
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing acid orange II degradation in ozonation processes with CaFe2O4 nanoparticles as a heterogeneous catalyst
Huu Tap Van ab,
Van Hung Hoang*cd,
Thi Cuc Luud,
Thuy Linh Vib,
Luong Thi Quynh Ngae,
Gio Serafin Ivan Jimenez Marcaidaf and
Truong-Tho Phamgh
ab,
Van Hung Hoang*cd,
Thi Cuc Luud,
Thuy Linh Vib,
Luong Thi Quynh Ngae,
Gio Serafin Ivan Jimenez Marcaidaf and
Truong-Tho Phamgh
aCenter for Advanced Technology Development, Thai Nguyen University, Tan Thinh Ward, Thai Nguyen City, Vietnam
bFaculty of Natural Resources and Environment, TNU – University of Sciences, Tan Thinh Ward, Thai Nguyen City, Vietnam
cThai Nguyen University, Tan Thinh Ward, Thai Nguyen City, Vietnam. E-mail: hoangvanhung@tnu.edu.vn
dFaculty of Agriculture and Forestry, TNU – Lao Cai Campus, Lao Cai City, Vietnam
eDepartment of Infectious Diseases, Faculty of Sub-Specialties, Thai Nguyen University of Medicine and Pharmacy (TNUMP), No. 284, Luong Ngoc Quyen Street, Thai Nguyen City, Vietnam
fDepartment of Environmental Science and Management, Advanced Education Program, TNU – University Agriculture and Forestry (TUAF), Quyet Thang Ward, Thai Nguyen City, Vietnam
gLaboratory of Magnetism and Magnetic Materials, Science and Technology Advanced Institute, Van Lang University, Ho Chi Minh City, Vietnam
hFaculty of Applied Technology, School of Technology, Van Lang University, Ho Chi Minh City, Vietnam
First published on 2nd October 2023
Abstract
This study used CaFe2O4 nanoparticles as a catalyst for ozonation processes to degrade Acid Orange II (AOII) in aqueous solution. The study compared heterogeneous catalytic ozonation (CaFe2O4/O3) with ozone treatment alone (O3) at different pH values (3–11), catalyst dosages (0.25–2.0 g L−1), and initial AOII concentrations (100–500 mg L−1). The O3 alone and CaFe2O4/O3 systems nearly completely removed AOII's color. In the first 5 min, O3 alone had a color removal efficiency of 75.66%, rising to 92% in 10 min, whereas the CaFe2O4/O3 system had 81.49%, 94%, and 98% after 5, 10, and 20 min, respectively. The O3 and CaFe2O4/O3 systems degrade TOC most efficiently at pH 9 and better with 1.0 g per L CaFe2O4. TOC removal effectiveness reduced from 85% to 62% when the initial AOII concentration increased from 100 to 500 mg L−1. The study of degradation kinetics reveals a pseudo-first-order reaction mechanism significantly as the solution pH increased from 3 to 9. Compared to the O3 alone system, the CaFe2O4/O3 system has higher k values. At pH 9, the k value for the CaFe2O4/O3 system is 1.83 times higher than that of the O3 alone system. Moreover, increasing AOII concentration from 100 mg L−1 to 500 mg L−1 subsequently caused a decline in the k values. The experimental data match pseudo-first-order kinetics, as shown by R2 values of 0.95–0.99. AOII degradation involves absorption, ozone activation, and reactive species production based on the existence of CaO and FeO in the CaFe2O4 nanocatalyst. This catalyst can be effectively recycled multiple times.
Introduction
Along with numerous industrial pollutants, textile dyes are highly toxic and potentially carcinogenic.1 Dye pollution is a significant environmental problem that can negatively impact human health and ecosystems. Discharging highly toxic and possibly carcinogenic textile dyes into water bodies causes water pollution.1–3 The textile industry significantly contributes to water pollution because of its high water consumption and discharge of coloring materials.2 Dye contamination can inhibit microalgae growth and disrupt trophic interactions.3 Synthetic dyes in the textile industry, dyeing and printing industry, tannery and paint industries, paper and pulp industry, and cosmetic and human health industries can have catastrophic effects on human health and the environment.2It has been determined that azo dyes are deleterious to aquatic organisms and carcinogenic and mutagenic for humans. As a result, it is of the utmost importance to develop efficient techniques for removing azo dyes from the effluent before discharge into the environment. Due to stringent environmental regulations, the significance of dye removal technology has increased. Physical adsorption, chemical coagulation, advanced oxidation, membrane filtration, adsorption, photocatalysis, and biologically based technology have all been investigated for dye removal.4–6 The technique of film-supported catalysts attracts considerable interest.7 Physical adsorption is generally regarded as an efficient technique for rapidly reducing the concentration of dissolved dyes, and activated carbon is the most commonly used adsorbent for dye removal.8 Dye removal technologies based on biopolymers can potentially become viable alternatives for the remediation of industrial dye effluents and contaminated water bodies.8 Using agro/industrial waste residues for dye adsorption has been highlighted as a possible alternative material for dye removal.4 Membrane technology incorporating an electric field has demonstrated good dye removal performance and is believed to have a promising future.5 Nonetheless, most of these methods are limited by the production of toxic sludge and high operational and maintenance costs.6 Advanced oxidation technology is one of the most effective methods for dye removal among these technologies.9
In the advanced oxidation processes (AOPs), ozonation is one of the techniques used to degrade azo dyes.10–12 Due to their complex structure, azo dyes resist ozone, light, biodegradation, and other environmental conditions.13 Nanocatalysts have been used to degrade azo dyes, including acid orange II, in catalytic ozonation. The application of nanocatalysts can significantly boost the degradation and mineralization of dyes. Fe3O4/multi-walled carbon nanotubes have been used to catalyze the ozonation of p-hydroxybenzoic acid.14 In another study, using iron oxide as a catalyst in ozonation to degrade sunset yellow dye was proposed.15 Nanocatalysts of the spinel-type ZnAl2O4 have also been used in catalytic ozonation for wastewater treatment.16 The nano-ZnO/perlite catalyst has been used for catalytic ozonation of azo dyes, significantly improving the dye's mineralization.17 In addition, Fe–Ni bimetallic nanoparticles have been utilized as a catalyst for the degradation of orange G dye by ozone.18 In a recent study conducted by Meng et al. (2022),19 an assessment was carried out regarding the advancements and obstacles encountered in the electro- and photocatalytic oxidation of hydroxymethylfurfural derived from biomass. This oxidation process aims to convert hydroxymethylfurfural into valuable chemicals, notably 2,5-formylfuran (DFF) and 2,5-furandicarboxylic acid (FDCA). Solid catalytic materials with bifunctional properties were employed in the sequential conversion of biomass into biofuels and associated chemicals.20 Also, a Mn/Mg/Ce ternary ozone catalyst was used to degrade printing and dyeing wastewater.21 It has been discovered that Ag and Au nanoparticles decorated on chitosan-functionalized graphene oxide exhibit excellent catalytic activity for reducing aromatic nitroarenes and degradation of azo dyes.22 Under ambient conditions, magnetically recyclable CoFe2O4/ZnO nanocatalysts have been utilized for the efficient catalytic degradation of acid blue 113.23 For the photocatalytic degradation of methyl orange, methylene blue, and rhodamine B, the AgCl nanocatalyst synthesized from its bulk material in the ionic liquid trihexyl(tetradecyl)phosphonium chloride has been used.24 These studies reveal the potential of nanocatalysts for the ozone-based treatment of wastewater containing azo dyes. Therefore, ozone catalytic oxidation improves dyeing wastewater treatment after catalyst addition. Consequently, nanocatalysts enhance the efficiency of ozone degradation of azo dyes.
CaFe2O4 nanoparticles are a novel catalyst for the ozone degradation of Acid Orange II (AOII). While other nanocatalysts, such as Fe3O4/multi-walled carbon nanotubes and CoFe2O4/ZnO, have been studied for the degradation of azo dyes using ozone, the use of CaFe2O4 nanoparticles for this purpose is uncommon. The mechanism of the catalytic ozonation process with the CaFe2O4 nanocatalyst to enhance the production of hydroxyl radicals, which are responsible for the degradation of the dye, was also investigated in this study. Consequently, using the CaFe2O4 nanocatalyst as a catalyst for the ozone degradation of acid orange II is a promising strategy that merits further investigation.
This research aims to synthesize CaFe2O4 nanocatalysts and examine their potential as a catalyst for the ozone-mediated degradation of AOII in aqueous solutions. This study aims to determine the effect of solution pH and nanoparticle amount on the degradation of AOII. In addition, the effect of initial AOII concentration on the degradation process will be investigated. In addition, the study will determine the reaction rate constant to evaluate the degradation process's efficacy. By examining these factors, this study aims to shed light on the usefulness of CaFe2O4 nanoparticles as a catalyst for the degradation of AOII. The findings will ultimately contribute to developing effective and sustainable methods for treating wastewater containing dyes.
Materials and methods
Chemicals
Acid Orange II (AOII) dye, with the formula 26H17ClN7Na3O10S3 (Fig. 1), was imported from Tianjin De Sheng Wang Chemical Trade Co., Ltd., China, and utilized without further purification. CaCO3, Fe2O3, H2SO4, and NaOH chemicals were obtained from Merck & Co., Inc. To prepare a laboratory-synthesized AOII dye solution, 1.0 g of AOII was dissolved in 1 L of deionized water to obtain a 1000 mg L−1 concentration. The stock AOII solution was diluted with distilled water to produce synthetic wastewater samples with AOII concentrations ranging from 100 to 500 mg L−1. Ozone (O3) was produced to remove AOII using the NextOzone ozone generator model Next 10P, which has a maximal ozone production capacity of 10 g O3 per h. The ozone was introduced through the bottom inlet of the reactor. | ||
| Fig. 1 Chemical structure of AOII. | ||
Synthesis of catalyst materials
Experimental setup
The ozone treatment system employed a tubular borosilicate glass reactor with a volume of 1.2 L (height = 450 mm, inner diameter = 60 mm).26 The reactor had a gas inlet, outlet ports, and sampling accessories. To investigate the effects of AOII solution pH and CaFe2O4 nanoparticle dosage (in fluidized status), CaFe2O4 nanoparticles were introduced into the reactor along with 500 mL of AOII solution at the desired concentration. Ozone was generated from pure oxygen using an ozone generator (NextOzone 20P, International Ozone Generator Company, Vietnam). The ozone production rate was maintained at 15 mL min−1, and regulated by a flow meter, and ozone was introduced into the reactor from the bottom through a diffuser. The reactor, containing 500 mL of AOII solution, ensured uniform distribution of CaFe2O4 nanoparticles throughout the solution using a porous glass diffuser. Each ozonation cycle lasted for 5 min, with a total treatment time of 50 min.Analytical methods
The degree of mineralization of AOII following degradation was assessed by measuring the concentration of total organic carbon (TOC) using a TOC analyzer (Multi N/C 3100, Germany). Additionally, the color change was analyzed using a double-beam UV/vis spectrophotometer (Shimadzu, Model Z2000, Japan) at a wavelength of 483 nm.The pore volume and surface area of CaFe2O4 nanoparticles were determined using the Brunauer–Emmet–Teller (BET) method on an SA 3000 instrument (Coulter, USA). The morphology of the CaFe2O4 nanoparticles was observed using a transmission electron microscope (SEM) equipped with TEM, JEOL JEM-2100 Plus, Hillsboro, USA. The X-ray diffraction (XRD) pattern of the CaFe2O4 nanoparticles was recorded using a Siemens D5005 X-ray diffractometer with Cu Kα radiation (λ = 1.5417 Å).
Results and discussion
Characteristics of CaFe2O4 nanoparticles
CaFe2O4 nanoparticles as a catalyst for the degradation of acid orange II dye with ozone have the following characteristics: average pore volume of 0.011 cm3 g−1, average surface area of 379 m2 g−1, and average particle size of 11.63 nm. The TEM analysis of CaFe2O4 nanoparticles from Fig. 2a reveals their rough surface texture and a variety of structures with varying sizes. These structures have an uneven distribution and a propensity to clump together. Amorphous CaFe2O4 nanoparticles are distinguished by their irregular shapes and lack of uniformity. The average diameter of CaFe2O4 particles ranges from 500 to 1000 nm, and they form clusters of varying sizes. The SEM analysis reveals a broad size distribution within these clustered structures, whose dimensions are relatively homogeneous. While maintaining their anisotropic shape, the particles exhibit interconnected irregular and porous structures. Individual particle sizes range from 70 to 300 nm, while the maximum length of the anisotropic nanostructures is 650 nm. Remarkably, the particles exhibit remarkable interconnectivity and cohesion while retaining an open structure, which enhances their capacity for efficient substance absorption. These porous structures provide favorable conditions for adsorption processes in aqueous environments.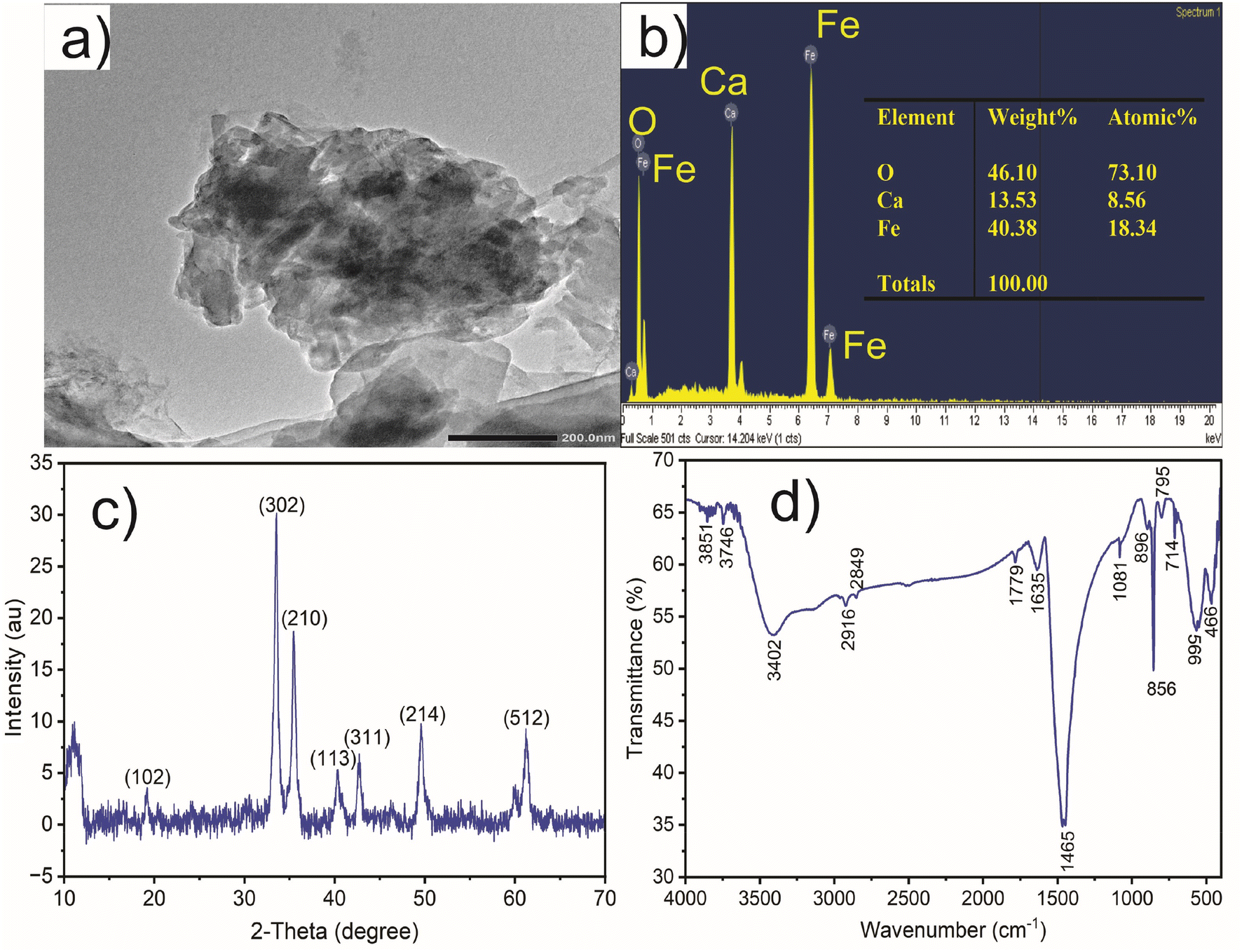 | ||
| Fig. 2 TEM (a), EDX (b), XRD (c) and FTIR (d) graph of CaFe2O4 nanoparticles. | ||
The EDS analysis of CaFe2O4 nanoparticles (Fig. 2b) verifies the presence of oxygen (O), calcium (Ca), and iron (Fe) elements, which account for 46.10%, 13.53.3%, and 40.30%, respectively, of the total weight. These elements have respective atomic weight percentages of 73.10%, 8.56%, and 18.34%. EDS analysis provides information about the percentage composition of chemical elements present in the material. In the case of the CaFe2O4 nanocatalyst, EDS results reveal the presence of calcium (Ca), iron (Fe), and oxygen (O). These elements play a crucial role in the ozone degradation process of azo dyes. Therefore, this demonstrates that CaFe2O4 is a potential catalyst for ozonation to degrade various azo dyes. Therefore, the nano CaFe2O4 nanoparticles can be utilized as a catalyst for the ozonation of AOII.
X-ray diffraction (XRD) analysis was used to examine the phase composition of the CaFe2O4 nanocatalyst material. Fig. 2c demonstrates the XRD analysis results of the CaFe2O4 nanocatalyst. The X-ray diffraction pattern provides insight into the nanoparticle crystallinity of the CaFe2O4 nanocatalyst. The nano CaFe2O4 nanocatalyst phase can be seen in Fig. 2c with major and low-intensity peaks. The crystalline structure of CaFe2O4 nanoparticles can be deduced using the widely employed XRD measurement technique. The (302) and (210) planes at 2θ angles of 33.6° and 35.41°, respectively, account for the former peak. Additionally, notable peaks (102) at 2θ = 19.25°, (113) at 2θ = 40.39°, (311) at 2θ = 42.1°, (214) at 2θ = 49.71°, and (512) at 2θ = 61.23° have been observed. These XRD patterns of the CaFe2O4 nanocatalyst display multiple peaks, each corresponding to a distinct crystallographic plane within the crystal structure. The XRD results of CaFe2O4 from this study are similar to the findings reported by Alhassan et al.,27 showing that the planes (102), (202), (302), (210), (113), (311), (214), and (512) correspond to the 2θ angles of 19.2°, 25.7°, 33.6°, 35.6°, 40.4°, 42.8°, 49.6°, and 61.4°, respectively. Understanding materials' physical and chemical characteristics using the data from XRD measurements is useful for creating new applications in sensing, energy storage, and catalysis.
The FTIR spectrum of CaFe2O4 nanoparticles exhibits various absorption peaks corresponding to distinct vibrational modes of the chemical bonds inherent in the nanoparticles.28 A broad absorption peak is evident within the 3402 to 3851 cm−1 range, indicative of the O–H stretching mode.29 Peaks at 1081 and 1635 cm−1 signify the elongation of C–O bonds.30 A peak at 1779 cm−1 suggests the existence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups, potentially resulting from the establishment of hydrogen bonds between water groups and silanols. The absorption peaks at 2849 and 2916 cm−1 exhibit lower intensity, implying the reduced stretching of C–H bonds. The FTIR spectrum offers insights into the calcium (Ca) and iron (Fe) bonds within the CaFe2O4 nanoparticles. Peaks at wavelengths of 466 and 566 cm−1 correspond to the Ca–O and Fe–O bonds, respectively.31 Moreover, peaks at 856 and 896 cm−1 emerge from bending Fe–O–H bonds, while peaks at 714 and 759 cm−1 arise from the elongation of Fe–O bonds.28
O functional groups, potentially resulting from the establishment of hydrogen bonds between water groups and silanols. The absorption peaks at 2849 and 2916 cm−1 exhibit lower intensity, implying the reduced stretching of C–H bonds. The FTIR spectrum offers insights into the calcium (Ca) and iron (Fe) bonds within the CaFe2O4 nanoparticles. Peaks at wavelengths of 466 and 566 cm−1 correspond to the Ca–O and Fe–O bonds, respectively.31 Moreover, peaks at 856 and 896 cm−1 emerge from bending Fe–O–H bonds, while peaks at 714 and 759 cm−1 arise from the elongation of Fe–O bonds.28
The effect of solution pH on AOII degradation
The details offered make it clear how pH affects the concentration and rate of disintegration of AOII during the ozonation process. This dramatically impacts how quickly azo dyes react and how well they degrade. The pH of the solution directly influences the rate at which hydroxyl radicals are produced and the availability of the iron catalyst inside the reactor.Experiments were carried out to determine the effect of solution pH on the decolorization and total organic carbon (TOC) degradation effectiveness of AOII. The pH ranged from 3 to 11, with variations. The catalyst dosage was 1.0 g L−1, the starting concentration of AOII was fixed at 200 mg L−1, and the contact times for AOII were 0–50 min, respectively (Fig. 3).
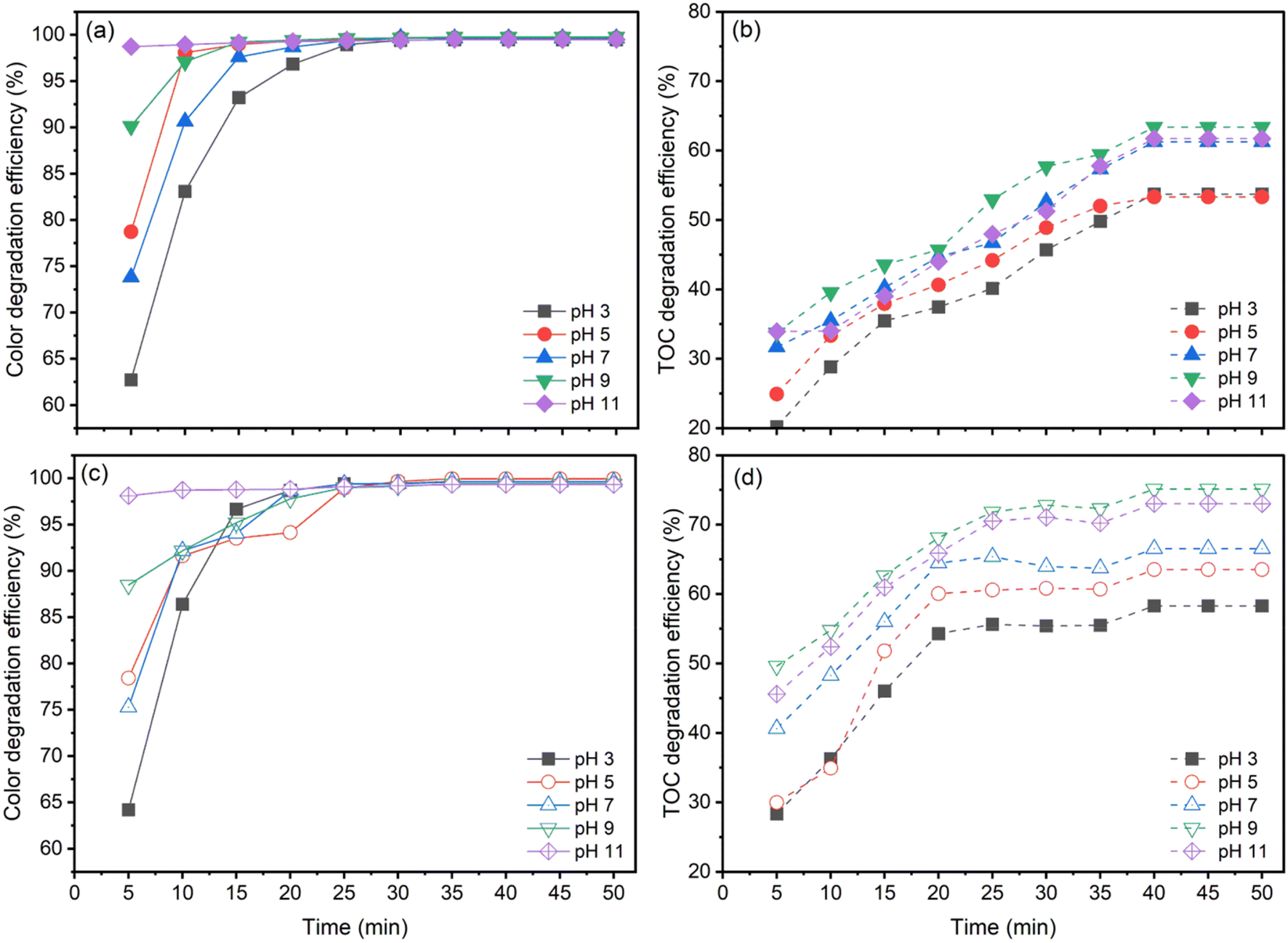 | ||
| Fig. 3 Color and TOC degradation efficiency for AOII by ozone alone (a and b) and CaFe2O4 nanocatalytic ozone (c and d) at various solution pH values. Conditions: [AOII] = 200 mg L−1, catalyst dosage = 1.0 g, contact time = 50 min, and reaction temperature = 25 ± 2 °C. | ||
According to the data shown in Fig. 3, both ozone alone (O3 alone) and CaFe2O4 nanocatalytic ozone (CaFe2O4/O3) systems demonstrate distinct patterns in the decolorization and total organic carbon (TOC) degradation of AOII at various pH levels. From pH 3 to pH 9, the decolorization and TOC degradation rates of AOII typically decrease, with a minor fall at pH 11 (Fig. 3a and c).
For both O3 alone and CaFe2O4/O3 systems, the decolorization rate of AOII advanced quickly, reaching approximately 100% in a short reaction time of 25 min. However, the efficiency of TOC decomposition varied. When the pH rose from 3 to 9, the efficiency of O3 alone ranged between 49% and 59%. It slightly decreased to 57% after 40 min of reaction time (Fig. 3b). The TOC degradation efficiency in the CaFe2O4/O3 system varied from 58% to 75% when the solution pH increased from 3 to 9. It dropped to 72% when the pH was further raised to 11 within the same 40 min reaction period (Fig. 3d). Therefore, the findings shown in Fig. 3 reveal that both O3 alone and CaFe2O4/O3 systems achieve their maximum AOII degradation efficiency under alkaline conditions (pH 9), with the CaFe2O4/O3 system demonstrating a higher efficiency than O3 alone by a margin of 10% to 16%. It is noteworthy that, compared to ozone alone (O3 alone), the CaFe2O4/O3 system's usage of CaFe2O4 nanocatalytic ozone exhibits the most effective AOII degradation. It is essential to note that pH levels over 11 did not improve AOII treatment efficiency in this investigation compared to pH 9.
Acid orange II can be directly attacked by ozone through electrophilic reactions in an acidic environment.32 Peroxynitrous acid attacks acid orange II using a radical cage solvent during this chemical process. Previous research33 has examined the oxidation of orange II with ozone and H2O2/O3 oxidation systems. In addition, orange II has been observed to undergo decolorization under various conditions.34 The production of isocyanates via the nucleophilic or electrophilic reaction of ozone with isonitriles has also been reported35 as a reaction involving ozone. In previous literature,36 the electrophilic attack of ozone on aromatic bonds has also been discussed.
According to the well-established Criegee mechanism of ozonation, the dominant mechanism for the oxidation of acid orange II by O3 molecules37 will be direct ozonation. However, due to the formation of aldehydes and carboxylic acids that do not react continuously with ozone, the reaction rate is sluggish and selective, resulting in incomplete degradation of organic matter.38 The reaction between hydroxide ions (–OH) and ozone (O3) results in the formation of superoxide (O2−) and hydroperoxide 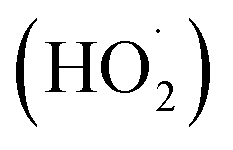 radicals, according to several investigations.39–41 The hydroxide ions catalyze ozone decomposition, and the concentration of hydroperoxide and superoxide radicals increases with increasing pH. Ozone then continuously reacts with superoxide radicals to produce highly reactive hydroxyl (˙OH) radicals, effectively oxidizing organic and inorganic compounds in water. Several studies39,40 corroborate that an increase in ozone degradation rates occurs as pH increases. Due to ozone's lower oxidation potential than hydroxyl radicals, direct oxidation by ozone is slower than radical oxidation.42
radicals, according to several investigations.39–41 The hydroxide ions catalyze ozone decomposition, and the concentration of hydroperoxide and superoxide radicals increases with increasing pH. Ozone then continuously reacts with superoxide radicals to produce highly reactive hydroxyl (˙OH) radicals, effectively oxidizing organic and inorganic compounds in water. Several studies39,40 corroborate that an increase in ozone degradation rates occurs as pH increases. Due to ozone's lower oxidation potential than hydroxyl radicals, direct oxidation by ozone is slower than radical oxidation.42
In one study, however, pH levels greater than 11 did not enhance the treatment efficiency of AOII compared to pH 9.43 The study examined the decomposition of AOII using ozone and hydrogen peroxide (H2O2) in a batch reactor under varying pH conditions. After 60 min of treatment, the highest degradation efficiency was observed at pH 9, with a rate of 98.5%. At pH levels greater than 11, however, the degradation efficiency dropped to 92.5% after 60 min of treatment. The study suggested that the decrease in degradation efficiency at high pH levels may be attributable to the formation of hydroxide ions, which can compete with AOII for ozone and H2O2, thereby decreasing the concentration of reactive species available for AOII degradation. According to additional studies, the optimal pH range for the degradation of various organic compounds using ozone and H2O2 is 7–9.44–46
The CaFe2O4 nanocatalyst is a variety of catalyst that has demonstrated great promise for enhancing the removal of AOII from aqueous solutions by catalytic ozonation. In catalytic ozonation systems, using the CaFe2O4 nanocatalyst as a heterogeneous catalyst has increased the generation of ˙OH radicals. The primary mechanism underlying the activation of catalytic ozonation is that the catalyst accelerates the production of hydroxyl radicals ˙OH, which result from the decomposition of ozone by the catalyst. This increases the oxidation potential, thereby accelerating the degradation of AOII. Due to its ability to support substantially as an ozonation catalyst (CaFe2O4/O3) and enhance the removal of AOII from aqueous solutions, the CaFe2O4 nanocatalyst is considered the most intriguing catalyst. Nanocatalysts were discovered to increase the production of ˙OH radicals in catalytic ozonation systems.47 Another study mentioned using a manganese catalyst in heterogeneous catalytic ozonation, indicating that hydroxyl radicals play a significant role in the oxidation process.48 Shokrollahzadeh et al.17 also reported that adding a nano-ZnO/perlite nanocatalyst increased the degradation rate during the catalytic ozonation of an azo dye. Overall, these references demonstrated that nanocatalysts improved the performance of catalytic ozonation systems by producing hydroxyl radicals.
The comparison between heterogeneous catalytic ozonation (CaFe2O4/O3) and O3 alone revealed that catalytic ozonation systems were more effective at TOC degradation than non-catalytic ozonation systems (O3 alone). Catalytic ozonation is a technology for the non-selective degradation of organic matter in water utilizing active free radicals produced by ozone degradation.49 The AOII degradation by CaFe2O4/O3 and O3 alone was more efficacious as the solution pH rose from 3 to 9. Notably, the AOII degradation efficiency, as measured by the removal of TOC, was lower than that of AOII decolorization. This disparity results from AOII's distinct contributions to color and TOC. The main source of color is dyed with complete chromophores, and the destruction of the dye chromophore through the cleavage of the –NN– group in AOII is more accessible than the degradation of AOII into CO2, H2O, and other byproducts. The color disappeared when the molecular structure of AOII was broken down by the direct or indirect reaction with O3, either through ˙OH radicals. Still, the complete conversion of organic matter into CO2 and H2O had not yet occurred. As a result, the efficiency of TOC degradation was lower than the decolorization rate. Ozonation resulted in a greater decolorization than TOC degradation efficiency.50 In phosphate- and bicarbonate-buffered solutions, the comparison between conventional ozonation and heterogeneous catalytic ozonation processes revealed that catalytic ozonation systems exhibited superior decolorization and TOC degradation efficiency compared to non-catalytic ozonation systems.51
The effect of CaFe2O4 nanocatalyst mass on AOII degradation
Experiments were conducted at pH 9 with an ozone generation rate of 3.038 g h−1 for both O3 alone and CaFe2O4/O3 systems. The study examined the decolorization and degradation of AOII via color and TOC elimination and varying the dosage of the CaFe2O4 nanocatalyst (0.25 to 2.0 g L−1) at an initial AOII concentration of 200 mg L−1, solution pH of 9 and contact time of 0–50 min. The results are shown in Fig. 4.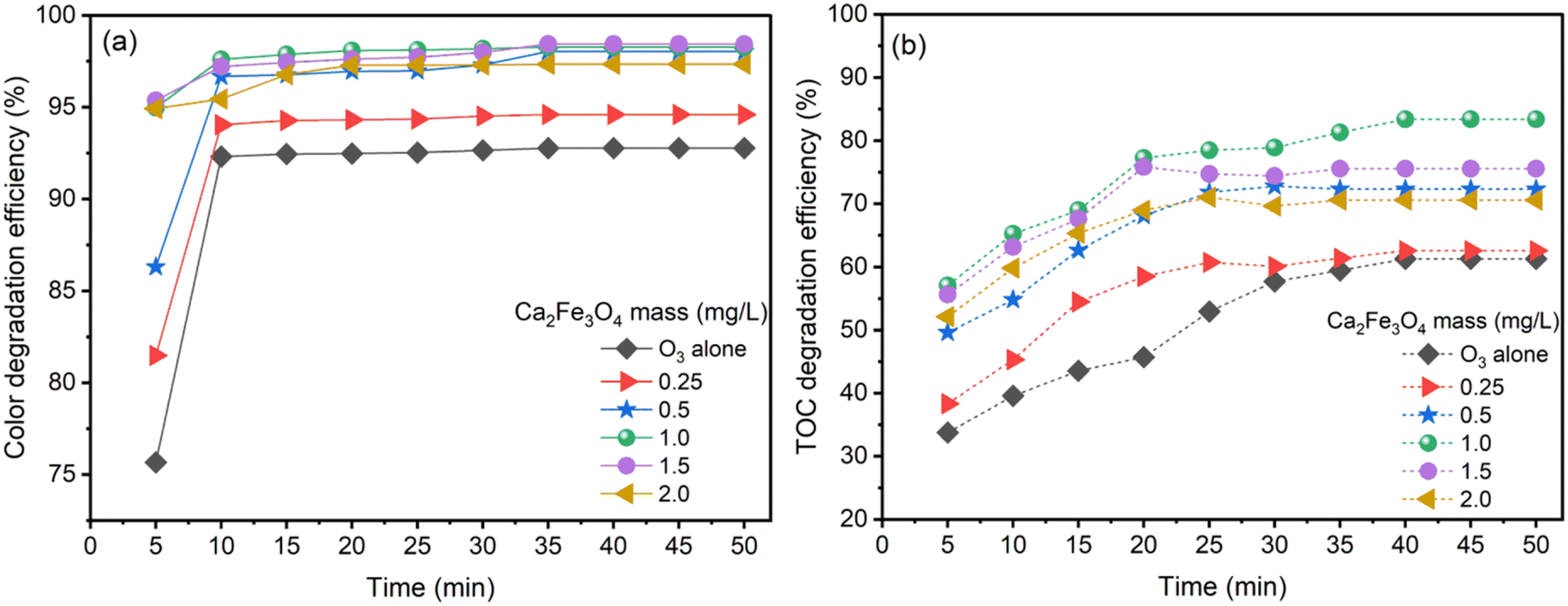 | ||
| Fig. 4 Color and TOC degradation efficiency by CaFe2O4 nanoparticle catalytic ozonation (a and b) at various dosages of CaFe2O4. Conditions: [AOII] = 200 mg L−1, initial pH = 9, contact time = 50 min, and reaction temperature = 25 ± 2 °C. | ||
Fig. 4a illustrates the color removal efficiency of AOII via ozone catalysis under various conditions: ozone alone and CaFe2O4/O3 with variable amounts of CaFe2O4 nanocatalyst. Both processes degraded AOII, resulting in rapid color reduction within five to ten minutes of the reaction. When considering ozone alone, the color removal efficiency for a 5 min contact time began at 75.66%. With extended contact times, the efficiency increased progressively, reaching 92% after 10 min of contact. The color removal efficacy remained constant at 92% until the 50 minute period. In the CaFe2O4/O3 system, adding 0.25 g per L CaFe2O4 nanocatalyst increased the color removal efficiency to 81.49% after 5 min of reaction, 94% after 10 to 20 min and 98% after 50 min. The color removal efficiency gradually improved as the dosage of the CaFe2O4 nanocatalyst increased from 0.5 g L−1 to 2 g L−1. After 20 min of reaction with the CaFe2O4 nanocatalyst ranging between 0.5 g L−1 and 2 g L−1, the color removal efficiency reached a maximum of 98%. The swift progression of AOII decolorization in the O3 and CaFe2O4/O3 systems arises from the catalytic breakdown of dye molecules.52 When AOII exists in its initial colored form, its molecular structure is disrupted, causing an instant loss of color. Particularly in the O3 system, O3 acted as a potent oxidizing agent, directly breaking down the structure of dye molecules and swiftly diminishing AOII color within a 10 minute period. The presence of CaFe2O4 nanoparticles in the CaFe2O4/O3 system enhanced decolorization further. These nanoparticles served as catalysts, promoting the degradation process of dye molecules by O3, aided by their high adsorption capacity, enabling effective interaction between dye molecules and O3, thereby accelerating the decolorization process. The synergistic strength between O3 and CaFe2O4 nanoparticles even accelerates the rate beyond that of O3 alone, manifested by a higher decolorization rate in the CaFe2O4/O3 system due to increased adsorption surface area, intensifying the catalytic degradation efficiency.
Fig. 4b displays the influence of CaFe2O4 nanocatalyst dosage on the TOC removal efficiency of AOII using O3 alone and CaFe2O4/O3 systems over time. For a contact time of 5 min, the TOC removal efficacy for O3 alone began at 34%. The efficiency progressively increased as the contact time increased, reaching 58% after 30 min. After 40 min and 50 min of reaction, the efficiency increased insignificantly. Regarding the CaFe2O4/O3 systems, when 0.25 g per L CaFe2O4 nanocatalyst was added, the TOC removal efficiency began at 38% after 5 min and increased steadily to 61% after 25 min, without further improvement with extended reaction times. Upon increasing the amount of CaFe2O4 nanocatalyst from 0.25 g L−1 to 1.0 g L−1, the efficiency of TOC treatment exhibited an ascending trend, reaching a maximum of 83.38% after 40 min of reaction, and then remained stable for the next 50 min. Increasing the CaFe2O4 nanocatalyst dosage to 1.5 g L−1 and 2.0 g L−1 leads to a minor decrease in efficiency. The results demonstrate that adding the CaFe2O4 nanocatalyst improves the efficiency of TOC elimination compared to O3 alone. The CaFe2O4 nanocatalyst at a dosage of 1.0 g L−1 provides the most outstanding efficiency, with a maximal efficiency of 83.38% after 40 min, which is maintained until 50 min. However, a minor decrease in efficiency is induced by surpassing the dosage of the CaFe2O4 nanocatalyst.
The CaFe2O4 nanocatalyst substantially enhanced the treatment efficiency of AOII from aqueous solution and processed 83.38% of the TOC in 40 min with CaFe2O4/O3 systems. At the same time, the TOC processing efficacy with O3 alone was only 58%. A synergistic combination of the gas phase (O3), liquid phase (AOII solution), and solid phase (CaFe2O4 nanocatalyst) occurred when the CaFe2O4 nanocatalyst was introduced into the reactor in the presence of ozone (O3).26 This configuration allowed for the simultaneous interaction of O3 and the azo dye (AOII) on the nanocatalyst's surface. Direct interactions between O3 molecules and AOII resulted in the oxidation of AOII. Moreover, the ˙OH radicals generated by catalytic ozonation processes contributed to the oxidation of AOII.
The CaFe2O4 nanocatalyst was indispensable to the system. First, it acted as an adsorbent, facilitating contact between the reactants and enhancing the overall process efficacy. It also provided sites where O3 and ˙OH radicals could react with AOII molecules, thereby enabling their oxidation. This cooperative action of the nanocatalyst, O3, and ˙OH radicals effectively removed color from the azo dye solution. There are also several plausible explanations for why the CaFe2O4/O3 system was able to process a higher percentage of TOC than O3 alone: (1) synergistic effect: the combination of the CaFe2O4 nanocatalyst and O3 may have a synergistic effect in which the two components enhance the treatment efficacy of AOII. This may result from CaFe2O4's ability to function as a catalyst to promote the decomposition of AOII by O3 alone. The CaFe2O4 nanocatalyst has a high surface area, which can provide more active sites for the AOII and O3 reactions. This can increase the reaction rate and enhance the treatment's efficacy. This may explain why CaFe2O4/O3 could metabolize a more significant percentage of TOC than O3 alone. However, it is essential to evaluate the optimal CaFe2O4 nanocatalyst dosage. Exceeding the optimal dosage may result in an inhibitory effect, reducing the decolorization and total organic carbon (TOC) removal effectiveness. Several factors can explain this phenomenon. For instance, an excessive quantity of the nanocatalyst may lead to the saturation of active sites on its surface, thereby reducing the availability of reactive sites for O3 and ˙OH radicals. In addition, at higher concentrations, the nanocatalyst may promote unintended side reactions, thereby reducing the efficiency of AOII oxidation and TOC removal.
According to previous investigations, an ozone (O3) catalyst can be more effective at degrading azo dyes than O3 alone. Using a catalyst in conjunction with ozone, catalytic ozonation is effective for degrading azo dyes. For instance, a study discovered that combining ozone and ZnO catalysts effectively degraded azo dyes.53 According to another study, using a Ni-containing layered double hydroxide nanocatalyst in conjunction with ozone effectively degrades an azo dye.54 The presence of Au/TiO2 increased the corona discharge's sterilization efficiency from 73% to 91%. It decreased the O3 concentration from 0.38 ppm to 0.04 ppm, whereas the presence of TiO2 decreased the sterilization efficiency and O3 concentration to 66% and 0.17 ppm, respectively.55
The effect of initial AOII concentration
Exhaustive experiments were conducted under tightly regulated conditions to evaluate the efficacy of the heterogeneous catalytic ozonation process (CaFe2O4/O3 system) for varying concentrations of AOII. Experiments were conducted at pH 9, ambient temperature (26 ± 2 °C), and a discharge rate of 3.038 g h−1. Adjusting the initial AOII concentration within the range of 100 to 500 mg L−1 corresponding to TOC concentrations ranging from 16.89 to 82.94 mg L−1 was required for the ozonation process. Fig. 5 depicts the effect of the initial AOII concentration on the removal of color and degradation graphically. As shown in Fig. 5, increasing the AOII concentration reduced the efficacy for color removal and TOC degradation, regardless of whether the CaFe2O4/O3 system was used. When the AOII concentration was increased from 100 mg L−1 to 500 mg L−1, a significant reduction in color was observed between 10 and 30 min later (Fig. 5a). When the AOII concentration increased from 100 to 500 mg L−1, the TOC removal efficiency decreased from 85% to 62% (Fig. 5b). However, extending the treatment duration did not result in a significant improvement in TOC removal efficiency. Previous studies have also demonstrated that the efficiency of azo dye degradation by nano-catalyzed ozone decreases as the initial concentration of azo dyes increases.56 Lowering the concentration of azo dyes, utilizing extreme pH conditions, and employing higher ozone concentrations have been identified as more advantageous for dye degradation.15 In addition, as previously mentioned, adding iron oxide as a catalyst does not result in a substantial reaction acceleration. Another study focusing on the degradation of acid red 18 dye using ZnO immobilized on a stone surface as a catalyst reveals a decrease in the rate constant as the initial dye concentration increases.57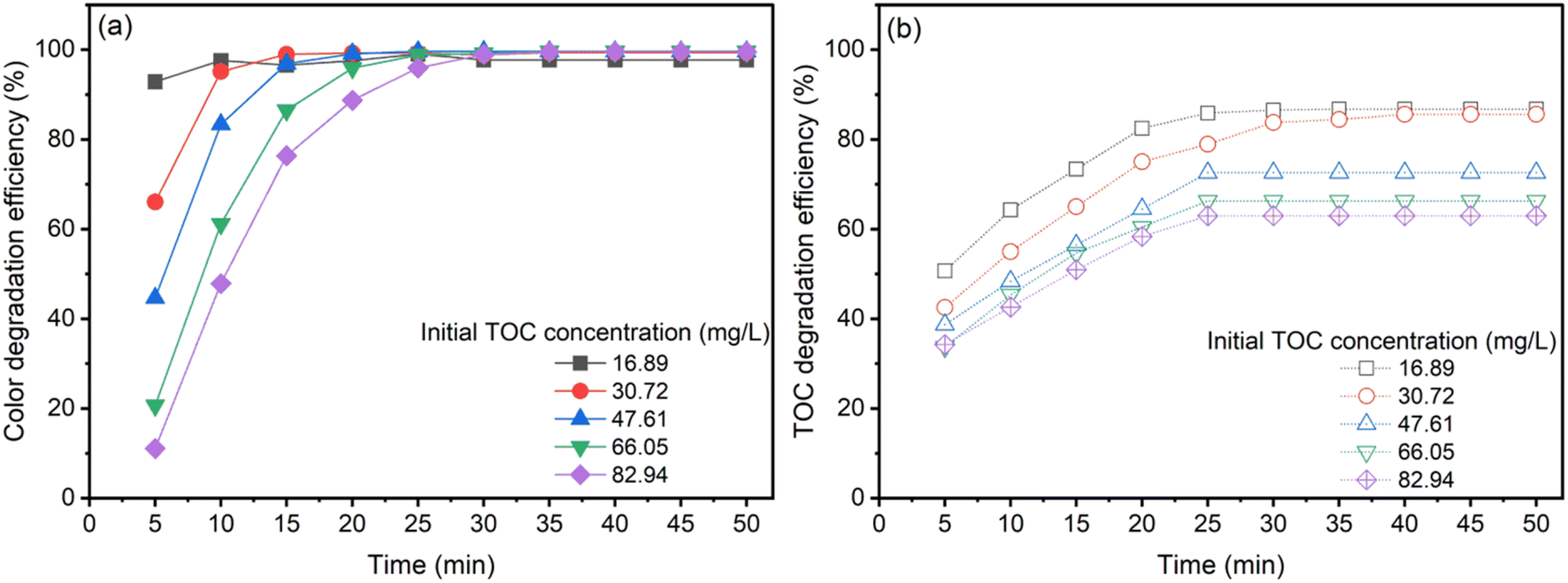 | ||
| Fig. 5 The effect of AOII concentration on color (a) and TOC (b) degradation by CaFe2O4 nanoparticle catalytic ozonation processes. Conditions: CaFe2O4 dosage = 1.0 g, initial pH = 9, contact time = 50 min, and reaction temperature = 25 ± 2 °C. | ||
The relative distribution of O3, CaFe2O4 nanocatalyst, and ˙OH radicals is significantly influenced by the concentration of AOII in the solution. With an increase in AOII concentration, the proportion of O3, nanocatalyst, and ˙OH radicals decreased compared to AOII. This observation revealed that as the concentration of AOII increased, the concentration of ˙OH radicals remained unchanged. This phenomenon can be attributed to more AOII molecules occupying active sites on the catalyst's surface, inhibiting the production of ˙OH radicals.58 The occupation of active sites by AOII ions inhibited the production of ˙OH radicals on the catalyst's surface, reducing their availability for degradation. The degradation efficiency of azo dyes also decreased due to the consumption of hydroxyl radicals by higher concentrations of organic dyes while the concentration of the oxidizing agent remained constant.59 This observation illuminates the complexities of the catalytic ozonation process and highlights the importance of contemplating concentration-dependent phenomena for efficient pollutant removal.
Reaction rate constant and mechanism in AOII degradation by catalytic ozonation processes
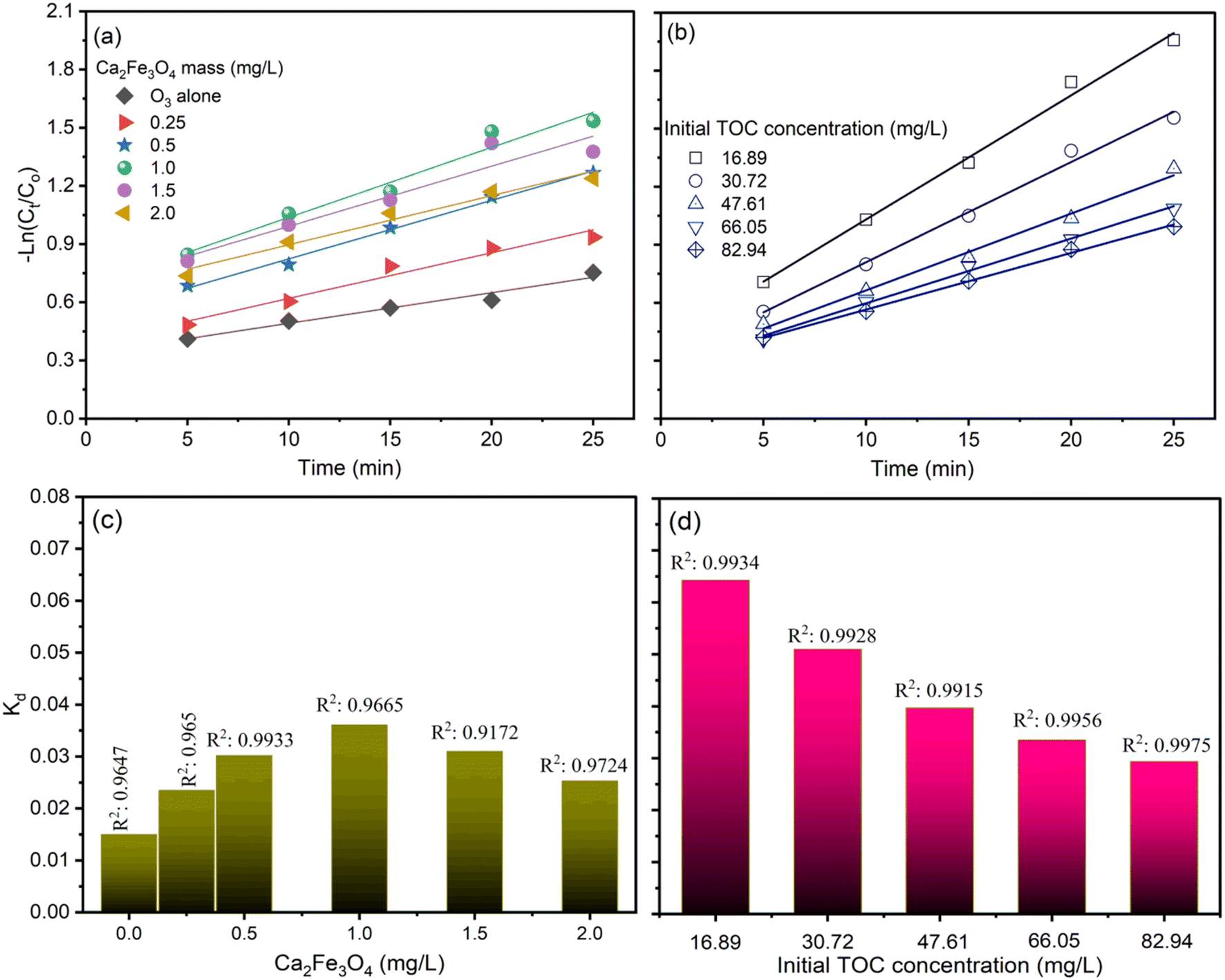
Considering a distinct first-order reaction, the degradation rate is proportional to the removal of TOC.26 In this regard, the following equation can be used to describe the kinetics of the reaction:Here, C0 and C represent the concentration of AOII in the aqueous solution at the start of the reaction and after t min, respectively. The data align with a straight line when ln(C/C0) is plotted against time, and k (the first-order rate constant) corresponds to the slope of this line. The rate constant was calculated by plotting ln(C/C0) against time (t). With a coefficient of determination (R2) ranging from 0.95 to 0.99, the linearity of these graphs validates the pseudo-first-order kinetics of the degradation process.
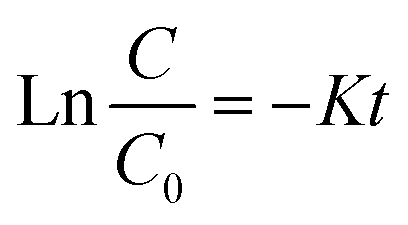
Fig. 6 and 7 illustrate the findings of plotting ln(C/C0) against time (t) and calculating k values. In determining the reaction kinetics for TOC degradation by CaFe2O4/O3 systems, the logarithmic transformation of the C/C0 ratio against time is commonly employed. The determined rate constant, k, is consistent with a pseudo-first-order reaction. Fig. 6 depicts the influence of pH on k values, indicating that the degradation rate of TOC increased with increasing pH from 3 to 9 and then reduced slightly as the pH rose to 11. The k values for the O3 alone system in AOII degradation ranged from 0.0146 min−1 to 0.017 min−1 as the pH of the solution increased from 3 to 9 and then decreased slightly to 0.0162 min−1 at pH 11 (Fig. 6a and c). As the pH of the AOII solution increased from 3 to 9, the k values for the CaFe2O4/O3 system increased from 0.025 min−1 to 0.0311 min−1 and then decreased to 0.0302 min−1 at pH 11 (Fig. 6b and d). The obtained reaction rate constants correspond to the TOC degradation efficiency influenced by the AOII solution's pH. In addition, the results indicate that the k values for the CaFe2O4/O3 system are substantially greater than those for O3 alone. At a solution pH of 9, the k value for the CaFe2O4/O3 system is 1.83 times that of O3 alone. This further demonstrates that the CaFe2O4 nanocatalyst significantly enhanced the ozone degradation process of AOII. Except for the pH 5 condition in the CaFe2O4/O3 system, where the R2 value reached 0.927, most R2 values fall between 0.9649 and 0.9959. Most treatment experiments exhibit linearity and are consistent with the pseudo-first-order kinetics of the degradation process, as indicated by the results.
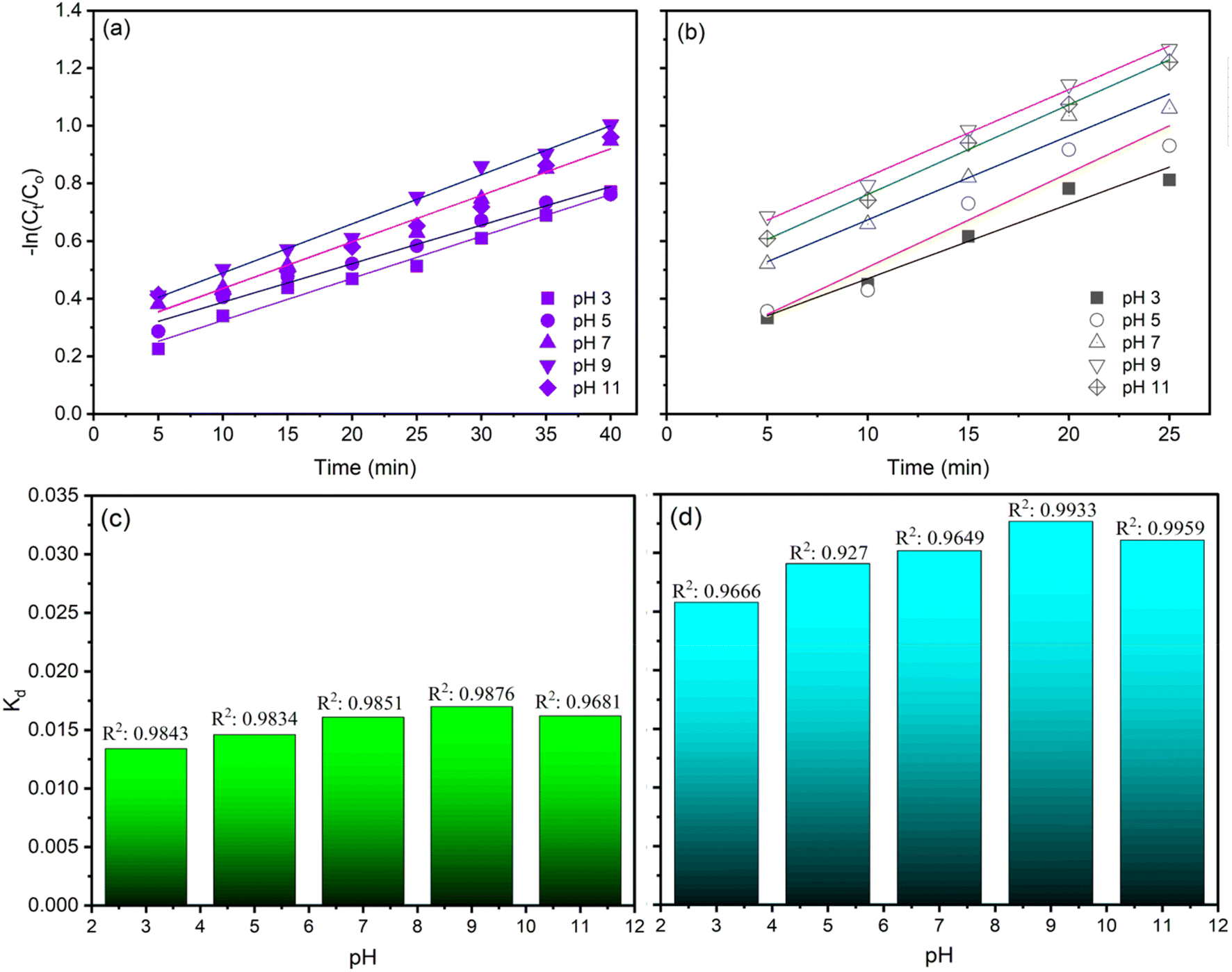 | ||
| Fig. 6 The plots of ln(C/C0) and reaction rate constant of TOC degradation by ozone alone (a and c) and CaFe2O4 nanoparticle catalytic ozonation processes (b and d) at various solution pH values. | ||
 | ||
| Fig. 7 The plots of ln(C/C0) and reaction rate constant of TOC degradation by CaFe2O4 nanoparticle catalytic ozonation processes at various amounts of CaFe2O4 (a and c) and AOII concentration (b and d). | ||
Similarly to the pH effect, the plot of ln(C/C0) and the reaction rate constant of TOC degradation using the CaFe2O4/O3 system exhibits an increase in k values from 0.015 min−1 to 0.0361 min−1 when the amount of CaFe2O4 increased from 0.025 g L−1 to 1.0 g L−1, followed by a decrease to 0.031 min−1 and 0.0253 min−1 when the amount of CaFe2O4 was further increased to 1.5 and 2.0 g L−1 (Fig. 7a and c). The R2 values were more significant than 0.95, except for the condition containing 1.5 g per L CaFe2O4. Fig. 7b and d illustrate a decreasing trend in the reaction rate constant (k) from 0.0643 min−1 to 0.0294 min−1 as the AOII concentration increased from 100 mg L−1 to 500 mg L−1, which corresponds to TOC concentrations from 16.89 mg L−1 to 82.94 mg L−1. The R2 values for the conditions evaluating the effect of AOII concentration on TOC removal efficiency using the CaFe2O4/O3 system were also greater than 0.99. The k values calculated correspond to the TOC treatment efficacy of the CaFe2O4/O3 system when the CaFe2O4 and AOII concentrations were varied. These findings reaffirm the applicability of the reaction rate constant (k) according to a pseudo-first-order equation and by the treatment model.
According to numerous investigations, most treatment experiments are linear and consistent with the pseudo-first-order kinetics of the degradation process. According to Olea-Mejia et al.,60 Fe nanoparticles can substantially accelerate the degradation of phenolphthalein. The degree of phenolphthalein mineralization was also determined by measuring the total organic carbon (TOC) concentration. Brito et al.61 demonstrated that using a continuous reactor for the catalytic ozonation process and devising a kinetic model can provide the required technical data to support the scaling up of the process for the degradation of organic carbon in landfill leachate. Franco et al.62 reported that the degradation rate of textile dyes evaluated by total organic carbon is substantially more influenced by pH and ozone exposure time than by dye composition.
Several stages compose the mechanism of AOII degradation by the CaFe2O4 nanocatalytic ozone process (Fig. 8). The initial phase was absorption: the CaFe2O4 nanocatalyst has a high affinity for organic molecules and a large surface area (379 m2 g−1). This nanocatalyst's high surface area was ascribed to its small size, which was typically on the order of tens of nanometers. This compact size increases the number of sites on the surface that can interact with reactant molecules, resulting in more significant activity. The subsequent step was ozone activation: ozone (O3) is introduced into the system and activated in the presence of the CaFe2O4 nanocatalyst. The catalyst's surface contains active sites where ozone molecules can endure partial decomposition to form reactive oxygen species, such as hydroxyl radicals (˙OH) and superoxide radicals (˙O2−). Phase three was the production of reactive species: the activated ozone and reactive species generated from the catalyst surface, such as ˙OH and ˙O2−,63 were essential for the degradation of AOII. These highly reactive species attacked the adsorbed AOII molecules, causing chemical bonds to dissolve and the dye to degrade. Phase 4 consists of degrading reactions: various chemical reactions, including oxidation, hydroxylation, and azo bond cleavage,31 contribute to the degradation of AOII. The reactive species attacked the molecular structure of AOII, resulting in the formation of organic compounds that are smaller and less complex. These degradation products are frequently less environmentally harmful than the original pigment molecule. As the degradation reactions proceed, the degraded molecules desorb from the catalyst's surface, allowing new AOII molecules to adsorb and continue the degradation process. In subsequent reactions, such as mineralization, the degradation products are transformed into simpler inorganic compounds, such as carbon dioxide (CO2), water (H2O), and intangible ions.64
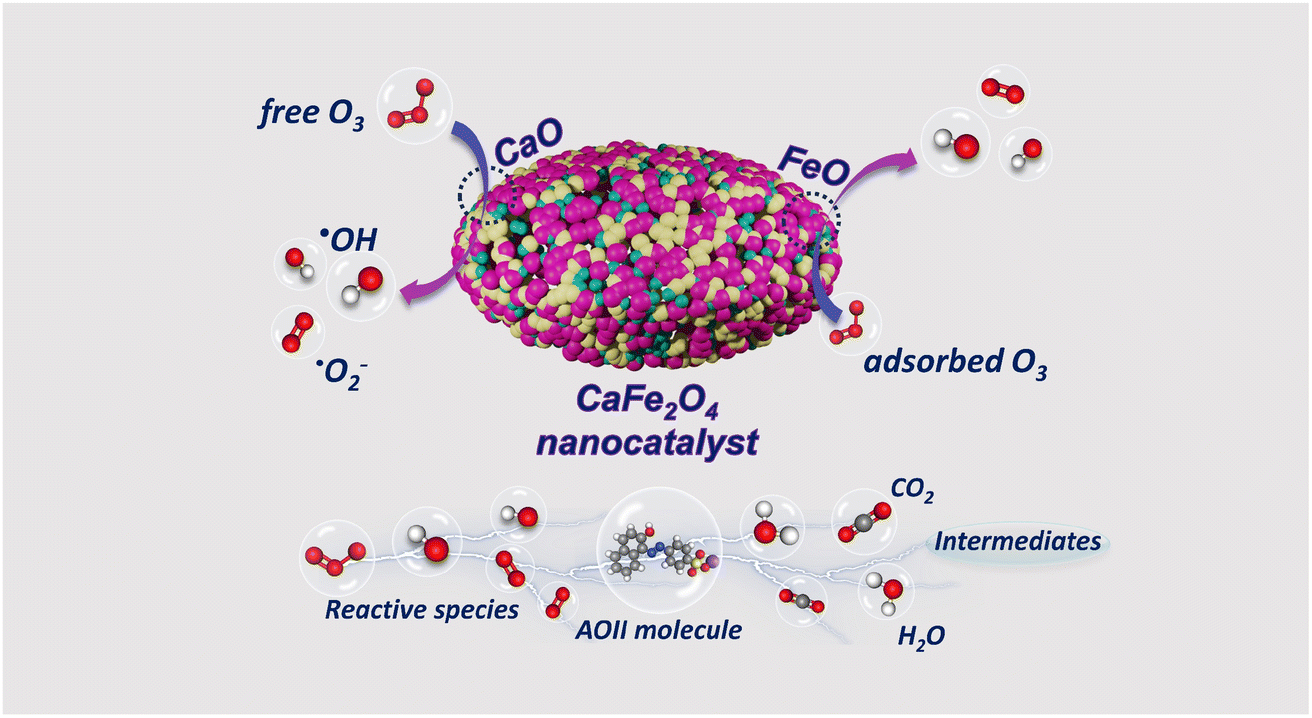 | ||
| Fig. 8 Schematic diagram of the degradation mechanism of AOII using the CaFe2O4 nanocatalyst. | ||
The CaFe2O4 nanocatalyst, composed of CaO and FeO, exhibits dual functionality as an adsorption and heterogeneous catalyst in ozone oxidation reactions. The material analysis results from EDX, XRD, and FTIR data (Fig. 2) confirm the presence of CaO and FeO on the surface of the nano CaFe2O4 nanocatalyst during the ozonation processes, which enhances the decomposition of O3 and leads to the generation of ˙OH radicals. The CaFe2O4 nanocatalyst provides active sites on its surface, allowing for the participation of both the catalyst and ˙OH radicals in the ozone oxidation reaction for the degradation of AOII. The responses that predominantly yield ˙OH radicals in the ozonation processes follow three mechanisms: H atom abstraction, ˙OH addition to CC, and ˙OH interaction with S and N atoms from AOII. The CaFe2O4 nanocatalyst, with its CaO and FeO structure, plays a crucial role in efficiently adsorbing O3 and surface pollutants, thereby accelerating the O3 decomposition process and increasing the production of hydroxyl (˙OH) radicals, which are essential for the degradation of AOII. The fragmentation of AOII through the intervention of ˙OH radicals is achieved through the heterogeneous catalysis mechanism inherent in the ozone oxidation process. The CaFe2O4 nanocatalyst, with its CaO and FeO composition, contributes to the catalytic performance of the catalyst in the ozone oxidation process. The activities of FeO and CaO are essential factors in the catalytic activity of the CaFe2O4 nanocatalyst. Previous studies showed that the Co2+–HCO3− system can efficiently degrade the cationic dye methylene blue (MB).65 However, when both AOII and MB are present in the solution, most of the Co2+ ions will form stable complexes with AOII, hindering the degradation of MB. The application of heterogeneous catalytic ozonation, such as using catalysts like alumina, has been explored for treating refractory organics in wastewater.66 This demonstrates the broader application of stimuli in the degradation of organic compounds, similar to the role of the CaFe2O4 nanocatalyst in the ozonation processes.
Comparing azo dye degradation using heterogeneous ozonation processes
Table 1 presents the AOII removal capacity comparison of the CaFe2O4/O3 system with other systems. Among the studies highlighted, the utilization of nano CaFe2O4 as the catalyst in conjunction with ozone for AOII degradation was undertaken in our ongoing research. The optimized parameters included maintaining a pH of 9, applying a catalyst dosage of 1.0 g per L CaFe2O4 nanocatalyst, and varying AOII initial concentrations from 100 to 500 mg L−1. This configuration resulted in the attainment of a substantial 85% efficiency in the removal of TOC. Notably, the study demonstrated significant prowess in the degradation of AOII.| Dyes | Catalyst | Optimal processing conditions | Degradation efficiency | References |
|---|---|---|---|---|
| AOII | CaFe2O4 nanoparticles | pH 9, CaFe2O4: 1.0 g L−1, AOII concentration: 100–500 mg L−1 | 85% TOC removal | This study |
| Methyl orange | Ni-based layered double hydroxides (Ni-LDHs) nanomaterials | Reaction time: 60 min, pH 9; ozone flow rate: 109 mg h−1; catalyst dosage: 1 g L−1; initial COD: 620 mg L−1 | 72% COD removal | 54 |
| Methyl orange | MnO2 nanoparticles | Initial pH 5.8, reaction time: 60 min, methyl orange concentration: 90 mg L−1, catalyst dosage: 20 mg L−1 | 98.37% methyl orange removal | 68 |
| AOII | ZnO nanoparticles | pH range: 4.6–9.6, ZnO nanoparticle: 1.0 g L−1, AOII: 50–200 mg L−1 | 90% TOC removal | 70 |
| Acid blue 113 | Fe2O3/MoS2 nanocomposite | Initial pH 6.4, nanocomposite: 20 mg, ozone flow: 0.2 g L−1 h−1, and reaction time: 45 min | 99.0% AOII removal | 69 |
| Reactive blue 5 | Nano-ZnO/perlite | pH 11, reaction time: 60 min, dye concentration: 100 mg L−1, initial TOC: 31.45 mg L−1, catalyst dosage: 12 mg L−1 | 92% TOC removal | 17 |
| Reactive red 24 | Iron slag | pH 11, catalyst dosage: 1.5 g L−1, initial reactive red 24 concentration: 300 mg L−1, inlet O3: 3.038 g h−1, H2O2 concentration: 100 mg L−1, reaction time: 40 min | 84% COD removal | 26 |
| Active brilliant red X-3B | Natural mineral brucite | pH 9.24, catalyst dosage: 0.5 g L−1, active brilliant red X-3B: 500 mg L−1, reaction time: 30 min | 32% COD removal | 67 |
The current study observed a significant efficiency enhancement compared to Hassani et al.'s (2019)54 findings for employing dual-layered nickel hydroxide (Ni-LDH) to degrade methyl orange. This improvement became particularly evident when contrasted with the reported 72% COD removal efficiency. The discoveries in this investigation also demonstrate superior effectiveness compared to the removal efficacy of active brilliant red X-3B using ozonation catalyzed by the natural mineral brucite, as conducted by Dong et al. (2007),67 achieving a COD degradation efficiency of 32% under experimental conditions of pH 9.24, catalyst dosage of 0.5 g L−1, active brilliant red X-3B concentration of 500 mg L−1 and a reaction time of 30 min (Table 1).
In the context of Shokrollahzadeh et al. (2019)17 research concerning nano-ZnO/pearl stone use for reactive blue 5 degradation, a higher efficacy was demonstrated, surpassing the 92% TOC removal efficiency. Compared to Zhang et al. (2022),68 who focused on methyl orange degradation by utilizing nano MnO2 particles, the current study showed slightly lower degradation efficiency. This is emphasized by the outstanding removal efficiency of 98.37% for methyl orange. Similarly, in contrast to the results of Bagheri et al. (2021),69 where Fe2O3/MoS2 nanocomposites were employed for acid blue 113 degradation, achieving a removal efficiency of 99.0%, our study exhibited slightly lower efficacy. Similarly, compared to Van et al. (2019),26 targeting reactive red 24 degradation through iron slag, the present study showed similar effectiveness. This achievement corresponds to a COD removal efficiency of 84%.
Reusability of the catalyst
The significant role of wastewater removal applications, encompassing general and specific instances such as textile dye effluent, is underscored by the vital importance of stability and reusability exhibited by the CaFe2O4 nanocatalyst in the catalytic ozonation processes. Consequently, after each reaction cycle, the solid residue underwent collection via filtration and thorough rinsing with distilled water and was subjected to drying at 80 °C for 12 h. This treated residue was redeployed in consecutive cycles to evaluate its stability and reusability. The experimental conditions entailed maintaining the pH of 9, utilizing the catalyst dosage of 1 g L−1, applying the initial AOII concentration of 200 mg L−1, and employing the reaction time of 40 min. Fig. 9 presents the results thus obtained. The findings illustrate insubstantial fluctuations in AOII degradation across five successive operations. The efficiency of AOII degradation experienced a slight reduction from 85% to 69% after five consecutive cycles, indicating the non-significant degradation of the nano CaFe2O4 catalyst during the ozonation process and the continual preservation of its notable stability. The diminished catalytic effectiveness of the CaFe2O4/O3 system potentially stems from catalyst attrition during the reusability experiments, aligning with prior research findings. Nguyen et al. (2023)71 documented a decline in treatment efficacy throughout five consecutive runs while employing a biochar-supported NiFe2O4 composite as a catalyst for the ozone/Fenton treatment of moxifloxacin in wastewater. Another study also revealed that the efficacy of the cyclic hydrothermal conversion of ethyl levulinate (EL) into γ-valerolactone (GVL), utilizing a layered Ov-WO3@CB catalyst, exhibited a marginal decline from 98% to 91% after undergoing five recycling cycles, thereby emphasizing the intricate nature of catalytic responses over repeated operations.72 The results indicated that the CaFe2O4 nanocatalyst could potentially eliminate azo dyes from wastewater.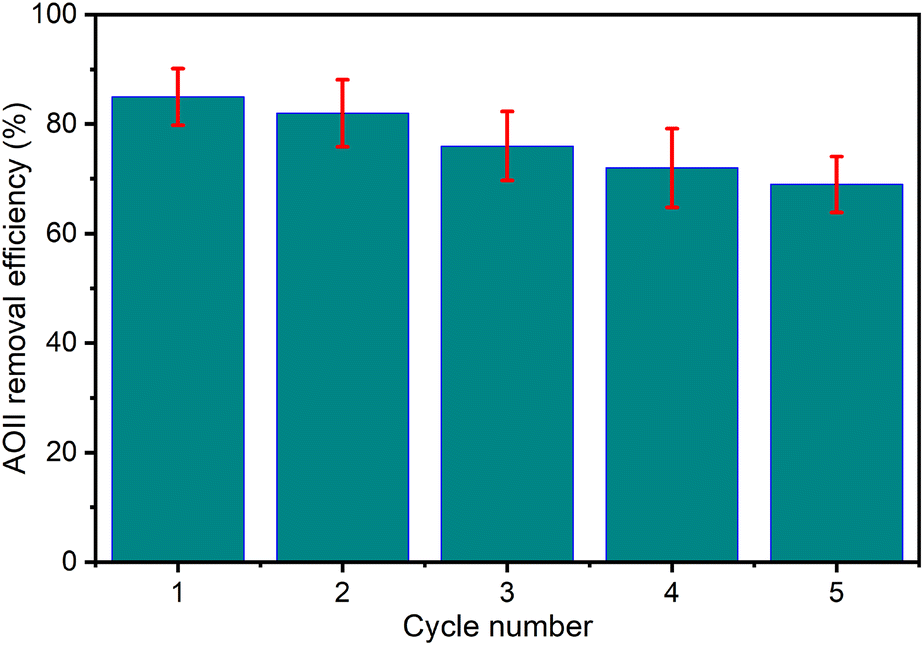 | ||
| Fig. 9 Reusability of CaFe2O4 nanoparticles for catalytic ozonation processes at solution pH 9, the catalyst dosage of 1.0 g L−1, initial AOII concentration of 200 mg L−1, reaction time of 40 min and temperature of 25 ± 2 °C. | ||
Conclusion
This study investigated the degradation of AOII via the CaFe2O4 nanocatalytic ozonation approach. Experimental results revealed the factors affecting AOII degradation efficiency and kinetics. O3 alone and CaFe2O4/O3 systems remove AOII color nearly perfectly in experiments. Degradation was found to depend on the solution pH. The degradation of AOII increased with pH 3 to 9, while pH 11 slightly decreased it. AOII concentration also affected the degradation efficiency. AOII degradation was also examined with CaFe2O4 nanocatalyst mass. The main catalyst portion was crucial to degradation, according to the results. As the amount of the nanocatalyst increased from 0.025 g L−1 to 1.0 g L−1, k values increased from 0.015 min−1 to 0.0361 min−1. However, increasing the amount of the catalyst to 1.5 and 2.0 g L−1 decreased the reaction rate constant to 0.031 min−1 and 0.0253 min−1, respectively. The findings also indicate that the efficiency of TOC removal decreased from 85% to 62% as the initial concentration of AOII increased from 100 to 500 mg L−1. Determining the reaction rate constant confirmed pseudo-first-order degradation kinetics. At pH 9, the rate constant (k) of the CaFe2O4/O3 system ranged from 0.025 min−1 to 0.0311 min−1, depending on catalyst quantity. Ozone alone had a rate constant of 0.0146 min−1 to 0.017 min−1 at the same pH. These results show that the CaFe2O4 nanocatalytic ozonation degrades AOII. Degradation efficiency and rate depended on pH, AOII concentration, and catalyst dosage. CaFe2O4 nanoparticles catalyzed degradation faster than ozone alone. The CaFe2O4/O3 system outperformed ozone at pH 9. The CaFe2O4 nanocatalytic ozonation process can remediate wastewater containing AOII and other azo dyes. Its use in water treatment and environmental remediation requires optimization and scaling-up research. This study illuminates catalytic ozonation kinetics and mechanisms and lays the groundwork for future research.Data availability
The data that has been used is confidential.Author contributions
Van Hung Hoang: conceptualization, methodology, software. Huu Tap Van: conceptualization, methodology, software. Huu Tap Van: data curation, writing – original draft. Thi Cuc Luu: data curation, writing – original draft. Thuy Linh Vi: visualization, investigation. Luong Thi Quynh Nga: visualization, investigation. Van Hung Hoang: supervision. Gio Serafin Ivan Jimenez Marcaida: conducted experiments, software, validation. Huu Tap Van: writing – review & editing. All authors provided critical feedback and helped shape the research, analysis, and manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Vietnam Ministry of Education and Training under project number: B2022-TNA-45.References
- B. Lellis, C. Z. Fávaro-Polonio, J. A. Pamphile and J. C. Polonio, Biotechnol. Res. Innov., 2019, 3, 275–290 CrossRef
.
- T. Islam, M. R. Repon, T. Islam, Z. Sarwar and M. M. Rahman, Environ. Sci. Pollut. Res. Int., 2023, 30, 9207–9242 CrossRef CAS PubMed
- R. Al-Tohamy, S. S. Ali, F. Li, K. M. Okasha, Y. A.-G. Mahmoud, T. Elsamahy, H. Jiao, Y. Fu and J. Sun, Ecotoxicol. Environ. Saf., 2022, 231, 113160 CrossRef CAS PubMed
- K. S. Bharathi and S. T. Ramesh, Appl. Water Sci., 2013, 3, 773–790 CrossRef
- Y. Ning, Y. Yang, C. Wang, T. Ngai and Z. Tong, Chem. Commun., 2013, 49, 8761–8763 RSC
- T. A. Aragaw and F. M. Bogale, Front. Environ. Sci., 2021, 9, 764958 CrossRef
- P. De Luca, P. Foglia, C. Siciliano, J. B. Nagy and A. Macario, Environments, 2019, 6, 101–129 CrossRef
- J. I. Mary Jency, L. Gowrisankar, J. Krishnaveni and N. Renugadevi, J. Environ. Treat. Tech., 2020, 9, 218–223 Search PubMed
- H. F. Liu, B. J. Zheng, A. Q. Dao, S. T. Yi, D. S. Jiang, C. Y. Fu and F. Xiao, Mater. Res. Innovations, 2014, 18, S2-707–S2-710 CrossRef
- I. Gültekin and N. H. Ince, Ultrason. Sonochem., 2006, 13, 208–214 CrossRef PubMed
- M. H. Beak, C. O. Ijagbemi and D. S. Kim, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 623–629 CrossRef CAS PubMed
- A. Chaturvedi, B. N. Rai, R. S. Singh and R. P. Jaiswal, Rev. Chem. Eng., 2022, 38, 617–639 CrossRef CAS
- M. Torun, D. Aracagök and M. Kabalak, Hittite J. Sci. Eng., 2018, 5, 307–311 CrossRef
- Z. Y. Bai, Q. Yang and J. L. Wang, Int. J. Environ. Sci. Technol., 2016, 13, 483–492 CrossRef CAS
- C. A. T. Toro, J. L. A. Dagostin, É. de Castro Vasques, M. R. Spier, L. Igarashi-Mafra and T. L. P. Dantas, Can. J. Chem. Eng., 2020, 98, 2530–2544 CrossRef CAS
- Q. Dai, Z. Zhang, T. Zhan, Z.-T. Hu and J. Chen, ACS Omega, 2018, 3, 6506–6512 CrossRef CAS PubMed
- S. Shokrollahzadeh, M. Abassi and M. Ranjbar, Environ. Eng. Res., 2019, 24, 513–520 CrossRef
- A. D. Bokare, R. C. Chikate, C. V. Rode and K. M. Paknikar, Environ. Sci. Technol., 2007, 41, 7437–7443 CrossRef CAS PubMed
- Y. Meng, S. Yang and H. Li, ChemSusChem, 2022, 15, e202102581 CrossRef CAS PubMed
- H. Li, Z. Fang, R. L. Smith and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef
- S. Zhang, H. Ren, K. Fu, W. Cheng, D. Wu, C. Luo, S. Jiang, J. Li and M. Zhang, Front. Energy Res., 2022, 9, 1–8 Search PubMed
- R. Rajesh, E. Sujanthi, S. S. Kumar and R. Venkatesan, Phys. Chem. Chem. Phys., 2015, 17, 11329–11340 RSC
- S. Krishna, P. Sathishkumar, N. Pugazhenthiran, K. Guesh, R. V. Mangalaraja, S. Kumaran, M. A. Gracia-Pinilla and S. Anandan, RSC Adv., 2020, 10, 16473–16480 RSC
- B. Rodríguez-Cabo, I. Rodríguez-Palmeiro, R. Corchero, R. Rodil, E. Rodil, A. Arce and A. Soto, Water Sci. Technol., 2016, 75, 128–140 CrossRef PubMed
- N. H. Sulaiman, M. J. Ghazali, J. Yunas, A. Rajabi, B. Y. Majlis and M. Razali, Ceram. Int., 2018, 44, 46–50 CrossRef CAS
- H. T. Van, L. H. Nguyen, T. K. Hoang, T. P. Tran, T. A. Vo, T. T. Pham and X. C. Nguyen, Sep. Purif. Technol., 2019, 224, 431–442 CrossRef CAS
- S. Alhassan, M. Alshammari, K. Alshammari, T. Alotaibi, A. H. Alshammari, Y. Fawaz, T. A. M. Taha and M. Henini, Polymers, 2023, 15(9), 2223 CrossRef PubMed
- M. I. ur Rahman, H. M. Khan, M. N. Ashiq, M. U. Islam, S. A. Buzdar, I. Sadiq, S. Honey, Z. Batool, R. Sheikh, M. Zahid, M. A. Assiri, M. Imran and T. Alshahrani, RSC Adv., 2023, 13, 14461–14471 RSC
- S. Altaf, H. Ajaz, M. Imran, A. Ul-Hamid, M. Naz, M. Aqeel, A. Shahzadi, A. Shahbaz and M. Ikram, Appl. Nanosci., 2020, 10, 2113–2127 CrossRef CAS
- M. Goodarzi, S. Joukar, D. Ghanbari and K. Hedayati, J. Mater. Sci.: Mater. Electron., 2017, 28, 12823–12838 CrossRef CAS
- M. M. Tauber, G. M. Guebitz and A. Rehorek, Appl. Environ. Microbiol., 2005, 71, 2600–2607 CrossRef CAS PubMed
- F. J. Poblete, P. Corrochano and B. Cabañas, J. Phys. Org. Chem., 2009, 22, 546–549 CrossRef CAS
- A. Lopez, H. Benbelkacem, J. -S. Pic and H. Debellefontaine, Environ. Technol., 2004, 25, 311–321 CrossRef CAS PubMed
- M. Tokumura, T. Katoh, H. Ohata and Y. Kawase, Ind. Eng. Chem. Res., 2009, 48, 7965–7975 CrossRef CAS
- E. J. Smutny, M. C. Caserio and J. D. Roberts, J. Am. Chem. Soc., 1960, 82, 1793–1801 CrossRef CAS
- M. Harfenist, J. Org. Chem., 1963, 28, 1834–1837 CrossRef CAS
- D. R. Corbin, W. S. Willis, E. N. Duesler and G. D. Stucky, J. Am. Chem. Soc., 1980, 102, 5969–5971 CrossRef CAS
- J. Santrock, R. A. Gorski and J. F. O'Gara, Chem. Res. Toxicol., 1992, 5, 134–141 Search PubMed
- C.-C. Wu, W.-J. Huang and B.-H. Ji, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50, 1116–1126 Search PubMed
- P. Van Aken, N. Lambert, R. den Broeck, J. Degrève and R. Dewil, Environ. Sci.: Water Res. Technol., 2019, 5, 444–481 RSC
- S. Khuntia, S. K. Majumder and P. Ghosh, Ind. Eng. Chem. Res., 2013, 52, 318–326 CrossRef CAS
- R. Zhang, C. Zhang, X. Cheng, L. Wang, Y. Wu and Z. Guan, J. Hazard. Mater., 2007, 142, 105–110 CrossRef CAS PubMed
- J. Kaushal, M. Khatri and S. K. Arya, Clean. Eng. Technol., 2021, 2, 100083 CrossRef
- Y. Ma, L. Sun, R. Wang, Y. Gu, H. Xu and P. Lei, Molecules, 2022, 27(21), 7209 CrossRef CAS PubMed
- J. Roussy, M. Van Vooren and E. Guibal, J. Appl. Polym. Sci., 2005, 98, 2070–2079 CrossRef CAS
- J. T. Marois-Fise, A. Carabin, A. Lavoie and C. C. Dorea, Appl. Environ. Microbiol., 2013, 79, 2107–2109 CrossRef PubMed
- M. Zhang, Q. Yao, W. Guan, C. Lu and J.-M. Lin, J. Phys. Chem. C, 2014, 118, 10441–10447 CrossRef CAS
- Advanced Ozonation Processes for Water and Wastewater Treatment: Active Catalysts and Combined Technologies, ed. H. Cao, Y. Xie, Y. Wang and J. Xiao, The Royal Society of Chemistry, 2022 Search PubMed
- X. Li, L. Fu, F. Chen, S. Zhao, J. Zhu and C. Yin, Catalysts, 2023, 13(2), 342 CrossRef CAS
- J. S. Park, H. Choi and K. H. Ahn, Water Sci. Technol., 2003, 47, 179–184 CrossRef CAS PubMed
- Y. Yuan, M. Mortazavi, S. Garg, J. Ma and T. D. Waite, ACS ES&T Engg, 2022, 2, 210–221 Search PubMed
- Y. Wang, L. Gai, W. Ma, H. Jiang, X. Peng and L.-C. Zhao, Ind. Eng. Chem. Res., 2015, 54, 2279–2289 CrossRef CAS
- R. B. Pachwarya, E. N. Hidayah, R. Kashyap, A. Ramanathan, R. S. Meena, P. K. Meena and S. Q. Z. Nisa, International Journal of Eco-Innovation in Science and Engineering, 2022, 1, 41–44 CrossRef
- K. El Hassani, D. Kalnina, M. Turks, B. H. Beakou and A. Anouar, Sep. Purif. Technol., 2019, 210, 764–774 CrossRef CAS
- B. Zhu, Q. Li, Y. Gao, Y. Yan, Y. Zhu and L. Xu, Plasma Sci. Technol., 2022, 25, 15505 CrossRef
- S. Liakou, S. Pavlou and G. Lyberatos, Water Sci. Technol., 1997, 35(4), 279–286 CrossRef CAS
- H. Mahdizadeh, A. Nasiri, M. A. Gharaghani and G. Yazdanpanah, MethodsX, 2020, 7, 101118 CrossRef CAS PubMed
- K. Okitsu, K. Iwasaki, Y. Yobiko, H. Bandow, R. Nishimura and Y. Maeda, Ultrason. Sonochem., 2005, 12, 255–262 CrossRef CAS PubMed
- V. Vaiano and I. De Marco, Separations, 2023, 10(4), 230 CrossRef CAS
- O. Olea-Mejia, M. Fernández-Mondragón, C. Barrera-Díaz and A. Cabral-Prieto, Int. J. Photoenergy, 2017, 9314764 Search PubMed
- R. P. de Brito, H. J. I. Filho, L. G. Aguiar, M. A. K. de Alcântara, A. A. G. Siqueira and P. C. M. Da Rós, Ind. Eng. Chem. Res., 2019, 58, 9855–9863 CrossRef CAS
- D. V. Franco, W. F. Jardim, J. F. C. Boodts and L. M. Da Silva, Clean: Soil, Air, Water, 2008, 36, 34–44 CAS
- G. Munuera, V. Rives-Arnau and A. Saucedo, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 736–747 RSC
- J. Wang and Z. Bai, Chem. Eng. J., 2017, 312, 79–98 CrossRef CAS
- X. Long, Z. Yang, H. Wang, K. Peng, Q. Zeng and X. Aihua, Ind. Eng. Chem. Res., 2012, 51, 11998–12003 CrossRef CAS
- B. Gao, H. Zhang, F. Wang, X. Xiong, K. Tian, Y. Sun and T. Yu, Catalysts, 2019, 9, 241 CrossRef
- Y. Dong, K. He, B. Zhao, Y. Yin, L. Yin and A. Zhang, Catal. Commun., 2007, 8, 1599–1603 CrossRef CAS
- X. Zhang, B. Zhou, S. Yin, Y. Wang, X. Zhang, Q. Meng, F. Meng, C. Wei and G. Wen, Vacuum, 2022, 206, 111495 CrossRef CAS
- F. Bagheri and N. Chaibakhsh, Sep. Sci. Technol., 2021, 56, 3022–3032 CrossRef CAS
- J. Lu, X. Wei, Y. Chang, S. Tian and Y. Xiong, J. Chem. Technol. Biotechnol., 2016, 91, 985–993 CrossRef CAS
- T. H. Nguyen, X. H. Nguyen, T. G. Do and L. H. Nguyen, Chem. Eng. J. Adv., 2023, 16, 100550 CrossRef CAS
- Y. Meng, Y. Jian, J. Li, H. Wu, H. Zhang, S. Saravanamurugan, S. Yang and H. Li, Chem. Eng. J., 2023, 452, 139477 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |