the ROS resulted from H2O2 treatment. It was also noticed that EGCG was unable to impart any significant antioxidant effect on H2O2 treatment (Fig. 4a and b and ESI Fig. S13†). This further provides the evidence that the TPP-linked mitochondrial-targeting EGCG derivatives protect cells from the ROS-induced cell damage by scavenging them.
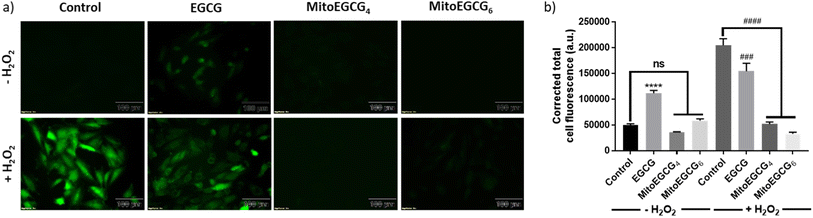 |
| | Fig. 4 (a) Fluorescence microscopic images of DCFH-DA stained H9c2 cells pretreated with DMSO (control), 50 μM of EGCG, MitoEGCG4, and MitoEGCG6 and then kept in media with and without H2O2 (50 μM). Images were acquired at an ex/em wavelength of 485/530 nm and (b) Corresponding relative fluorescence intensity calculated using ImageJ software and statistical analysis was done using GraphPad Prism software. Data are represented as a mean ± SEM, n = 10. ns represents non significance and ****p < 0.0001 as compared to control (−H2O2). ###p < 0.001, and ####p < 0.0001 as compared to control (+H2O2) (one-way ANOVA with Dunnett's multiple-comparison post-test). | |
3.6. MitoEGCGn protected H9c2 cells from H2O2-induced damage by preventing the loss of mitochondrial outer membrane potential (Ψm)
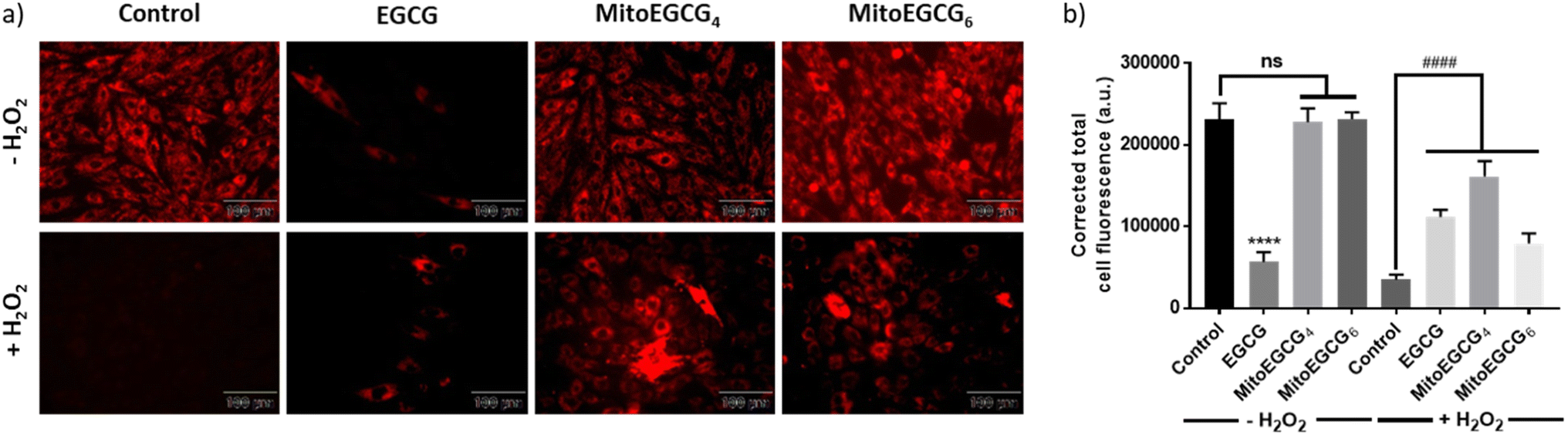
To examine the effect of MitoEGCG4 and MitoEGCG6 on the H2O2-induced mitochondrial potential disruption, we measured the mitochondrial potential of MitoEGCG4 and MitoEGCG6 treated cells in the presence and absence of H2O2 by staining the cells with tetramethylrhodamine ethyl ester (TMRE). TMRE is a cell-permeant cationic dye that can accumulate inside the mitochondria due to the higher negative potential of the mitochondria.34 Mitochondrial function is highly regulated by the mitochondrial outer membrane potential (Ψm) and it plays an important role in mitochondrial biogenesis and metabolism.35–37 Pretreatment of MitoEGCG4 and MitoEGCG6 attenuated the loss of Ψm induced by H2O2 treatment in H9c2 cells in comparison to EGCG. It was also observed that EGCG was reducing the Ψm of cells at concentrations above 50 μM and therefore acting as a pro-oxidant to cardiomyocyte cells (Fig. 5a and b and ESI Fig. S14†). The disruption of Ψm in cardiomyocyte cells is ascribed to the initiation of oxidative stress in the physiology of cardiomyopathies.20,38–40 EGCG has been extensively demonstrated for regulating mitochondrial health, biogenesis, ATP production, mitochondrial metabolism, and bioenergetics. Furthermore, EGCG has also been known to regulate mitochondrial redox levels by modulating antioxidant enzymes, and Ca2+ influx system in the cell etc.41–43 Hence, targeting EGCG directly inside the mitochondria which is the central hub for ROS species generation results in loss in Ψm may prove effective in protecting the cells from oxidative stress. As expected, our results targeted EGCG into the mitochondrial matrix and protected the mitochondrial functions and thereby, helped the fight against the injury induced through H2O2 treatment.
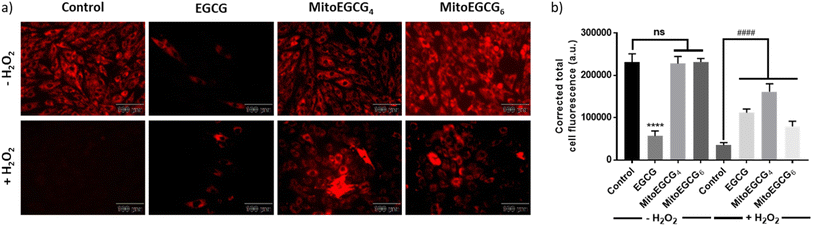 |
| | Fig. 5 (a) Fluorescence microscopic images of TMRE stained H9c2 cells pretreated with DMSO (control), 50 μM of EGCG, MitoEGCG4, and MitoEGCG6 and then kept in media with and without H2O2 (400 μM). Images were acquired at an ex/em range of 552/574 nm and (b) Corresponding relative fluorescence intensity was calculated using ImageJ software and statistical analysis was done using GraphPad Prism software. Data are represented as a mean ± SEM, n = 10. ns represents non significance and ****p < 0.0001 as compared to control (−H2O2). Wereas ####p < 0.0001 as compared to control (+H2O2) (one-way ANOVA with Dunnett's multiple-comparison post-test). | |
3.7. MitoEGCGn ameliorated H2O2-induced apoptosis in H9c2 cell line
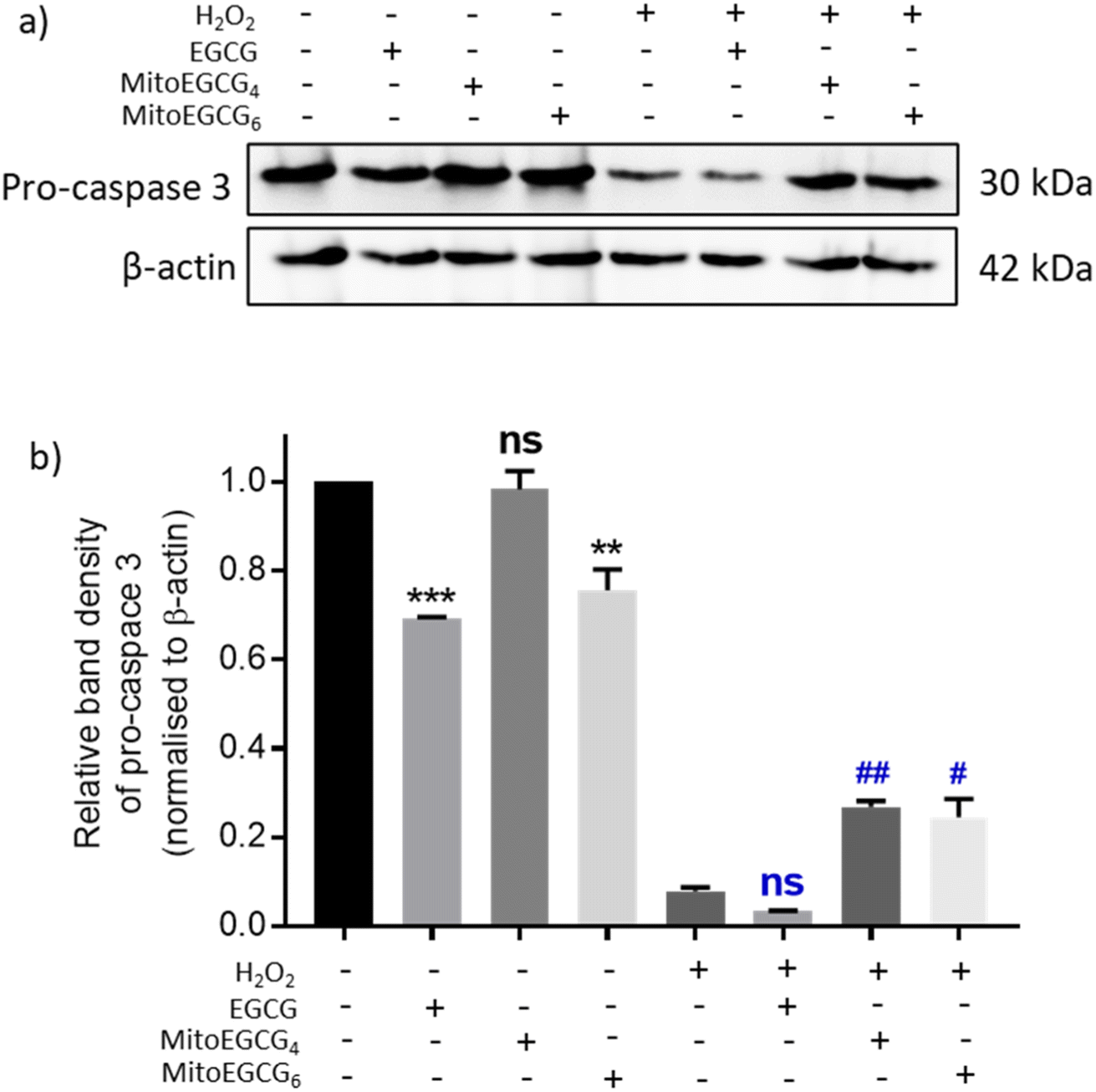
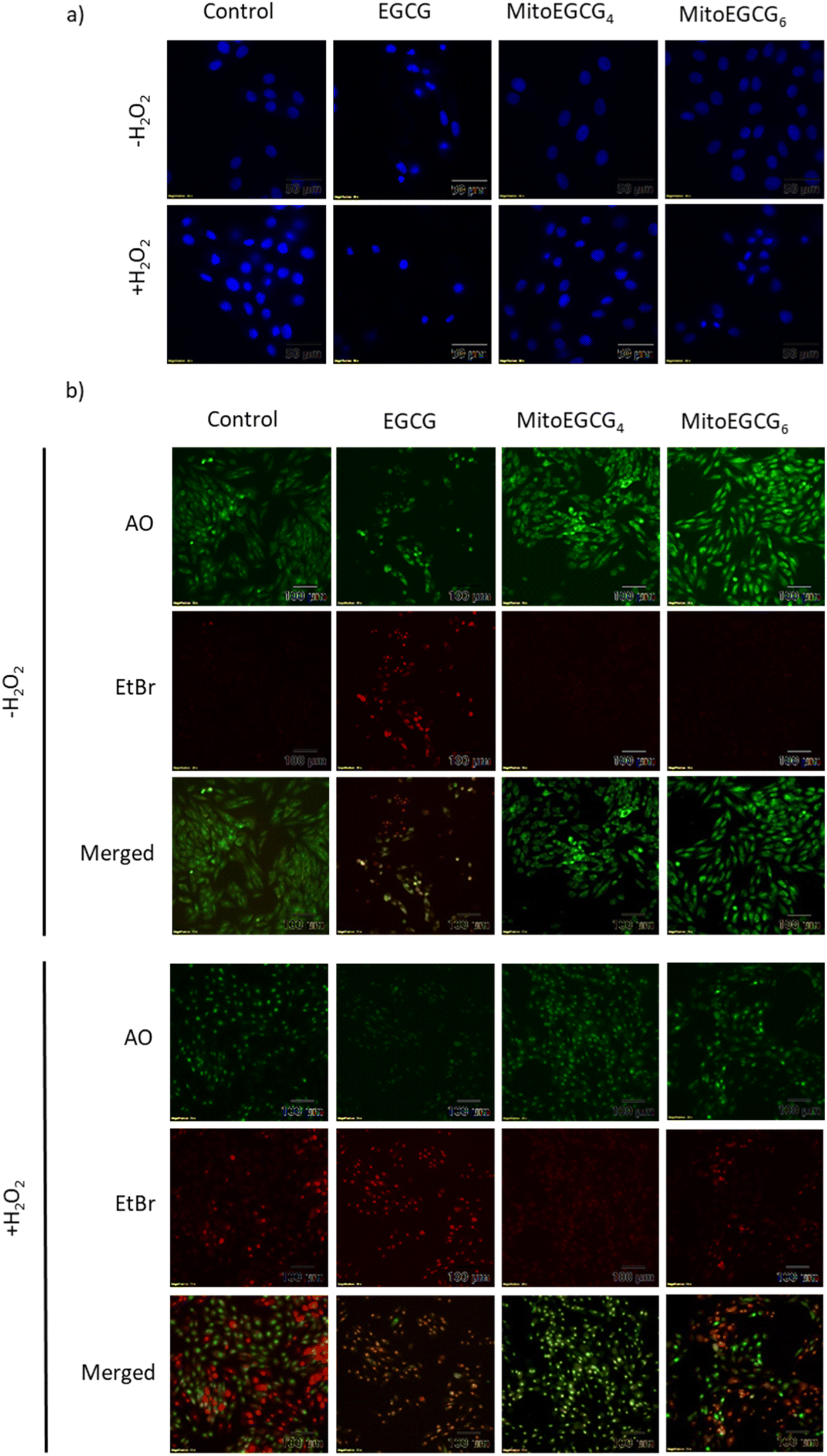
Disruption in mitochondrial outer membrane potential is associated with the activation of intrinsic apoptosis pathway. H2O2 treatment in many cases has been well documented in inducing mitochondria-dependent apoptosis pathway which involves a loss in mitochondrial membrane potential followed by release of cytochrome c, which ultimately results in the activation of caspase.44 In order to assess the capability of MitoEGCG4 and MitoEGCG6 in protecting the cells from H2O2-induced apoptosis activation, we performed the western blot analysis to assess the pro-caspase 3 level. The data clearly show that MitoEGCG4 and MitoEGCG6 effectively protected the cells from H2O2-induced caspase 3 activation in comparison to EGCG (Fig. 6a and b and ESI Fig. S15†). It was observed that EGCG in the tested concentration exhibited pro-oxidant activity in H9c2 cells and as a result, EGCG-treated cells completely succumbed to cell death in the presence of H2O2 treatment (Fig. 3d and ESI Fig. S13 and S14†). Activation of caspase 3 in cells further leads to downstream cleavage of oligonucleosomal DNA which can easily be detected using Hoechst staining of cells. As shown in Fig. 7a, MitoEGCG4 and MitoEGCG6-pretreated cells retained the status of healthy nucleus in the presence of H2O2 treatment. This result indicated the antioxidant ability of MitoEGCG4 and MitoEGCG6 in protecting the cells from H2O2-induced cell injury. We also examined the integrity of cell membrane of H9c2 cells with similar treatment. It was observed that H2O2 was unable to induce loss in membrane integrity in the cells pretreated with MitoEGCG4 and MitoEGCG6 to the same extent (Fig. 7b). The aforementioned results demonstrated that sequestering EGCG into the mitochondria by pretreating cells with MitoEGCGn protected them from H2O2-induced cell death.
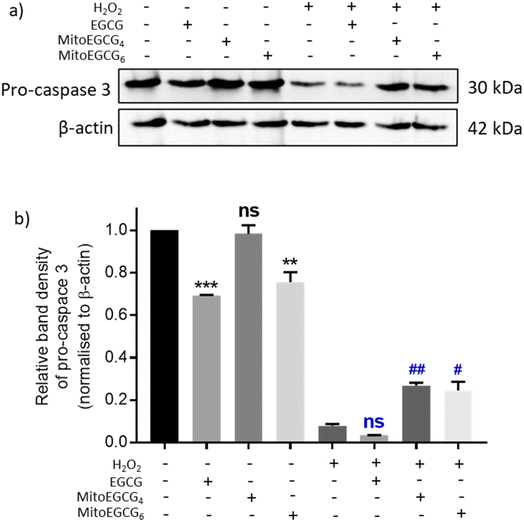 |
| | Fig. 6 MitoEGCG4 protected H9c2 cell lines from H2O2-induced activation of pro-caspase 3. (a) Western blot analysis depicting pro-caspase 3 protein expression in cells pretreated with EGCG, MitoEGCG4 and MitoEGCG6 and further exposed to 400 μM of H2O2 for 4 h and (b) density gradient graph prepared by measuring the band density of pro-caspase 3 in each lane and normalizing against the loading control. All the data were represented as mean ± standard error of the mean (SEM) from at least 3 independent experiments. ns represents non significance, **p < 0.01 and ***p < 0.001 as compared to control (−H2O2). Whereas ns represents non significance (p > 0.05) and ##p < 0.01 as compared to control (+H2O2) (one-way ANOVA with Dunnett's multiple-comparison post-test). | |
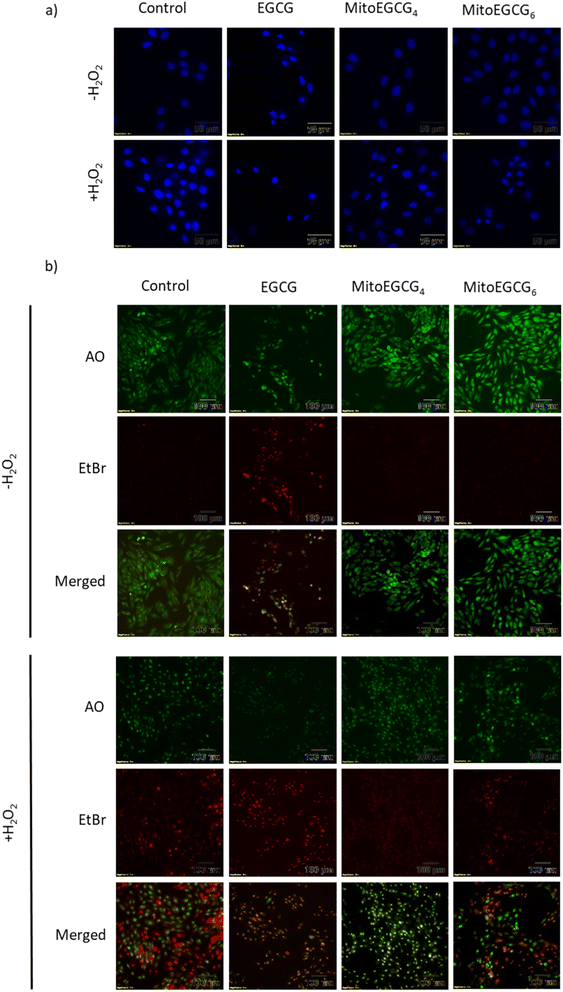 |
| | Fig. 7 MitoEGCG4 protected H9c2 cell lines from H2O2-induced activation of apoptosis (a) Hoechst staining of cells pretreated with EGCG, MitoEGCG4 and MitoEGCG6 in the absence and presence of H2O2 (400 μM) and (b) AO/EtBr staining of cells pretreated with EGCG, MitoEGCG4 and MitoEGCG6 in the presence and absence of H2O2 (400 μM). | |
4. Conclusion
A series of mitochondrial-targeting EGCG probes, namely MitoEGCGn, were synthesized via conjugating a triphenylphosphonium ion with EGCG at its 4′′ position using alkyl chain linkers of different hydrophobicity. MitoEGCG4 and MitoEGCG6 were found to be effective in protecting the cells from H2O2-induced cell injury as revealed by the MTT analysis, unlike EGCG that was unable to exhibit any such cytoprotection. On further probing the mechanism of the protective effect rendered by MitoEGCG4 and MitoEGCG6 against H2O2 treatment in rat cardiomyocyte cells, we observed that these compounds imparted an antioxidant effect to the cells by reducing the production of ROS produced by H2O2 treatment in cells. We also demonstrated that MitoEGCG4 and MitoEGCG6 protected the mitochondria of the cells from H2O2-induced loss in the outer mitochondrial membrane potential, thereby also blocking the activation of intrinsic apoptotic pathway. Cells pretreated with MitoEGCG4 and MitoEGCG6 did not show caspase activation as well as other apoptotic features like nuclear fragmentation and loss in membrane integrity. Thereby, our findings unequivocally demonstrate that MitoEGCG4 and MitoEGCG6 both functioned as antioxidants within the mitochondria, preventing H2O2-induced cell damage. However, further investigation is warranted to fully comprehend the mechanisms behind the protective benefits of MitoEGCGn compounds against H2O2-induced cardiomyocyte mitochondrial damage.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We sincerely acknowledge the Indian Institute of Technology (IIT) Palakkad, India for the facilities and support provided to carry out this work. We also acknowledge the funding from the Council of Scientific & Industrial Research (CSIR), India (02(0434)/21/EMR-II and 09/1282(0003)/2019-EMR-I) and the National Post-doctoral Fellowship (NPDF), Science and Engineering Research Board (SERB), India (PDF/2020/001950). We greatly acknowledge the Central Instrumentation Facility (CIF) of IIT Palakkad for the technical support and the Central Instrumentation Centre (CIC), Bharathiar University supported by DST (PURSE Phase II program), New Delhi for the NMR analysis.
References
- G. Siasos, V. Tsigkou, M. Kosmopoulos, D. Theodosiadis, S. Simantiris, N. M. Tagkou, A. Tsimpiktsioglou, P. K. Stampouloglou, E. Oikonomou, K. Mourouzis, A. Philippou, M. Vavuranakis, C. Stefanadis, D. Tousoulis and A. G. Papavassiliou, Mitochondria and cardiovascular diseases from pathophyiology to treatment, Ann. Transl. Med., 2018, 6, 256 CrossRef PubMed.
- K. Chaudhari, B. Hamad and B. A. Syed, Antithrombotic drugs market, Nat. Rev. Drug Discovery, 2014, 13, 571–572 CrossRef CAS PubMed.
- C. L. Li, M. H. Dong, Y. J. Ren and L. H. Li, Design, synthesis, biological evaluation and molecular docking of novel dabigatran derivatives as potential thrombin inhibitors, RSC Adv., 2015, 5, 23737–23748 RSC.
- X. Zhu and L. Zuo, Characterization of oxygen radical formation mechanism at early cardiac ischemia, Cell Death Dis., 2013, 4, e787 CrossRef CAS PubMed.
- C. Y. Mao, H. Bin Lu, N. Kong, J. Y. Li, M. Liu, C. Y. Yang and P. Yang, Levocarnitine protects H9c2 rat cardiomyocytes from H2O2-induced mitochondrial dysfunction and apoptosis, Int. J. Med. Sci., 2014, 11, 1107–1115 CrossRef CAS PubMed.
- M. Chiong, Z. V. Wang, Z. Pedrozo, D. J. Cao, R. Troncoso, M. Ibacache, A. Criollo, A. Nemchenko, J. A. Hill and S. Lavandero, Cardiomyocyte death: Mechanisms and translational implications, Cell Death Dis., 2011, 2, e244 CrossRef CAS PubMed.
- K. Konstantinidis, R. S. Whelan and R. N. Kitsis, Mechanisms of cell death in heart disease, Arterioscler., Thromb., Vasc. Biol., 2012, 32, 1552–1562 CrossRef CAS PubMed.
- G. S. Gorman, P. F. Chinnery, S. DiMauro, M. Hirano, Y. Koga, R. McFarland, A. Suomalainen, D. R. Thorburn, M. Zeviani and D. M. Turnbull, Mitochondrial diseases, Nat. Rev. Dis. Prim., 2016, 2, 16080 CrossRef PubMed.
- P. Martín-Maestro, R. Gargini, E. García, G. Perry, J. Avila and V. García-Escudero, Slower dynamics and aged mitochondria in Sporadic Alzheimer's disease, Oxid. Med. Cell. Longevity, 2017, 2017, 9302761 Search PubMed.
- L. Basiricò, P. Morera, D. Dipasquale, R. Bernini, L. Santi, A. Romani, N. Lacetera and U. Bernabucci, (−)-Epigallocatechin-3-gallate and hydroxytyrosol improved antioxidative and anti-inflammatory responses in bovine mammary epithelial cells, Animal, 2019, 13, 2847–2856 CrossRef PubMed.
- S. Bettuzzi, M. Brausi, F. Rizzi, G. Castagnetti, G. Peracchia and A. Corti, Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: A preliminary report from a one-year proof-of-principle study, Cancer Res., 2006, 66, 1234–1240 CrossRef CAS PubMed.
- Y. Du, H. Ding, K. Vanarsa, S. Soomro, S. Baig, J. Hicks and C. Mohan, Low dose epigallocatechin gallate alleviates experimental colitis by subduing inflammatory cells and cytokines, and improving intestinal permeability, Nutrients, 2019, 11, 1743 CrossRef CAS PubMed.
- Q. Y. Eng, P. V. Thanikachalam and S. Ramamurthy, Molecular understanding of Epigallocatechin gallate (EGCG) in cardiovascular and metabolic diseases, J. Ethnopharmacol., 2018, 210, 296–310 CrossRef CAS PubMed.
- A. Binoy, D. Nedungadi, N. Katiyar, C. Bose, S. A. Shankarappa, B. G. Nair and N. Mishra, Plumbagin induces paraptosis in cancer cells by disrupting the sulfhydryl homeostasis and proteasomal function, Chem.-Biol. Interact., 2019, 310, 108733 CrossRef CAS PubMed.
- R. Sahadevan, S. Singh, A. Binoy and S. Sadhukhan, Chemico-biological aspects of (−)-epigallocatechin-3-gallate (EGCG) to improve its stability, bioavailability and membrane permeability: Current status and future prospects, Crit. Rev. Food Sci. Nutr., 2022, 1–30 CrossRef PubMed.
- R. Sahadevan, A. Binoy, S. K. Vechalapu, P. Nanjan and S. Sadhukhan, In situ global proteomics profiling of EGCG targets using a cell-permeable and Click-able bioorthogonal probe, Int. J. Biol. Macromol., 2023, 237, 123991 CrossRef CAS PubMed.
- S. Singh, R. Sahadevan, R. Roy, M. Biswas, P. Ghosh, P. Kar, A. Sonawane and S. Sadhukhan, Structure-based design and synthesis of a novel long-chain 4′′-alkyl ether derivative of EGCG as potent EGFR inhibitor: in vitro and in silico studies, RSC Adv., 2022, 12, 17821–17836 RSC.
- C. Minnelli, L. Cianfruglia, E. Laudadio, M. Giovanna, R. Galeazzi and T. Armeni, Effect of epigallocatechin-3-gallate on egfr signaling and migration in non-small cell lung cancer, Int. J. Mol. Sci., 2021, 22, 11833 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- J. S. Bhatti, G. K. Bhatti, P. H. Reddy, U. States, U. States, U. States, H. S. Departments, U. States, S. W. Campus and U. States, Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies, Biochim. Biophys. Acta, 2018, 1863, 1066–1077 CrossRef PubMed.
- L. Zhang, Y. Liu, J. Y. Li, L. Z. Li, Y. L. Zhang, H. Y. Gong and Y. Cui, Protective effect of rosamultin against H2O2 -induced oxidative stress and apoptosis in H9c2 cardiomyocytes, Oxid. Med. Cell. Longevity, 2018, 8415610 Search PubMed.
- E. Son, D. Lee, C. W. Woo and Y. H. Kim, The optimal model of reperfusion injury in vitro using H9c2 transformed cardiac myoblasts, Korean J. Physiol. Pharmacol., 2020, 24, 173–183 CrossRef CAS PubMed.
- M. Maeda-Yamamoto, N. Inagaki, J. Kitaura, T. Chikumoto, H. Kawahara, Y. Kawakami, M. Sano, T. Miyase, H. Tachibana, H. Nagai and T. Kawakami, O-methylated catechins from tea leaves inhibit multiple protein kinases in mast cells, J. Immunol., 2004, 172, 4486–4492 CrossRef CAS PubMed.
- M. Suzuki, K. Yoshino, M. Maeda-Yamamoto, T. Miyase and M. Sano, Inhibitory effects of tea catechins and O-methylated derivatives of (−)-epigallocatechin-3-O-gallate on mouse type IV allergy, J. Agric. Food Chem., 2000, 48, 5649–5653 CrossRef CAS PubMed.
- M. P. Murphy, How mitochondria produce reactive oxygen species, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed.
- Q. Chen, E. J. Vazquez, S. Moghaddas, C. L. Hoppel and E. J. Lesnefsky, Production of reactive oxygen species by mitochondria: Central role of complex III, J. Biol. Chem., 2003, 278, 36027–36031 CrossRef CAS PubMed.
- Y. Kayama, U. Raaz, A. Jagger, M. Adam, I. N. Schellinger, M. Sakamoto, H. Suzuki, K. Toyama, J. M. Spin and P. S. Tsao, Diabetic cardiovascular disease induced by oxidative stress, Int. J. Mol. Sci., 2015, 16, 25234–25263 CrossRef CAS PubMed.
- G. Pizzino, N. Irrera, M. Cucinotta, G. Pallio, F. Mannino, V. Arcoraci, F. Squadrito, D. Altavilla and A. Bitto, Oxidative stress: Harms and benefits for human health, Oxid. Med. Cell. Longevity, 2017, 8416763 Search PubMed.
- H. C. Chou, Y. W. Chen, T. R. Lee, F. S. Wu, H. T. Chan, P. C. Lyu, J. F. Timms and H. L. Chan, Proteomics study of oxidative stress and Src kinase inhibition in H9C2 cardiomyocytes: a cell model of heart ischemia-reperfusion injury and treatment, Free Radicals Biol. Med., 2010, 49, 96–108 CrossRef CAS PubMed.
- M. F. Ross, T. A. Prime, I. Abakumova, A. M. James, C. M. Porteous, R. A. J. Smith and M. P. Murphy, Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells, Biochem. J., 2008, 411, 633–645 CrossRef CAS PubMed.
- W. Zhang, L. Chen, Y. Xiong, A. C. Panayi, A. Abududilibaier, Y. Hu, C. Yu, W. Zhou, Y. Sun, M. Liu, H. Xue, L. Hu, C. Yan, X. Xie, Z. Lin, F. Cao, B. Mi and G. Liu, Antioxidant therapy and antioxidant-related bionanomaterials in diabetic wound healing, Front. Bioeng. Biotechnol., 2021, 9, 707479 CrossRef PubMed.
- S. Shaban, M. W. A. El-Husseny, A. I. Abushouk, A. M. A. Salem, M. Mamdouh and M. M. Abdel-Daim, Effects of antioxidant supplements on the survival and differentiation of stem cells, Oxid. Med. Cell. Longevity, 2017, 2017, 5032102 Search PubMed.
- H. S. Kim, M. J. Quon and J. Kim, New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate, Redox Biol., 2014, 2, 187–195 CrossRef CAS PubMed.
- L. C. Crowley, M. E. Christensen and N. J. Waterhouse, Measuring mitochondrial transmembrane potential by TMRE staining, Cold Spring Harb. Protoc., 2016, 2016, 1092–1096 Search PubMed.
- M. R. De Oliveira, S. F. Nabavi, M. Daglia, L. Rastrelli and S. M. Nabavi, Epigallocatechin gallate and mitochondria - A story of life and death, Pharmacol. Res., 2016, 104, 70–85 CrossRef PubMed.
- J. Ren, P. Lakshmi, A. Whaley-Connell and J. R. Sowers, Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease, J. Mol. Med., 2010, 88, 993–1001 CrossRef CAS PubMed.
- A. Braczko, B. Kutryb-Zajac, A. Jedrzejewska, O. Krol, P. Mierzejewska, M. Zabielska-Kaczorowska, E. M. Slominska and R. T. Smolenski, Cardiac mitochondria dysfunction in dyslipidemic mice, Int. J. Mol. Sci., 2022, 23, 11488 CrossRef CAS PubMed.
- C. Y. Guo, L. Sun, X. P. Chen and D. S. Zhang, Oxidative stress, mitochondrial damage and neurodegenerative diseases, Neural Regener. Res., 2013, 8, 2003–2014 CAS.
- E. Marzetti, A. Csiszar, D. Dutta, G. Balagopal, R. Calvani and C. Leeuwenburgh, Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics, Am. J. Physiol. Heart Circ. Physiol., 2013, 305, H459–H476 CrossRef CAS PubMed.
- T. Autumn, E. K. Quarles, N. Basisty, L. Gitari and P. S. Rabinovitch, Mitochondrial dysfunction in cardiac ageing, Biochim. Biophys. Acta, 2015, 1847, 1424–1433 Search PubMed.
- W. Zhu, J. Xu, Y. Ge, H. Cao, X. Ge, J. Luo, J. Xue, H. Yang, S. Zhang and J. Cao, Epigallocatechin-3-gallate (EGCG) protects skin cells from ionizing Radiation via heme oxygenase-1 (HO-1) overexpression, J. Radiat. Res., 2014, 55, 1056–1065 CrossRef CAS PubMed.
- I. N. Zelko, T. J. Mariani and R. J. Folz, Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression, Free Radicals Biol. Med., 2002, 33, 337–349 CrossRef CAS PubMed.
- J. Oyama, A. Shiraki, T. Nishikido, T. Maeda, H. Komoda, T. Shimizu, N. Makino and K. Node, EGCG, a green tea catechin, attenuates the progression of heart failure induced by the heart/muscle-specific deletion of MnSOD in mice, J. Cardiol., 2017, 69, 417–427 CrossRef PubMed.
- J. Xiang, C. Wan, R. Guo and D. Guo, Is hydrogen peroxide a suitable apoptosis inducer for all cell types?, Biomed Res. Int., 2016, 2016, 7343965 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab