
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable poly(lauryl methacrylate) surface grafting via SI-ATRP on a one-pot synthesized cellulose nanofibril macroinitiator core as a shear-thinning rheology modifier and drag reducer†
Mengzhe Guoa and
You-Lo Hsieh *ab
*ab
aChemical Engineering, University of California at Davis, Davis, California 95616-8722, USA. E-mail: ylhsieh@ucdavis.edu; Tel: +1 530 752 084
bBiological and Agricultural Engineering, University of California at Davis, Davis, California 95616-8722, USA
First published on 1st September 2023
Abstract
The optimally one-pot synthesized 2-bromoproponyl esterified cellulose nanofibril (Br-CNF) has been validated as a robust macroinitiator for self-surface-initiated atom transfer radical polymerization (SI-ATRP) of lauryl methacrylate (LMA) in tunable graft lengths and high conversions of up to 92.7%. SI-ATRP of LMA surface brushes on Br-CNF followed first order kinetics in lengths at up to 46 degree of polymerization (DP) based on mass balance or 31 DP by solution-state 1H NMR in DMSO-d6. With increasing PLMA graft lengths, Br-CNF-g-PLMA cast films exhibited increasing hydrophobicity with water contact angles from 80.9° to 110.6°. The novel Br-CNF-g-PLMA exhibited dual shear thinning behavior of the Br-CNF core as evident by n < 1 flow behavior index and drag reducing properties of PLMA grafts with increased viscosity at up to 21 071×. Br-CNF-g-PLMA with 46 DP could be fully dispersed in silicon pump oil to function as a drag reducer to enhance viscosity up to 5× at 25, 40, and 55 °C. The novel macroinitiator capability of Br-CNF in SI-ATRP of vinyl monomers and the bottlebrush-like LMA surface grafted Br-CNF as highly effective viscosity modifier and drag reducer further demonstrate the versatile functionality of Br-CNF beyond hydrophobic coatings and reactive polyols previously reported.
1 Introduction
Nanocelluloses are unique renewable one dimensional (1D) nanomaterials derived from cellulose, the most abundant natural polymer with annual production of up to 100 billion tons.1 The most reported cellulose nanocrystals (CNC) and cellulose nanofibrils (CNF) have been produced by top-down methods, such as acid hydrolysis,2–6 oxidation,6–12 mechanical forces,6–11,13–16 or a combination of the latter two.7–11 All these nanocelluloses contain crystalline cores and hydrophilic surfaces of which some are anionic,2–12 making them highly water dispersible. To acquire compatibility with organic liquids and most synthetic polymers, nanocelluloses have been chemically modified to render hydrophobicity by a variety of reactions, including esterification, silanation, and amidation.17Surface grafting of polymers to nanocellulose is another approach that has been extensively reported for CNCs18–41 and CNFs42–49 by ring opening polymerization (ROP),20,42 reversible-deactivation radical polymerization (RDRP),18,45 and cerium free radical graft polymerization.19,21,43,44 Surface grafting by atom transfer radical polymerization (ATRP) is particularly attractive due to its advantage of synthesizing polymers in uniform lengths or low polydispersity from a wider range of monomers and suitable solvents.50
ATRP has been most extensively reported on CNCs22–41 and a few on CNFs46–49 to produce stimuli-responsive materials,22–25,39 polymer fillers,26–33,40,46 gold nanoparticle stabilizers,34 binders for ionic35,36 or organic47 pollutants, and hard domain in thermoplastic polybutyl acrylate elastomer37 (Table 1). To generate ATRP initiators on nanocellulose surfaces, however, aqueously dispersed nanocelluloses have to be freeze-dried to be dispersed in or solvent exchanged into organic liquids to react with 2-bromoisobutyryl bromide (2-BIB), 2-bromopropionic acid (2-BPA), or 2-bromo-2-methylpropionic acid (2-BMPA). While ATRP of nanocelluloses have focused on applications, many fundamental questions regarding the immobilization of ATRP initiators on nanocelluloses and the polymerization thereof remain. First, the extents of immobilized 2-BIB initiator, when reported, were relatively low, i.e., 1.4–9.5 wt% Br by elementary analysis or 26 to 43% hydroxyl to Br ester conversion by NMR. Also, the monomer conversion of ATRP on CNC with surface initiators from 2-BIB was less than 35%,22,30,32 possibly due to the lower accessibility of surface immobilized initiators than free initiators. While incorporating additional ethyl 2-bromoisobutyrate (EBIB) and 2-hydroxyethyl 2-bromoisobutyrate (HEBIB) initiators could significantly increase monomer conversions to 85% for CNC33 and 70% for CNF,47 it was unknown whether the enhanced polymerization was grafted on the nanocelluloses or as unattached homopolymers. Furthermore, the grafted polymer chain length and their distribution have only been estimated by adding sacrificial initiators, under the assumption of equal accessibility and reactivity for both sacrificial and immobilized initiators, thus not directly measured. To date, ATRP on nanocelluloses has not only required lengthy preparation of organic reactions to immobilize initiators, but also the surface immobilized initiators and conversion to polymers were low or not fully characterized. Furthermore, the grafted polymer chain lengths were indirectly estimated.
| Reference | Nanocellulosea | Pretreatment | Solvent | Initiator reagent | Br content/conversion | Monomerb | DP | Conversion (%) | Sacrifier initiatorc | PDI |
|---|---|---|---|---|---|---|---|---|---|---|
| a “SH” for sulfuric acid hydrolysis; “HH” for hydrochloric acid hydrolysis.b “DMAEMA” for dimethylaminoethyl acrylate; “NIPAM” for N-isopropylacrylamide.c “EBIB” for ethyl 2-bromoisobutyrate; “HEBIB” for 2-hydroxyethyl 2-bromoisobutyrate. | ||||||||||
| X. Zhang22 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | Ethylene glycol methacrylate | 16.4 | — | — | |
| W. Yuan23 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | azoC6MA-co-DMAEMA | — | — | — | — |
| J. Zhang24 | CNC (SH) | Solvent exchange | DMF | 2-BIB | 26% OH to Br | N-Vinylcaprolactam | — | — | — | — |
| B. Risteen25 | CNC (SH) | Solvent exchange | Anisole | 2-BPA | — | Styrene, NIPAM | — | — | — | — |
| A. Boujemaoui26 | CNC (SH) | Freeze-drying | toluene | 2-BIB | — | Butyl acylate | 110, 486 | 70 | EBIB | 1.09 |
| Y. Yin27 | CNC (SH) | Freeze-drying | Anisole | 2-BIB | — | Styrene | — | — | — | — |
| G. Morandi28 | CNC (SH) | Freeze-drying | Anisole | 2-BIB | 9.5 Br% | Styrene | Up to 173 | 40 | EBIB | 1.1 |
| Z. Zhang29 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | Styrene | Up to 160 | 26.5 | EBIB | 1.05–1.15 |
| H. D. Wang30 | CNC (SH) | Freeze-drying | DMSO | 2-BIB | 1.4 Br% | Methyl acylate | — | 30.3 | — | 1.46–2.06 |
| S. A. Kedzior31 | CNC (SH) | Freeze-drying | Toluene | 2-BIB | 3.5 Br% | Methyl methacylate | — | — | EBIB | 1.1, 1.2 |
| Z. Zhang32 | CNC (unknown) | Freeze-drying | DMF | 2-BIB | — | Cinnamoyloxy ethyl methacrylate | — | 35 | — | |
| M. Le Gars33 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | Glycidyl methacrylate | — | 85 | EBIB | Up to 1.4 |
| Z. Zhang34 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | 4-Vinylpyridine | — | 23.1 | EBIB | Up to 1.6 |
| H. Rosilo35 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | DMAEMA | 35 | EBIB | ||
| Z. Abousalman-Rezvani36 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | DMAEMA | 13–26 | — | — | |
| J. Yu37 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | Methyl methacylate | — | 61.4 | EBIB | 1.18, 1.21 |
| M. V. Kiriakou38 | CNC (SH) | Freeze-drying | DMF | 2-BIB | 7.44 Br % | Butyl acylate | 47–101 | — | EBIB | Less than |
| J. Majoinen39 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | t-Butyl acylate | 36 | EBIB | — | |
| M. Morits40 | CNC (SH) | Freeze-drying | DMF | 2-BIB | — | DMAEMA | — | — | — | 1.09–1.19 |
| Z. Zhang41 | CNC (SH) | Solvent exchange | DMF | 2-BIB | — | Styrene | — | 25 | EBIB | 1.06 |
| J. R. Navarro & U. Edlund46 | CNF (enzymatic) | Solvent exchange | DMSO/toluene | 2-BPMA | — | Stearyl acrylate | — | 80 | 2-ethA-bromo-2-methylpropionate | 1.09–1.17 |
| C.-F. Huang47 | CNF (TEMPO) | Solvent exchange | Anisole | 2-BIB | — | Styrene | 135–278 | 70 | HEBIB | 1.06 |
| J.-K Chen48 | CNF (TEMPO) membrane | Casting film | toluene | 2-BIB- | Styrene | 150 | 50 | HEBIB- | — | |
| M. Morits49 | CNF(homogenized) | Freeze-drying | DMF | 2-BIB | 43% OH to Br | Butyl acylate | Up to 800 | 11.8 | EBIB | — |
We have successfully synthesized 2-bromopropionyl esterified CNF (Br-CNF) via facile one-pot esterification of cellulose with the more stable 2-bromopropionyl bromide (BPB) and disintegration via in situ ultrasonication.51 This robust esterification-ultrasonication approach is tunable to convert cellulose hydroxyls to organically compatible Br-esters at varying extents while also permits the Br-esterified cellulose to be directly disintegrated into Br-CNF in the same media. These Br-CNFs were versatile in creating hydrophobic surfaces simply by dilute solution dipping (0.005 w/v%) to form coatings, forming thin films, or blade coating of gel (2.5 w/v%).51 With tunable Br-ester on their surfaces, Br-CNFs are compatible in various organic liquids for organic syntheses. The remaining surface hydroxyls enable Br-CNF to function as reactive polyols either as prepolymers or chain extenders in the syntheses of thermoplastic polyurethanes with significantly improved modulus (3.2×) and strength (3.9×) and strain-to-failure (1.5×).52
The 2-bromopropionyl ester on Br-CNF surfaces provides the alkyl bromine characteristic of Br-bearing ATRP initiator that these Br-CNFs are hypothesized to function as macroinitiators for self surface initiated ATRP (SI-ATRP) directly on CNF. Furthermore, the optimally synthesized Br-CNF contains 3.2 mmol Br per g of Br-CNF,52 that is significantly higher than the 1.4–9.5 wt% Br contents or 0.44–1.19 mmol Br g−1 cellulose reported to date.28,30,31,49 The robust one-pot esterification and in situ ultrasonication approach to the synthesis of versatile Br-CNFs represents a significantly streamlined strategy to the previously reported multi-step preparation of already fabricated nanocelluloses via freeze-drying and/or organic solvent exchange then surface initiator immobilization by separate reactions.22–41,46–49
This study aims to explore the unique potential of Br-CNFs to serve as macroinitiators for SI-ATRP of vinyl monomers on Br-CNF directly. Lauryl methacrylate (LMA), a 12C vinyl monomer that can be derived from sustainable fatty acids, was selected to produce defined lengths of poly(lauryl methacrylate) (PLMA) bottle brush-like grafts on CNF or Br-CNF-g-PLMA and to exploit the synergistic coupling of the properties of the Br-CNF core and PLMA surface graft. PLMA homopolymer has shown to be an excellent oil-soluble drag reducer, by reducing 68% drag with only 0.06 w% added in kerosene.53 Aqueous CNFs, being mechanical treated,54 TEMPO55,56 or periodate57 oxidized, are known to exhibit shear thinning rheological behaviors desired for coating,57 thickening,54 and 3D printing/bioprinting.55,56 By coupling the shear-thinning behavior of the CNF core and the drag reducing characteristics of the PLAM graft, these bottle brush-like Br-CNF-g-PLMA may present both characters synergistically to become novel drag reducers with shear-thinning behaviors in organic media.
SI-ATRP of LMA directly on Br-CNF was investigated using copper bromide (CuBr) catalyst and N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA) ligand (Scheme 1). PMDETA was selected to yield the more stable copper(I) to mediate ATRP comparable to aliphatic amine ligand like 2,2′-bipyridine.58 Conversion of LMA into PLMA were studied by sequentially varying Br-CNF macroinitiator concentrations [I] (9.6 or 16 mM), LMA monomer concentrations [M] (800 or 1600 mM), and reaction times (1–24 h). The morphology of Br-CNF-g-PLMA was imaged by atom force microscopy (AFM) and their structures were characterized by attenuated total reflection (ATR) and solution phase proton nuclear magnetic resonance (1H NMR) spectroscopy. Thermal properties were characterized by thermogravimetric analysis (TGA). Surface hydrophobicity of Br-CNF-g-PLMA copolymer was characterized by WCA measurements of their cast films. Br-CNF-g-PLMA with varied DPs were further investigated as rheology modifier in toluene and drag reducer in pump oil under varied shear rates and temperatures.
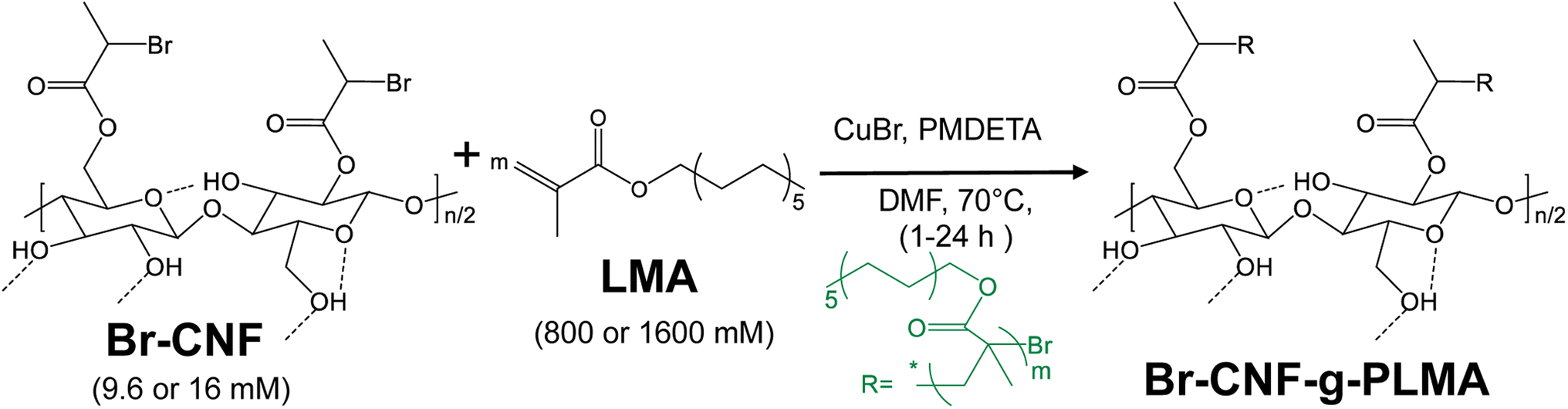 | ||
| Scheme 1 Reaction scheme for SI-ATRP of LMA on Br-CNF to Br-CNF-g-PLMA. | ||
2 Experimental section
2.1 Materials
Cellulose was isolated from rice straw by a previously reported three-step 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v toluene/ethanol extraction, acidified NaClO2 (1.4%, pH 3–4, 70 °C, 5 h) delignification, and alkaline hemicellulose dissolution (5% KOH, 90 °C, 2 h) process then lyophilized (Labconco Lyophilizer).59 Br-CNF was prepared by combined esterification with 2-Bromopropionyl bromide (BPB) at 5:1 BPB to anhydroglucose unit (AGU) (6 h, 23 °C) and ultrasonication (Qsonica Q700, 50/60 Hz; 50% amplitude, 30 min) method previously reported51 to 5.7 mmol surface Br esters per g of cellulose based on 80 wt% mass gain, i.e., equivalent to 3.2 mmol g−1 Br-CNF. Cuprous bromide (CuBr, Spectrum Chemical), N,N,N,N′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%, TCI America), N,N-dimethylformamide (DMF, certified grade, Fisher Scientific), toluene (ACS grade, Spectrum Chemical), methanol (ACS grade, Sigma Aldrich), tetrahydrofuran (THF, ACS grade, Alfa Aesar), deuterated dimethyl sulfoxide-d6 (DMSO-d6, ≥99.5% isotropic, Thermo Scientific), acetone (certified grade, Fisher Scientific), silicone (high temperature, Thermo Scientific), and vacuum pump oil (Welch® DuoSeal®) were used as received without further purification. Lauryl methacrylate (LMA, 97%, TCI America) was flushed by 5 M sodium hydroxide solution to remove inhibitor then dried by molecule sieves overnight. Highly oriented pyrolytic graphite (HOPG, grade ZYB) was used for AFM characterization. For UV-vis spectrophotometry, UV-vis standard cell quartz cuvettes (Fisher Scientific, 10 mm path length) were used.
:1 v/v toluene/ethanol extraction, acidified NaClO2 (1.4%, pH 3–4, 70 °C, 5 h) delignification, and alkaline hemicellulose dissolution (5% KOH, 90 °C, 2 h) process then lyophilized (Labconco Lyophilizer).59 Br-CNF was prepared by combined esterification with 2-Bromopropionyl bromide (BPB) at 5:1 BPB to anhydroglucose unit (AGU) (6 h, 23 °C) and ultrasonication (Qsonica Q700, 50/60 Hz; 50% amplitude, 30 min) method previously reported51 to 5.7 mmol surface Br esters per g of cellulose based on 80 wt% mass gain, i.e., equivalent to 3.2 mmol g−1 Br-CNF. Cuprous bromide (CuBr, Spectrum Chemical), N,N,N,N′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%, TCI America), N,N-dimethylformamide (DMF, certified grade, Fisher Scientific), toluene (ACS grade, Spectrum Chemical), methanol (ACS grade, Sigma Aldrich), tetrahydrofuran (THF, ACS grade, Alfa Aesar), deuterated dimethyl sulfoxide-d6 (DMSO-d6, ≥99.5% isotropic, Thermo Scientific), acetone (certified grade, Fisher Scientific), silicone (high temperature, Thermo Scientific), and vacuum pump oil (Welch® DuoSeal®) were used as received without further purification. Lauryl methacrylate (LMA, 97%, TCI America) was flushed by 5 M sodium hydroxide solution to remove inhibitor then dried by molecule sieves overnight. Highly oriented pyrolytic graphite (HOPG, grade ZYB) was used for AFM characterization. For UV-vis spectrophotometry, UV-vis standard cell quartz cuvettes (Fisher Scientific, 10 mm path length) were used.
2.2 SI-ATRP of Br-CNF with LMA
The Br-CNF macroinitiator at a 9.6 mM initiator concentration [I] was prepared by transferring 25 mL 0.3 w/v% Br-CNF (3.2 mmol g−1) in DMF to a Schlenk flask to which catalyst CuBr (0.034 g, 0.24 mmol) was dissolved under constant stirring. The mixture was degassed by 5 min sonication (Branson 2510) and purged with nitrogen for 10 min then capped with a rubber septum. The PMDETA (50.1 μL, 0.24 mmol) complexing ligand was dissolved in LMA (5.1 g, 20.0 mmol) monomer and sonicated (1 min). The prepared LMA at [M]o = 800 mM was then injected through a syringe into flask to initiate polymerization at 70 °C silicone oil bath for 1, 3, 4.5, 6 or 24 h and terminated by adding 5 mL THF. Each final mixture was washed by cold methanol and centrifugated (Eppendorf 5804R, 5k rpm, 10 min) to decant supernatant, then repeated two more times to remove all catalyst and unreacted monomer. The final precipitate was vacuum dried (0.5 atm) at 50 °C overnight to obtain Br-CNF-g-PLMA in the form of an elastic gel. SI-ATRP of LMA was also performed with Br-CNF macroinitiator [I] at 16 mM and LMA [M]o at 800 and 1600 mM, with 0.4 mmol of both CuBr and PMDETA for up to 24 h (Table S1†).The conversion (%) of LMA to PLMA was determined by PLMA mass gain on Br-CNF-g-PLMA over initial LMA mass. According ATRP unity polydispersity or equal chain lengths of PLMA, the degree of polymerization (DPmass) of PLMA based on mass gain was calculated as
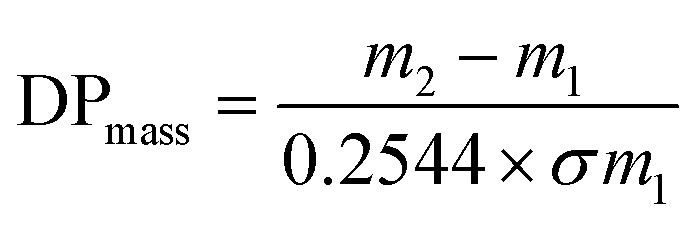 | (1) |
2.3 Characterizations
For imaging, Br-CNFs (10 μL, 0.0005 w/v%) in DMF and Br-CNF-g-PLMA (10 μL, 0.0005 w/v%) in toluene were deposited on highly oriented pyrophoric graphite (HOPG), air-dried in fume hood for 6 h, and profiled by AFM (Asylum-Research MFP-3D) in the tapping mode in 5 μm × 5 μm scan size and at rate of 512 Hz.For solution-state 1H NMR (Bruker AVIII 800 MHz 1H NMR spectrometer), Br-CNF was solvent exchanged to acetone then to DMSO-d6 followed by vacuum evaporation (0.5 atm, 50 °C, 1 h) as reported.51 Br-CNF-g-PLMA (ca. 10 mg) was added into 1 mL DMSO-d6, bath sonicated (1 h), and centrifuged (5k rpm, 10 min) to collect the supernatant for 1H NMR. The substitution (ρ) of Br-CNF surface OHs to 2-bromopropoinyl groups was quantified by solution state 1H NMR for calculation of percent OH converted to Br initiating sites, detailed previously.51 Crystallinity index (CrI) of Br-CNF was determined as previously described.51
Br-CNF-g-PLMA elastic gel was oven-dried (56 °C, overnight) for attenuated total reflectance (ATR) Fourier transform infrared (FTIR) spectroscopy and thermogravimetric analysis (TGA) characterization. For ATR characterization, each Br-CNF-g-PLMA was scanned by Thermo Nicolet 6700 FTIR spectrometer under ambient conditions from an accumulation of 128 scans at a 4 cm−1 resolution from 4000 to 400 cm−1. TGA was performed on each sample (10 mg) at 10 °C min−1 from 25 to 500 °C under purging N2 (50 mL min−1) using a TGA-50 thermogravimetric analyzer (Shimadzu, Japan). Moisture content (%) was the mass loss at 140 °C and char residue (%) was the mass at 500 °C.
Viscosities of Br-CNF and Br-CNF-g-PLMA were determined in their most compatible liquids. Viscosities of Br-CNF were determined in DMF at 0.5, 0.3 and 0.1 w/v% and Br-CNF-g-PLMA in toluene at 4, 6, 8 and 10 w/v% at 25 °C in shear rates from 1 to 220 s−1 using a Brookfield DV3T rheometer. Similarly, viscosities of Br-CNF-g-PLMA in toluene (4 w/v%) or oil (1, 2 and 4 w/v%) were measured at elevated temperatures of 40 °C and 55 °C. Power law model60 was used to calculate the flow behavior index (n) of Br-CNF-g-PLMA in toluene as follows:
| η = aγn−1 | (2) |
Br-CNF-g-PLMA dispersions in toluene (1 w/v%) were scanned by UV-vis spectroscopy (Thermo Scientific, Evolution 600) from 325 to 800 cm−1 at 4 cm s−1. Thin films were prepared by depositing ca. 1 mL Br-CNF-g-PLMA in toluene (1 w/v%) on clean glass slides and dried overnight in fume hood. Water contact angle (WCA) measurements on both sides of sessile drops Milli-Q water (5 μL) on Br-CNF-g-PLMA films were measured on a total of 5 images (n = 5). Using the ImageJ Analyzer and the average values reported.
2.4 Rheology of Br-CNF-g-PLMA dispersions in toluene and pump oil
Br-CNF-g-PLMA (1.5 g) with DPmass = 16, 32, 40 and 46 was added in 10 mL toluene then sonicated (Branson 2510) 1 h to prepare homogeneous dispersions at 15 w/v% for viscosity measurements at varied shear rates from 1 to 220 s−1 shear rates at 25 °C. The same procedure was repeated for 10, 8, 7, 6, 4, 2, 1 and 0.5 w/v% Br-CNF-g-PLMA in toluene. For Br-CNF-g-PLMA with DPmass = 3, 0.2 g was added in 5 mL toluene then sonicated 1 h to prepare 4 w/v% homogeneous dispersion and 2, 1 and 0.5 w/v% serial dilutions for the same rheology measurements. To prepare pump oil dispersions, 10 mL vacuum pump oil was added to 4 w/v% Br-CNF-g-PLMA toluene dispersion, sonicated for 10 min, then was vacuum oven dried (0.5 atm, 50 °C, 24 h) to evaporate toluene to 4 w/v% Br-CNF-g-PLMA oil dispersion. This 4 w/v% was then serial diluted to 2 and 1 w/v% Br-CNF-g-PLMA oil dispersions.3 Results and discussion
3.1 SI-ATRP on Br-CNF
The 2-bromoproponyl esterified CNF (Br-CNF) macroinitiator was facilely synthesized via one-pot esterification and in situ ultrasonication in DMF to carry 3.2 mmol g−1 Br ester in average 4.6 nm thickness, 29.3 nm width, ca. 1 μm length, and 0.48 CrI. SI-ATRP of LMA on Br-CNF was conducted at 70 °C with 9.6 or 16 mM Br-CNF macroinitiator concentration [I], 800 or 1600 mM LMA monomer concentration [M]o for 1 to 24 h (Table S1†). The overall LMA to PMLA conversion and semilogarithmic ln[M]o/[M] monomer consumption increased with polymerization reaction time at up to 6 h, then leveled (Fig. 1a and b). The exception was the continuing increase of LMA consumption at 16 mM [I] and 800 mM [M]. At the lower 800 mM [M]o, higher apparent LMA consumption at a rate constant of 0.1829 h−1 was observed at higher [I], 41% higher than the 0.1295 h−1 rate constant for the lower [I], as expected. However, at the higher 16 mM [I], the apparent rate constant with the higher 1600 mM [M]o, or 100 [M]o/[I] ratio, was 0.0401 h−1, only about one fifth of that at 800 mM, half of [M]o. The lower rate constant at high [M]o might be attributed to pre-termination at early stage (t < 1 h) as reported before.61 At 9.6 mM [I] and 800 mM [M], or 83 [M]o/[I] ratio, LMA to PLMA conversion raised most rapidly with polymerization time to 74.8% at 6 h, then further increased to reach 92.7% at 24 h. Clearly, higher conversion was achieved at the highest macroinitiator [I] of 16 mM, but optimized at 83 [M]o to [I] ratio.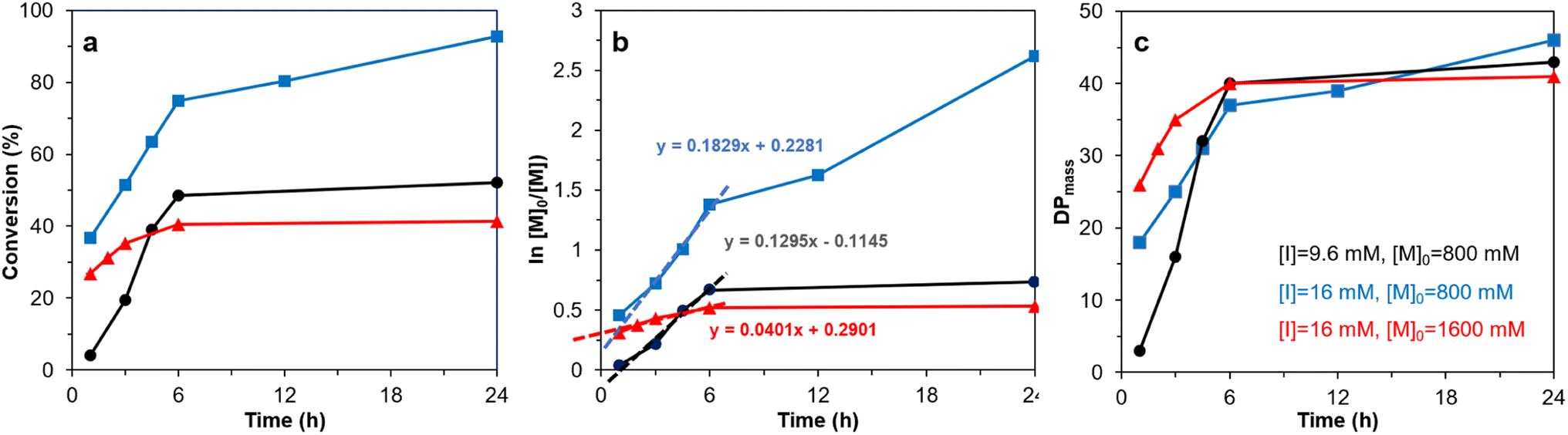 | ||
| Fig. 1 SI-ATRP of LMA ([M]o = 800 or 1600 mM) on Br-CNF ([I] = 9.6 or 16 mM) at 70 °C: (a) conversion, (b) ln ([M]o/[M]), (c) DPmass. Same sample legends in (c) apply to (a) and (b). | ||
Under the equal chain length assumption for ATRP, the PLMA graft length on Br-CNF-g-PLMA surfaces increased dramatically from 3, 18, and 26 to the similar 37 and 40 DPmass with increasing polymerization time from 1 to 6 h (Fig. 1c), then only slightly increased to the respective 43, 41, and 46 DPmass at 24 h. The negligible chain growth beyond ca. 40 DPmass suggests chain termination beyond 6 h. Thus, 6 h is the optimal propagation time for preparing Br-CNF-g-PLMA with ca. 40 repeating units under all three scenarios. Taking highest monomer conversion, polymerization rate, and achievable graft chain length into consideration, 800 mM [M]o, 16 mM [I], and 24 h were deemed the optimal condition to reach the 92.7% conversion, significantly higher than all previously reported conversions, i.e., 15 to 35% on as-is CNCs22,31,33 and 23 to 85% even with added sacrificial initiators26,27,29,30,34,35,37,39,41,46–49 (Table 1). The robust polymerization and significantly higher monomer to PLMA conversion reflect the unique charactertistics of Br-CNF including the advantageously high Br ester contents and the excellent compatibility of Br-CNF in the reaction media while also confirm the superior accessibility of initiating cites on the Br-CNF macroinitiator surfaces. All supports the superior characteristics of Br-CNF macroinitiator and the advantage of this one-pot synthesis via esterification of cellulose with 2-bromopropionyl bromide (BPB) and in situ ultrasonication. Clearly, Br-CNF has demonstrated to be a highly effective macroinitiator capable of SI-ATRP of LMA of controlled lengths of 3 to 46 DPmass at high conversions of up to 92.7% (Table S2†).
3.2 Mv of Br-CNF-g-PLMA by viscosity
SI-ATRP of LMA on Br-CNF surfaces was highly effective to produce substantial surface PLMA grafts that the Br-CNF core only amounted to 2.7 to 7.4 w% of Br-CNF-g-PLMA, except for 28.1 w% from one with the shortest 3 DPmass graft and lowest 4% conversion (Table S1†). These consistently low CNF core contents of Br-CNF-g-PLMA are highly unusual, with only two ATRP grafted CNC22,37 having less than 10% core mass. With predominant 92.7 to 97.3% surface PLMA grafts, Br-CNF-g-PLMA can be considered analogous to copolymers to derive molecular mass by their solution viscosity or Mv. First, the most compatible solvent was determined by suspending 5 w/v% Br-CNF-g-PLMA with longest graft in pyridine, ethyl acetate, chloroform, toluene, and hexane. Toluene was found to give transparent Br-CNF-g-PLMA dispersion, thus most compatible to PLMA surface graft, while all others were translucent (Fig. S1†). The viscosities of four Br-CNF-g-PLMA with 16, 32, 40, and 46 DPmass in toluene at varied concentrations were thus measured at 25 °C to estimate Mv of Br-CNF-g-PLMA using the Mark–Houwink equation:62| Mv = ([η]/K)1/α | (3) |
ηr) over concentration (C) or inherent viscosity 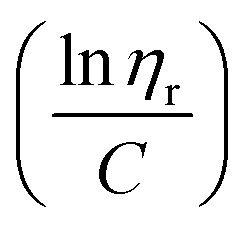 to the y axis, i.e.,
to the y axis, i.e., 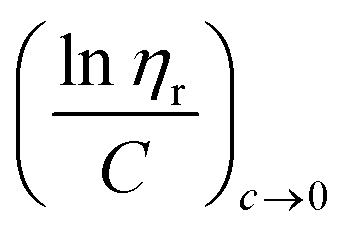 (Fig. 2a). To meet sufficiently dilute concentration criteria64 for accurate intrinsic viscosity determination, only viscosities of Br-CNF-g-PLMA at concentrations below 0.15 g mL−1 were used. In addition, only the linear regions for each sample were included. The inherent viscosity
(Fig. 2a). To meet sufficiently dilute concentration criteria64 for accurate intrinsic viscosity determination, only viscosities of Br-CNF-g-PLMA at concentrations below 0.15 g mL−1 were used. In addition, only the linear regions for each sample were included. The inherent viscosity 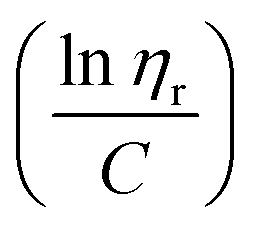 was considered more reliable in deriving Mv due to their higher linear relationships, than the reduced viscosity
was considered more reliable in deriving Mv due to their higher linear relationships, than the reduced viscosity 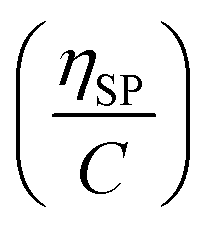 (Fig. S2†).
(Fig. S2†).
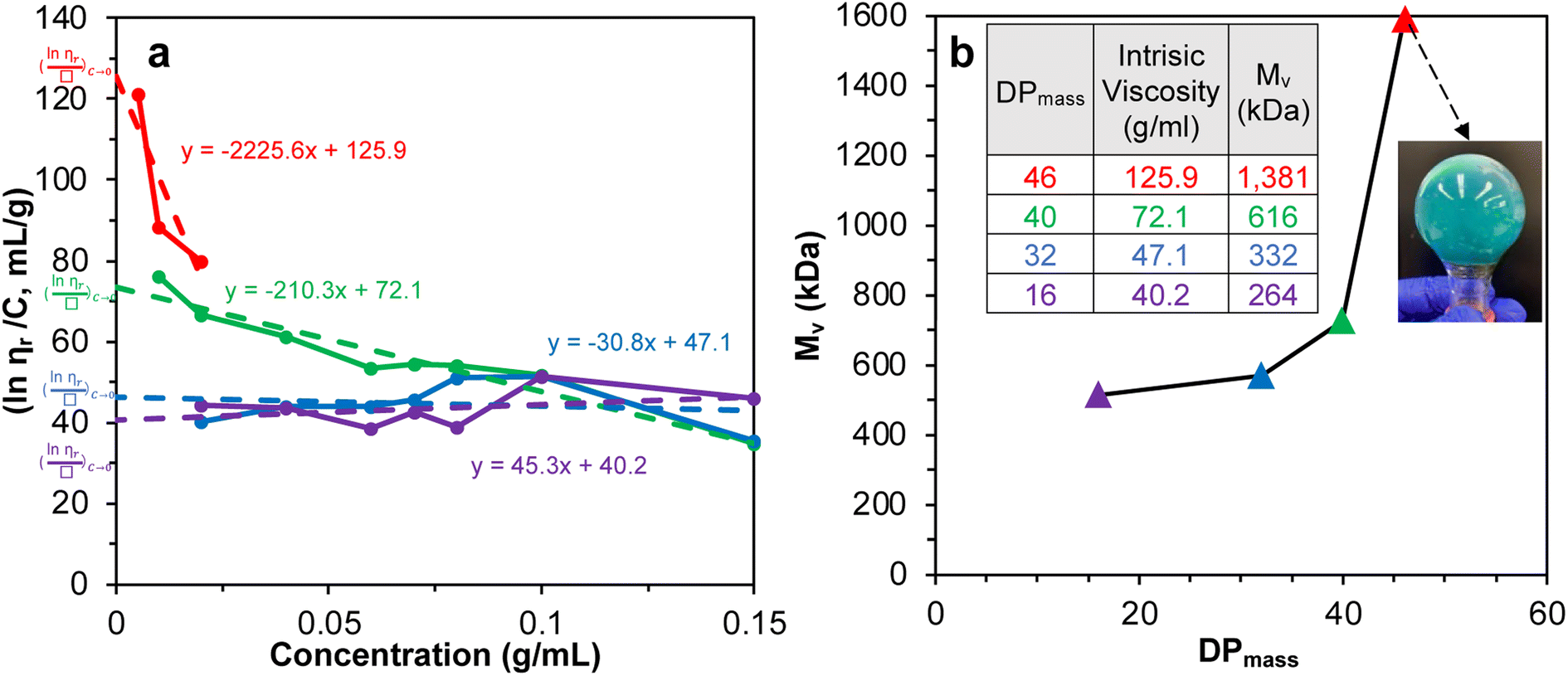 | ||
Fig. 2 Plot of (a) inherent 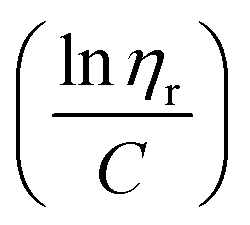 versus concentration (C) for Br-CNF-g-PLMA in toluene; (b) Mv versus estimated DPmass by eqn (1) with calculated intrinsic viscosities. Inset image shows gel in an inverted round-bottom flask. versus concentration (C) for Br-CNF-g-PLMA in toluene; (b) Mv versus estimated DPmass by eqn (1) with calculated intrinsic viscosities. Inset image shows gel in an inverted round-bottom flask. | ||
The Mv derived from the intrinsic viscosity (h), i.e., the intercept of inherent viscosity 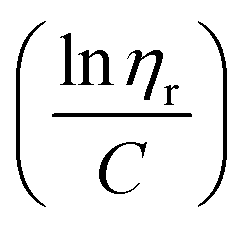 vs. concentration plots, moderately increased from 264 to 616 kDa, corresponding to increasing DPmass from 16 to 40, then more than doubled to 1381 kDa (2.2×) or 46 DPmass (Fig. 2b). With increasing grafting lengths of the hydrophobic LMA on the relatively polar Br-CNF surface, Br-CNF-g-PLMA dispersed in DMF was transparent initially, became milky at 1 h, then phase separated at 3 h, and finally reached gelation (800 mM [M]o, 16 mM [I]) at 24 h. This observation is consistent with the expectation that Br-CNF-g-PLMA with longer PLMA chain lengths became less compatible to the polar DMF to coalesce and the surface grafted chains contracted around the Br-CNF core. One possible reason for significantly higher Mv corresponding to the slight 40 to 46 DPmass increase (Fig. 2b) may be attributed to termination by coupling that was supported by the gelation observed in DMF noted earlier. It should also be mentioned that these Mv may be underestimated since K and α values used in these derivations were from the more polar THF, thus likely higher for toluene (Fig. S1†). Nevertheless, the robustly synthesized Br-CNF-g-PLMA with PLMA surface grafts in tunable lengths have estimated Mv of 264 to 1381 kDa (Table S2†).
vs. concentration plots, moderately increased from 264 to 616 kDa, corresponding to increasing DPmass from 16 to 40, then more than doubled to 1381 kDa (2.2×) or 46 DPmass (Fig. 2b). With increasing grafting lengths of the hydrophobic LMA on the relatively polar Br-CNF surface, Br-CNF-g-PLMA dispersed in DMF was transparent initially, became milky at 1 h, then phase separated at 3 h, and finally reached gelation (800 mM [M]o, 16 mM [I]) at 24 h. This observation is consistent with the expectation that Br-CNF-g-PLMA with longer PLMA chain lengths became less compatible to the polar DMF to coalesce and the surface grafted chains contracted around the Br-CNF core. One possible reason for significantly higher Mv corresponding to the slight 40 to 46 DPmass increase (Fig. 2b) may be attributed to termination by coupling that was supported by the gelation observed in DMF noted earlier. It should also be mentioned that these Mv may be underestimated since K and α values used in these derivations were from the more polar THF, thus likely higher for toluene (Fig. S1†). Nevertheless, the robustly synthesized Br-CNF-g-PLMA with PLMA surface grafts in tunable lengths have estimated Mv of 264 to 1381 kDa (Table S2†).
3.3 Characterization of Br-CNF-g-PLMA by ATR spectroscopy and thermal analysis
The FTIR of Br-CNF showed prominent 3400 cm−1 O–H and 1040 cm−1 C–O and C–C–O (1035 cm−1)65 stretching peaks, characteristics cellulose, whereas the appearance of ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak at 1740 cm−1 confirmed the successful conversion of cellulose OHs to 2-bromopropionyl esters (Fig. 3a). This 1040 cm−1 peak intensity dramatically reduced for Br-CNF-g-PLMA with 3 and 16 DPmass, then disappeared for those with higher DPmass of 32, 40 and 46, constant with their very low respective 3.8, 3.1 and 2.7 w% cellulose contents, whereas the O–H stretching peak at 3400 cm−1 disappeared for all Br-CNF-g-PLMA irrespective of their DPs, corresponding to absence of moisture. The ester CO stretching at 1740 cm−1 from Br-CNF and PLMA grafts remained similar while both sp3 ester and sp2 C–C stretching peaks at 2860 cm−1 and 2930 cm−1 were slightly more intense for Br-CNF-g-PLMA. With increasing PLMA graft lengths from 0 to 46 repeating units, the corresponding Br-CNF contents decreased significantly from 100 to 3.1 w%. Consequently, the moisture contents proportionally reduced from 8.5% to 0.05% and the char residue (%) lowered from 9.9 to 1.7%. Intriguingly, the increasing PLMA grafting on Br-CNF elevated the respective onset and maximum decomposition temperatures by a few oC to up to 24 °C and 59 °C, respectively (Fig. 3b–d). This is in contrast to the observed degradation of CNF backbone with high density of surface grafts.49 All Br-CNF-g-PLMAs showed 2nd, even 3rd max degradation at and above 317 °C, where the second is close to the 327 °C depolymerization temperature reported for PLMA (Mn = 29 kDa).66 The significantly improved thermal stability of Br-CNF-g-PLMAs presents the evidence of another unique advantage.
O stretching peak at 1740 cm−1 confirmed the successful conversion of cellulose OHs to 2-bromopropionyl esters (Fig. 3a). This 1040 cm−1 peak intensity dramatically reduced for Br-CNF-g-PLMA with 3 and 16 DPmass, then disappeared for those with higher DPmass of 32, 40 and 46, constant with their very low respective 3.8, 3.1 and 2.7 w% cellulose contents, whereas the O–H stretching peak at 3400 cm−1 disappeared for all Br-CNF-g-PLMA irrespective of their DPs, corresponding to absence of moisture. The ester CO stretching at 1740 cm−1 from Br-CNF and PLMA grafts remained similar while both sp3 ester and sp2 C–C stretching peaks at 2860 cm−1 and 2930 cm−1 were slightly more intense for Br-CNF-g-PLMA. With increasing PLMA graft lengths from 0 to 46 repeating units, the corresponding Br-CNF contents decreased significantly from 100 to 3.1 w%. Consequently, the moisture contents proportionally reduced from 8.5% to 0.05% and the char residue (%) lowered from 9.9 to 1.7%. Intriguingly, the increasing PLMA grafting on Br-CNF elevated the respective onset and maximum decomposition temperatures by a few oC to up to 24 °C and 59 °C, respectively (Fig. 3b–d). This is in contrast to the observed degradation of CNF backbone with high density of surface grafts.49 All Br-CNF-g-PLMAs showed 2nd, even 3rd max degradation at and above 317 °C, where the second is close to the 327 °C depolymerization temperature reported for PLMA (Mn = 29 kDa).66 The significantly improved thermal stability of Br-CNF-g-PLMAs presents the evidence of another unique advantage.
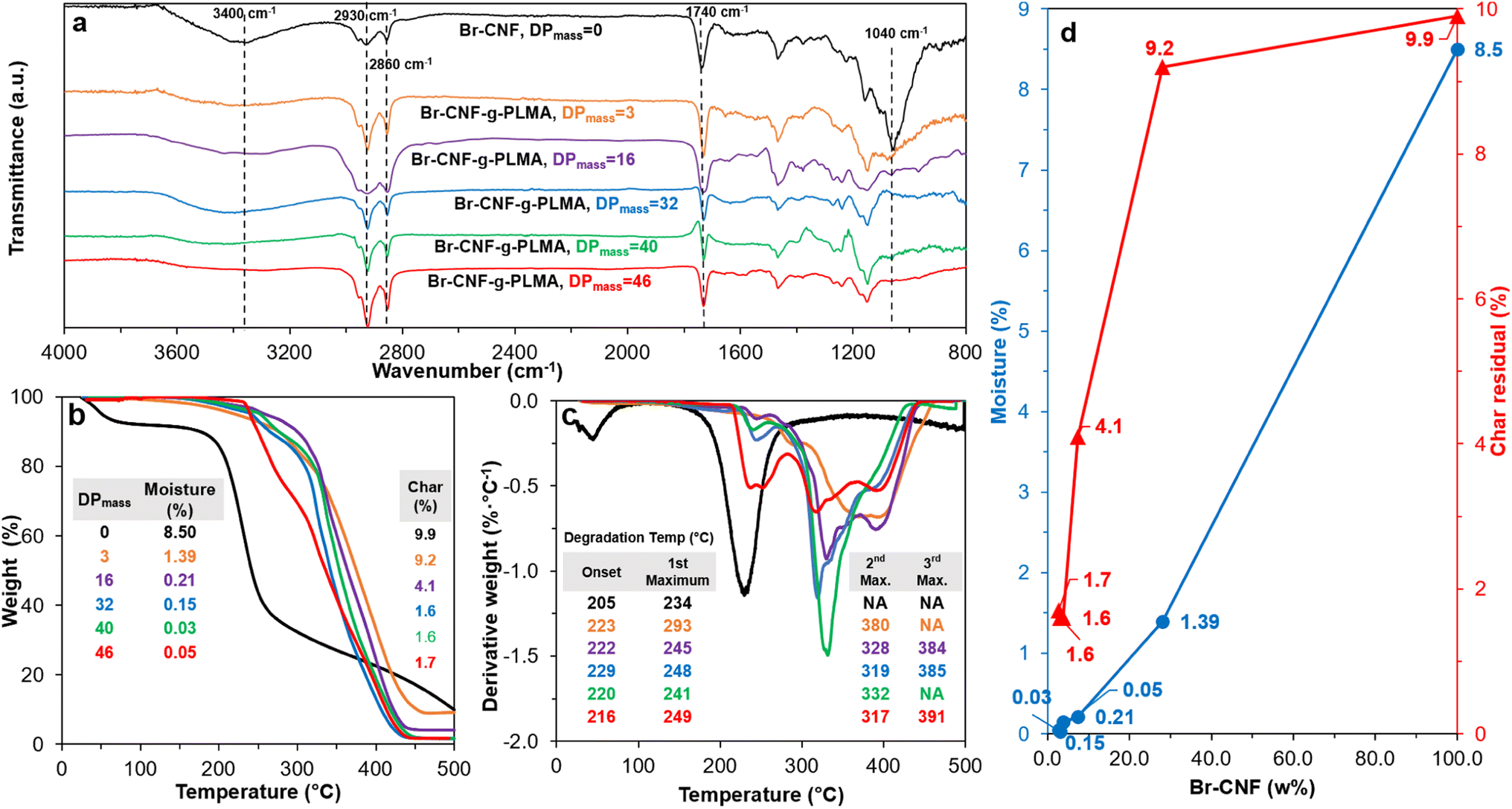 | ||
| Fig. 3 Characterization of Br-CNF and Br-CNF-g-PLMA: (a) ATR spectra; (b) TGA; (c) DTGA curves; and (d) moisture and char residue in relationship to Br-CNF contents. | ||
Degree of polymerization (DP) of PLMA graft by solution-state 1H-NMR.
The 1H-NMR spectra of Br-CNF and Br-CNF-g-PLMA with varied DPs (Fig. 4a) were displayed with corresponding protons (Fig. 4b). The 1H-NMR spectra of all five Br-CNF-g-PLMA spectra showed the furthest downfield H6 and H6′ peaks of the Br-CNF protons at δ 3.63–3.89, consistent with those at δ 3.71–4.06 for Br-CNF ref. 51 and δ 3.65–3.88 for dissolved MCC in NaOD/D2O.67 Multiple overlapping peaks between δ 3.29–3.58 were assigned to H2, H3, H4 and H5, matching those at δ 3.16–3.70 of Br–CNF51 and δ 3.27–3.66 of TEMPO-CNF in D2O.68 The theoretical furthest downfield cellulosic H1 proton peak at δ 4.20–4.52 in Br-CNF disappeared upon grafting with PLMA due to potential overlapping with broad PLMA methylene He at δ 4.10. For proton peaks on grafted PLMA chains, chemical shift of He, Hc + Hf, Hd + Hh, and Hg were assigned to δ 4.05, δ 1.41–1.58, δ 0.81, and δ 1.13–1.27, corresponding to δ 3.96, δ 1.65–1.84, δ 0.93 and δ 1.32 of homopolymer PLMA in chloroform-d1.69 The average ratios of integrated protons He (methylene, –CH2–O–):Hc + Hf (methylene, –CH2–):Hd + Hh (methyl, –CH3):Hg (methylene, –(CH2)9-peaks were 1:1.9:2.3:12.8 for all five Br-CNF-g-PLMAs (Fig. 4c), close to the theoretical 1:2:3:9 proton ratio, thus confirming these proton assignment for the PLMA grafts.
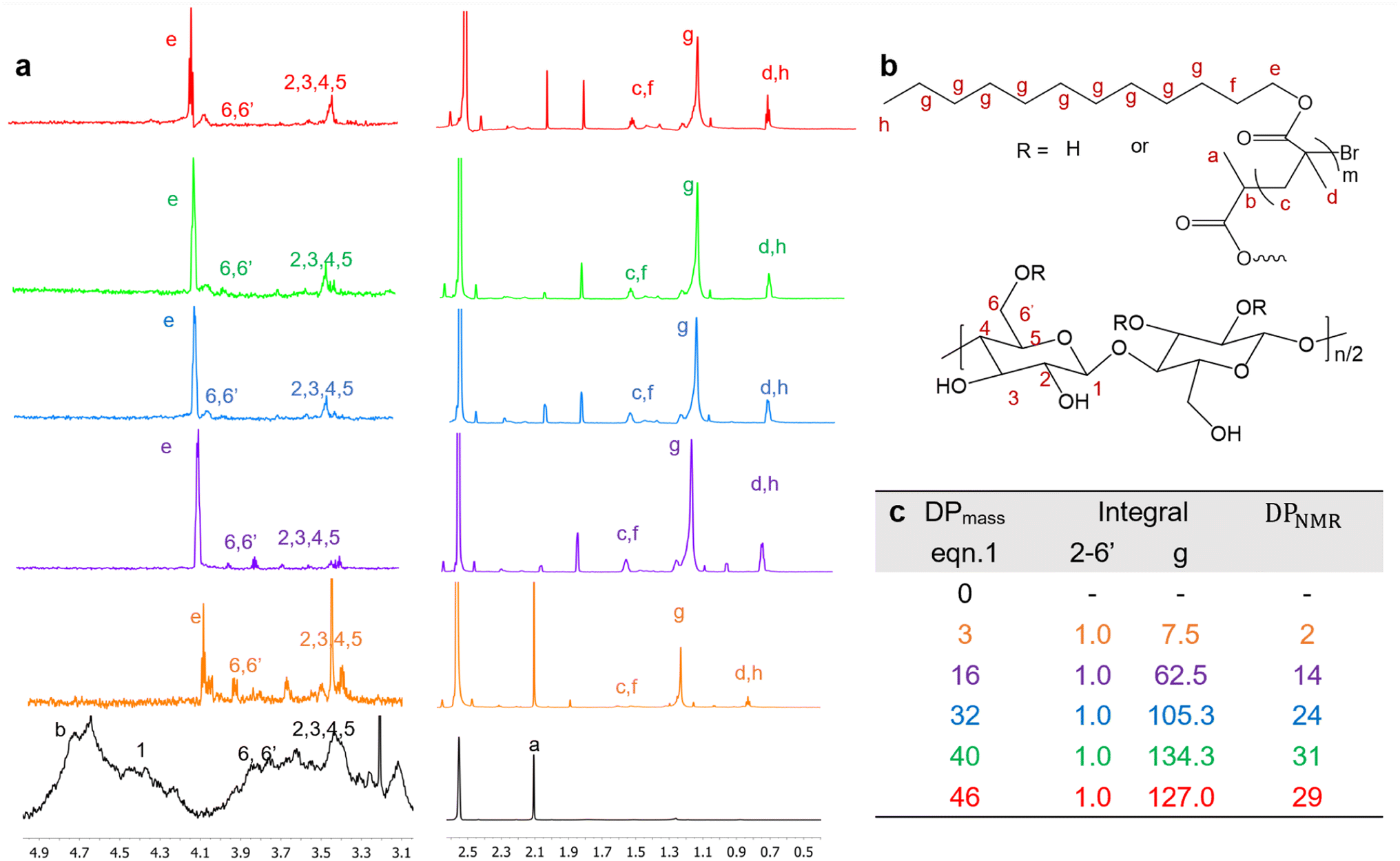 | ||
| Fig. 4 1H NMR spectra of Br-CNF and Br-CNF-g-PLMA in DMSO-d6 (color coded in c): (a) spectra with assigned protons; (b) proton assignment for Br-CNF backbone and PLMA chains; (c) DPNMR integral of Br-CNF 2–6′ protons and PLMA methylene g protons. | ||
Assuming all anomeric protons of amorphous and surface AGUs of Br-CNF are detectable by 1H NMR, surface AGUs was the sum of the integrated areas for anomeric H2 to H6′ proton peaks, averaged by 6 anomic protons for amorphous or 3 anomic protons from the half exposed on the surface. H1 proton peak was excluded due to overlapping with methylene proton (Hb). LMA units could be estimated by integration of the areas of methylene Hg divided by 18 respective protons. LMA units per surface AGU was determined mathematically by the area ratio of LMA calculated from Hg over surface AGUs calculated from H2 to H6′. The DPNMR could then be calculated from DPs in the amorphous region or crystalline surfaces as follows.
For amorphous Br-CNF, each AGU has 3 exposed OHs, DPNMR,amorphous representing the # of LMA per initiating sites, was calculated by dividing # of LMA by 3 OHs per AGU and level of substitution (ρ = 0.48) as
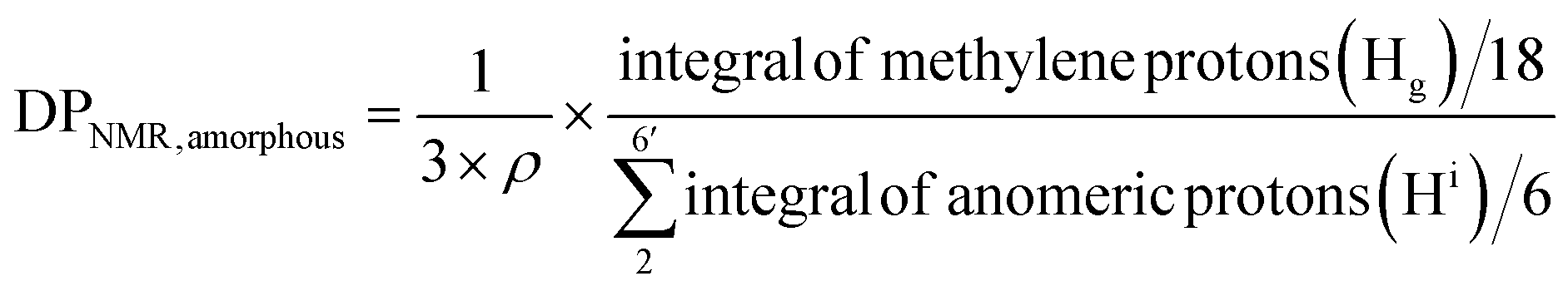 | (4) |
For crystalline surfaces of Br-CNF, each cellobiose (two AGUs) has three exposed OHs, DPNMR,crystalline, representing the # of LMA per initiating sites, was calculated by dividing # of LMA by 1.5 OHs per surface AGU and level of substitution (ρ = 0.48) according to eqn (5):
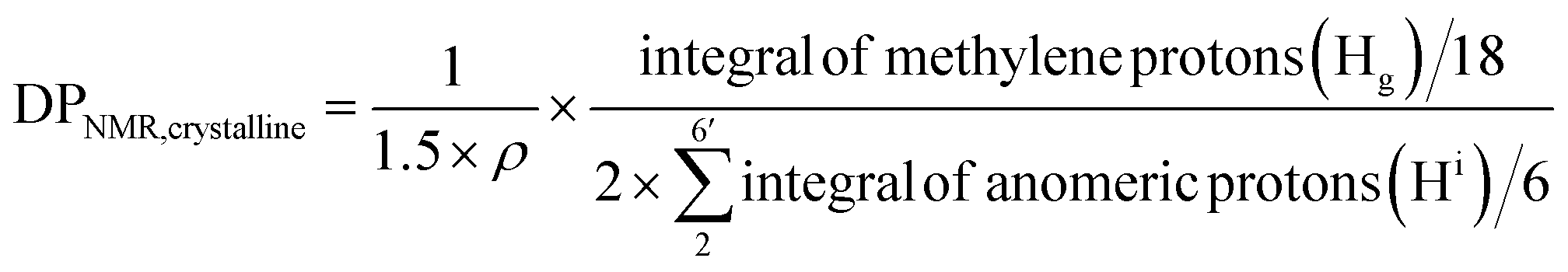 | (5) |
Since DP derived from either amorphous regions or crystalline surfaces gave the same by either eqn (4) or (5), DPNMR can be obtained as DPNMR,amorphous = DPNMR,crystalline. Where ρ is 0.48, or 48% OHs on surface AGUs of Br-CNF were converted to Br initiating sites. DPNMR calculated by 1H NMR were 2, 14, 24, 31, and 29 for Br-CNF-g-PLMA, lower than the DPmass estimated from mass gain (eqn. (1)) by 12.5% to up to 37% (Fig. 4c). In the polar DMSO-d6 used for solution-state NMR, the more hydrophilic Br-CNFs were dispersed but the increasingly more hydrophobic Br-CNF-g-PLMA with longer and more hydrophobic PLMA became less dispersible to be fully detected by NMR thus might underestimate Hg and DPNMRthan the DP derived by mass balance. Never-the-less, this is the first successful demonstration of direct chain length determination (without chain cleavage) of grafted polymers on nanocellulose surfaces via 1H NMR. This direct analytical approach is significant, in particular for such opposing solvent dispersibility of hydrophilic Br-CNF backbone and hydrophobic surface PLMA grafts.
3.4 Surface compatibility of Br-CNF-g-PLMA
Br–CNF and Br-CNF-g-PLMA (DPmass = 3 and 46) were imaged by AFM on freshly exfoliated graphite. Br-CNF spread evenly and appeared as interconnecting nanofibrils with 4.7 nm average thickness and varying lengths at the order of ca. 1 μm (Fig. 5a). Br-CNF-g-PLMA with 3 DPmass grafts agglomerated into particulates with some nanofibrils (Fig. 5b) whereas Br-CNF-g-PLMA with the longest 46 DPmass grafts appeared as larger particulates only (Fig. 5c). With increasing lengths of hydrophobic PLMA grafts, Br-CNF-g-PLMA became increasingly incompatible to the moderately hydrophobic graphite surface (WCA = 71.8°) to coil and aggregate. All 1 w/v% Br-CNF-g-PLMA (DPmass = 3, 16, 32, 40 and 46) dispersions in toluene appeared transparent, but transmitted less visible light from 400 to 800 cm−1 wavelength with increasing graft lengths from 16 to 46 DPmass (Fig. 5d). Br-CNF-g-PLMA with longer grafts led to higher molar attenuation coefficient in Beer–Lambert law,70 causing increased absorbance at same concentration and path length. The Br-CNF-g-PLMA with shortest 3 DPmass graft was least compatible with the nonpolar toluene to aggregate, increasing molar attenuation coefficient to higher absorbance than those with 16 and 32 DPmass. The WCAs of casted films increased from 69.8° for Br-CNF to 80.9° and 86.2° for Br-CNF-g-PLMA with respective 3 and 40 DPmass, then significantly increased to 110.6° for Br-CNF-g-PLMA with 46 DPmass (Fig. 5e). The slightly increased surface hydrophobicity of Br-CNF-g-PLMA than Br-CNF reflect the slightly more hydrophobic PLMA than 2-bromopropionyl ester of Br-CNF as reported,52 but the hydrophobicity was less dependent on the PLMA chain lengths until reaching 46 DPmass, the gelation point (Fig. 2b).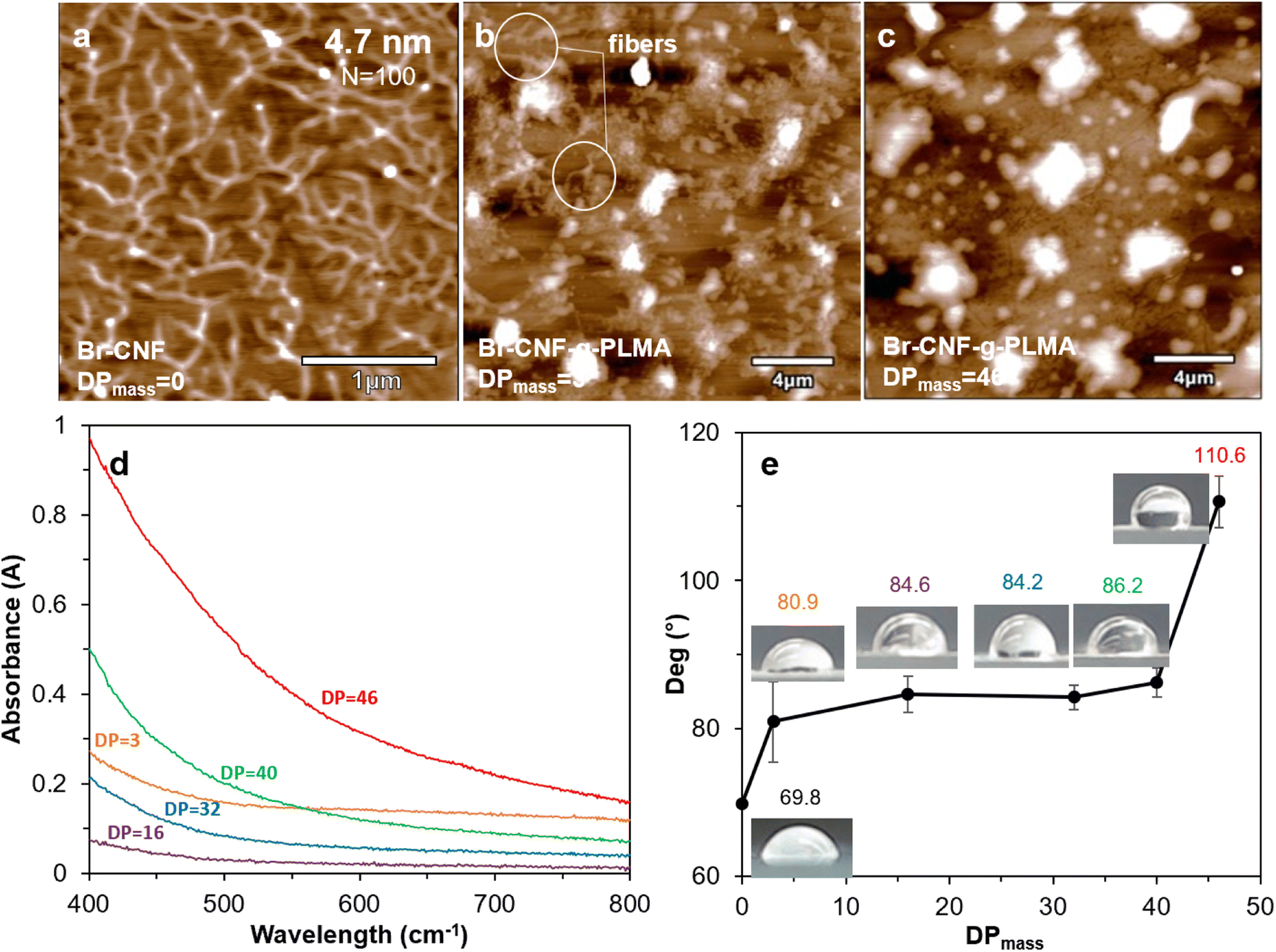 | ||
| Fig. 5 AFM images on HOPG: (a) Br-CNF in DMF; Br-CNF-g-PLMA in toluene (b) DPmass = 3, (c) DPmass = 46, all from 10 μL at 0.0005 w/v%; (d) UV-vis spectra of Br-CNF-g-PLMA (1 w/v%, toluene); (e) WCAs on films cast from Br-CNF-g-PLMA with varied DPmass. | ||
3.5 Viscosities of Br-CNF-g-PLMA at varied shear rates
The viscosities (η) of Br-CNF-g-PLMA with varied graft lengths and in varying concentrations (4, 6, 8 and 10 w/v%) in toluene were measured at 1 to 220 s−1 shear rates (γ) at 25 °C. The flow behavior index (n) was derived from the slope (n − 1) of natural logarithm η vs. γ plot according to the Power law model η = aγn−1 (eqn (2)). Theoretically, n < 1 indicates pseudoplastic or shear-thinning behavior of a liquid. For Br-CNF-PLMA with short 16 DPmass grafts, Newtonian behaviors were observed at lower concentrations and shear thinning behavior became apparent only at 10 w/v% (n = 0.72) (Fig. 6a and S3a†). As PLMA grafts lengthened to 32 and 40 DPmass, shear-thinning behaviors were observed at 10 and 8 w/v% with corresponding n values near 0.71 and 0.84–0.87, respectively (Fig. S3b and c†). Only Br-CNF-g-PLMA with longest 46 DPmass grafts exhibited pseudoplastic behaviors at all concentrations from 4 to 10 w/v% and lower n values between 0.38 and 0.47 (Fig. 6b). The Newtonian behavior Br-CNF-g-PLMA at lower concentrations are in similar trend as Br-CNF in DMF.52 Br-CNF dispersions in DMF exhibited Newtonian behaviors, i.e., their viscosities were independent of shear rates from 0.06 to 0.25 w/v%, but exhibited a shear thinning region at low shear rates below 150 s−1 and a Newtonian region above at 0.5 w/v%. The shear-rate dependent viscosity thresholds of Br-CNF-g-PLMA concentrations in toluene are, however, ca. one magnitude higher, attributing to the much greater effects of PLMA grafts. At any given concentration between 4 and 10w/v% Br-CNF-g-PLMA, those with proportionally more Br-CNF (red, blue, green) exhibited several magnitude higher viscosity enhancement than that with far less CNF (purple), demonstrating Br-CNF-g-PLMA copolymers to be better drag reducers than PLMA alone. These observations confirm that the shear thinning phenomenon of Br-CNF core and viscosity enhanced effects of PLMA grafts were successfully combined in Br-CNF-g-PLMA produced via ATRP of PLMA on Br-CNF macroinitiator.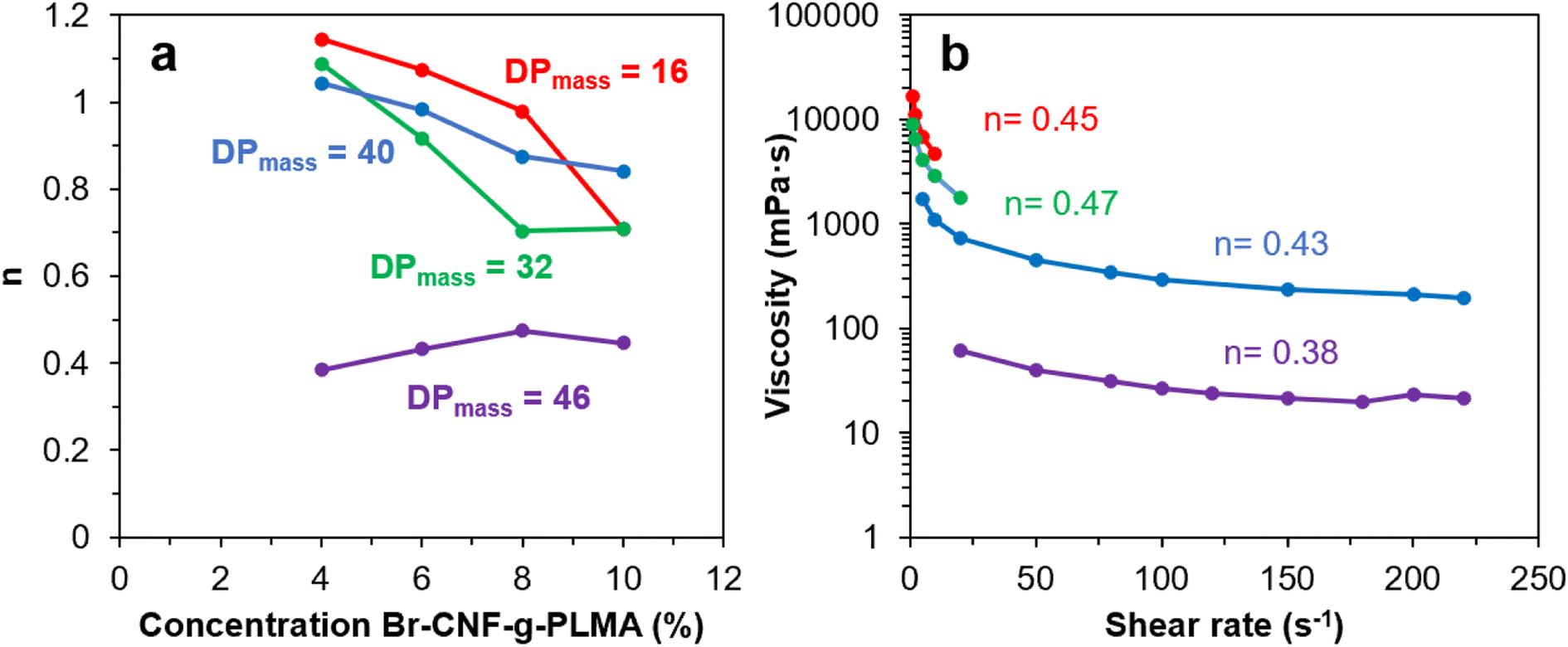 | ||
| Fig. 6 Br-CNF-g-PLMA toluene dispersions (16, 32, 40, 46 DPmass): (a) flow behavior index (n) at varied concentrations; (b) viscosity (10 w/v%) at varied shear rates. | ||
3.6 Br-CNF-g-PLMA rheology at expanded concentrations and elevated temperatures
Rheology of Br-CNF-g-PLMA in toluene were further evaluated to include lower concentrations (0.5 to 10 w/v%) and elevated temperatures (25, 40 and 55 °C) (Fig. 7). Viscosities of Br-CNF-g-PLMA in toluene at the lowest 0.5 w/v% concentration were all below 1 mPa·s, only slightly higher than the 0.464 mPa·s of toluene alone (Fig. 7a). Viscosities of Br-CNF-g-PLMA with shorter chain lengths (DPmass = 3, 16 and 32) moderately increased to 1.51 (3.3×), 3.16 (6.8×) and 3.24 (7.0×) mPa·s as concentration increased to 4 w/v%, while viscosities of those with longer chains (DPmass = 40 and 46) significantly increased to 6.93 (15×) and 29.7 (64×) mPa·s at 4 w/v% concentrations. As concentration increased to 10 w/v%, viscosities of Br-CNF-g-PLMA (DPmass = 16, 32 and 40) significantly increased (153 to 211×), while that with longest side chain (DPmass = 46) appeared as a viscous gel with dramatically increased 9777 mPa·s viscosity (21071×). At constant 4 w/v%, the viscosities of all Br-CNF-g-PLMA with varied DPmass decreased with increasing temperatures from 25 to 55 °C (Fig. 7b), as expected. The viscosities reduced slightly more to 0.61×, 0.64× and 0.67× for Br-CNF-g-PLMA with longer 32, 40 and 46 DPmass grafts than to 0.71× and 0.84× for those with shorter 3 and 16 DPmass grafts, respectively. The enhanced viscosity improvement with longer side chain was also observed (Fig. S4†), in which viscosity significantly increased to 9.3× or 307× with increase of DPmass from 16 to 46 at respective 4 or 8 w/v%. The longer PLMA grafts on Br-CNF surface are expected to increase inter Br-CNF-g-PLMA attractions at any given concentration to resist flow, causing more significant viscosity enhancement than those with shorter grafts, for application as viscosity modifiers in paints and coatings.
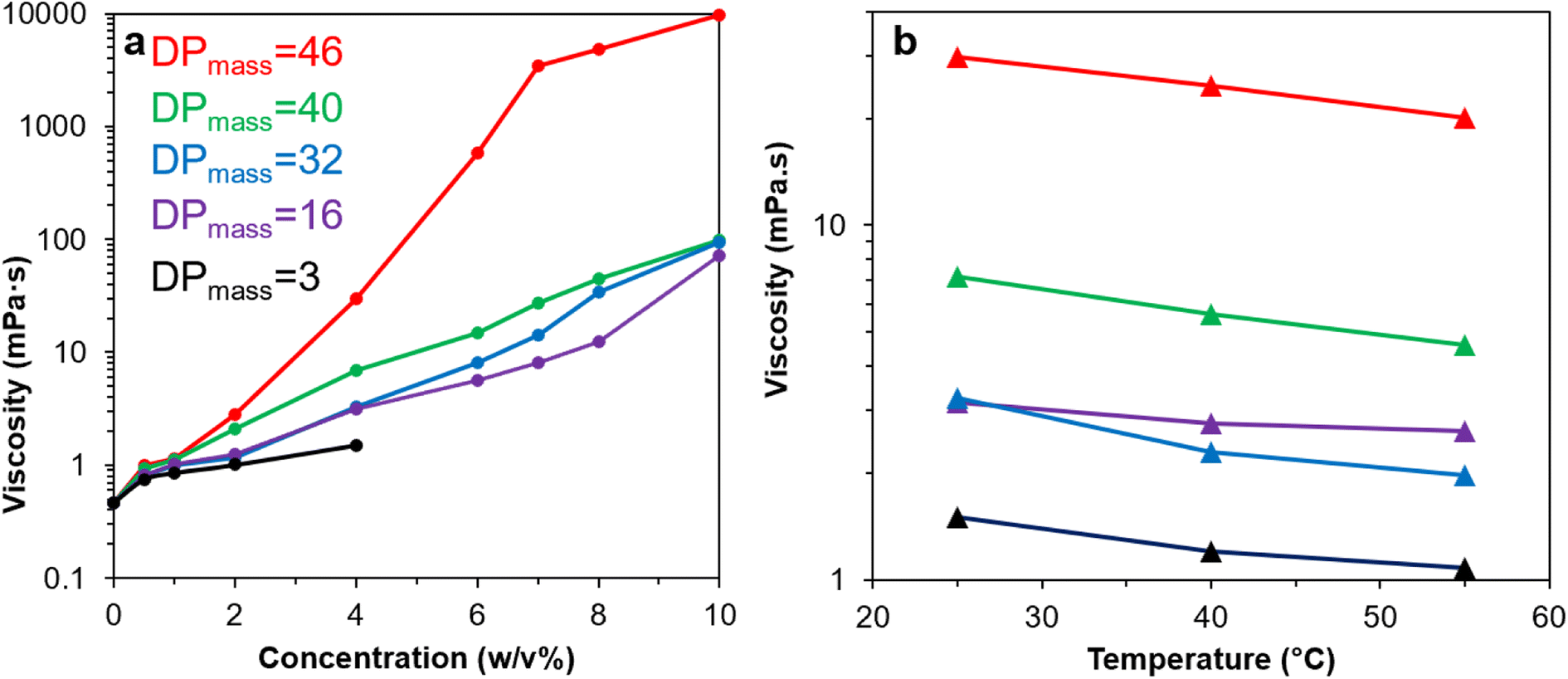 | ||
| Fig. 7 Viscosity of Br-CNF-g-PLMA with varied DPmass in toluene in relationships to: (a) concentration at 25 °C; (b) temperature at 4 w/v%. Average viscosity at shear rates from 1 to 220 s−1 were used. | ||
3.7 Br-CNF-g-PLMA as drag reducer in pump oil
Br-CNF-g-PLMA with the longest PLMA graft (46 DPmass) exhibited the most (least n values) and consistent shear-thinning behaviors at 4 to 10 w/v% and was used to evaluate their drag reducing effects in oil-based fluid. Br-CNF-g-PLMA was solvent exchanged from toluene to pump oil at 0.1, 0.3, and 0.5 w/v% to measure their viscosities over varied shear rates (Fig. 8a). Viscosity decreased to ¾, ⅖ and ⅕ for respective 0.1, 0.3, and 0.5w/v% Br-CNF with increasing shear rates from 1 to 220 s−1, showing more significant shear-thinning behaviors with increasing concentrations for original Br-CNF. The PLMA grafts significantly increased the dispersibility of Br-CNF-g-PLMA in pump oil to 1, 2 and 4 w/v% while also appearing translucent (Fig. 8b). At 25 °C, viscosities slightly increased from 102.2 to 110.9 mPa·s with up to 2 w/v% Br-CNF-g-PLMA, then significantly increased to 256.0 mPa·s at 4 w/v%. Similar trends were also observed at elevated temperatures of 40 and 55 °C, indicating 4 w/v% was the adequate concentration for Br-CNF-g-PLMA to function as a viscosity transducer for pump oil. Pure pump oil exhibited Newtonian behavior at all three temperatures, while shear-thinning phenomenon occurred with the addition of 4 w/v% Br-CNF-g-PLMA (Fig. 8c). Viscosity at 25 °C significantly increased from 104.2 mPa·s to 406.3 mPa·s (3.9×) at 1 s−1 and only slightly increased to 178.2 mPa·s (1.7×) at 220 s−1. At elevated temperature of 55 °C, viscosity dramatically increased to 5.0× and 2.2× at the respective 1 and 220 s−1 shear rate. The more significant viscosity enhancing effect thus validates the capability of Br-CNF-g-PLMA as a highly effective oil drag reducer at low temperatures. Furthermore, the addition of 4% Br-CNF-g-PLMA also converted the turbulent flow to laminar flow that requires less energy in processing.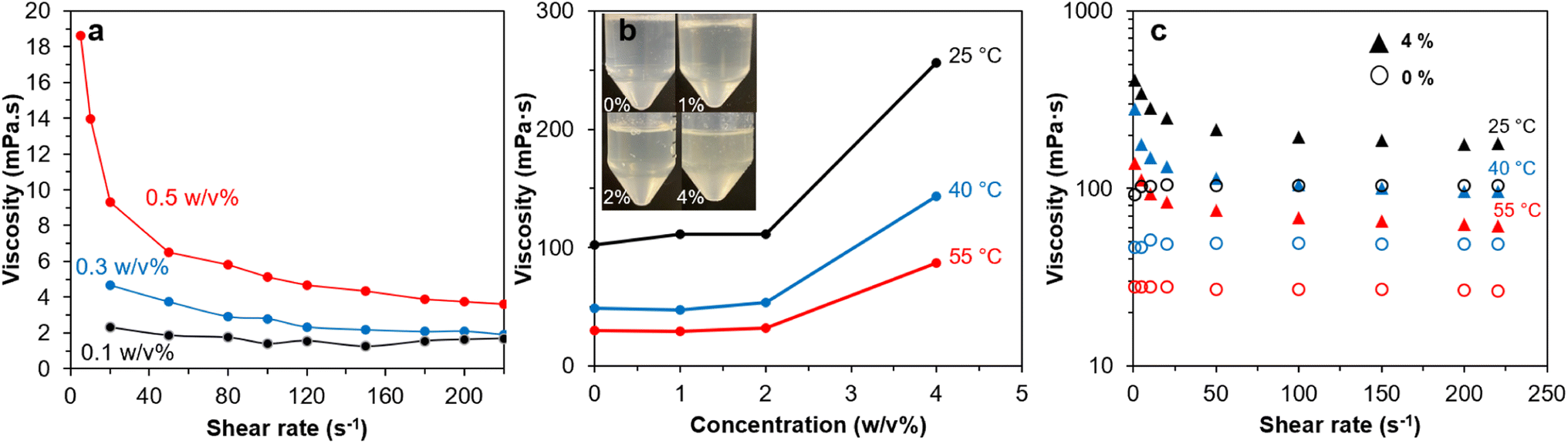 | ||
| Fig. 8 Rheology of: (a) Br-CNF in DMF, (b) Br-CNF-g-PLMA (DPmass = 46) in pump oil, and (c) shear rates of pump oil with 0% and 4% Br-CNF-g-PLMA at temperatures of 25 °C, 40 °C and 55 °C. | ||
4 Conclusions
This work has demonstrated that the one-pot synthesized Br-CNF could function as highly effective macroinitiators for surface-initiated atom transfer polymerization (SI-ATRP) of vinyl monomer lauryl methacrylate (LMA) in controlled graft lengths with excellent conversions up to 92.7%, significantly higher than all previous reported initiators immobilized on nanocelluloses by multiple step preparation and many even aided by added sacrificial initiators. SI-ATRP of Br-CNF was robust, following first order kinetics, evident by linear semilogarithmic monomer consumption vs. time plots, in high apparent rate constants of 0.1295 h−1 and 0.1829 h−1 at respective 9.7 mM and 16 mM Br-CNF macroinitiator concentrations. The Br-CNF-g-PLMA synthesized contained significant surface PLMA grafts in tunable lengths to contain only 2.7 to 7.4 w% Br-CNF core. The molecular mass of Br-CNF-g-PLMA derived by inherent viscosity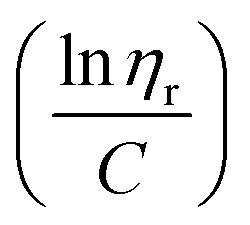 ranged from 264 to 1381 kDa whereas the surface PLMA grafts directly quantified by solution-state 1H NMR in DMSO-d6 gave 2–31 DPNMR, 12.5 to 37% lower than the 3–46 DPmass based on mass balance. These Br-CNF-g-PLMA with controlled graft lengths have proven to be highly effective viscosity modifiers in organic media. Especially, Br-CNF-g-PLMA (DP = 46, 4 w/v%) could be fully dispersed in silicon pump oil to function as a drag reducer to enhance viscosity up to 5 times at 25 to 55 °C. These findings validated Br-CNF as a novel macroinitiator for SI-ATRP grafting of vinyl monomer LMA on CNF and demonstrated that Br-CNF-g-PLMA couples synergistically the thinning behavior of Br-CNF core and the viscosity modifying and drag reducing properties of surface PLMA grafts, expanding their flow improving and lubricating oil additive role to many other coating, varnish, adhesive, and sealant applications. That LMA can be derived from renewable fatty acids makes Br-CNF-g-PLMA holistic bioderived. The ability of Br-CNF to function as macroinitiator for SI-ATRP grafting further demonstrated its versatility as functional CNF for novel applications beyond hydrophobic coatings and polyols for polyurethane syntheses previously documented. The one-pot synthesized Br-CNF macroinitiator is tunable to carry varied levels of surface initiating sites for SI-ATRP of other vinyl monomers to offer further rationally designed strategies to couple nanocellulose core for diverse bottle brush characteristics in potentially broader applications.
ranged from 264 to 1381 kDa whereas the surface PLMA grafts directly quantified by solution-state 1H NMR in DMSO-d6 gave 2–31 DPNMR, 12.5 to 37% lower than the 3–46 DPmass based on mass balance. These Br-CNF-g-PLMA with controlled graft lengths have proven to be highly effective viscosity modifiers in organic media. Especially, Br-CNF-g-PLMA (DP = 46, 4 w/v%) could be fully dispersed in silicon pump oil to function as a drag reducer to enhance viscosity up to 5 times at 25 to 55 °C. These findings validated Br-CNF as a novel macroinitiator for SI-ATRP grafting of vinyl monomer LMA on CNF and demonstrated that Br-CNF-g-PLMA couples synergistically the thinning behavior of Br-CNF core and the viscosity modifying and drag reducing properties of surface PLMA grafts, expanding their flow improving and lubricating oil additive role to many other coating, varnish, adhesive, and sealant applications. That LMA can be derived from renewable fatty acids makes Br-CNF-g-PLMA holistic bioderived. The ability of Br-CNF to function as macroinitiator for SI-ATRP grafting further demonstrated its versatility as functional CNF for novel applications beyond hydrophobic coatings and polyols for polyurethane syntheses previously documented. The one-pot synthesized Br-CNF macroinitiator is tunable to carry varied levels of surface initiating sites for SI-ATRP of other vinyl monomers to offer further rationally designed strategies to couple nanocellulose core for diverse bottle brush characteristics in potentially broader applications.
Author contributions
MG and YLH conceptualized the framework. MG conducted experiments and wrote the first draft of manuscript. YLH revised. MG and YLH edited.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the California Rice Research Board (RU-9) and the USDA National Institute of Food and Agriculture (CA-D-6706) is greatly appreciated.References
- L. Chen, J. Zhu, C. Baez, P. Kitin and T. Elder, Green Chem., 2016, 18, 3835–3843, 10.1039/C6GC00687F.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500, DOI:10.1021/cr900339w.
- D. Bondeson, A. Mathew and K. Oksman, Cellulose, 2006, 13, 171, DOI:10.1007/s10570-006-9061-4.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J.-L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2007, 9, 57–65, DOI:10.1021/bm700769p.
- A. Dufresne, Mater. Today, 2013, 16, 220–227, DOI:10.1016/j.mattod.2013.06.004.
- F. Jiang and Y.-L. Hsieh, Carbohydr. Polym., 2013, 95, 32–40, DOI:10.1016/j.carbpol.2013.02.022.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691, DOI:10.1021/bm060154s.
- A. Isogai, T. Saito and H. Fukuzumi, nanoscale, 2011, 3, 71–85, 10.1039/C0NR00583E.
- M. S. Wang, F. Jiang, Y.-L. Hsieh and N. Nitin, J. Mater. Chem. B, 2014, 2, 6226–6235, 10.1039/C4TB00630E.
- F. Jiang, S. Han and Y.-L. Hsieh, RSC Adv., 2013, 3, 12366–12375, 10.1039/c3ra41646a.
- F. Jiang and Y.-L. Hsieh, ACS Sustain. Chem. Eng., 2016, 4, 1041–1049, DOI:10.1021/acssuschemeng.5b01123.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989, DOI:10.1021/bm0497769.
- J. Li, X. Wei, Q. Wang, J. Chen, G. Chang, L. Kong, J. Su and Y. Liu, Carbohydr. Polym., 2012, 90, 1609–1613, DOI:10.1016/j.carbpol.2012.07.038.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson and O. Ikkala, Biomacromolecules, 2007, 8, 1934–1941, DOI:10.1021/bm061215p.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795, DOI:10.1021/la702481v.
- C. Aulin, E. Johansson, L. Wågberg and T. Lindström, Biomacromolecules, 2010, 11, 872–882, DOI:10.1021/bm100075e.
- L. Sun, X. Zhang, H. Liu, K. Liu, H. Du, A. Kumar, G. Sharma and C. Si, Curr. Org. Chem., 2021, 25, 417–436, DOI:10.2174/1385272824999201210191041.
- F. L. Hatton, S. A. Kedzior, E. D. Cranston and A. Carlmark, Carbohydr. Polym., 2017, 157, 1033–1040, DOI:10.1016/j.carbpol.2016.10.064.
- S. A. Kedzior, L. Graham, C. Moorlag, B. M. Dooley and E. D. Cranston, Can. J. Chem. Eng., 2016, 94, 811–822, DOI:10.1002/cjce.22456.
- E. Lizundia, E. Fortunati, F. Dominici, J. L. Vilas, L. M. León, I. Armentano, L. Torre and J. M. Kenny, Carbohydr. Polym., 2016, 142, 105–113, DOI:10.1016/j.carbpol.2016.01.041.
- K. H. Kan, J. Li, K. Wijesekera and E. D. Cranston, Biomacromolecules, 2013, 14, 3130–3139, DOI:10.1021/bm400752k.
- X. Zhang, J. Zhang, L. Dong, S. Ren, Q. Wu and T. Lei, Cellulose, 2017, 24, 4189–4203, DOI:10.1007/s10570-017-1414-7.
- W. Yuan, C. Wang, S. Lei, J. Chen, S. Lei and Z. Li, Polym. Chem., 2018, 9, 3098–3107, 10.1039/C8PY00613J.
- J. Zhang, Q. Wu, M.-C. Li, K. Song, X. Sun, S.-Y. Lee and T. Lei, ACS Sustain. Chem. Eng., 2017, 5, 7439–7447, DOI:10.1021/acssuschemeng.7b02033.
- B. Risteen, M. McBride, M. Gonzalez, B. Khau, G. Zhang and E. Reichmanis, ACS Appl. Mater. Interfaces, 2019, 11, 25338–25350, DOI:10.1021/acsami.9b06072.
- A. Boujemaoui, S. Mongkhontreerat, E. Malmström and A. Carlmark, Carbohydr. Polym., 2015, 115, 457–464, DOI:10.1016/j.carbpol.2014.08.110.
- Y. Yin, X. Tian, X. Jiang, H. Wang and W. Gao, Carbohydr. Polym., 2016, 142, 206–212, DOI:10.1016/j.carbpol.2016.01.014.
- G. Morandi, L. Heath and W. Thielemans, Langmuir, 2009, 25, 8280–8286, DOI:10.1021/la900452a.
- Z. Zhang, K. C. Tam, G. Sèbe and X. Wang, Carbohydr. Polym., 2018, 199, 603–609, DOI:10.1016/j.carbpol.2018.07.060.
- H. D. Wang, R. D. Roeder, R. A. Whitney, P. Champagne and M. F. Cunningham, J. Polym. Sci., Part A-1: Polym. Chem., 2015, 53, 2800–2808, DOI:10.1002/pola.27754.
- S. A. Kedzior, M. Kiriakou, E. Niinivaara, M. A. Dube, C. Fraschini, R. M. Berry and E. D. Cranston, ACS Macro Lett., 2018, 7, 990–996, DOI:10.1021/acsmacrolett.8b00334.
- Z. Zhang, G. Sèbe, X. Wang and K. C. Tam, ACS Appl. Nano Mater., 2018, 1, 632–641, DOI:10.1021/acsanm.7b00126.
- M. Le Gars, J. Bras, H. Salmi-Mani, M. Ji, D. Dragoe, H. Faraj, S. Domenek, N. Belgacem and P. Roger, Carbohydr. Polym., 2020, 234, 115899, DOI:10.1016/j.carbpol.2020.115899.
- Z. Zhang, G. Sèbe, X. Wang and K. C. Tam, Carbohydr. Polym., 2018, 182, 61–68, DOI:10.1016/j.carbpol.2017.10.094.
- H. Rosilo, J. R. McKee, E. Kontturi, T. Koho, V. P. Hytönen, O. Ikkala and M. A. Kostiainen, Nanoscale, 2014, 6, 11871–11881, 10.1039/C4NR03584D.
- Z. Abousalman-Rezvani, P. Eskandari, H. Roghani-Mamaqani, H. Mardani and M. Salami-Kalajahi, Polymer, 2019, 182, 121830, DOI:10.1016/j.polymer.2019.121830.
- J. Yu, C. Wang, J. Wang and F. Chu, Carbohydr. Polym., 2016, 141, 143–150, DOI:10.1016/j.carbpol.2016.01.006.
- M. V. Kiriakou, R. M. Berry, T. Hoare and E. D. Cranston, Biomacromolecules, 2021, 22, 3601–3612, DOI:10.1021/acs.biomac.1c00692.
- J. Majoinen, A. Walther, J. R. McKee, E. Kontturi, V. Aseyev, J. M. Malho, J. Ruokolainen and O. Ikkala, Biomacromolecules, 2011, 12, 2997–3006, DOI:10.1021/bm200613y.
- M. Morits, V. Hynninen, A. Niederberger, O. Ikkala, A. H. Gröschel and M. Müllner, Polym. Chem., 2018, 9, 1650–1657, 10.1039/C7PY01814B.
- Z. Zhang, X. Wang, K. C. Tam and G. Sebe, Carbohydr. Polym., 2019, 205, 322–329, DOI:10.1016/j.carbpol.2018.10.050.
- M. Lalanne-Tisné, M. A. Mees, S. Eyley, P. Zinck and W. Thielemans, Carbohydr. Polym., 2020, 250, 116974, DOI:10.1016/j.carbpol.2020.116974.
- K. Littunen, U. Hippi, L.-S. Johansson, M. Österberg, T. Tammelin, J. Laine and J. Seppälä, Carbohydr. Polym., 2011, 84, 1039–1047, DOI:10.1016/j.carbpol.2010.12.064.
- X. Zhang, Y. Wang, J. Zhao, M. Xiao, W. Zhang and C. Lu, ACS Sustain. Chem. Eng., 2016, 4, 4321–4327, DOI:10.1021/acssuschemeng.6b00814.
- S. Chen, Z.-L. Zhang, F. Song, X.-L. Wang and Y.-Z. Wang, Macromolecules, 2021, 54, 7409–7420, DOI:10.1021/acs.macromol.1c00903.
- J. R. Navarro and U. Edlund, Biomacromolecules, 2017, 18, 1947–1955, DOI:10.1021/acs.biomac.7b00398.
- C.-F. Huang, J.-K. Chen, T.-Y. Tsai, Y.-A. Hsieh and K.-Y. A. Lin, Polymer, 2015, 72, 395–405, DOI:10.1016/j.polymer.2015.02.056.
- J.-K. Chen, H.-Y. Huang, C.-W. Tu, L.-T. Lee, T. Jamnongkan and C.-F. Huang, Polymers, 2022, 14, 946, DOI:10.3390/polym14050946.
- M. Morits, J. R. McKee, J. Majoinen, J.-M. Malho, N. Houbenov, J. Seitsonen, J. Laine, A. H. Gröschel and O. Ikkala, ACS Sustain. Chem. Eng., 2017, 5, 7642–7650, DOI:10.1021/acssuschemeng.7b00972.
- A. Khabibullin, E. Mastan, K. Matyjaszewski and S. Zhu, in Controlled Radical Polymerization at and from Solid Surfaces, Springer, 2015, pp. 29–76. DOI:10.1007/12_2015_311.
- M. Guo and Y.-L. Hsieh, RSC Adv., 2022, 12, 15070–15082, 10.1039/D2RA00722C.
- M. Guo and Y.-L. Hsieh, Biomacromolecules, 2022, 23, 4574–4585, DOI:10.1021/acs.biomac.2c00747.
- M. D. Holtmyer and J. Chatterji, Polym. Eng. Sci., 1980, 20, 473–477, DOI:10.1002/pen.760200706.
- F. Hoeng, A. Denneulin, N. Reverdy-Bruas, G. Krosnicki and J. Bras, Appl. Surf. Sci., 2017, 394, 160–168, DOI:10.1016/j.apsusc.2016.10.073.
- A. Rees, L. C. Powell, G. Chinga-Carrasco, D. T. Gethin, K. Syverud, K. E. Hill and D. W. Thomas, BioMed Res. Int., 2015 Search PubMed.
- A. Tang, Y. Liu, Q. Wang, R. Chen, W. Liu, Z. Fang and L. Wang, Carbohydr. Polym., 2016, 148, 29–35, DOI:10.1016/j.carbpol.2016.04.034.
- T. Salo, K. Dimic-Misic, P. Gane and J. Paltakari, Nord. Pulp Pap. Res. J., 2015, 30, 165–178, DOI:10.3183/npprj-2015-30-01-p165-178.
- J. Xia and K. Matyjaszewski, Macromolecules, 1997, 30, 7697–7700, DOI:10.1021/ma971009x.
- P. Lu and Y.-L. Hsieh, Carbohydr. Polym., 2012, 87, 564–573, DOI:10.1016/j.carbpol.2011.08.022.
- J. Liao, K. A. Pham and V. Breedveld, Cellulose, 2020, 27, 3741–3757, DOI:10.1007/s10570-020-03048-2.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990, DOI:10.1021/cr940534g.
- J. Voeks, J. Polym. Sci., 1959, 36, 333–339, DOI:10.1002/pol.1959.1203613029.
- H. Mahabadi and K. O'driscoll, J. Appl. Polym. Sci., 1977, 21, 1283–1287, DOI:10.1002/app.1977.070210511.
- K. Nishida, K. Kaji, T. Kanaya and N. Fanjat, Polymer, 2002, 43, 1295–1300, DOI:10.1016/S0032-3861(01)00682-6.
- D. L. Sills and J. M. Gossett, Biotechnol. Bioeng., 2012, 109, 353–362, DOI:10.1002/bit.23314.
- M. Wu, M. Wu, M. Pan, F. Jiang, B. Hui and L. Zhou, Int. J. Biol. Macromol., 2022, 207, 522–530, DOI:10.1016/j.ijbiomac.2022.02.169.
- A. Isogai, Cellulose, 1997, 4, 99–107, DOI:10.1023/A:1018471419692.
- F. Jiang, J. L. Dallas, B. K. Ahn and Y.-L. Hsieh, Carbohydr. Polym., 2014, 110, 360–366, DOI:10.1016/j.carbpol.2014.03.043.
- A. Iborra, G. Díaz, D. López, J. M. Giussi and O. Azzaroni, Eur. Polym. J., 2017, 87, 308–317, DOI:10.1016/j.eurpolymj.2016.12.027.
- D. F. Swinehart, J. Chem. Educ., 1962, 39, 333, DOI:10.1021/ed039p333.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04610a |
| This journal is © The Royal Society of Chemistry 2023 |