
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted chemoselective synthesis and photophysical properties of 2-arylazo-biphenyl-4-carboxamides from hydrazonals†
Abdulrahman M. Alazemi *a,
Kamal M. Dawood*b,
Hamad M. Al-Matara and
Wael M. Tohamyac
*a,
Kamal M. Dawood*b,
Hamad M. Al-Matara and
Wael M. Tohamyac
aChemistry Department, Faculty of Science, University of Kuwait, P.O. Box 5969, Safat 13060, Kuwait. E-mail: a.alazmi@ku.edu.kw; Fax: +965 24816482
bDepartment of Chemistry, Faculty of Science, Cairo University, Giza 12613, Egypt. E-mail: kmdawood@sci.cu.edu.eg; Fax: +202 35727556
cOrganometallic and Organometalloid Chemistry Department, National Research Centre, Cairo, Egypt
First published on 22nd August 2023
Abstract
The reaction of 3-oxo-2-arylhydrazonopropanals with acetoacetanilide in an equimolar ratio, under DBU/1,4-dioxane/microwave irradiation reaction conditions, resulted in chemoselective formation of 4-arylazo-5-hydroxy-benzamide derivatives. The structures of the obtained biphenyl-4-carboxamides were characterized by several spectroscopic techniques including IR, 1H- and 13C-NMR, MS and HRMS, and X-ray single crystals of three examples. The photophysical properties of the new products were also evaluated, with a particular focus on their absorption and emission spectra, which provided valuable information regarding their optical properties. The new compounds emitted 513–549 nm green fluorescence in acetone solution under UV irradiation.
1. Introduction
Salicylanilides (2-hydroxy-N-phenylbenzamides) have attracted substantial research interest from the field of medicinal chemistry owing to their exceptional bioactivities. They exhibit anti-inflammatory, antibacterial, antifungal activities,1–9 are antiviral agents against various viral pathogens such as coronaviruses (MERS-CoV & SARS-CoV), West Nile virus (WNV), Hepatitis-C virus (HCV), Japanese encephalitis virus (JEV), human rhinovirus (HRV), dengue virus (DENV), yellow fever virus (YFV)10–12 and are used for treatment of tuberculosis.13–15The Salicylanilide family is useful in veterinary and human medicines. For example, as shown in Fig. 1, Oxyclozanide,16 Closantel17 was discovered to be a powerful antistaphylococcal,18–21 Rafoxanide22 was successful for the treatment of F. hepatica infection in cattle and sheep, and Niclosamide, was approved by FDA as an antihelmintic drug,23,24 highly potent for treatment of COVID-19 and SARS-CoV-2 (ref. 25–28) and has high anti-cancer activity.29–32
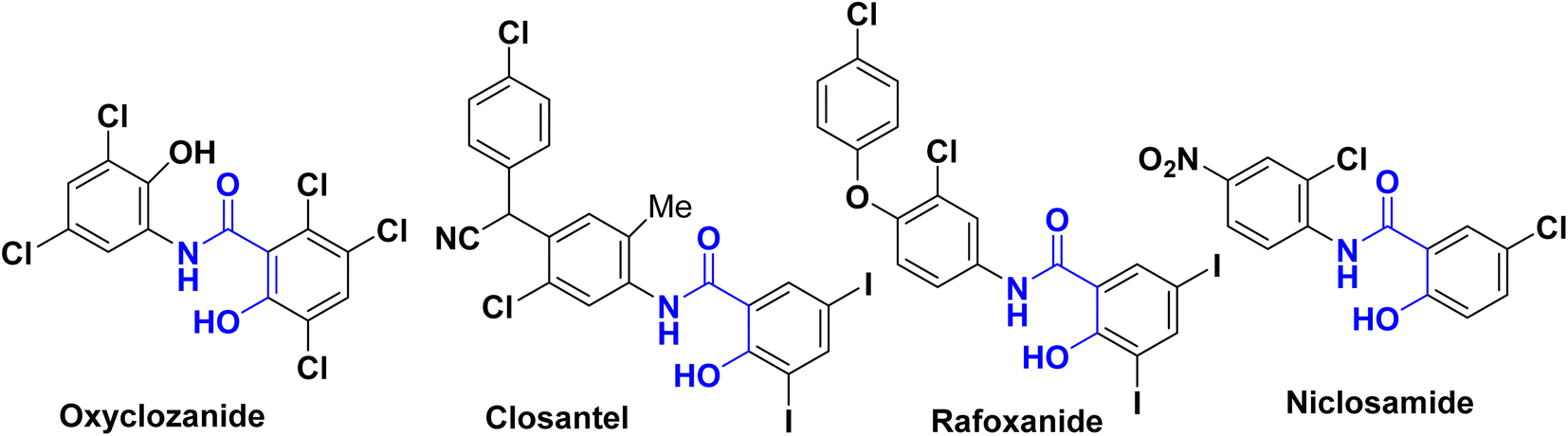 | ||
| Fig. 1 Examples of bioactive salicylanilides. | ||
The classical synthesis of salicylanilides was achieved by condensing the appropriate salicylic acid derivatives with aromatic amines in the presence of dehydrating agents such as phosphorus oxychloride,33 phosphorus trichloride,34–36 carbonyldiimidazole (CDI),37 1-(3-dimethylamino-propyl)-3-ethylcarbodiimide (EDC),38 thionyl chloride,39,40 P2O5,41 or N,N′-dicyclohexylcarbodiimide (DCC).42 High boiling solvents such as xylene, chlorobenzene, or toluene were used or solvent-free conditions were used. These processes were fairly difficult and lengthy due to the low reactivity of the carboxylic acids.43 Aminolysis of phenyl salicylate in chlorobenzene or xylene was also used when the solvent offered a significant challenge in the separation process of the product.4,13,36 Thus, we concluded that these procedures had many obstacles, such as complicated operations, expensive reagents, low productivity and long reaction times. On the other hand, aryl azobenzene systems were of valuable importance in various fields, such as dye industries,44 liquid crystals,45,46 material science,47,48 chemosensors,49,50 polymers,51 photochemical switches52 and pharmaceutical products.53–55 Microwave radiation produces a rapid intense heating of polar compounds, resulting in significantly shorter reaction times, high reaction yields and a cleaner technique.
The fundamental advantage of microwave heating is its significant energy savings through its instantaneous “in-core” heating of substances in a selective and homogeneous manner with a significantly shorter response time.56–58 The straightforward amidation of carboxylic acids was successfully modified by microwave activation.59–65 Therefore, in continuation of our research interest in microwave-assisted organic synthesis,66–77 herein we re-investigated our previous work on a supplemental reaction of 3-oxo-2-arylhydrazonopropanals 1 with acetoacetanilide 2,68 by optimization of reaction conditions (components' molar ratios, solvents, bases and activation modes) towards chemoselective construction of the 2-(arylazo)-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide derivatives 3 instead of the cinnoline derivatives 4 (Scheme 1) and the photophysical properties of the obtained biphenyl-4-carboxamides were evaluated. The structures of the new products were characterized by IR, 1H- and 13C-NMR, MS and HRMS spectral data as well as X-ray single crystallography of selected examples. The photoluminescent spectrum and UV-Vis absorption were also studied.
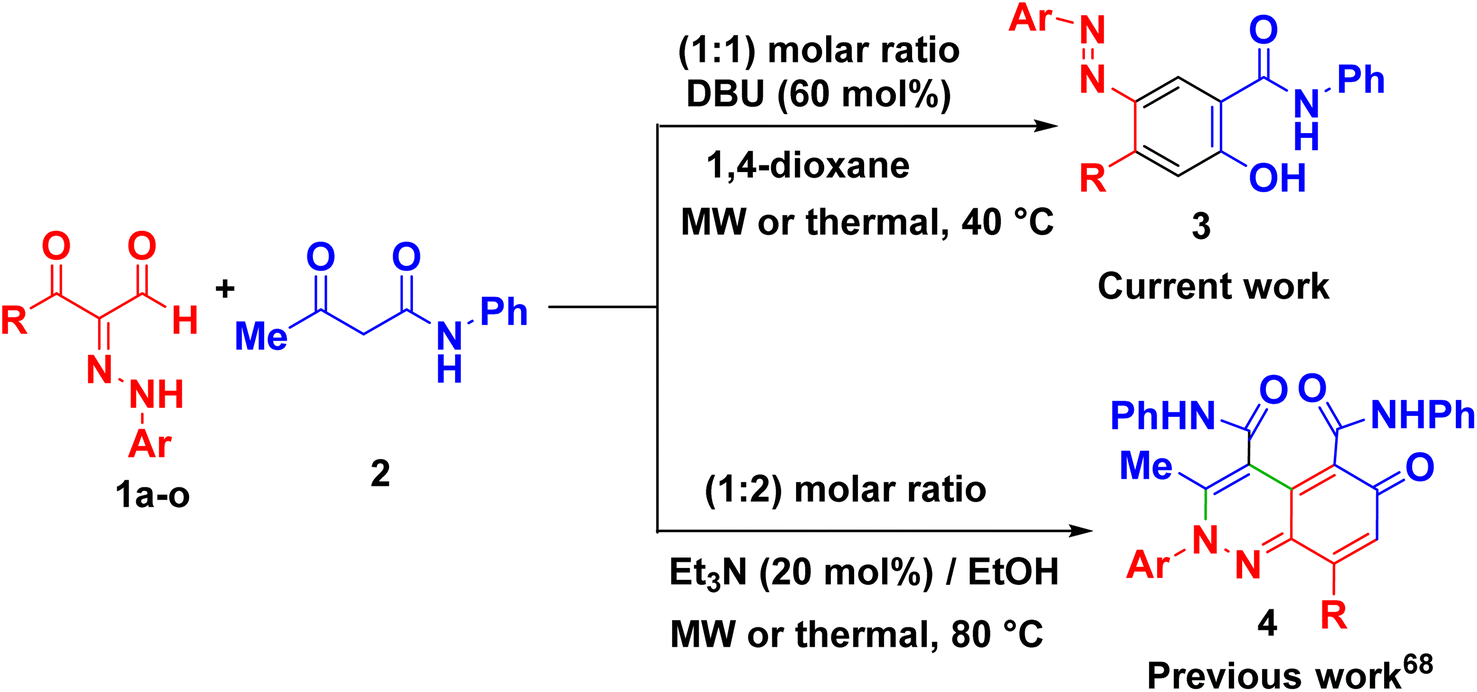 | ||
| Scheme 1 Current and previous work on reaction of 3-oxo-2-arylhydrazonopropanals 1 with acetoacetanilide 2. | ||
2. Results and discussion
The key starting substrates 3-oxo-2-arylhydrazonopropanals 1a–o were prepared following the literature procedures.78,79 A representative reaction example was conducted by heating an equimolar ratio of 3-oxo-2-(4-bromophenyl)hydrazonopropanal 1a and acetoacetanilide 2 under various reaction conditions (different solvents, different bases and different heating modes) as shown in Table 1 and Scheme 2. Thus, the effect of solvents (such as methanol, ethanol, hexane, DMF, chloroform, toluene, 1,4-dioxane) on the reaction between 1a and 2 using DBU (60 mol%) as an organic base, under thermal heating and microwave irradiation at 40 °C was carefully studied and the reaction path was checked by TLC. Conducting the reaction in methanol under thermal heating for 2 h afforded a mixture of the 4-bromphenylazo-5-hydroxy-benzamide derivative 3a and the 2,6-dihydrocinnoline-4,5-dicarboxamide 4a in 50% and 40% yields, respectively, compared with 65% and 28% yields after for 3 min of microwave irradiation (entry 1, Table 1). Repeating the same reaction using ethanol, hexane, DMF, chloroform, toluene, 1,4-dioxane, as reaction solvents, led to the formation of a mixture of 3a and 4a in 45–60% and 36–42%, respectively, under thermal condition compared with 52–68% (3a) and 25–32% (4a) yields under microwave condition (entries 2–6, Table 1). However, 1,4-dixane was found to be the proper reaction solvent (at 40 °C and DBU (0.6 mmol)) where it exclusively directed the reaction towards the formation of 3e chemoselectively (95% yield) under microwave irradiation compared with a mixture of 3a (in 84% yield) and 4a (in 10% yield), respectively, under thermal heating (entry 7, Table 1). When the same reaction was repeated under typical reaction factors but at room temperature a mixture of the products 3a and 4a were obtained in 48% and 40% yields under conventional condition and in 60% and 25% yields under microwave, respectively (entry 8, Table 1). Then, the same reaction condition was repeated using different concentrations of DBU (50 mol% and 80 mol%), at 40 °C, interestingly only 3a was chemoselectively isolated in 88 and 90% yields, respectively, under microwave conditions compared with about 80% (3a) and 14% (4a) under thermal heating (entries 9 and 10, Table 1).| Entry | Base (mol%) | Solvent | Temp. (°C) | Thermal (2 h) | MW (3 min) | ||
|---|---|---|---|---|---|---|---|
| Yieldb | Yieldb | ||||||
| 3a | 4a | 3a | 4a | ||||
| a Reaction condition: 1a (0.5 mmol), acetoacetanilide (0.5 mmol), solvent (6 mL) and base (50–80 mol%), (6 mL), at 40 °C for 2 h, microwave irradiations at 40 °C (200 W) for 3 min.b Isolated yields of 3a and 4a. | |||||||
| 1 | DBU (60%) | Methanol | 40 | 50 | 40 | 65 | 28 |
| 2 | DBU (60%) | Ethanol | 40 | 55 | 38 | 67 | 25 |
| 3 | DBU (60%) | Hexane | 40 | 45 | 42 | 52 | 32 |
| 4 | DBU (60%) | DMF | 40 | 60 | 36 | 65 | 25 |
| 5 | DBU (60%) | Chloroform | 40 | 58 | 38 | 68 | 25 |
| 6 | DBU (60%) | Toluene | 40 | 50 | 40 | 60 | 28 |
| 7 | DBU (60%) | 1,4 dioxane | 40 | 84 | 10 | 95 | Trace |
| 8 | DBU (60%) | 1,4 dioxane | rt | 48 | 40 | 60 | 25 |
| 9 | DBU (50%) | 1,4 dioxane | 40 | 80 | 14 | 88 | Trace |
| 10 | DBU (80%) | 1,4 dioxane | 40 | 82 | 14 | 90 | Trace |
| 11 | Pyridine (60%) | 1,4 dioxane | 40 | 33 | 54 | 42 | 45 |
| 12 | DABCO (60%) | 1,4 dioxane | 40 | 38 | 50 | 45 | 42 |
| 13 | Et3N (60%) | 1,4 dioxane | 40 | Trace | 25 | Trace | 34 |
| 14 | KOH (60%) | 1,4 dioxane | 40 | 48 | 40 | 55 | 35 |
| 15 | Cs2CO3 (60%) | 1,4 dioxane | 40 | 52 | 30 | 68 | 22 |
| 16 | Na2CO3 (60%) | 1,4 dioxane | 40 | 50 | 40 | 60 | 28 |
| 17 | NaHCO3 (60%) | 1,4 dioxane | 40 | 65 | 20 | 72 | 18 |
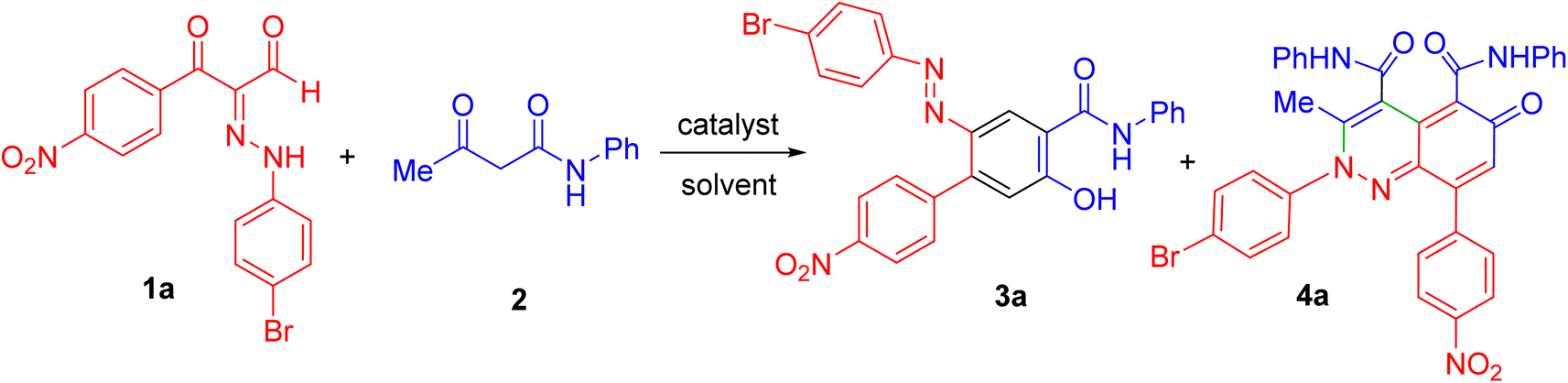 | ||
| Scheme 2 Annulation of the arylhydrazonopropanal 1a with acetoacetanilide 2. | ||
Changing the catalytic base also affected the chemoselectivity, where replacing DBU with pyridine and DABCO (60 mol% all), in dioxane at 40 °C resulted in the formation of a mixture, in all cases, of the carboxamide 3a and cinnoline 4a in yields varied between 33–48% and 40–54% under thermal heating and between 42–55% and 35–45% under microwave condition, respectively (entries 11, 12, Table 1). Using Et3N instead of DBU (60 mol%) in dioxane at 40 °C led to the selective formation of the cinnoline derivative 4a in low yields; 25% and 34% under thermal and microwave conditions, respectively (entries 13, Table 1). Some inorganic bases were also tested for this reaction by applying the same condition as above. Thence, KOH, Cs2CO3, Na2CO3 and NaHCO3 (60 mol% each) was employed separately where in all cases a mixture of the two products 3a and 4a were obtained in variable yields; 48–65% (3a) and 30–40% (4a), respectively under a thermal mode but in 55–72% (3a) and 18–35% (4a) under microwave mode, respectively (entries 14–17, Table 1). Therefore, the highest chemoselectivity towards the formation of the pure 3a structure was established using the reaction system; 1a/2/base/solvent/temperature/heating mode; 1 mmol/1 mmol/0.6 mmol DBU/1,4-dioxane/40 °C/microwave irradiation.
It was concluded that the outcomes of microwave irradiation were established by high productivity and chemoselectivity compared with thermal heating mode. The chemical constitution of the reaction product 3a was confirmed from its microanalytical and all possible spectroscopic analyses (IR, 1H- and 13C-NMR, MS and HRMS). Spectral data and the melting point of structure of 4a were typical of that previously published by our group.68 Microwave works faster in polar solvents where it is well known that polar solvents absorb microwave energy faster and convert it into heat leading to bulk heating and simultaneously elevating the reaction temperature, in contrast with thermal conductive heating mode.
In the following, the best-optimized condition above was applied in the assembly of a library of 5-arylazo-2-hydroxybenzamides 3a–o as described in Table 2 and Scheme 3. Thus, conducting the reaction of various 3-oxo-2-arylhydrazonopropanal 1a–o and acetoacetanilide 2 and molar ratios of the components and reaction conditions were as follows; 1a–o/2/base/solvent/temperature; 0.5 mmol/0.5 mmol/0.3 mmol DBU/1,4-dioxane (6 mL)/40 °C, both under thermal as well as microwave irradiating conditions. Again, heating under microwave conditions led to an increase in the chemoselectivity towards the formation of the products 3a–o compared with conventional heating, where the yields dramatically enhanced when conventional heating was switched to microwave heating and compounds 3a–c as well as 3i–j were obtained as the sole products (entries 1, 2, 3, 9, 10, Table 2). In these examples, the R groups of the respective hydrazonal substrates 1 had highly electron-withdrawing substituents (NO2 or F). In the other examples, microwave irradiation of compounds 1 having groups other than NO2 or F resulted in the formation of a mixture of compounds 3 and 4 mostly in 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratios of total >90% yields. In contrast, conventional heating led, in all cases, to the production of a mixture of 3a–c and 4a–c in 8:1 ratios of total >90% yields. The presence of the electron-donating (MeO) group led to the least chemoselectivity, where products 3 and 4 were obtained in almost 2:1 ratio under both thermal as well as microwave conditions (entries 11, 12, Table 2). Thus, chemoselectivity was encountered only under microwave for the hydrazonals 1 having highly electron-withdrawing substituents on the R moiety. All structures of the isolated products were thoroughly characterized using spectral analyses as well as X-ray single crystals of selected compounds (3f, 3h and 3n) (Fig. 2).80 Mechanistically, formation of the 4-arylazo-5-hydroxybenzamide derivatives 3a–o and the 2,6-dihydrocinnoline-4,5-dicarboxamides 4a–o proceeded in a tandem pathway as described in Scheme 4.
:1 ratios of total >90% yields. In contrast, conventional heating led, in all cases, to the production of a mixture of 3a–c and 4a–c in 8:1 ratios of total >90% yields. The presence of the electron-donating (MeO) group led to the least chemoselectivity, where products 3 and 4 were obtained in almost 2:1 ratio under both thermal as well as microwave conditions (entries 11, 12, Table 2). Thus, chemoselectivity was encountered only under microwave for the hydrazonals 1 having highly electron-withdrawing substituents on the R moiety. All structures of the isolated products were thoroughly characterized using spectral analyses as well as X-ray single crystals of selected compounds (3f, 3h and 3n) (Fig. 2).80 Mechanistically, formation of the 4-arylazo-5-hydroxybenzamide derivatives 3a–o and the 2,6-dihydrocinnoline-4,5-dicarboxamides 4a–o proceeded in a tandem pathway as described in Scheme 4.
| Entry | Products | R | Ar | Thermal (2 h) | MW (3 min) | ||
|---|---|---|---|---|---|---|---|
| Yieldb | Yieldb | ||||||
| 3 | 4 | 3 | 4 | ||||
| a Reaction condition: 1a–o (0.5 mmol), acetoacetanilide 2 (0.5 mmol), DBU (0.3 mmol) in 1,4-dioxane (6 mL), at 40 °C for 2 h (thermal heating), and 40 °C (200 W) for 3 min (microwave irradiation).b Isolated yields of 3 and 4. | |||||||
| 1 | 3a/4a | 4-NO2C6H4 | 4-BrC6H4 | 84 | 10 | 95 | Trace |
| 2 | 3b/4b | 4-NO2C6H4 | 4-ClC6H4 | 80 | 13 | 93 | Trace |
| 3 | 3c/4c | 4-NO2C6H4 | 3-ClC6H4 | 78 | 16 | 90 | Trace |
| 4 | 3d/4d | 4-NO2C6H4 | C6H5 | 72 | 20 | 83 | 10 |
| 5 | 3e/4e | 4-ClC6H4 | C6H5 | 62 | 28 | 70 | 22 |
| 6 | 3f/4f | 4-ClC6H4 | 4-BrC6H4 | 74 | 18 | 86 | 8 |
| 7 | 3g/4g | 4-BrC6H4 | 4-ClC6H4 | 66 | 25 | 75 | 16 |
| 8 | 3h/4h | 4-BrC6H4 | 4-BrC6H4 | 67 | 25 | 75 | 18 |
| 9 | 3i/4i | 4-FC6H4 | 4-ClC6H4 | 75 | 18 | 88 | Trace |
| 10 | 3j/4j | 4-FC6H4 | 4-BrC6H4 | 76 | 18 | 88 | Trace |
| 11 | 3k/4k | 4-OMeC6H4 | 4-BrC6H4 | 52 | 38 | 60 | 28 |
| 12 | 3l/4l | 4-OMeC6H4 | o-NO2C6H4 | 55 | 35 | 64 | 24 |
| 13 | 3m/4m | C6H5 | 4-ClC6H4 | 62 | 27 | 70 | 20 |
| 14 | 3n/4n | C6H5 | 4-BrC6H4 | 60 | 30 | 70 | 18 |
| 15 | 3o/4o | C6H5 | o-NO2C6H4 | 70 | 20 | 83 | 10 |
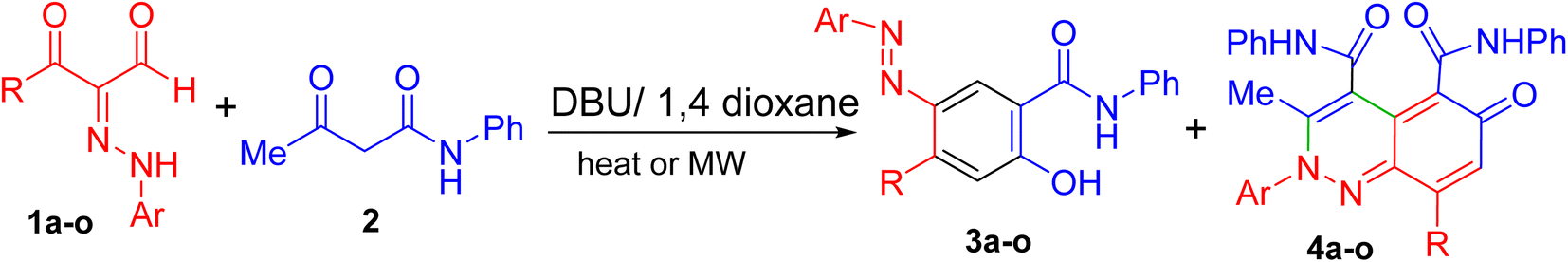 | ||
| Scheme 3 Annulation of arylhydrazonopropanals 1a–o with acetoacetanilide 2. | ||
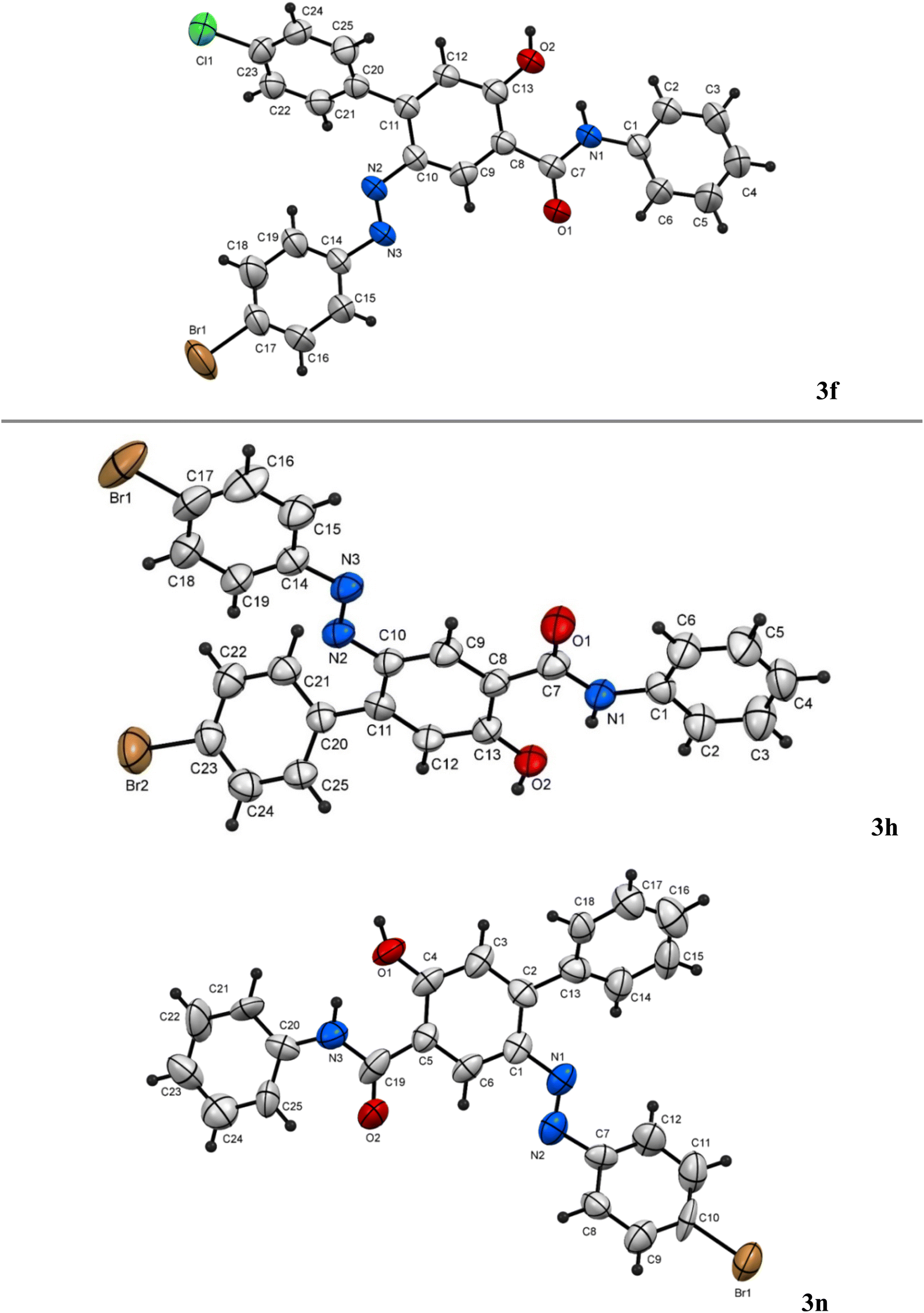 | ||
| Fig. 2 X-ray single crystals of compounds 3f, 3h and 3n obtained from diffraction data. | ||
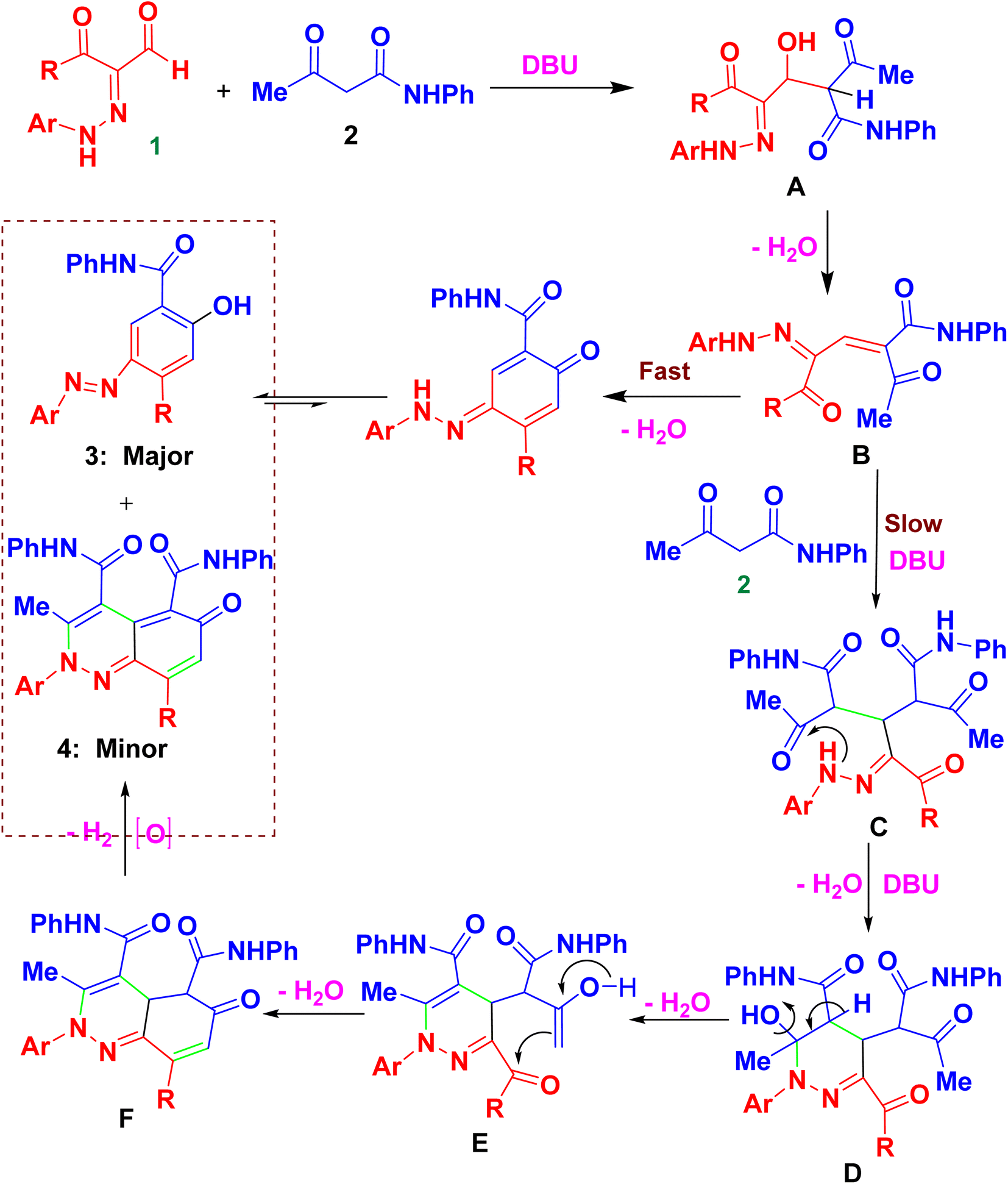 | ||
| Scheme 4 A proposed mechanism for the chemoselective formation of 3 and 4. | ||
2.1. Photophysical studies
The photophysical properties of the synthesized azodye derivatives 3a–o revealed distinct absorption and emission characteristics. Fig. 3 showed how the substituents (R1 and R2) in the molecular structure affected the absorption and emission bands. We studied the azodye photophysical behaviors by systematically changing these substituents. Understanding these structure–property correlations allows organic molecules to be used in optoelectronic devices and sensor platforms by changing their optical properties. | ||
| Fig. 3 Molecular structures of the azodyes 3a–o with various substituents (R1 and R2). | ||
All photophysical characteristic data were displayed and applied to determine the maximum wavelength absorption (λmax/abs), maximum molar absorptivity (εmax), and maximum wavelength emission (λmax/em) for all the azodyes as described in Table 3. The Ultraviolet-Visible (UV-Vis) spectra (Fig. 4a) indicated absorption bands ranging between 444 and 476 nm. Notably, compound 3o had the largest absorption maxima in this range, indicating that it has superior absorption properties in comparison to the other derivatives. The molar absorption coefficients for the azodyes 3a, 3b, 3i, 3j, 3f, 3m, and 3o were calculated as 20569, 30036, 22908, 25639, 30432, 12852, and 240212 M−1 cm−1, respectively. The greater molar absorptivity (ε) obtained for the azodye 3o (240212 M−1 cm−1) with substituents R1 = 2-NO2 and R2 = H compared to the other azodyes, suggested that this particular chemical configuration possessed superior light absorption capabilities. The inclusion of the nitro group (NO2) at the R1 in the ortho-position was likely to introduce strong electron-withdrawing effects due to the inductive effect (–I), which resulted in a more effective interaction between the dye and the incident light. This increased number implies that the azodye 3o had a better capacity to absorb photons and transform them into the excited states, making it more appropriate for the applications that require a strong light absorption, such as photovoltaic devices and light-harvesting systems.
| Azodyes | Absorptiona, λmax/abs (nm) | εmax (M−1 cm−1) | Emissionb, λmax/em (nm) | Stokes shiftsc (nm) |
|---|---|---|---|---|
| a Determined in acetone at room temperature when the concentration of each compound is 1.0 × 10−5 M.b Excited at the longest wavelength of the absorption maxima.c Stokes shifts are provided as wavelength differences, Δλmax = λmax(em) − λmax(ex). | ||||
| 3a | 475 | 20569 |
551 | 76 |
| 3b | 472 | 30036 |
549 | 77 |
| 709 | 237 | |||
| 3f | 476 | 30432 |
552 | 76 |
| 3i | 475 | 22908 |
549 | 74 |
| 3j | 476 | 25639 |
553 | 77 |
| 3m | 444 | 12852 |
513 | 69 |
| 3o | 494 | 240212 |
576 | 82 |
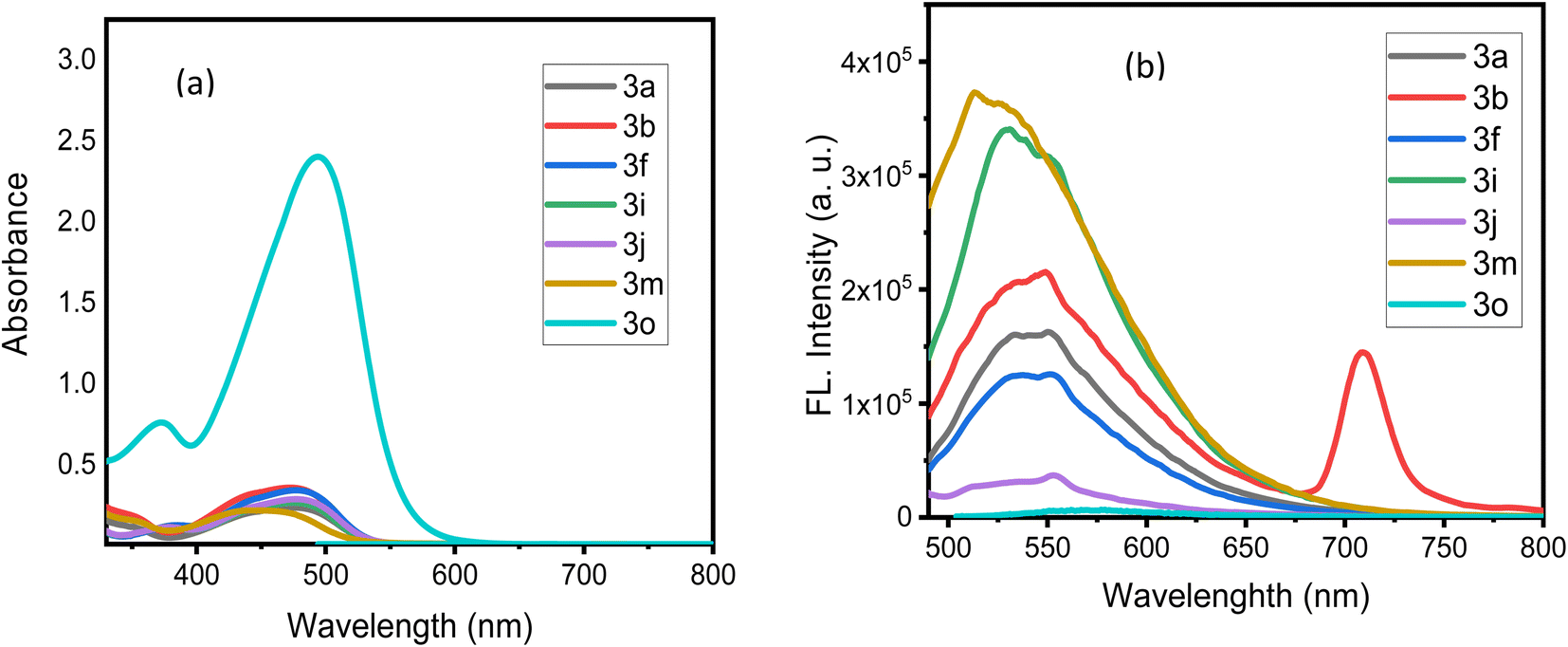 | ||
| Fig. 4 (a) UV-Vis spectra of the azodye derivatives 3a–o; (b) emission spectra of the azodye derivatives 3a–o recorded in acetone. | ||
In the emission spectra (Fig. 4b), compounds 3a, 3f, 3i, 3j, 3m, and 3o had comparable emission bands between 513–549 nm. This broad range showed that the emission properties of these compounds were comparable, with emission maxima between 520 and 540 nm. These emission bands were probably a result of similar photophysical processes that occurred within these substances. Notably, compound 3b, which contains an electron-withdrawing NO2-group, exhibited a unique emission behavior. It displayed a bathochromic (red) shift with an emission maximum at 709 nm, and a Stokes shift that was notably large at 237 nm (Table 3). The presence of the electron-withdrawing group, which greatly affected the electronic structure and energy levels of the molecule, was responsible for the dramatic red shift observed in compound 3b.
Overall, the presence of the electron-withdrawing NO2-group in compound 3b resulted in an extremely large Stokes shift due to a considerable red shift in the emission wavelength. These results demonstrated the significance of molecular structure and substitution patterns in modifying the photophysical characteristics of the azodyes.
We examined the absorption and emission spectra of the azodye 3o in various organic solvents, such as acetone, dimethyl sulfoxide (DMSO), ethyl acetate (EtOAc), tetrahydrofuran (THF), acetonitrile (MeCN), and chloroform (CHCl3). The results obtained are shown in Table 4 and Fig. 5 and 6. The broad band detected between 483 and 494 nm was due to an n–π* electronic transition in the azoaromatic chromophore and intramolecular charge transfer. In addition, a shoulder in the region of 371–379 nm was noticed, which was indicative of the azobenzene molecule's easier π–π* absorption.
| Solvent | Absorptiona, λmax/abs | εmax (M−1 cm−1) | Emissionb, (λmax/em) |
|---|---|---|---|
| a Concentration of 3o 1.0 × 10−5 M in different solvents.b Excitations were executed at or near the wavelength position of absorption maxima. | |||
| DMSO | 494 | 225212 |
580 |
| CH3CN | 489 | 227258 |
574 |
| Acetone | 494 | 239836 |
576 |
| EtOAc | 486 | 73652 |
569 |
| CHCl3 | 483 | 157062 |
567 |
| THF | 488 | 162157 |
569 |
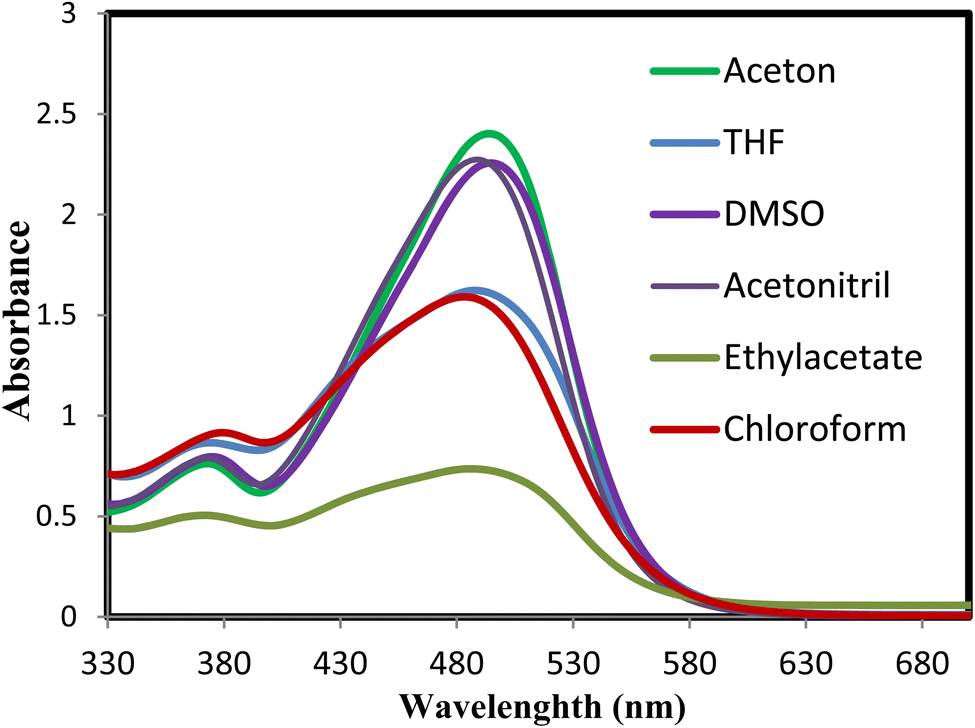 | ||
| Fig. 5 Absorption and emission spectra of dye 3o in different organic solvents (acetone, DMSO, EtOAc, THF, MeCN, and CHCl3). | ||
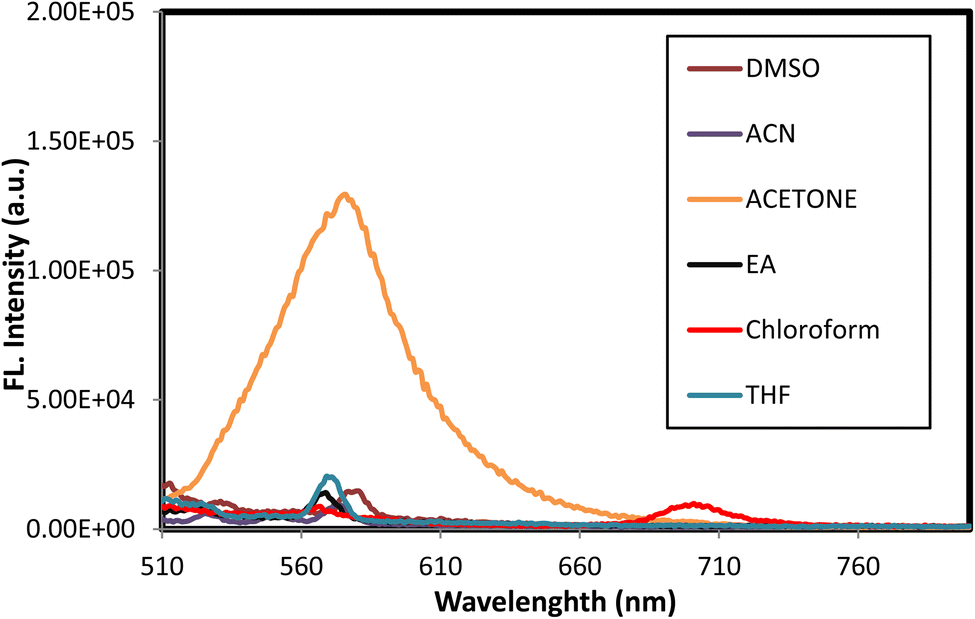 | ||
| Fig. 6 Effect of solvent polarity on the emission spectra of the azodye 3o. The emission maxima were shown for different solvents, including acetone, DMSO, EtOAc, THF, MeCN, and chloroform. | ||
Moreover, the effect of solvents on the azodye 3o was examined. The 11 nm red shift in the absorption maxima when changing from DMSO to chloroform illustrated the influence of solvent polarity on the photophysical parameters. Variations in dipole moment in the excited state or changes in hydrogen bonding strength in polar liquids could account for this shift. The existence of hydrogen bonding could greatly influence numerous photophysical properties.81 Additionally, excited-state intermolecular proton transfer (ESIPT)82 might also influence the photophysical characteristics of the azodye 3o. In contrast, the lower molar absorptivity values reported in other solvents such as chloroform (157062 M−1 cm−1), ethyl acetate (73652 M−1 cm−1), and THF (162157 M−1 cm−1) showed a relatively lower light absorption efficiency than acetone. These solvents might have weaker molecular connections or less suitable conditions for the absorption process of the azodye.
Regarding fluorescence characteristics, all the fluorescence maxima mentioned in Table 4 displayed a slight bathochromic shift as the solvent polarity increased, with the maximum emission wavelength in DMSO measured at 580 nm when stimulated at 494 nm. In the presence of DMSO, the extinction coefficient (ε) is approximately 1.4 times greater than that in the presence of chloroform, demonstrating that the polarity of the solvent influenced the intensity of emission. Higher intensities were recorded in polar solvents, as shown in Fig. 6.
The difference in molar absorptivity values between solvents implied that the choice of solvent could greatly affect the optical properties and absorption characteristics of the azodye 3o. This information was useful for comprehending the solvatochromic effect and customizing the photophysical properties of the azodye for applications requiring precise control of light absorption and energy conversion processes.
These results illustrated the effect of solvent polarity on the photophysical properties of the azodye 3o and scaled light on the unique absorption and emission features of the synthesized azodye derivatives.
3. Experimental section
3.1. Materials and methods
All starting substrates were highly pure and purchased from Sigma-Aldrich. Griffin apparatus was used for measuring the melting points and were uncorrected. Thin layer chromatography (TLC) was established using Polygram SIL G/UV 254 TLC plates, and visualization was done under ultraviolet light at 254 and 365 nm. IR spectra were conducted using KBr disks in PerkinElmer System 2000 FTIR spectrophotometer. 1H- and 13C-NMR spectra were recorded at 600 and 150 MHz, respectively, on a Bruker DPX 400 or 600 super-conducting NMR spectrometer, at ambient temperature using DMSO-d6 as a deuterated solvent and TMS as the internal standard (with chemical shifts given in parts per million (ppm)). Low-resolution electron impact mass spectrometry [MS (EI)] and high-resolution electron impact mass spectrometry [HRMS (EI)] were carried out using a high-resolution thermos spectrometer [GC-MS (DFS)] using a magnetic sector mass analyzer at 70.1 eV. Microwave (MW) experiments were conducted using a Discover LabMate CEM microwave instrument (300 W with CHEMDRIVER software; Matthews, NC). MW irradiation reactions were carried out in equipped closed pressured Pyrex tubes. The X-ray single-crystal data were performed involving a Bruker X8 Prospector and a Rigaku RAXISRAPID diffractometer, and the single-crystal data collection was conducted using Cu Kα radiation at ambient temperature. Solving and refining the structures was performed using the Bruker SHELXTL software package (refinement program-SHELXL97 and structure solution program-SHELXS-97). UV-Vis experiments were carried out on a Varian Cary 5 spectrometer from Agilent. Fluorescence assessments were conducted with Horiba Jobin Yvon-Fluoromax-4 equipped with a Time-Correlated Single Photon Counting (TCSPC) module.3.2. Synthesis of the biphenyl-carboxamides 3a–o and cinnoline derivatives 4a–o
:petroleum ether 40–60, 2:8 v/v).:petroleum ether 40–60 °C, 2:8 v/v) to give the corresponding biphenyl-carboxamides 3a–o and the cinnoline derivatives 4a–o.Notice: the cinnoline derivatives 4a–b and 4e–m were already characterized previously68 and all data of the of currently prepared cinnolines 4a–b and 4e–m were in complete accordance with the reported ones. However, the newly synthesized herein, cinnoline derivatives 4c–d and 4n–o are fully characterized at the end of this part.
3.2.2.1. 2-(4-Bromophenylazo)-5-hydroxy-4′-nitro-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3a). Orange solid, mp 266 °C; IR (KBr): ν/cm−1 3247, 3080, 1635, 1578, 1474, 1385; 1H NMR (DMSO-d6): δ = 6.49 (s, 1H, Ar-H), 7.01 (t, J = 7.5 Hz, 1H, Ar-H), 7.31 (t, J = 7.8 Hz, 2H, Ar-H), 7.46 (d, J = 9 Hz, 2H, Ar-H), 7.60 (d, J = 8.4 Hz, 2H, Ar-H), 7.70 (d, J = 7.8 Hz, 2H, Ar-H), 7.78–7.71 (m, 2H, Ar-H), 8.28 (d, J = 8.4 Hz, 2H, Ar-H), 8.61 (s, 1H, Ar-H), 9.60 (b, 1H, NH), 14.43 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.47, 119.27, 119.50, 120.87, 122.31, 122.96, 123.37, 128.80, 131.35, 132.02, 134.88, 140.08, 146.28, 146.85, 165.21, 165.69 (Ar-C), 176.09 (CO). MS (EI): 518.10 [M]+. HRMS: calcd for C25H17BrN4O4: 517.04331, found 516.0428.
3.2.2.2. 2-(4-Chlorophenylazo)-5-hydroxy-4′-nitro-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3b). Brown solid, mp 272–273 °C; IR (KBr): ν/cm−1 3246, 3044, 2922, 1636, 1578, 1548, 1475, 1455, 1287; 1H NMR (DMSO-d6): δ = 6.54 (s, 1H, Ar-H), 7.01 (t, J = 7.2 Hz, 1H, Ar-H), 7.33 (t, J = 7.8 Hz, 2H, Ar-H), 7.34–7.31 (m, 2H, Ar-H), 7.56–7.54 (m, 2H, Ar-H), 7.73 (d, J = 7.8 Hz, 2H, Ar-H), 7.78 (d, J = 9 Hz, 2H, Ar-H), 8.28 (d, J = 9 Hz, 2H, Ar-H), 8.65 (s, 1H, Ar-H), 9.43 (b, 1H, NH), 14.45 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.45, 119.29, 119.49, 122.32, 122.46, 122.97, 123.06, 128.82, 129.12, 131.37, 132.25, 134.87, 140.10, 144.54, 146.29, 146.88, 151.89, 165.28, 165.71 (Ar-C), 176.04 (CO). MS (EI): 472.1 [M]+. HRMS: calcd for C25H17ClN4O4: 472.0938, found 472.0933.
3.2.2.3. 2-(3-Chlorophenylazo)-5-hydroxy-4′-nitro-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3c). Orange solid, mp 235–236 °C; IR (KBr): ν/cm−1 3249, 3047, 1637, 1578, 1475, 1388; 1H NMR (DMSO-d6): δ = 6.46 (s, 1H, Ar-H), 7.01 (t, J = 7.5 Hz, 1H, Ar-H), 7.80 (t, J = 7.2 Hz, 2H, Ar-H), 7.50–7.47 (m, 2H, Ar-H), 7.56–7.54 (m, 2H, Ar-H), 7.72–7.71 (m, 2H, Ar-H), 7.80–7.77 (m, 2H, Ar-H), 8.29–8.27 (m, 2H, Ar-H), 8.62 (s, 1H, Ar-H), 9.53 (br. s, 1H, NH), 14.46 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.44, 119.28, 119.46, 122.30, 122.45, 122.97, 123.06, 123.38, 128.38, 128.82, 129.12, 131.37, 132.20, 134.80, 140.10, 144.53, 146.28, 146.89, 151.89, 165.36, 165.71 (Ar-C), 176.10 (CO) MS (EI): 472.11 [M]+. HRMS: calcd for C25H17ClN4O4: 472.0938, found 472.0941.
3.2.2.4. 5-Hydroxy-2-(4-nitrophenylazo)-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3d). Orange solid, mp 210–211 °C; IR (KBr): ν/cm−1 3247, 3067, 1639, 1594, 1578, 1446, 1325; 1H NMR (DMSO-d6): δ = 6.50 (s, 1H, Ar-H), 7.02 (t, J = 10.8 Hz, 1H, Ar-H), 7.31–7.35 (m, 3H, Ar-H), 7.44 (t, J = 11.4 Hz, 2H, Ar-H), 7.57–7.55 (m, 2H, Ar-H), 7.73–7.71 (m, 2H, Ar-H), 7.81–7.79 (m, 2H, Ar-H), 8.30–8.28 (m, 2H, Ar-H), 8.61 (s, 1H, Ar-H), 9.59 (b, 1H, NH) 14.55 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.19, 119.27, 119.33, 121.58, 122.25, 122.42, 122.86, 128.21, 128.81, 129.06, 131.39, 134.83, 140.18, 144.31, 146.26, 147.04, 153.18, 165.35, 165.85 (Ar-C), 175.71 (CO). MS (EI): 438.18 [M]+. HRMS: calcd for C25H18N4O4: 438.1323, found 438.1323.
3.2.2.5. 4′-Chloro-5-hydroxy-N-phenyl-2-(phenylazo)-[1,1′-biphenyl]-4-carboxamide (3e). Orange solid, mp 253–254 °C; IR (KBr): ν/cm−1 3266, 3090, 1640, 1585, 1476, 1389; 1H NMR (DMSO-d6): δ = 6.43 (s, 1H, Ar-H), 6.99 (t, J = 7.2 Hz, 1H, Ar-H), 7.32 (t, J = 7.8 Hz, 3H, Ar-H), 7.48–7.42 (m, 4H, Ar-H), 7.56–7.51 (m, 4H, Ar-H), 7.69 (d, J = 7.8 Hz, 2H, Ar-H), 8.56 (s, 1H, Ar-H), 9.51 (b, 1H, NH), 14.59 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.03, 118.70, 119.23, 121.50, 122.13, 122.62, 127.27, 128.02, 128.79, 129.03, 131.62, 131.95, 135.04, 138.60, 140.28, 145.21, 153.28, 165.36, 166.03 (Ar-C), 175.86 (CO). MS (EI): 427.10 [M]+. HRMS: calcd for C25H18ClN3O2: 427.1087, found 427.1083.
3.2.2.6. 2-(4-Bromophenylazo)-4′-chloro-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3f). Orange solid, mp 253–254 °C; IR (KBr): ν/cm−1 3252, 3050, 2945, 1638, 1590, 1579, 1453, 1386; 1H NMR (DMSO-d6): δ = 6.44 (s, 1H, Ar-H), 6.99 (t, J = 7.8 Hz, 1H, Ar-H), 7.32 (t, J = 7.8 Hz, 2H, Ar-H), 7.51–7.50 (m, 4H, Ar-H), 7.53–7.52 (m, 2H, Ar-H), 7.63–7.61 (m, 2H, Ar-H), 7.31 (d, J = 7.8 Hz, 2H, Ar-H), 8.59 (s, 1H, Ar-H), 9.54 (b, 1H, OH) 14.48 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 118.32, 118.89, 119.24, 120.66, 122.21, 123.29, 127.31, 128.79, 131.71, 131.92, 132.01, 135.10, 138.43, 140.18, 145.45, 152.30, 165.22, 165.86 (Ar-C), 176.24 (CO). MS (EI): 507.1 [M]+. HRMS: calcd for C25H17BrClN3O2: 505.01926, found 505.01933. Single crystals of 3f were obtained by the slow evaporation of a CHCl3/CH3OH solution of 3f. Crystal data: C25H17BrClN3O2, triclinic, a = 8.4153 Å, b = 12.6081 Å, c = 14.8911 Å, α = 92.688°, β = 97.266°, γ = 94.691°, V = 1559.4 Å3, T = 293 K, space group: P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, calculated density = 1.403 g cm−3, no. of reflection measured 5471, θmax = 24.987, R1 = 0.0597.80
, Z = 2, calculated density = 1.403 g cm−3, no. of reflection measured 5471, θmax = 24.987, R1 = 0.0597.80
3.2.2.7. 4′-Bromo-2-(4-chlorophenylazo)-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3g). Yellow solid, mp 221–223 °C; IR (KBr): ν/cm−1 3252, 3050, 1637, 1577, 1483, 1387; 1H NMR (DMSO-d6): δ = 6.42 (s, 1H, Ar-H), 6.99 (t, J = 7.2 Hz, 1H, Ar-H), 7.31 (t, J = 7.8 Hz, 2H, Ar-H), 7.44 (dd, J = 2.1, 6.3 Hz, 2H, Ar-H), 7.48 (dd, J = 1.8, 6.6 Hz, 2H, Ar-H), 7.53 (dd, J = 1.8, 6.6 Hz, 2H, Ar-H), 7.58 (dd, J = 1.8, 6.6 Hz, 2H, Ar-H), 7.69 (d, J = 7.8 Hz, 2H, Ar-H), 8.57 (s, 1H, Ar-H), 9.45 (s, 1H, OH), 14.49 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.33, 118.89, 119.28, 120.38, 122.26, 122.71, 126.83, 128.83, 129.13, 130.26, 132.31, 135.07, 138.84, 140.23, 145.64, 151.99, 165.37, 165.89 (Ar-C), 176.18 (CO). MS (EI): 507.10 [M]+; HRMS: calcd for C25H17BrClN3O2: 505.01926, found 505.0265.
3.2.2.8. 4′-Bromo-2-(4-bromophenylazo)-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3h). Yellow solid, mp 159–160 °C; IR (KBr): ν/cm−1 3250, 3055, 2970, 1660, 1590, 1562, 1475, 1295; 1H NMR (DMSO-d6): δ = 6.42 (s, 1H, Ar-H), 6.99 (t, J = 6.6 Hz, 2H, Ar-H), 7.31 (t, J = 7.8 Hz, 2H, Ar-H), 7.48–7.44 (m, 4H, Ar-H), 7.72–7.68 (m, 5H, Ar-H), 8.57 (s, 1H, Ar-H), 9.53 (s, 1H, NH), 14.47 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.20, 118.70, 119.29, 120.50, 122.14, 122.84, 123.27, 126.84, 127.37, 128.89, 130.19, 131.86, 135.19, 139.64, 140.33, 146.97, 152.38, 164.66, 165.98 (Ar-C), 176.43 (CO). MS (EI): 551.10 [M]+. HRMS: calcd for C25H17Br2N3O2: 548.9687, found 548.9681. Single crystals of 3h were obtained by the slow evaporation of a CHCl3/CH3OH solution of 3h. Crystal data: C25H17Br2N3O2, monoclinic, a = 11.5200 Å, b = 12.9090 Å, c = 21.274 Å, α = 90°, β = 93.818°, γ = 90°, V = 315.7 Å3, T = 150 K, space group: P21/c1, Z = 4, calculated density = 1.480 g cm−3, no. of reflection measured 5947, θmax = 25.630, R1 = 0.0672.80
3.2.2.9. 2-(4-Chlorophenylazo)-4′-fluoro-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3i). Orange solid, mp 269–270 °C; IR (KBr): ν/cm−1 3436, 3266, 2936, 1637, 1597, 1578, 1419, 1390; 1H NMR (DMSO-d6): δ = 6.43 (s, 1H, Ar-H), 7.00 (t, J = 7.5 Hz, 1H, Ar-H), 7.25–7.22 (m, 2H, Ar-H), 7.32 (t, J = 7.8 Hz, 2H, Ar-H), 7.54–7.46 (m, 6H, Ar-H), 7.70 (d, J = 8.4 Hz, 2H, Ar-H), 8.57 (s, 1H, Ar-H), 9.50 (b, 1H, NH),14.49 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 114.04, 114.18, 118.24, 118.72, 119.25, 122.20, 122.76, 122.97, 128.81, 129.08, 131.99, 132.04, 135.22, 135.88, 135.90, 140.22, 145.74, 152.03, 160.64, 162.26, 165.36, 165.94 (Ar-C), 176.21 (CO). MS (EI): 445.20 [M]+. HRMS: calcd for C25H17ClFN3O2: 445.09933, found 445.0988.
3.2.2.10. 2-(4-Bromophenylazo)-4′-fluoro-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3j). Orange solid, mp 257–258 °C; IR (KBr): ν/cm−1 3253, 2935, 1637, 1595, 1548, 1417, 1385; 1H NMR (DMSO-d6): δ = 6.42 (s, 1H, Ar-H), 7.00 (t, J = 7.2 Hz, 1H, Ar-H), 7.23 (t, J = 7.8 Hz, 2H, Ar-H), 7.29–7.32 (m, 2H, Ar-H), 7.46 (d, J = 8.64 Hz, 2H, Ar-H), 7.50–7.52 (m, 2H, Ar-H), 7.60 (d, J = 8.64 Hz, 2H, Ar-H), 7.69 (d, J = 7.56 Hz, 2H, Ar-H), 8.57 (s, 1H, Ar-H), 9.50 (b, 1H, NH), 14.49 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 114.12, 114.26, 118.30, 118.81, 119.35, 120.77, 122.34, 122.84, 123.38, 128.88, 132.07, 132.11, 135.42, 135.89, 140.81, 152.81, 152.36, 160.72, 162.34, 165.96, (Ar-C), 176.16 (CO). MS (EI): 491.0 [M + 2]+. HRMS: calcd for C25H17BrFN3O2: 489.0488, found 489.0483.
3.2.2.11. 2-(4-Bromophenylazo)-5-hydroxy-4′-methoxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3k). Orange solid, mp 255–257 °C; IR (KBr): ν/cm−1; 3270, 3053, 1657, 1543, 1474, 1391; 1H NMR (DMSO-d6): δ = 3.85 (s, 3H, OCH3), 6.54 (s, 1H, Ar-H), 6.97 (t, J = 7.8 Hz, 1H, Ar-H), 7.14–7.00 (m, 4H, Ar-H), 7.26–7.19 (m, 4H, Ar-H), 7.49–7.30 (m, 1H, Ar-H), 7.83–7.69 (m, 1H, Ar-H), 8.27 (d, J = 9 Hz, 2H, Ar-H), 8.55 (s, 1H, Ar-H), 9.75 (b, 1H, NH), 14.50 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 55.69 (OCH3), 114.00, 115.73, 119.99, 120.28, 123.56, 127.80, 128.61, 128.68, 128.89, 132.00, 133.26, 134.90, 139.28, 139.97, 141.90, 142.29, 143.43, 152.36, 160.21 (Ar-C), 179.45 (CO). MS (EI): 500.1 [M]+. HRMS: calcd for C26H20BrN3O3: 501.0688, found 501.0674.
3.2.2.12. 5-Hydroxy-4′-methoxy-2-(2-nitrophenylazo)-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3l). Orange solid, mp 235–237 °C; IR (KBr): ν/cm−1 3270, 3069, 2924, 1685, 1601, 1542, 1473, 1309; 1H NMR (DMSO-d6): δ = 3.82 (s, 3H, OCH3), 7.06 (dd, J = 1.8, 6.6 Hz, 2H, Ar-H), 7.39–7.37 (m, 2H, Ar-H), 7.45 (d, J = 8.4 Hz, 3H, Ar-H), 7.70–7.67 (m, 1H, Ar-H), 7.73 (d, J = 7.8 Hz, 2H, Ar-H) 7.81–7.78 (m, 2H, Ar-H), 8.05 (dd, J = 1.2, 8.4 Hz, 2H, Ar-H), 8.19 (s, 1H, Ar-H), 9.57 (b, 1H, NH), 14.50 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 55.18, 113.53, 118.06, 118.11, 118.72, 119.68, 120.36, 124.00, 124.11, 128.76, 129.24, 130.68, 131.66, 133.58, 138.34, 141.79, 144.73, 146.19, 146.77, 159.42, 164.34 (Ar-C), 176.42 (CO). MS (EI): 468.1 [M]+. HRMS: calcd for C26H20N4O5: 468.1433, found 468.1428.
3.2.2.13. 2-(4-Chlorophenylazo)-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3m). Gray yellow solid, mp 156–158 °C; IR (KBr): ν/cm−1 3252, 3050, 2928, 1638, 1598, 1582, 1434, 1389; 1H NMR (DMSO-d6): δ = 6.46 (s, 1H, Ar-H), 7.00 (t, J = 7.5 Hz, 1H, Ar-H), 7.25 (t, J = 7.2 Hz, 2H, Ar-H), 7.38–7.35 (m, 1H, Ar-H), 7.42 (t, J = 7.5 Hz, 2H, Ar-H), 7.50–7.46 (m, 4H, Ar-H), 7.56–7.54 (m, 2H, Ar-H), 7.72 (d, J = 8.4 Hz, 2H, Ar-H), 8.58 (s, 1H, Ar-H), 9.60 (s, 1H, NH), 14.50 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.15, 118.59, 119.24, 122.17, 122.76, 122.95, 126.85, 127.28, 128.79, 129.03, 130.19, 131.94, 140.23, 146.84, 152.04, 165.36, 165.97 (Ar-C), 176.01(CO). MS (EI): 427.25 [M]+. HRMS: calcd for C25H18ClN3O2: 427.1087, found 427.1082.
3.2.2.14. 2-(4-Bromophenylazo)-5-hydroxy-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3n). Orange solid, mp 252–253 °C; IR (KBr): ν/cm−1 3250, 3048, 2932, 1639, 1582, 1569, 1473, 1432, 1299; 1H NMR (DMSO-d6): δ = 6.43 (s, 1H, Ar-H), 7.00 (t, J = 7.2 Hz, 1H, Ar-H), 7.36–7.30 (m, 3H, Ar-H), 7.41 (t, J = 6.6 Hz, 2H, Ar-H), 7.48–7.45 (m, 4H, Ar-H), 7.60–7.58 (m, 2H, Ar-H), 7.69 (d, J = 7.2 Hz, 2H, Ar-H), 8.57 (s, 1H, Ar-H), 9.70 (b, 1H, NH), 14.53 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.19, 118.60, 119.23, 120.50, 122.14, 122.84, 123.27, 126.84, 127.27, 128.79, 130.19, 131.94, 135.29, 139.64, 140.26, 146.87, 152.38, 164.66, 165.98 (Ar-C), 176.31 (CO). MS (EI): 472.10 [M]+. HRMS: calcd for C25H18BrN3O2: 471.05823, found 471.0577. Single crystals of 3n were obtained by the slow evaporation of a CHCl3/CH3OH solution of 3n. Crystal data: C25H18BrN3O2, monoclinic, a = 12.653 Å, b = 8.433 Å, c = 28.374 Å, α = 90°, β = 96.333°, γ = 90°, V = 3009.2 Å3, T = 293 K, space group: P21/c1, Z = 4, calculated density = 1.379 g cm−3, no. of reflection measured 5304, θmax = 25.036, R1 = 0.0830.80
3.2.2.15. 5-Hydroxy-2-(2-nitrophenylazo)-N-phenyl-[1,1′-biphenyl]-4-carboxamide (3o). Orange solid, mp 213–214 °C; IR (KBr): ν/cm−1 3270, 3053, 2922, 1657, 1543, 1474, 1474, 1391; 1H NMR (DMSO-d6): δ = 6.47 (s, 1H, Ar-H), 7.03 (t, J = 7.5 Hz, 1H, Ar-H), 7.43–7.32 (m, 7H, Ar-H),7.51–7.49 (m, 2H, Ar-H), 7.60 (t, J = 6.9 Hz, 1H, Ar-H), 7.73–7.72 (m, 2H, Ar-H), 7.87 (d, J = 7.8 Hz, 1H, Ar-H), 8.55 (s, 1H, Ar-H), 14.28 (s, 1H, OH); 13C NMR (DMSO-d6): δ = 118.17, 119.04, 119.18, 119.26, 122.35, 123.04, 123.44, 126.99, 127.37, 127.45, 128.81, 130.09, 132.48, 136.10, 139.34, 140.04, 145.47, 146.77, 147.82, 164.99, 165.56 (Ar-C), 177.31 (CO). MS (EI): 437.13 [M]+. HRMS: calcd for C25H18N4O4: 438.13280, found 438.1323.
3.2.2.16. 2-(4-Bromophenyl)-3-methyl-8-(4-nitrophenyl)-6-oxo-N4,N5,diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4a). Orange solid, Lit.68 mp 302–304 °C; 1H NMR (DMSO-d6): d = 2.36 (s, 3H, CH3), 6.96–7.26 (m, 7H, Ar-H), 7.47 (d, J = 7.2 Hz, 2H, Ar-H), 7.56 (d, J = 7.8 Hz, 2H, Ar-H), 7.60–7.65 (m, 2H, Ar-H), 7.86–7.95 (m, 4H, Ar-H), 8.27 (d, J = 7.8 Hz, 1H, Ar-H), 8.34 (d, J = 7.8 Hz, 1H, Ar-H), 10.45 (s, 1H, NH), 10.57 (s, 1H, NH); 13C NMR (DMSO-d6): d = 19.39 (CH3), 115.70, 115.81, 119.52, 119.82, 121.76, 122.60, 122.98, 123.16, 123.21, 123.60, 124.03, 124.69, 127.20, 128.12, 128.40, 130.78, 131.08, 131.48, 132.81, 135.92, 136.85, 138.74, 139.44, 140.35, 140.45, 141.66, 142.28, 143.46, 147.41 (Ar-C), 162.28, 164.18, 178.68 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
3.2.2.17. 2-(4-Chlorophenyl)-3-methyl-8-(4-nitrophenyl)-6-oxo-N4,N5,diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4b). Orange crystals, Lit.68 mp 249–251 °C; 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.95 (t, J = 7.5 Hz, 1H, Ar-H), 7.07 (t, J = 7.5 Hz, 1H, Ar-H), 7.12 (t, J = 7.5 Hz, 2H, Ar-H), 7.22–7.25 (m, 3H, Ar-H), 7.47 (d, J = 7.8 Hz, 2H, Ar-H), 7.55 (d, J = 7.8 Hz, 2H, Ar-H), 7.68–7.72 (m, 4H, Ar-H), 7.94 (d, J = 8.4 Hz, 2H, Ar-H), 8.26 (d, J = 8.4 Hz, 2H, Ar-H), 10.41 (s, 1H, NH), 10.56 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.87 (CH3), 116.44, 119.99, 120.30, 123.09, 123.47, 124.09, 124.59, 127.61, 128.40, 128.61, 128.89, 130.37, 131.98, 135.07, 136.70, 139.18, 139.92, 140.66, 141.72, 142.35, 142.64, 147.86 (Ar-C), 162.92, 164.55, 178.84 (C
O).
3.2.2.18. 2-(3-Chlorophenyl)-3-methyl-8-(4-nitrophenyl)-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4c). Orange crystals, mp 281.283 °C; IR (KBr): ν/cm−1 3246 (NH), 1675 (C
O), 1601 (CC); 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3), 6.96 (t, J = 7.32 Hz, 1H, Ar-H), 7.01–7.02 (m, 2H, Ar-H), 7.07–7.09 (m, 2H, Ar-H), 7.55–7.59 (m, 2H, Ar-H), 7.13 (t, J = 7.86 Hz, 2H, Ar-H), 7.25 (t, J = 7.89 Hz, 2H, Ar-H), 7.47 (d, J = 7.56 Hz, 2H, Ar-H), 7.55 (d, J = 7.56 Hz, 2H, Ar-H), 7.60–7.64 (m, 6H, Ar-H), 7.87 (d, J = 8.76 Hz, 2H, Ar-H), 10.50 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.36 (CH3), 113.51, 115.22, 119.50, 119.80, 122.54, 123.06, 123.52, 123.91, 127.31, 128.13, 128.40, 131.50, 132.77, 134.40, 138.78, 139.47, 140.69, 142.95, 159.71 (Ar-C) 162.29, 164.28, 178.96 (CO). MS (EI): 629.1 [M−1]+. HRMS: calcd for C35H24ClN5O5: 629.1465, found 629.1423.
3.2.2.19. 3-Methyl-2-(4-nitrophenyl)-6-oxo-N4,N5,8-triphenyl-2,6-dihydrocinnoline-4,5-dicarbox-amide (4d). Yellow crystals, mp 246–247 °C; IR (KBr): ν/cm−1 3251 (NH), 1668 (C
O), 1599 (CC); 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3), 6.96 (t, J = 7.35 Hz, 1H, Ar-H), 7.07–7.14 (m, 3H, Ar-H), 7.22–7.26 (m, 3H, Ar-H), 7.47–7.48 (m, 2H, Ar-H), 7.55–7.59 (m, 3H, Ar-H), 7.63–7.66 (m, 4H, Ar-H), 7.94–7.95 (m, 2H, Ar-H), 8.27–8.29 (m, 2H, Ar-H), 10.45 (s, 1H, NH), 10.60 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.41 (CH3), 115.66, 119.51, 119.82, 122.59, 122.97, 123.58, 124.28, 125.97, 127.23, 128.13, 128.40, 129.85, 130.00, 131.50, 136.25, 138.73, 139.45, 140.08, 141.95, 142.26, 142.52, 147.36 (Ar-C) 162.51, 164.14, 178.25 (CO). MS (EI): 592.85 [M−2]+. HRMS: calcd for C35H25N5O5: 595.1855, found 595.1857.
3.2.2.20. 8-(4-Chlorophenyl)-3-methyl-6-oxo-N4,N5,2-triphenyl-2,6-dihydrocinnoline-4,5-dicarbox-amide (4e). Orange crystals, Lit.68 mp 268–270 °C; 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.95 (t, J = 6.9 Hz, 1H, Ar-H), 7.06 (t, J = 6.6 Hz, 1H, Ar-H), 7.11 (m, 3H, Ar-H), 7.24 (t, J = 7.8 Hz, 2H, Ar-H), 7.46–7.47 (m, 2H, Ar-H), 7.49–7.50 (m, 2H, Ar-H), 7.55–7.62 (m, 3H, Ar-H), 7.64 (d, J = 4.2 Hz, 4H, Ar-H), 7.66–7.68 (m, 2H, Ar-H), 10.48 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.35 (CH3), 115.17, 119.48, 119.79, 123.50, 124.20, 125.94, 127.33, 127.97, 128.09, 128.36, 129.78, 131.90, 133.46, 134.43, 135.41, 138.74, 139.43, 141.73, 142.56 (Ar-C) 162.59, 164.20, 178.96 (C
O).
3.2.2.21. 2-(4-Bromophenyl)-8-(4-chlorophenyl)-3-methyl-6-oxo-N4,N5,diphenyl-2,6-dihydro-cinnoline-4,5-dicarboxamide (4f). Pale yellow crystals, Lit.68 mp 274–276 °C; 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3); 6.95 (t, J = 7.25 Hz, 1H, Ar-H), 7.06 (t, J = 7.2 Hz, 1H, Ar-H), 7.10–7.13 (m, 3H, Ar-H), 7.24 (t, J = 7.8 Hz, 2H, Ar-H), 7.45 (d, J = 7.2 Hz, 2H, Ar-H), 7.50 (d, J = 8.4 Hz, 2H, Ar-H), 7.53 (d, J = 7.8 Hz, 2H, Ar-H), 7.61 (d, J = 8.4 Hz, 2H, Ar-H), 7.67 (d, J = 8.4 Hz, 2H, Ar-H), 7.85 (d, J = 8.4 Hz, 2H, Ar-H), 10.44 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 115.56, 119.52, 119.82, 122.59, 123.56, 123.16, 123.58, 124.04, 127.22, 128.03, 128.13, 128.15, 128.41, 131.94, 132.80, 133.55, 134.36, 135.39, 138.74, 139.45, 140.40, 141.63, 141.73, 142.03 (Ar-C), 162.55, 164.18, 178.68 (C
O).
3.2.2.22. 8-(4-Bromophenyl)-2-(4-chlorophenyl)-3-methyl-6-oxo-N4,N5,diphenyl-2,6-dihydro-cinnoline-4,5-dicarboxamide (4g). Orange crystals, Lit.68 mp 276–278 °C; 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 6.95 (t, J = 7.2 Hz, 1H, Ar-H), 7.06 (t, J = 7.2 Hz, 1H, Ar-H), 7.11 (m, 3H, Ar-H), 7.24 (t, J = 8.1 Hz, 2H, Ar-H), 7.46 (d, J = 7.8 Hz, 2H, Ar-H), 7.54 (d, J = 7.8 Hz, 2H, Ar-H), 7.60–7.65 (m, 4H, Ar-H), 7.67–7.73 (m, 4H, Ar-H), 10.44 (s, 1H, NH), 10.54 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.37 (CH3), 115.54, 119.49, 119.79, 122.24, 122.56, 123.55, 124.00, 127.22, 128.11, 129.85, 130.94, 132.21, 134.55, 134.73, 135.36, 138.73, 139.44, 140.31, 141.29, 141.67, 142.07 (Ar-C), 162.53, 164.15, 179.43 (C
O).
3.2.2.23. 2,8-Di(4-bromophenyl)-3-methyl-6-oxo-N4,N5,diphenyl-2,6-dihydrocinnoline-4,5-dicarbox-amide (4h). Pale yellow solid, Lit.68 mp 268–270 °C; 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 6.95 (t, J = 7.5 Hz, 1H, Ar-H), 7.06 (t, J = 7.5 Hz, 1H, Ar-H), 7.10–7.13 (m, 3H, Ar-H), 7.24 (t, J = 7.8 Hz, 2H, Ar-H), 7.46 (d, J = 7.8 Hz, 2H, Ar-H), 7.54 (d, J = 7.8 Hz, 2H, Ar-H), 7.59–7.65 (m, 6H, Ar-H), 7.86 (d, J = 9 Hz, 2H, Ar-H), 10.44 (s, 1H, NH), 10.54 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.37 (CH3), 115.55, 119.49, 119.79, 122.24, 122.56, 123.15, 123.55, 124.01, 127.20, 128.11, 128.14, 128.39, 130.94, 132.20, 132.79, 134.72, 135.35, 138.72, 139.44, 140.31, 141.60, 141.71, 142.05 (Ar-C), 162.52, 164.15, 178.65 (C
O).
3.2.2.24. 2-(4-Chlorophenyl)-8-(4-fluorophenyl)-3-methyl-6-oxo-N4,N5,diphenyl-2,6-dihydro-cinnoline-4,5-dicarboxamide (4i). Yellow solid, Lit.68 mp 271–273 °C; 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.95 (t, J = 7.5 Hz, 1H, Ar-H), 7.06 (t, J = 7.2 Hz, 1H, Ar-H), 7.09 (s, 1H, Ar-H), 7.12 (t, J = 7.8 Hz, 2H, Ar-H), 7.22–7.29 (m, 4H, Ar-H), 7.45 (d, J = 7.2 Hz, 2H, Ar-H), 7.55 (d, J = 7.2 Hz, 2H, Ar-H), 7.67–7.72 (m, 6H, Ar-H), 10.46 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.38 (CH3), 114.86, 115.00, 115.44, 119.51, 119.80, 122.57, 123.56, 124.01, 127.25, 127.92, 128.12, 128.41, 129.83, 131.87, 131.89, 132.26, 132.31, 134.51, 135.26, 138.75, 139.46, 140.54, 141.33, 141.61, 142.20 (Ar-C), 162.59, 164.20, 178.75 (C
O).
3.2.2.25. 2-(4-Bromophenyl)-8-(4-fluorophenyl)-3-methyl-6-oxo-N4,N5,diphenyl-2,6-dihydro-cinnoline-4,5-dicarboxamide (4j). Yellow crystals, Lit.68 mp 267–269 °C; 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3); 6.95 (t, J = 7.2 Hz, 1H, Ar-H), 7.06 (t, J = 7.5 Hz, 1H, Ar-H), 7.09 (s, 1H, Ar-H), 7.12 (t, J = 7.8 Hz, 2H, Ar-H), 7.22–7.29 (m, 4H, Ar-H), 7.47 (d, J = 7.2 Hz, 2H, Ar-H), 7.55 (d, J = 7.2 Hz, 2H, Ar-H), 7.61 (d, J = 8.4 Hz, 2H, Ar-H), 7.68–7.71 (m, 2H, Ar-H), 7.85 (d, J = 9 Hz, 2H, Ar-H), 10.46 (s, 1H, NH), 10.57 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.38 (CH3), 114.85, 114.99, 115.45, 119.50, 122.55, 123.10, 123.53, 124.02, 127.24, 128.11, 128.14, 128.39, 131.87, 132.24, 132.29, 132.76, 135.25, 138.75, 139.45, 140.54, 141.54, 141.74, 142.19, 161.54, 162.27 (Ar-C) 163.17, 164.19, 178.75 (C
O).
3.2.2.26. 8-(4-Anisyl)-2-(4-bromophenyl)-3-methyl-6-oxo-N4,N5,diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4k). Orange crystals, Lit.68 mp 252–254 °C; 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 6.97 (t, J = 7.5 Hz, 1H, Ar-H), 7.02 (d, J = 8.4 Hz, 2H, Ar-H), 7.07 (t, J = 7.5 Hz, 2H, Ar-H), 7.12 (t, J = 7.8 Hz, 2H, Ar-H), 7.24 (t, J = 7.8 Hz, 2H, Ar-H), 7.46 (d, J = 7.8 Hz, 2H, Ar-H), 7.55–7.62 (m, 6H, Ar-H), 7.86 (d, J = 8.4 Hz, 2H, Ar-H), 10.49 (s, 1H, NH), 10.54 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 55.20 (OCH3), 113.52, 115.24, 119.51, 119.80, 122.56, 123.08, 123.54, 123.92, 127.32, 127.74, 128.14, 128.19, 128.42, 131.52, 132.78, 134.41, 138.80, 139.49, 140.70, 141.42, 141.80, 142.95, 159.72 (Ar-C), 162.68, 164.30, 178.96 (C
O).
3.2.2.27. 8-(4-Anisyl)-3-methyl-2-(4-nitrophenyl)-6-oxo-N4,N5,diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4l). Orange crystals, Lit.68 mp 248–250 °C; 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 3.78 (s, 3H, OCH3), 6.92–7.02 (m, 5H, Ar-H), 7.13 (t, J = 7.8 Hz, 2H, Ar-H), 7.22–7.31 (m, 4H, Ar-H), 7.47 (m, 2H, Ar-H), 7.54–7.58 (m, 3H, Ar-H), 7.89 (t, J = 7.8 Hz, 1H, Ar-H), 8.01 (s, 1H, Ar-H), 8.36 (d, J = 7.8 Hz, 1H, Ar-H), 10.40 (br. s, 1H, NH), 10.64 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 55.19 (OCH3), 113.40, 113.61, 119.43, 119.52, 119.78, 119.93, 122.46, 122.69, 123.59, 123.54, 126.68, 127.54, 128.07, 128.10, 128.32, 128.41, 128.73, 130.91, 131.52, 131.94, 134.53, 134.85, 135.37, 138.63, 139.54, 141.43, 141.95, 142.95, 144.05, 159.58, 159.64 (Ar-C), 162.20, 164.64, 179.15 (C
O).
3.2.2.28. 2-(4-Chlorophenyl)-3-methyl-6-oxo-N4,N5,8-triphenyl-2,6-dihydrocinnoline-4,5-dicarbox-amide (4m). Yellow crystals, Lit.68 mp 270–271 °C; 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3); 6.97 (t, J = 7.2 Hz, 1H, Ar-H), 7.07–7.15 (m, 2H, Ar-H), 7.13 (t, J = 7.8 Hz, 2H, Ar-H), 7.25 (t, J = 8.1 Hz, 2H, Ar-H), 7.42–7.49 (m, 5H, Ar-H), 7.56 (d, J = 7.8 Hz, 2H, Ar-H), 7.65 (d, J = 8.4 Hz, 2H, Ar-H), 7.63–7.72 (m, 4H, Ar-H), 10.48 (s, 1H, NH), 10.56 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.33 (CH3), 115.41, 119.49, 119.79, 122.54, 123.53, 123.95, 127.23, 127.92, 127.98, 128.11, 128.39, 128.59, 129.79, 130.11, 134.49, 135.23, 135.58, 138.75, 139.47, 140.59, 141.32, 141.59, 143.32 (Ar-C), 162.55, 164.23, 178.80 (C
O).
3.2.2.29. 2-(4-Bromophenyl)-3-methyl-6-oxo-N4,N5,8-triphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4n). Orange crystals, mp 233–235 °C; IR (KBr): ν/cm−1 3266 (NH), 1677, 1657 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.97 (t, J = 7.32 Hz, 1H, Ar-H), 7.06–7.15 (m, 4H, Ar-H), 7.25 (t, J = 7.8 Hz, 2H, Ar-H), 7.42.7.49 (m, 5H, Ar-H), 7.56–7.66 (m, 6H, Ar-H), 7.84–7.86 (m, 2H, Ar-H), 10.46 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.33 (CH3), 115.43, 119.48, 119.77, 122.52, 123.07, 123.52, 123.93, 127.19, 127.97, 128.09, 128.15, 128.38, 128.57, 130.09, 132.73, 135.21, 135.56, 138.74, 139.46, 140.58, 141.51, 141.73 (Ar-C) 162.58, 164.22, 178.78 (CO). MS (EI): 627.03 [M−1]+. HRMS: calcd for C35H25BrN4O3: 628.1110, found 630.1083.
3.2.2.30. 3-Methyl-2-(2-nitrophenyl)-6-oxo-N4,N5,8-triphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (4o). Orange crystals, mp 251–252 °C; IR (KBr): ν/cm−1 3272 (NH), 1661, 1633 (C
O); 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3), 6.95–6.96 (m, 1H, Ar-H), 7.03–7.05 (m, 2H, Ar-H), 7.11–7.14 (m, 3H, Ar-H), 7.21–7.26 (m, 2H, Ar-H), 7.46–7.47 (m, 2H, Ar-H), 7.37–7.39 (m, 4H, Ar-H), 7.46–7.47 (m, 2H, Ar-H), 7.53–7.57 (m, 2H, Ar-H), 7.88 (t, J = 7.86 Hz, 1H, Ar-H), 8.33 (d, J = 7.98 Hz, 1H, Ar-H), 10.28 (br s, 1H, NH), 10.67 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 18.54 (CH3), 119.41, 119.50, 119.91, 122.57, 123.60, 127.93, 128.33, 128.60, 130.18, 131.93, 134.81, 135.10, 135.36, 138.63, 139.55, 141.33, 141.83, 143.34, 144.06, 152.64 (Ar-C) 162.14, 163.96, 178.95 (CO). MS (EI): 593.75 [M−1]+, HRMS: calcd for C35H25N5O5: 595.1855, found 595.1835.
4. Conclusions
In conclusion, a highly chemoselective synthesis of 4-arylazo-5-hydroxy-1,1′-biphenyl-4-carboxamide derivatives was carried out employing 1:1 molar ratio of 3-oxo-2-arylhydrazonopropanals with acetoacetanilide using the optimized condition; DBU/1,4-dioxane under microwave irradiation. Spectroscopic analyses (IR, 1H- and 13C-NMR, MS and HRMS) and X-ray single crystals of three model examples were used in characterization of the newly isolated compounds. The photophysical properties of the new products were also investigated to assess their optical behavior. Remarkably, compound 3b displayed a distinctive emission pattern. It had a bathochromic (red) shift with a maximum emission at 709 nm and a noticeable significant Stokes shift at 237 nm. It is anticipated that the currently reported strategy opens a new research avenue in the field of synthetic organic chemistry.
Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This research work was funded by the University of Kuwait, grant number (SC01/21). The RSP unit general facilities of the Faculty of Science GFS supported by research grants GS01/05, GS01/03, GS03/01, GS02/01 and GS03/08 are greatly appreciated.References
- M. J. Macielag, J. P. Demers, S. A. Fraga-Spano, D. J. Hlasta, S. G. Johnson, R. M. Kanojia, R. K. Russell, Z. Sui, M. A. Weidner-Wells and H. Werblood, Substituted salicylanilides as inhibitors of two-component regulatory systems in bacteria, J. Med. Chem., 1998, 41, 2939–2945 CrossRef CAS PubMed.
- M. Krátký and J. Vinšová, Antifungal activity of salicylanilides and their esters with 4-(trifluoromethyl) benzoic acid, Molecules, 2012, 17, 9426–9442 CrossRef PubMed.
- G. Paraskevopoulos, S. Monteiro, R. Vosátka, M. Krátký, L. Navrátilová, F. Trejtnar, J. Stolaříková and J. Vinšová, Novel salicylanilides from 4,5-dihalogenated salicylic acids: Synthesis, antimicrobial activity and cytotoxicity, Bioorg. Med. Chem., 2017, 25, 1524–1532 CrossRef CAS PubMed.
- A. Imramovský, J. Vinšová, J. M. Férriz, V. Buchta and J. Jampílek, Salicylanilide esters of N-protected amino acids as novel antimicrobial agents, Bioorg. Med. Chem. Lett., 2009, 19, 348–351 CrossRef PubMed.
- R. De La Fuente, N. D. Sonawane, D. Arumainayagam and A. S. Verkman, Small molecules with antimicrobial activity against E. coli and P. aeruginosa identified by high-throughput screening, Br. J. Pharmacol., 2006, 149, 551–559 CrossRef CAS PubMed.
- M. K. Dahlgren, A. M. Kauppi, I.-M. Olsson, A. Linusson and M. Elofsson, Design, synthesis, and multivariate quantitative structure–activity relationship of salicylanilides potent inhibitors of type III secretion in Yersinia, J. Med. Chem., 2007, 50, 6177–6188 CrossRef CAS PubMed.
- J. Vinsova, A. Imramovsky, V. Buchta, M. Ceckova, M. Dolezal, F. Staud, J. Jampilek and J. Kaustova, Salicylanilide acetates: Synthesis and antibacterial evaluation, Molecules, 2006, 12, 1–12 CrossRef PubMed.
- G. Daidone, D. Raffa, S. Plescia, M. Matera, A. Caruso, V. Leone and M. Amico-Roxas, Synthesis and evaluation of the analgesic and antiinflammatory activities of N-substituted salicylamides, Farmaco, 1989, 44, 465–473 CAS.
- K. Waisser, J. Matyk, H. Divišová, P. Husakova, J. Kuneš, V. Klimešová, J. Kaustova, U. Möllmann, H. M. Dahse and M. Miko, The oriented development of antituberculotics: salicylanilides, Arch. Pharm., 2006, 339, 616–620 CrossRef CAS PubMed.
- A. Jurgeit, R. McDowell, S. Moese, E. Meldrum, R. Schwendener and U. F. Greber, Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects, PLoS Pathog., 2012, 8, e1002976 CrossRef CAS PubMed.
- Y.-W. Chang, T.-K. Yeh, K.-T. Lin, W.-C. Chen, H.-T. Yao, S.-J. Lan, Y.-S. Wu, H.-P. Hsieh, C.-M. Chen and C.-T. Chen, Pharmacokinetics of anti-SARS-CoV agent niclosamide and its analogs in rats, J. Food Drug Anal., 2006, 14, 15 Search PubMed.
- J. Xu, P.-Y. Shi, H. Li and J. Zhou, Broad spectrum antiviral agent niclosamide and its therapeutic potential, ACS Infect. Dis., 2020, 6, 909–915 CrossRef CAS PubMed.
- Z. Baranyai, M. Krátký, R. Vosátka, E. Szabó, Z. Senoner, S. Dávid, J. Stolaříková, J. Vinšová and S. Bősze, In vitro biological evaluation of new antimycobacterial salicylanilide-tuftsin conjugates, Eur. J. Med. Chem., 2017, 133, 152–173 CrossRef CAS PubMed.
- G. Piccaro, F. Giannoni, P. Filippini, A. Mustazzolu and L. Fattorini, Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions, Antimicrob. Agents Chemother., 2013, 57, 1428–1433 CrossRef CAS PubMed.
- M. Krátký, J. Vinšová, E. Novotná, J. Mandíková, V. Wsól, F. Trejtnar, V. Ulmann, J. Stolaříková, S. Fernandes and S. Bhat, Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase, Tuberculosis, 2012, 92, 434–439 CrossRef PubMed.
- W. C. Campbell, The chemotherapy of parasitic infections, J Parasitol., 1986, 72, 45–61 CrossRef CAS PubMed.
- J. Bogan and J. Armour, Anthelmintics for ruminants, Int. J. Parasit., 1987, 17, 483–491 CrossRef CAS PubMed.
- R. Rajamuthiah, B. B. Fuchs, E. Jayamani, Y. Kim, J. Larkins-Ford, A. Conery, F. M. Ausubel and E. Mylonakis, Whole animal automated platform for drug discovery against multi-drug resistant Staphylococcus aureus, PLoS One, 2014, 9, e89189 CrossRef PubMed.
- R. Rajamuthiah, B. B. Fuchs, A. L. Conery, W. Kim, E. Jayamani, B. Kwon, F. M. Ausubel and E. Mylonakis, Repurposing salicylanilide anthelmintic drugs to combat drug resistant Staphylococcus aureus, PLoS One, 2015, 10, e0124595 CrossRef PubMed.
- H. Mohammad, A. AbdelKhalek, N. S. Abutaleb and M. N. Seleem, Repurposing niclosamide for intestinal decolonization of vancomycin-resistant enterococci, Int. J. Antimicrob. Agents, 2018, 51, 897–904 CrossRef CAS PubMed.
- I. E. Escobar, A. White, W. Kim and E. Mylonakis, New antimicrobial bioactivity against multidrug-resistant gram-positive bacteria of kinase inhibitor IMD0354, Antibiotics, 2020, 9, 665 CrossRef CAS PubMed.
- V. Srivastava, B. Singh and V. Deorani, Cestodical activity of some compounds against echinococcosis in dogs, Indian Vet. J., 1989, 66, 807–809 Search PubMed.
- Z. Wang, J. Ren, J. Du, H. Wang, J. Liu and G. Wang, Niclosamide as a Promising Therapeutic Player in Human Cancer and Other Diseases, Int. J. Mol. Sci., 2022, 23, 16116 CrossRef CAS PubMed.
- W. Chen, R. A. Mook Jr, R. T. Premont and J. Wang, Niclosamide: Beyond an antihelminthic drug, Cell. Signalling, 2018, 41, 89–96 CrossRef CAS PubMed.
- K. Shamim, M. Xu, X. Hu, E. M. Lee, X. Lu, R. Huang, P. Shah, X. Xu, C. Z. Chen, M. Shen, H. Guo, L. Chen, Z. Itkin, R. T. Eastman, P. Shinn, C. Klumpp-Thomas, S. Michael, A. Simeonov, D. C. Lo, G.-l. Ming, H. Song, H. Tang, W. Zheng and W. Huang, Application of niclosamide and analogs as small molecule inhibitors of Zika virus and SARS-CoV-2 infection, Bioorg. Med. Chem. Lett., 2021, 40, 127906 CrossRef CAS PubMed.
- Y.-W. Chang, T. K. Yeh, K.-T. Lin, W.-C. Chen, H.-T. Yao, S. J. Lan, Y.-S. Wu, H. P. Hsieh, C.-m. Chen and C. T. Chen, Pharmacokinetics of anti-SARS-CoV agent niclosamide and its analogs in rats, J. Food Drug Anal., 2006, 14, 329–333 CAS.
- J. Xu, P. Y. Shi, H. Li and J. Zhou, Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential, ACS Infect. Dis., 2020, 6, 909–915 CrossRef CAS PubMed.
- N. C. Gassen, D. Niemeyer, D. Muth, V. M. Corman, S. Martinelli, A. Gassen, K. Hafner, J. Papies, K. Mösbauer, A. Zellner, A. S. Zannas, A. Herrmann, F. Holsboer, R. Brack-Werner, M. Boshart, B. Müller-Myhsok, C. Drosten, M. A. Müller and T. Rein, SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection, Nat. Commun., 2019, 10, 5770 CrossRef CAS PubMed.
- T. Osada, M. Chen, X. Y. Yang, I. Spasojevic, J. B. Vandeusen, D. Hsu, B. M. Clary, T. M. Clay, W. Chen, M. A. Morse and H. K. Lyerly, Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations, Cancer Res., 2011, 71, 4172–4182 CrossRef CAS PubMed.
- U. Sack, W. Walther, D. Scudiero, M. Selby, D. Kobelt, M. Lemm, I. Fichtner, P. M. Schlag, R. H. Shoemaker and U. Stein, Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer, J. Natl. Cancer Inst., 2011, 103, 1018–1036 CrossRef CAS PubMed.
- Y. Li, P. K. Li, M. J. Roberts, R. C. Arend, R. S. Samant and D. J. Buchsbaum, Multi-targeted therapy of cancer by niclosamide: A new application for an old drug, Cancer Lett., 2014, 349, 8–14 CrossRef CAS PubMed.
- R. A. Mook Jr, J. Wang, X. R. Ren, M. Chen, I. Spasojevic, L. S. Barak, H. K. Lyerly and W. Chen, Structure-activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure, Bioorg. Med. Chem., 2015, 23, 5829–5838 CrossRef PubMed.
- A. F. Bekhli, M. B. Braude, Z. G. Vorob'eva and V. I. Shvedova, [Synthesis of phenasal], Med. Prom-st. SSSR, 1965, 19, 25–27 CAS.
- E. Schraufstätter, W. Meiser and R. Gönnert, Untersuchungen über ein neues Molluscicid, Z. Naturforsch., 1961, 16b, 95–108 CrossRef.
- J. Lal, G. Kaul, A. Akhir, S. B. Ansari, S. Chopra and D. N. Reddy, Bio-evaluation of fluoro and trifluoromethyl-substituted salicylanilides against multidrug-resistant S. aureus, Med. Chem. Res., 2021, 30, 2301–2315 CrossRef CAS PubMed.
- A. Fomovska, R. D. Wood, E. Mui, J. P. Dubey, L. R. Ferreira, M. R. Hickman, P. J. Lee, S. Leed, E. Leed, J. M. Auschwitz, W. J. Welsh, C. Sommerville, S. Woods, C. Roberts and R. McLeod, Salicylanilide Inhibitors of Toxoplasma gondii, J. Med. Chem., 2012, 55, 8375–8391 CrossRef CAS PubMed.
- A. Kumar, B. Narasimhan and D. Kumar, Synthesis, antimicrobial, and QSAR studies of substituted benzamides, Bioorg. Med. Chem., 2007, 15, 4113–4124 CrossRef CAS PubMed.
- Z.-W. Zhu, L. Shi, X.-M. Ruan, Y. Yang, H.-Q. Li, S.-P. Xu and H.-L. Zhu, Synthesis and antiproliferative activities against Hep-G2 of salicylanide derivatives: potent inhibitors of the epidermal growth factor receptor (EGFR) tyrosine kinase, J. Enzyme Inhib. Med. Chem., 2011, 26, 37–45 CrossRef CAS PubMed.
- X. Bao and Y. Zhou, Synthesis and recognition properties of a class of simple colorimetric anion chemosensors containing OH and CONH groups, Sens. Actuators, B, 2010, 147, 434–441 CrossRef CAS.
- E. Aki-Sener, K. Bingol, I. Oren, O. Temiz-Arpaci, I. Yalcin and N. Alatanlar, Synthesis and microbiological activity of some N-(o-hydroxylphenyl) benzamides and phenylacetamides as the possible metabolites of antimicrobial active benzoxazoles: part II, Farmaco, 2000, 55, 469–476 CrossRef PubMed.
- D. M. Stout, W. Matier, C. Barcelon-Yang, R. D. Reynolds and B. S. Brown, Synthesis and antiarrhythmic and parasympatholytic properties of substituted phenols. 3. Modifications to the linkage region (region 3), J. Med. Chem., 1985, 28, 295–298 CrossRef CAS PubMed.
- M. Gargantilla, L. Persoons, T. Kauerová, N. Del Río, D. Daelemans, E.-M. Priego, P. Kollar and M.-J. Pérez-Pérez, Hybridization Approach to Identify Salicylanilides as Inhibitors of Tubulin Polymerization and Signal Transducers and Activators of Transcription 3 (STAT3), Pharmaceuticals, 2022, 15, 835 CrossRef CAS PubMed.
- A. Islam, I. Hannout, E. Hassan and A. Ihsan, Synthesis of some substituted salicylanilides of expected biological activity, J. Prakt. Chem./Chem.-Ztg., 1972, 314, 727–734 CrossRef CAS.
- K. Hunger, Industrial dyes: chemistry, properties, applications, John Wiley & Sons, 2007 Search PubMed.
- Y. Zhao and T. Ikeda, Smart light-responsive materials: azobenzene-containing polymers and liquid crystals, John Wiley & Sons, 2009 Search PubMed.
- L. W. Giles, J. B. Marlow, C. S. Butler, G. A. Turpin, L. De Campo, S. T. Mudie, C. F. Faul and R. F. Tabor, Structural relationships for the design of responsive azobenzene-based lyotropic liquid crystals, Phys. Chem. Chem. Phys., 2020, 22, 4086–4095 RSC.
- F. A. Pennacchio, C. Fedele, S. De Martino, S. Cavalli, R. Vecchione and P. A. Netti, Three-dimensional microstructured azobenzene-containing gelatin as a photoactuable cell confining system, ACS Appl. Mater. Interfaces, 2018, 10, 91–97 CrossRef CAS PubMed.
- J. García-Amorós and D. Velasco, Recent advances towards azobenzene-based light-driven real-time information-transmitting materials, Beilstein J. Org. Chem., 2012, 8, 1003–1017 CrossRef PubMed.
- N. DiCesare and J. R. Lakowicz, New color chemosensors for monosaccharides based on Azo dyes, Org. Lett., 2001, 3, 3891–3893 CrossRef CAS PubMed.
- K. C. Chang, I. H. Su, Y. Y. Wang and W. S. Chung, A bifunctional chromogenic calix [4] arene chemosensor for both cations and anions: a potential Ca2+ and F– switched INHIBIT logic gate with a YES logic function, Eur. J. Org. Chem., 2010, 24, 4700–4704 CrossRef.
- F. Puntoriero, P. Ceroni, V. Balzani, G. Bergamini and F. Vögtle, Photoswitchable dendritic hosts: a dendrimer with peripheral azobenzene groups, J. Am. Chem. Soc., 2007, 129, 10714–10719 CrossRef CAS PubMed.
- V. Ferri, M. Elbing, G. Pace, M. D. Dickey, M. Zharnikov, P. Samorì, M. Mayor and M. A. Rampi, Light-powered electrical switch based on cargo-lifting azobenzene monolayers, Angew. Chem., Int. Ed., 2008, 120, 3455–3457 CrossRef.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Analysis of the reactions used for the preparation of drug candidate molecules, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- S. D. Roughley and A. M. Jordan, The medicinal chemist's toolbox: an analysis of reactions used in the pursuit of drug candidates, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- S.-P. Wang, C. W. Cheung and J.-A. Ma, Direct amidation of carboxylic acids with nitroarenes, J. Org. Chem., 2019, 84, 13922–13934 CrossRef CAS PubMed.
- B. K. Banik and D. Bandyopadhyay, Advances in Microwave Chemistry, CRC Press, 2018 Search PubMed.
- S. C. Ameta, P. B. Punjabi, R. Ameta and C. Ameta, Microwave-assisted organic synthesis: a green chemical approach, CRC Press, 2014 Search PubMed.
- D. Bogdal, Microwave-assisted organic synthesis: one hundred reaction procedures, Elsevier, 2005 Search PubMed.
- L. Perreux, A. Loupy and F. Volatron, Solvent-free preparation of amides from acids and primary amines under microwave irradiation, Tetrahedron, 2002, 58, 2155–2162 CrossRef CAS.
- E. Gelens, L. Smeets, L. A. Sliedregt, B. J. van Steen, C. G. Kruse, R. Leurs and R. V. Orru, An atom efficient and solvent-free synthesis of structurally diverse amides using microwaves, Tetrahedron Lett., 2005, 46, 3751–3754 CrossRef CAS.
- A. Khalafi-Nezhad, A. Parhami, M. N. S. Rad and A. Zarea, Efficient method for the direct preparation of amides from carboxylic acids using tosyl chloride under solvent-free conditions, Tetrahedron Lett., 2005, 46, 6879–6882 CrossRef CAS.
- R. R. Poondra and N. J. Turner, Microwave-assisted sequential amide bond formation and intramolecular amidation: a rapid entry to functionalized oxindoles, Org. Lett., 2005, 7, 863–866 CrossRef CAS PubMed.
- C. Ferroud, M. Godart, S. Ung, H. Borderies and A. Guy, Microwaves-assisted solvent-free synthesis of N-acetamides by amidation or aminolysis, Tetrahedron Lett., 2008, 49, 3004–3008 CrossRef CAS.
- M. P. Vázquez-Tato, Microwave-mediated synthesis of amides, Synlett, 1993, 7, 506 CrossRef.
- G. Paraskevopoulos, M. Krátký, J. Mandíková, F. Trejtnar, J. Stolaříková, P. Pávek, G. Besra and J. Vinšová, Novel derivatives of nitro-substituted salicylic acids: Synthesis, antimicrobial activity and cytotoxicity, Bioorg. Med. Chem., 2015, 23, 7292–7301 CrossRef CAS PubMed.
- A. M. Alazemi, K. M. Dawood, H. M. Al-Matar and W. M. Tohamy, Efficient and Recyclable Solid-Supported Pd(II) Catalyst for Microwave-Assisted Suzuki Cross-Coupling in Aqueous Medium, ACS Omega, 2022, 7, 28831–28848 CrossRef CAS PubMed.
- H. M. Al-Matar, K. M. Dawood, W. M. Tohamy and M. A. Shalaby, Facile Assembling of Novel 2,3,6,7,9-pentaazabicyclo-[3.3.1] nona-3,7-diene Derivatives under Microwave and Ultrasound Platforms, Molecules, 2019, 24, 1110 CrossRef PubMed.
- H. M. Al-Matar, K. M. Dawood and W. M. Tohamy, Tandem one-pot synthesis of 2-arylcinnolin-6-one derivatives from arylhydrazonopropanals and acetoacetanilides using sustainable ultrasound and microwave platforms, RSC Adv., 2018, 8, 34459–34467 RSC.
- K. M. Dawood and M. Alaasar, Transition-metal-catalyzed Heteroannulation Reactions in Aqueous Medium, Asian J. Org. Chem., 2022, 11, e202200331 CrossRef CAS.
- K. M. Dawood, Microwave-assisted Suzuki–Miyaura and Heck–Mizoroki cross-coupling reactions of aryl chlorides and bromides in water using stable benzothiazole-based palladium(II) precatalysts, Tetrahedron, 2007, 63, 9642–9651 CrossRef CAS.
- K. M. Dawood, M. B. Elamin and A. M. Farag, Microwave-Assisted Synthesis of Arylated Pyrrolo [2,1-a] Isoquinoline Derivatives via Sequential [3 + 2] Cycloadditions and Suzuki-Miyaura Cross-Couplings in Aqueous Medium, J. Heterocycl. Chem., 2016, 53, 1928–1934 CrossRef CAS.
- K. M. Dawood and M. M. El-Deftar, Microwave-assisted CC cross-coupling reactions of aryl and heteroaryl halides in water, Arkivoc, 2010, 9, 319–330 Search PubMed.
- K. M. Dawood and M. M. El-Deftar, Microwave-assisted synthesis of 2-substituted 4-biarylyl-1,3-thiazoles by carbon-carbon cross-coupling in water, Synthesis, 2010, 6, 1030–1038 CrossRef.
- K. M. Dawood and A. Kirschning, Combining enabling techniques in organic synthesis: solid-phase-assisted catalysis under microwave conditions using a stable Pd(II)-precatalyst, Tetrahedron, 2005, 61, 12121–12130 CrossRef CAS.
- K. M. Dawood, W. Solodenko and A. Kirschning, Microwave-accelerated Mizoroki-Heck and Sonogashira cross-coupling reactions in water using a heterogeneous palladium(II)-precatalyst, Arkivoc, 2007, 7, 104–124 Search PubMed.
- K. M. Dawood, M. B. Elamin and A. M. Farag, Microwave-assisted synthesis of 2-acetyl-5-arylthiophenes and 4-(5-arylthiophen-2-yl) thiazoles via Suzuki coupling in water, Arkivoc, 2015, 7, 50–62 Search PubMed.
- K. M. Dawood, A. M. Farag, M. M. El-Deftar, M. Gardiner and H. A. Abdelaziz, Microwave-assisted synthesis of 5-arylbenzofuran-2-carboxylates via Suzuki coupling using a 2-quinolinealdoxime-Pd(II)-complex, Arkivoc, 2013, 3, 210–226 Search PubMed.
- M. A. Al-Shiekh, H. Y. Medrassi, M. H. Elnagdi and E. A. Hafez, Substituted hydrazonals as building blocks in heterocyclic synthesis: A new route to arylhydrazonocinnolines, J. Chem. Res., 2007, 7, 432–436 Search PubMed.
- H. M. Hassaneen and I. A. Abdelhamid, Acetylacetaldehyde Dimethyl Acetal as Versatile Precursors for the Synthesis of Arylazonicotinic Acid Derivatives: Green Multicomponent Syntheses of Bioactive Poly-Heteroaromatic Compounds, J. Heterocycl. Chem., 2017, 54, 1048–1053 CrossRef CAS.
- The Crystallographic Data for compounds 3f (ref. CCDC 2270147), 3h (ref. CCDC 2270148), and 3n (ref. CCDC 2270149†).
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2270147–2270149: crystallographic data for compounds 3f (2270147), 3h (2270148) and 3n (2270149). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04558g |
| This journal is © The Royal Society of Chemistry 2023 |