
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of cellulose materials on the mechanochemical-assisted reaction system with oleic acid†
Jacqueline Leasea,
Tessei Kawanoa and
Yoshito Andou *ab
*ab
aDepartment of Life Science and Systems Engineering, Graduate School of Life Science and Systems Engineering, Kyushu Institute of Technology, 2-4 Hibikino, Wakamatsu-ku, Kitakyushu, Fukuoka 808-0196, Japan. E-mail: yando@life.kyutech.ac.jp
bCollaborative Research Centre for Green Materials on Environmental Technology, Kyushu Institute of Technology, 2-4 Hibikino Wakamatsu-ku, Kitakyushu, Fukuoka 808-0196, Japan
First published on 15th September 2023
Abstract
As the most abundant natural polymer in nature, cellulose has become the promising alternative raw material to replace fossil-based polymer. Owing to the presence of innumerable hydroxyl groups, various approaches are employed to render processability of cellulose. Herein, a sustainable esterification strategy, mechanochemical-assisted esterification, was developed to produce cellulose oleate (CO) with only a small amount of solvent. The differences in reactivity between all types of cellulose were elucidated. According to thermal stability analysis, the degradation temperature decreased after modification due to the substitution of the long oleoyl group. High degree of substitution (DS) of CO also possessed glass transition temperature (Tg) based on differential scanning calorimetry (DSC) analysis. Herewith, the processability of cellulose was introduced after modification. In this study, bamboo waste cellulose nanofiber oleate (BW CNF-OA) showed the highest DS (2.28) among the COs. Its higher surface reactivity due to the high surface aspect ratio led to a higher quantity of fatty acids attached to the cellulose. For the mechanical properties, low DS of COs exhibited higher tensile strength values. In a nutshell, this greener approach is more favorable than conventional chemical esterification in terms of reduced solvent dosage and improved sustainability.
1. Introduction
Growing environmental concerns have advocated the development of sustainable processes and raw materials to replace fossil-based commodity plastics. The massive pollution and climate changes have led to switching the paradigm of a petroleum-based economy to a sustainable bio-based model. Cellulose, as the most abundant bio-based polymer in nature, offers intriguing unique characteristics, such as excellent mechanical properties, chemical uniqueness, shape flexibility,1 biodegradability, biocompatibility, and low cytotoxicity.2With decades of research and investigation, micro and nano-sized cellulose, including microcrystalline cellulose (MCC), cellulose nanofibers (CNF), and cellulose microfibers (MCF) could be segregated from cellulosic sources easily. MCC is partially depolymerized cellulose that is normally prepared by treating cellulose with an excessive amount of mineral acids, followed by purification. This process is used to degrade the amorphous regions of cellulose and downsize the fiber to a micron level.3 It typically has higher crystallinity and lower specific surface area,4 making it suitable for applications where mechanical strength is important, such as pharmaceutical tablets.5 In addition, CNF and MCF are generally extracted from wood fibers by mechanical, chemical, and enzymatic processes. The differences between them are the diameters of the cellulose fibrils and high energy consumption of CNF extraction.6
Besides, MCF has a highly fibrillated structure and varying levels of crystallinity and specific surface area depending on the production process.7 It is usually utilized in composites and coating industries.8,9 Further, CNF tends to have lower crystallinity compared to MCC, but its specific surface area is higher due to the nanoscale structure.10 This makes CNF useful in applications that require high surface area, such as reinforcement in materials or as a rheology modifier.7
Although cellulose has numerous applications in various fields, its inherently strong inter- and intramolecular hydrogen bonding makes it insoluble in organic solvents as well as water. Besides, the absence of thermal transition limits its processability, causing difficulties in utilization and development.11 Thus, in contemplation of solubility and processability, the chemical modification of the hydroxyl group of cellulose appears as a captivating approach.
Generally, the modification of cellulose can be carried out through homogeneous or heterogeneous reactions. Heterogeneous modification of cellulose is extensively employed in industries for the synthesis of cellulose derivatives, as it is a simple method. Nevertheless, the degree of substitution (DS) of cellulose is hard to control throughout the reaction.12 Conventional chemical esterification involves the use of large amounts of hazardous solvents, high temperatures, and long reaction times to weaken the interconnected hydrogen bonds.13,14
Heretofore, effective solvents of cellulose dissolution, such as ionic liquids (IL),15 deep eutectic solvents (DESs),16 N,N-dimethylacetamide/lithium chloride (DMAc/LiCl),15–17 or N-methylmorpholine-N-oxide (NMMO)18 have been used to prepare various cellulose esters. Despite the fact that these solvent systems are excellent in controlling the molecular structures, large amounts of expensive solvents are still required due to the low solubility of cellulose, making these strategies impractical in the industry sector. Hence, developing sustainable and effective ways is still highly desirable to replace conventional ways of synthesizing cellulose esters.
In recent years, mechanochemistry encompassing a set of processes employing mechanical forces to perform chemical reactions has imposed itself as a greener process.19 Mechanical force alters the cellulose supramolecular structure by interrupting the interchain hydrogen bonds while preserving the original powder form to improve the accessibility of hydroxyl groups from the cellulose backbone. Concurrently, the chemical thermodynamics changes on a molecular scale can also promote the esterification reaction.20 Therefore, only small amounts of solvent are needed in the mechanochemical esterification with shorter reaction times.
In this work, cellulose esterification employing a mechanochemical method in p-toluenesulfonyl chloride/pyridine (TsCl/Py) system has been studied. Different types of cellulose (MCC, CNF, and MCF) were modified with long-chain oleic acid (OA). OA acts as an esterifying agent,21 with excellent properties, such as hydrophobicity, non-toxic, good flexibility, wide thermal process window, and renewability.22,23 Furthermore, the degree of substitution (DS), mechanical properties, thermal stability, biodegradability, and chemical structure of the COs were identified.
2. Materials and methods
2.1. Materials
Microcrystalline cellulose (MCC) powder with a particle size of 20 μm was obtained from Sigma-Aldrich. Standard and ultra-long cellulose nanofibers (WFo and IMa) (5 wt% aqueous dispersion, DP 650, and DP 800) were obtained from Sugino Machine Limited, Toyama, Japan. Powderized Bamboo and bamboo waste microcellulose fiber (BW MCF) were obtained from Sanwa Company, Osaka, Japan, and sisal was acquired from Colombo, Sri Lanka. All the celluloses were dried under vacuum conditions at 50 °C overnight before use. Pyridine (Py) and oleic acid (OA) were purchased from Wako Pure Chemical Industry, Osaka, Japan. Toluenesulfonyl chloride (p-TsCl), which served as an activating agent, was supplied by Tokyo Chemical Industry (Tokyo, Japan) and stored in a desiccator cabinet. All the chemicals were used as received without further purification.2.2. Preparation of cellulose nanofibers (CNF) powder
Cellulose nanofiber suspension (50 g) was placed in a round bottom flask and frozen in a regular freezer at −30 °C for 24 h before the freeze-drying process. Then, the ice in the frozen CNF suspension was sublimated at −50 °C for 3 days using a benchtop freeze dryer (Eyela FDU-1200, Tokyo, Japan). After the freeze-drying process, the CNF was ground with a power mill (PM-2005, Osaka Chemical Co. Ltd, Osaka, Japan), and the size of CNF was confirmed using scanning electron microscopy (SEM) analysis.2.3. Preparation of cellulose oleates (COs)
Preparation of cellulose oleates in Py/TsCl system through mechanochemical esterification was implemented (Fig. 1). 5 g of cellulose (MCC, CNF, or sisal fiber) (30 mmol AGU – anhydroglucose unit) was added to a magnetic mortar containing a solution of 25 ml of Py (300 mmol) and 35 g of TsCl (180 mmol). The mixture was kneaded for 1 h for tosylation with a constant speed of 150 rpm (100 VAC, 15 W). Then, 25 g of OA (90 mmol) was slowly added to the mixture to give a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 cellulose/OA ratio. The mixture was continuously kneaded, and the reaction temperature was maintained at 50 °C for 4 h. After the esterification process, the mixture was precipitated and washed with methanol several times in order to remove the unwanted substances. Lastly, cellulose oleate was vacuum-dried at 50 °C overnight and stored at ambient conditions.
:3 cellulose/OA ratio. The mixture was continuously kneaded, and the reaction temperature was maintained at 50 °C for 4 h. After the esterification process, the mixture was precipitated and washed with methanol several times in order to remove the unwanted substances. Lastly, cellulose oleate was vacuum-dried at 50 °C overnight and stored at ambient conditions.
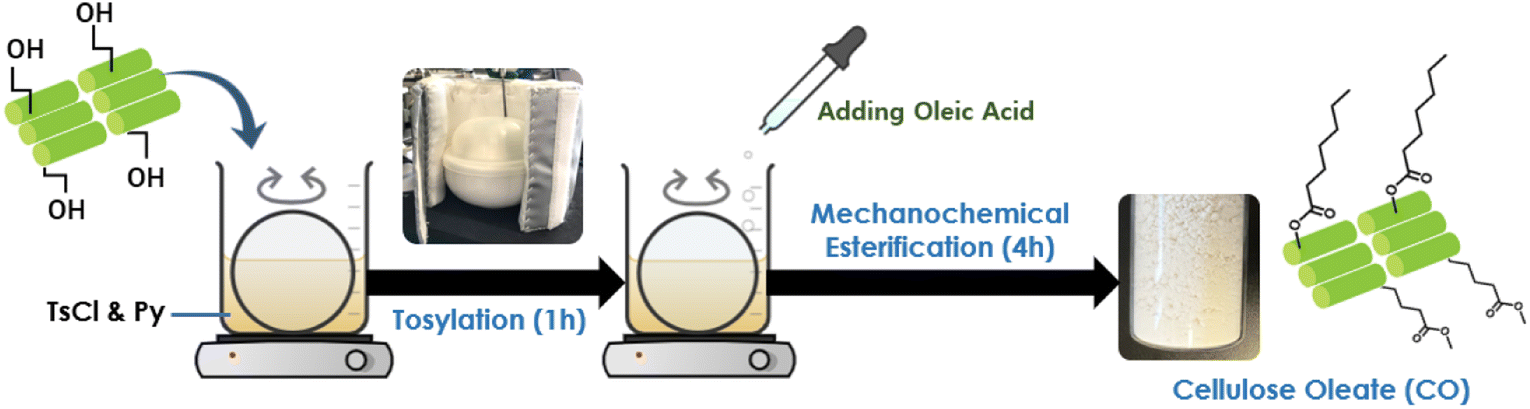 | ||
| Fig. 1 Preparation of cellulose oleate by mechanochemical method with oleic acid. | ||
2.4. Characterizations
:50. The mixture was then pressed into a pellet until a translucent film was obtained. Sixteen scans were collected from 4000 to 400 cm−1 at a resolution of 4 cm−1.| n = [(V0 − V1) × CHCl]/1000 | (1) |
| DS = 162n/m − [(M − 1) × n] | (2) |
3. Results and discussion
3.1. Effects of types of cellulose on the DS of cellulose oleates (COs)
In this study, the main targets were synthesizing COs with various celluloses and to perceive the differences between reactivity and availability of hydroxyl groups in MCC, MCF, and CNF. The neutralization titration method was used to determine the DS of the COs.As shown in Table 1, the DS of cellulose oleates synthesized from cellulose nanofibers attained higher DS when compared to micro-sized cellulose esters. The reason for this phenomenon was that nano-sized celluloses have a larger specific surface area and high aspect ratio, which improved the accessibility for the fatty acid to react with the O–H groups. The higher reactivity of the CNF leads to an increase in DS value. Therefore, the DS of the three micro-sized cellulose oleates (MCC-OA, sisal-OA, and BW MCF-OA) is lower than CNF oleates under the same conditions.
| Sample | Reagents | Py (ml) | Kneading speed (rpm) | DS | |||
|---|---|---|---|---|---|---|---|
| Cellulose (g, mmol) | OA (g, mmol) | TsCl (g, mmol) | [AGU]:[OA]:[TsCl] (mol%) |
||||
| Microcrystalline cellulose (MCC) | |||||||
| Microcrystalline cellulose oleate (MCC-OA) | 5, 30 | 25, 90 | 35, 180 | 1:3:6 |
25 | 150 | 1.44 |
|
|||||||
| Micro-cellulose fiber (MCF) | |||||||
| Sisal oleate (Sisal-OA) | 5, 30 | 25, 90 | 35, 180 | 1:3:6 |
25 | 150 | 1.93 |
| Bamboo waste micro-cellulose fiber oleate (BW MCF-OA) | 5, 30 | 25, 90 | 35, 180 | 1:3:6 |
25 | 150 | 1.79 |
|
|||||||
| Cellulose nanofiber (CNF) | |||||||
| Standard cellulose nanofiber oleate (WFo-OA) | 5, 30 | 25, 90 | 35, 180 | 1:3:6 |
25 | 150 | 1.93 |
| Ultra-long cellulose nanofiber oleate (IMa-OA) | 5, 30 | 25, 90 | 35, 180 | 1:3:6 |
25 | 150 | 1.93 |
| Bamboo waste cellulose nanofiber oleate (BW CNF-OA) | 5, 30 | 25, 90 | 35, 180 | 1:3:6 |
25 | 150 | 2.28 |
3.2. Structural analysis of cellulose oleate (COs)
The success of the modification reaction was first elucidated with structural analysis. The chemical structures of the obtained COs were characterized by FTIR and 1H NMR analyses.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching). This indicated that fatty chains had been directly grafted onto cellulose by ester junction. The obvious characteristic peaks corresponding to aliphatic chains of the oleic acid were also recorded at 2924 cm−1 and 2854 cm−1. These peaks were the anti-symmetric and symmetric stretching vibrations of aliphatic C–H in CH2 and terminal CH3 groups, respectively.24,25
O stretching). This indicated that fatty chains had been directly grafted onto cellulose by ester junction. The obvious characteristic peaks corresponding to aliphatic chains of the oleic acid were also recorded at 2924 cm−1 and 2854 cm−1. These peaks were the anti-symmetric and symmetric stretching vibrations of aliphatic C–H in CH2 and terminal CH3 groups, respectively.24,25
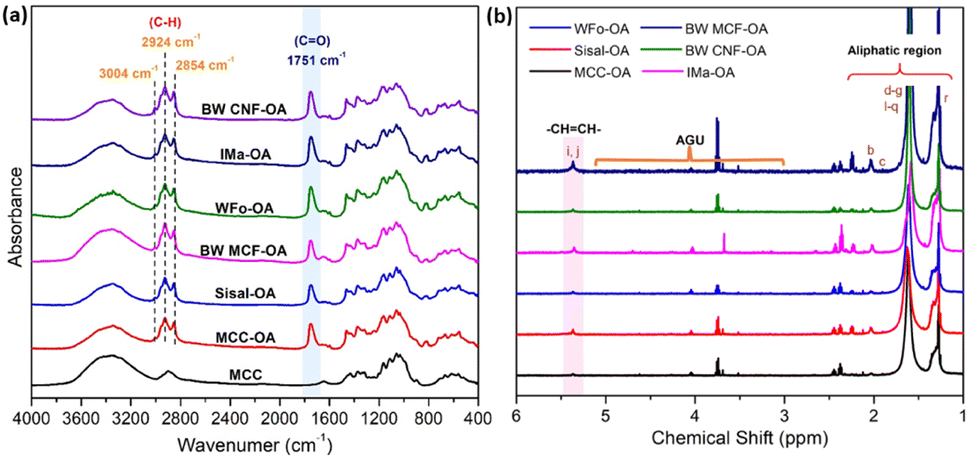 | ||
| Fig. 2 Images of (a) FTIR spectra of native MCC and various COs, (b) 1H NMR spectrum of various COs in d-chloroform. | ||
In addition, a new vibration at 3004 cm−1 was also observed due to C–H axial deformation vibration in the olefinic double bonds (H–CC) of oleic chains.12,26 These phenomena proved the successful esterification of cellulose with oleic acid by the proposed protocol. Furthermore, differences between the frequencies of carbonyl peaks may result from different degrees of substitution (DS). According to the previous literature,20,27,28 the higher the DS, the higher the intensity for CO ester absorption. Herein, the visibility of the CO peak implied the rate of mechanochemical esterification.
The vinylic protons of the oleic acid as side chains were observed at a chemical shift of 5.4 ppm in 1H NMR.14 The peaks between 5.0 and 3.0 ppm represented the protons of the cellulose backbone.29 From Fig. 2(b), cellulose backbone peaks were definitely due to the low solubility of the obtained COs in chloroform. Besides, the new peaks attributed to methyl protons from the grafted oleoyl group appeared at δ = 1.3–2.0 ppm, representing the methyl protons from C6, C2, and C3 positions from the glucose, showing the successful formation of COs.20
3.3. Crystal structure of cellulose oleate (COs)
The crystalline structures of the COs were investigated using XRD analysis. The diffraction patterns of raw celluloses and COs are displayed in Fig. S1† and 3. The native celluloses presented the typical XRD pattern of cellulose I, with the main diffraction angle at around 14.9, 16.3, 22.5, and 34.6°, which are related to the diffraction of planes (101), (10−1), (002), and (040), respectively.30,31 Based on Fig. S1,† MCC exhibited higher crystallinity as the amorphous region of cellulose has been removed, and the MCF and CNF showed lower crystallinity due to the fabrication processes.32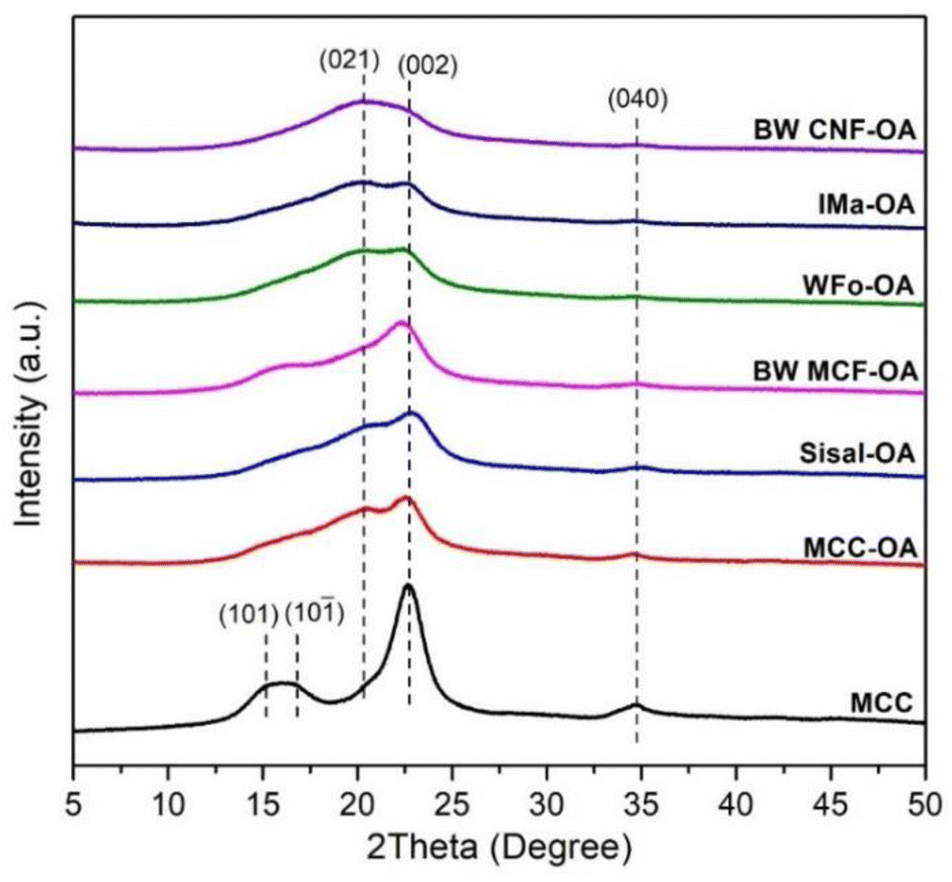 | ||
| Fig. 3 XRD spectra of native cellulose and various COs. | ||
After esterification, the broadening of the peaks at 22.5° (002) and 34.6° (040) could be noticed. Moreover, there was an increase in diffraction intensity at the (021) plane, which was attributed to the amorphous region of disordered cellulose chains or an aliphatic chain from the oleoyl group.33,34 These results showed that the esterification process leads to an increase in the amorphous regions of the cellulose. Broadening of the diffraction peaks suggests structural changes within the cellulose.
In general, the changes in the crystalline structure of COs corresponding to the esterification depend on the DS values. The diffraction pattern of BW-CNF OA with the highest DS value was quite different from that of the starting substrate. It showed a completely amorphous structure and has the highest peak at around 19.8°.
3.4. Surface morphologies of cellulose oleates (COs)
FESEM was used to study the morphological changes of the cellulose associated with mechanochemical esterification. The morphologies of cellulose before and after oleic acid esterification are presented in Fig. 4. It can be seen that neat CNF (WFo, Ima, and BW), BW MCF, and sisal possessed fibrous structures while MCC exhibited a large variety of sizes and shapes. The majority of them were rod-like and platelet-like particles.35 | ||
| Fig. 4 Surface morphology of cellulose and its derivatives: (a) & (b) BW CNF, (c) & (d) IMa, (e) & (f) WFo, (g) & (h) BW MCF, (i) & (j) Sisal, (k) & (l) MCC. | ||
When CNF was modified with oleic acid, the original fibrous structure was lost. Instead, IMa-OA and WFo-OA resembled semi-crystalline Cos, while BW CNF-OA displayed a more amorphous and sheet-like structure. In addition, sisal and MCC also changed from cellulose I to a semi-crystalline structure, but the rod-like particles of the sisal and MCC could still be noticed. Similar observations were also reported in the literature,36,37 in which cellulose acetate was produced by the acetylation of bacterial cellulose. This observation is consistent with the high DS and intensity of the (002) peak in the XRD spectra, which points towards significant bulk modification during the esterification reaction.
For BW-MCF, the intrinsic morphology and structure of the cellulose ester were nearly preserved. The result is in agreement with the XRD analysis that showed BW-MCF retained the cellulose I structure after the modification. Thus, high DS of cellulose (BW CNF-OA) turned from crystalline structure to amorphous after the substitution of the long fatty acid side chain.
3.5. Thermal stabilities of cellulose oleates (COs)
The thermal stabilities of COs were determined using thermogravimetric analysis (TGA) and differential thermogravimetric (DTG) analysis.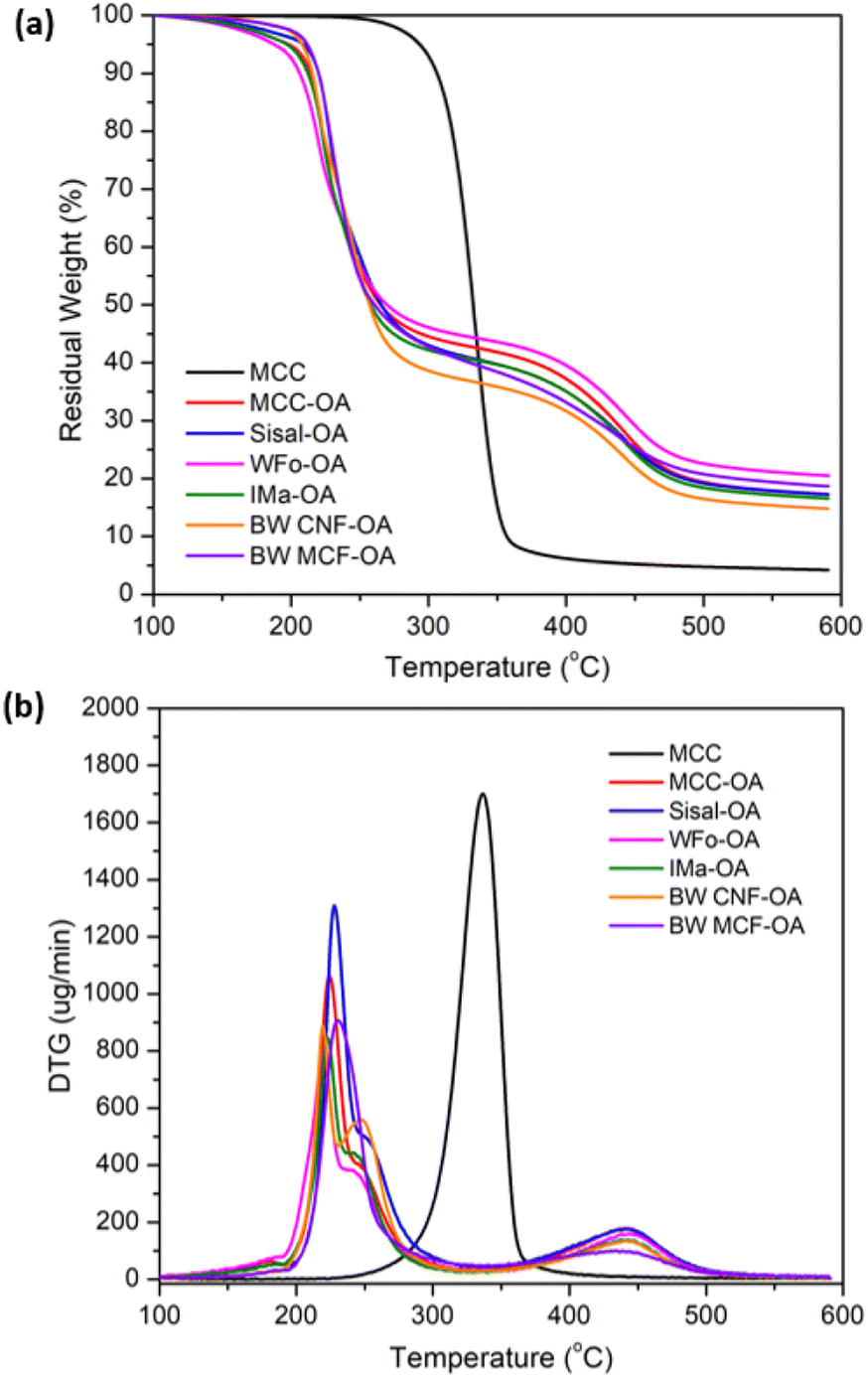 | ||
| Fig. 5 Graph of (a) TGA and (b) DTG curves of pristine MCC and COs. | ||
| Sample | Td1 (°C) | Td2 (°C) | Residual weight at 600 °C (%) | DS |
|---|---|---|---|---|
| MCC | 337 | — | 4.24 | — |
| MCC-OA | 224 | 440 | 17.32 | 1.44 |
| WFo-OA | 220 | 444 | 20.50 | 1.93 |
| IMa-OA | 224 | 443 | 16.58 | 1.93 |
| Sisal-OA | 228 | 442 | 17.25 | 1.93 |
| BW CNF-OA | 220/250 | 441 | 14.85 | 2.28 |
| BW MCF-OA | 231 | 439 | 18.67 | 1.79 |
The decreased thermal stability of surface-modified cellulose was due to the changes in crystallinity in the surface structure owing to esterification, with the strong inherent hydrogen bonding disrupted by the long fatty side chain. Besides, the lower decomposition temperature of pure oleic acid was another reason the thermal stabilities of the COs decreased after mechanochemical esterification.38
In addition, two thermal degradation transitions of the COs were detected. The first decomposition stage was attributed to the breakdown of ester groups and the unsaturated bonds from the oleoyl side chain. Meanwhile, the second degradation stage was assigned to the degradation of the alkyl chain attached to the cellulose backbone.28,30
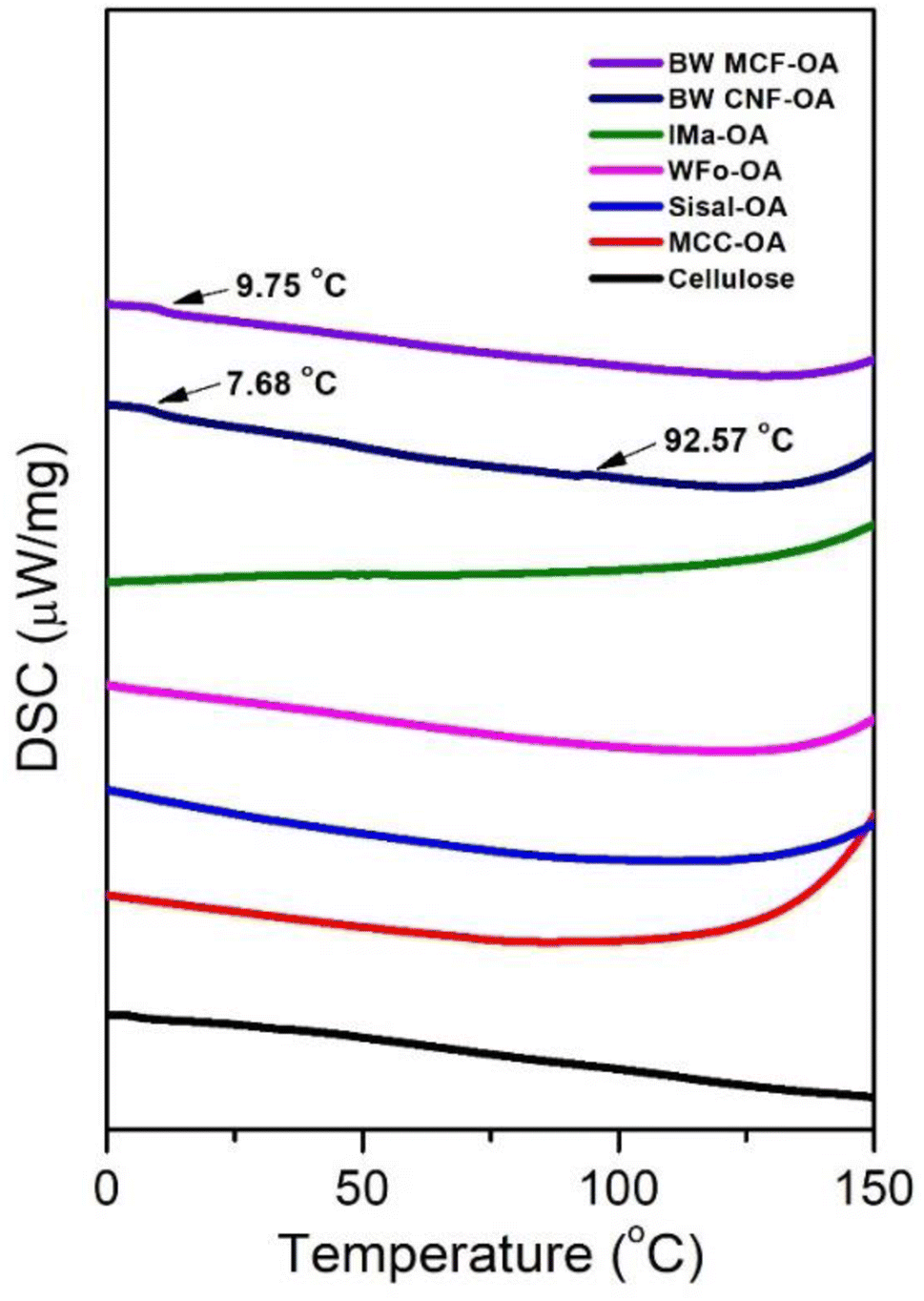 | ||
| Fig. 6 DSC thermograms of various COs. | ||
Native cellulose and COs had no obvious glass transition in the experimental temperature range except for BW MCF-OA and BW CNF-OA. Two major glass transitions of BW CNF-OA were detected at 7.68 °C and 92.57 °C, corresponding to the motion of oleic chains and cellulose backbones, respectively. Soft segments can act as internal plasticizers to improve the mobility of the cellulose chain. The oleoyl group disrupted the hydrogen bonding interaction within the cellulose. Herein, higher DS values of COs can improve the mobility of the cellulose backbones by increasing the distance between cellulose chains and improving the free volume of macromolecules. To detect the glass transition temperature (Tg) of the COs in this study, it was necessary to graft oleyl side chains to certain degrees. The structural arrangement is also one of the important factors in observing macromolecular motions.
3.6. Contact angle test
The water repellency of the cellulose oleate films was investigated using the water contact angle meter. Contact angles indicated the degree of wetting when liquid and solid phases were in interaction. A surface with contact angle values below 90° possessed hydrophilic characteristics, whereas one with contact angles above 90° was considered hydrophobic.41 According to the literature,20 the neat cellulose exhibited a high wettability as the water droplet was observed to spread and form a low contact angle with the cellulose surface around 30° to 40°. The polar behavior of the neat cellulose is due to the large number of hydroxyl groups on the surface of the film.Fig. 7 and Table 3 present the water contact angles of the cellulose oleate films. All the cellulose oleate samples showed hydrophilic characteristics, which is about 100°. Among all, IMa-OA shows the highest contact angle, which is 105.2°.
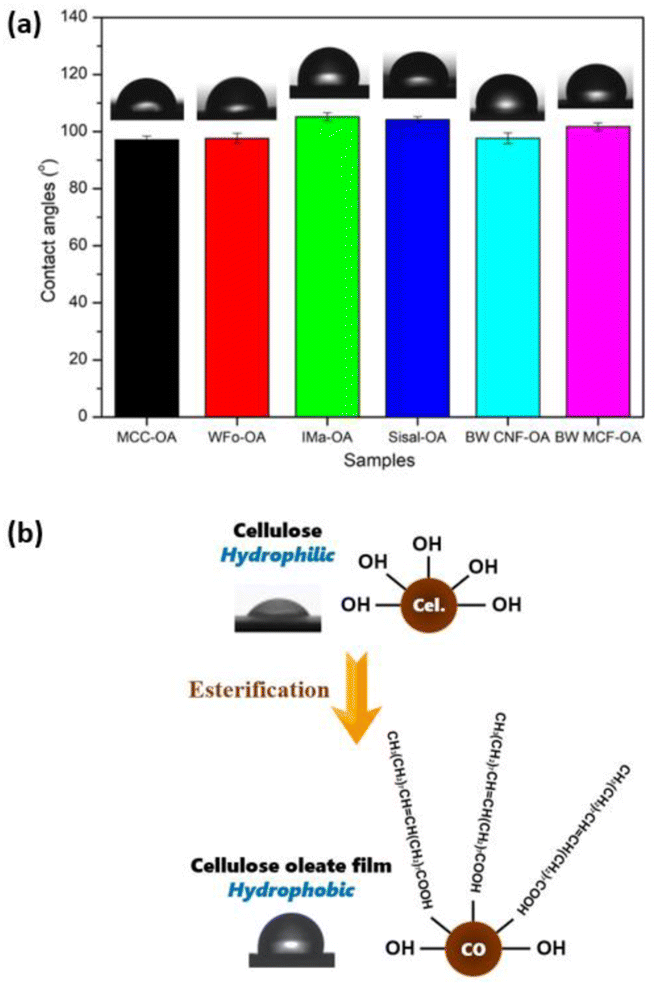 | ||
| Fig. 7 (a) Water contact angles of various CO films, (b) illustration of cellulose oleate film with hydrophobic characteristics. | ||
| Sample | Contact angles (°) | DS |
|---|---|---|
| MCC-OA | 97.1 ± 1.3 | 1.44 |
| WFo-OA | 97.6 ± 1.8 | 1.93 |
| IMa-OA | 105.2 ± 1.4 | 1.93 |
| Sisal-OA | 104.2 ± 1.0 | 1.93 |
| BW CNF-OA | 97.7 ± 1.9 | 2.28 |
| BW MCF-OA | 101.7 ± 1.4 | 1.79 |
The hydrophobic characteristic of the CO films was ascribed to the long hydrophobic tails of the oleic acid, which radially outwards from the surface of the COs.42 The long fatty chains grafted perpendicular to the cellulose, thereby forming a nano-pin structure. This surface morphology would highlight their non-polar property antagonist with the highly polar neat cellulose, increasing the free volume and consequently decreasing wettability and surface energy.43
3.7. Mechanical properties of cellulose oleate (CO) films
The mechanical properties of COs were evaluated by uniaxial tension tests. For each sample, three measurements were performed, and the average values of tensile strength, Young's modulus, and elongation at break, including their standard deviations, were calculated and are shown in Table 4 and Fig. 8(a). The stress–strain curves of the various COs are also displayed in Fig. 8(b).| Sample | Tensile strength (MPa) | Young's modulus (GPa) | Elongation at break (%) | DS |
|---|---|---|---|---|
| MCC-OA | 11.09 ± 0.65 | 0.62 ± 0.09 | 3.87 ± 0.41 | 1.44 |
| WFo-OA | 4.20 ± 1.79 | 0.54 ± 0.14 | 0.81 ± 0.53 | 1.93 |
| IMa-OA | 5.87 ± 1.42 | 0.62 ± 0.19 | 1.57 ± 1.15 | 1.93 |
| Sisal-OA | 4.55 ± 2.16 | 0.62 ± 0.18 | 1.24 ± 0.73 | 1.93 |
| BW CNF-OA | 4.50 ± 0.99 | 0.71 ± 0.63 | 2.97 ± 1.41 | 2.28 |
| BW MCF-OA | 5.50 ± 0.11 | 0.55 ± 0.09 | 1.01 ± 0.14 | 1.79 |
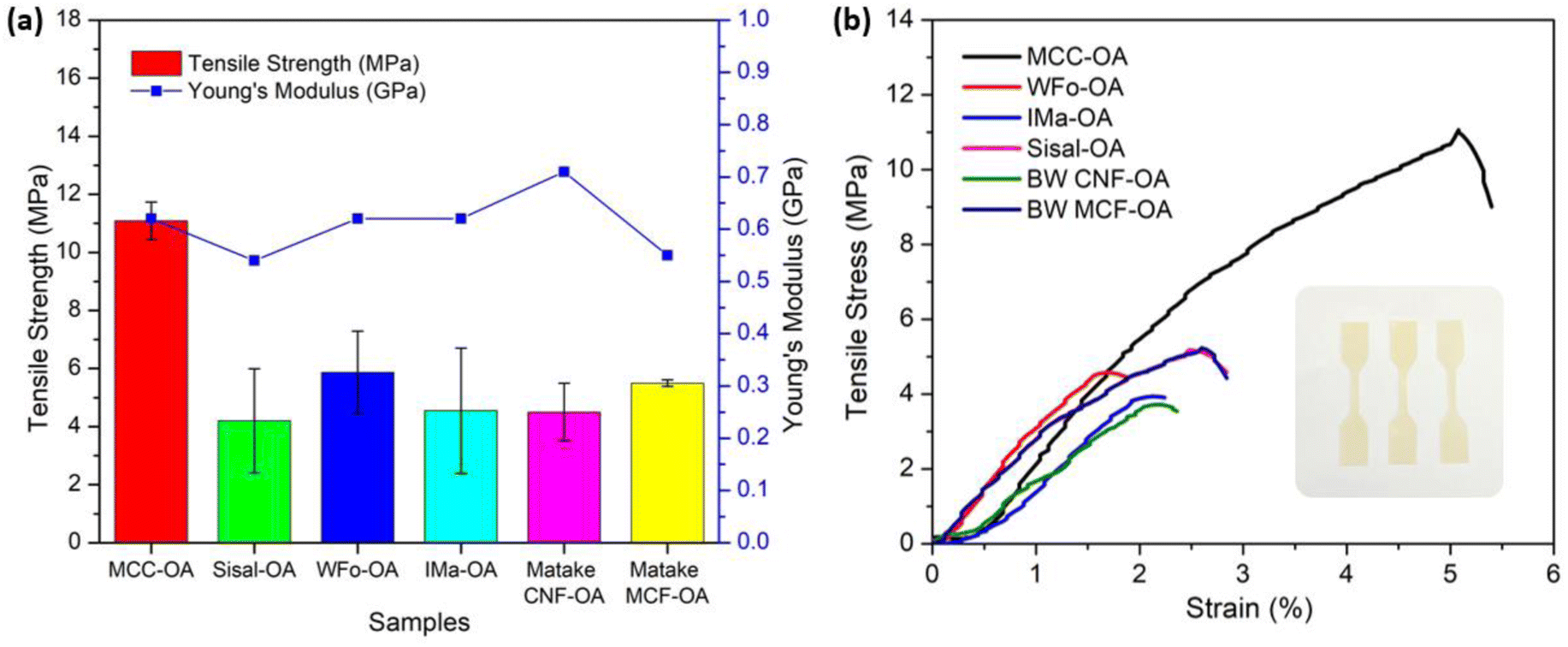 | ||
| Fig. 8 Graph of (a) tensile properties of CO films, (b) stress–strain curves of CO films. | ||
MCC-OA showed the highest value of tensile strength of 11.09 ± 0.65 MPa. Cellulose oleates with a lower DS exhibited greater ductility.
This scenario was posited to the higher remaining amount of free hydroxyl groups, which are able to form a strong hydrogen bond network.12 Thus, MCC-OA had a higher ability against the forces before break. The tensile strength of the MCC-OA film was only slightly lower in comparison with commercial polyolefin films, such as polyethylene and polypropylene, which show tensile strengths in the range of 15–40 MPa.44
Theoretically, CO films with higher DS obtained higher elongation at break percentages. This could be correlated with the plasticizer effect of oleoyl groups due to the intercalation of these molecules between cellulose chains and the reduction of the hydrogen bond network.45 However, in this work, BW CNF-OA, which obtained the highest DS displayed slightly lower elongation value when compared to MCC-OA. The films of nano-sized CO samples showed non-uniform surfaces. Due to the inhomogeneity and infusible aggregation, the ductility or flexibility of BW CNF-OA could be affected.
3.8. Biodegradation test
Cellulose derivatives have a high risk of ending up in wastewater and thereafter, in sewage sludge. It is, therefore, vital to understand their behavior and potential biodegradability in these environments. The biodegradation of CO films was tested by immersion of samples in sewage sludge for 70 days. Only MCC-OA, BW MCF-OA, and sisal-OA films were utilized to perform this test. The CO films were also added to water as a positive control.Aerobic biodegradation of CO films was observed based on the weight loss against the time graph, as shown in Fig. 9(a) and Table 5. A higher degree of degradability of CO films could be noticed at the initial 10 days, then decreased over 40 and 70 days, respectively. It was posited that the activated sewage sludge evaporated as the time increased. Therefore, the biodegradation rate diminished over time. The highest weight loss was found for the MCC-OA sample, which showed 14.7% over 40 days and 12.55% over 70 days. Basically, small changes were observed for BW MCF-OA and sisal-OA films.
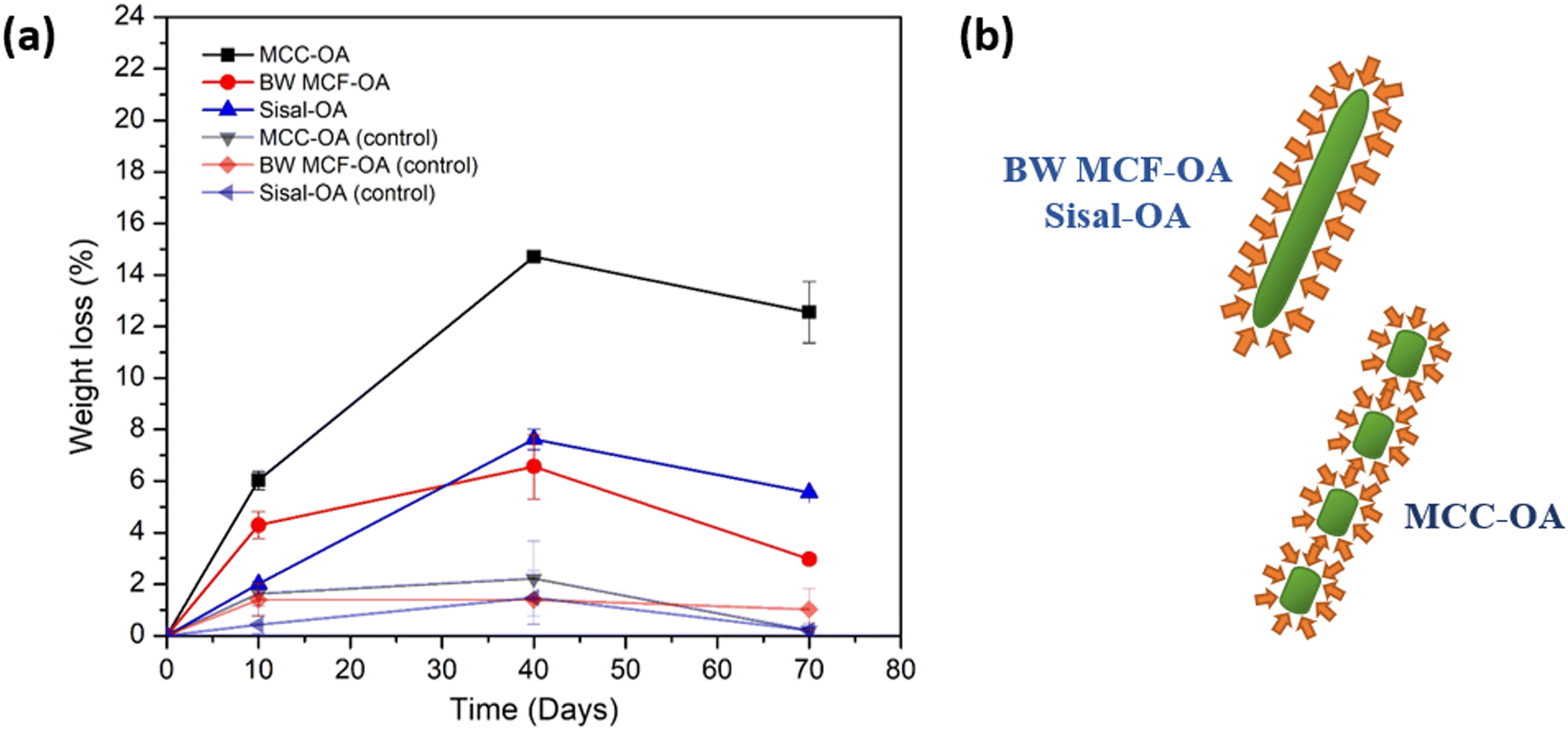 | ||
| Fig. 9 Graph of (a) weight loss versus time of CO films in respective days, and graphic of (b) surface aspect ratio of the enzyme subjected to the COs. | ||
| Days | MCC-OA (%) | BW MCF-OA (%) | Sisal-OA (%) | MCC-OA control (%) | BW MCF-OA control (%) | Sisal-OA control (%) |
|---|---|---|---|---|---|---|
| 10 | 6.03 ± 0.36 | 4.29 ± 0.53 | 2.01 ± 0.08 | 1.62 ± 0.46 | 1.38 ± 0.63 | 0.42 ± 0.35 |
| 40 | 14.70 ± 0.20 | 6.56 ± 1.25 | 7.62 ± 0.39 | 2.21 ± 1.45 | 1.39 ± 0.07 | 1.47 ± 1.03 |
| 70 | 12.55 ± 1.19 | 2.97 ± 0.19 | 5.556 ± 0.04 | 0.19 ± 0.28 | 1.01 ± 0.80 | 0.20 ± 0.09 |
The properties of cellulose ester films are associated with their biodegradability. The surface condition (hydrophilic and hydrophobic properties) is one of the determining factors of biodegradability because it affects the accessibility of the enzyme to the material polymer. MCC-OA film showed higher hydrophilicity owing to its susceptibility to enzyme degradation. Besides, the morphology of the cellulose ester films also affected their biodegradation rates. More organized regions of CO tend to hinder enzymatic hydrolysis since catalytic proteins diffuse with greater difficulty.46 BW MCF-OA has a higher degree of crystallinity when compared to sisal-OA and MCC-OA based on the XRD results, so it demonstrated the lowest biodegradation rate after 40 days. However, the particle size of MCC-OA was the smallest and with less fiber entanglement, which provided a high surface area for the enzyme to attack (Fig. 9(b)).46,47 The enzymes will attack the amorphous domain of the CO film first because the molecules in the amorphous region are loosely packed. Therefore, the higher crystallinity of the film was more resistant than the amorphous region.
Fig. 10 shows the SEM top-view images of the samples. The 0 day CO films displayed a smooth structure. After 70 days, the irregularities observed on the surfaces increased. MCC-OA film exhibited rough and porous surface morphology. BW MCF-OA and sisal-OA films appeared to have a less rough surface. This result was aligned with the weight loss or biodegradation rate that is discussed in the above section.
 | ||
| Fig. 10 SEM images of 0 and 70 days of (a) & (b) MCC-OA film; (c) & (d) BW MCF-OA film; (e) & (f) Sisal-OA film in sewage sludge. | ||
4. Conclusion
A green mechanochemical-assisted method to synthesize various kinds of cellulose oleates was developed in this study. Corresponding to the benefits of mechanical shearing force, COs with various DS values (1.44–2.28) were obtained by using only a low dosage of oleic acid and solvent. The celluloses showed different reactivities, and BW CNF eventually possessed the most reactive surface. The different reactivities of celluloses were caused by their different morphological structures. A larger specific surface area and high aspect ratio of cellulose as the starting material are vital in mechanochemical esterification. However, some aggregation was posited to occur due to these characteristics. In addition, the thermal stability of COs decreased after esterification due to the interruption of the hydrogen bonding interaction by the long-chain oleic acid. Based on the thermal stability test, the degradation temperature is proportional to the DS of the COs. Besides, the mechanochemical esterification endowed high CO with two major glass transitions at 7.68 °C and 92.57 °C by breaking the hydrogen bonding interaction of cellulose. MCC-OA with the low DS value showed the highest tensile strength among the Cos, and the grafting of the long fatty chains increased the water repellencies of celluloses. These phenomena presented promising properties for applications, which will be explored in our following work in order to optimize the reaction conditions and properties of the cellulose esters. In short, this work provides an alternative approach for preparing cellulose esters in a simple way, which will probably shed light on the fabrication of cellulose derivatives in the industry sectors.Author contributions
Jacqueline Lease: conceptualization, methodology, data curation, investigation, validation, visualization, writing-original draft, writing-review and editing. Tessei Kawano: conceptualization and formal analysis. Yoshito Andou: conceptualization, supervision, project administration, writing-review & editing, and funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by JST, the establishment of university fellowships towards the creation of science and technology innovation, Grant Number JPMJFS2133.References
- D. Zhao, Y. Zhu, W. Cheng, W. Chen, Y. Wu and H. Yu, Adv. Mater., 2021, 33, 2000619 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, C. Liu, Y. Liu, Y. Li, M. Wu, Z. Li and B. Li, Int. J. Biol. Macromol., 2021, 170, 397–405 CrossRef CAS PubMed.
- J. Du, S. Yang, Q. Zhu, Y. Wu, J. Guo and J. Jiang, Biomass Convers. Biorefin., 2023, 13, 12105–12114 CrossRef CAS.
- J.-A. Lee, M.-J. Yoon, E.-S. Lee, D.-Y. Lim and K.-Y. Kim, Macromol. Res., 2014, 22, 738–745 CrossRef CAS.
- S. J. Kulkarni, Bionanoscience, 2023, 13, 784–794 CrossRef.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- H. Kangas, P. Lahtinen, A. Sneck, A.-M. Saariaho, O. Laitinen and E. Hellén, Nord. Pulp Pap. Res. J., 2014, 29, 129–143 CrossRef CAS.
- S. Iwamoto, S. Yamamoto, S. H. Lee and T. Endo, Composites, Part A, 2014, 59, 26–29 CrossRef CAS.
- M. Iotti, Ø. Eriksen, Ø. Gregersen and M. Lenes, Int. Conf. Nanotechnol. For. Prod. Ind., 2010, vol. 2010, pp. 1297–1320 Search PubMed.
- F. Fahma, I. Febiyanti, N. Lisdayana, I. W. Arnata and D. Sartika, Arch. Mater. Sci. Surf. Eng., 2021, 2, 49–64 Search PubMed.
- L. Lin and K. Tsuchii, Carbohydr. Res., 2022, 511, 108490 CrossRef CAS PubMed.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustain. Chem. Eng., 2018, 6, 8826–8835 CrossRef CAS.
- L. Huang, Q. Wu, Q. Wang and M. Wolcott, Composites, Part A, 2020, 130, 105765 CrossRef CAS.
- D.-F. Hou, M.-L. Li, C. Yan, L. Zhou, Z.-Y. Liu, W. Yang and M.-B. Yang, Green Chem., 2021, 23, 2069–2078 RSC.
- M. Kostag, M. Gericke, T. Heinze and O. A. El Seoud, Cellulose, 2019, 26, 139–184 CrossRef CAS.
- Y.-L. Chen, X. Zhang, T.-T. You and F. Xu, Cellulose, 2019, 26, 205–213 CrossRef CAS.
- J. Kerwald, C. F. de Moura Junior, E. D. Freitas, J. de D. P. de Moraes Segundo, R. S. Vieira and M. M. Beppu, Cellulose, 2022, 29, 25–54 CrossRef CAS.
- C. Verma, A. Mishra, S. Chauhan, P. Verma, V. Srivastava, M. A. Quraishi and E. E. Ebenso, Sustainable Chem. Pharm., 2019, 13, 100162 CrossRef.
- L. Douard, M. N. Belgacem and J. Bras, ACS Sustain. Chem. Eng., 2022, 10, 13017–13025 CrossRef CAS.
- L. Huang, Q. Wu, Q. Wang and M. Wolcott, ACS Sustain. Chem. Eng., 2019, 7, 15920–15927 CrossRef CAS.
- W. Zhang, N. Zhou, Y. Zhang, Z. Huang, H. Hu, J. Liang and Y. Qin, J. Appl. Polym. Sci., 2021, 138, 50677 CrossRef CAS.
- G. Tedeschi, S. Guzman-Puyol, U. C. Paul, M. J. Barthel, L. Goldoni, G. Caputo, L. Ceseracciu, A. Athanassiou and J. A. Heredia-Guerrero, Chem. Eng. J., 2018, 348, 840–849 CrossRef CAS.
- Z. Wu, P. Liu, Z. Wu and G. Cravotto, Processes, 2021, 9, 391 CrossRef CAS.
- T. Huang, K. D. Li and M. Ek, Carbohydr. Polym., 2021, 257, 117615 CrossRef CAS PubMed.
- T. Kulomaa, J. Matikainen, P. Karhunen, M. Heikkilä, J. Fiskari and I. Kilpeläinen, RSC Adv., 2015, 5, 80702–80708 RSC.
- H. Nosal, K. Moser, M. Warzała, A. Holzer, D. Stańczyk and E. Sabura, J. Polym. Environ., 2021, 29, 38–53 CrossRef CAS.
- G. Zhao, F. Wang, X. Lang, B. He, J. Li and X. Li, RSC Adv., 2017, 7, 27017–27023 RSC.
- P. Uschanov, L. S. Johansson, S. L. Maunu and J. Laine, Cellulose, 2011, 18, 393–404 CrossRef CAS.
- Q. Xu, L. Song, L. Zhang, G. Hu, Q. Chen, E. Liu, Y. Liu, Q. Zheng, H. Xie and N. Li, Cellulose, 2018, 25, 205–216 CrossRef CAS.
- C. S. R. Freire, A. J. D. Silvestre, C. P. Neto, M. N. Belgacem and A. Gandini, J. Appl. Polym. Sci., 2006, 100, 1093–1102 CrossRef CAS.
- P. Jandura, B. V. Kokta and B. Riedl, J. Appl. Polym. Sci., 2000, 78, 1354–1365 CrossRef CAS.
- W. H. Wan Ishak, N. A. Rosli and I. Ahmad, Sci. Rep., 2020, 10, 11342 CrossRef CAS PubMed.
- M. Jebrane, N. Terziev and I. Heinmaa, Biomacromolecules, 2017, 18, 498–504 CrossRef CAS PubMed.
- Y. Guo, X. Wang, D. Li, H. Du, X. Wang and R. Sun, Polym. Bull., 2012, 69, 389–403 CrossRef CAS.
- L. El Hamdaoui, A. Es-said, M. El Marouani, M. El Bouchti, R. Bchitou, F. Kifani-Sahban and M. El Moussaouiti, ChemistrySelect, 2020, 5, 7695–7703 CrossRef CAS.
- H. S. Barud, A. M. de Araújo Júnior, D. B. Santos, R. M. N. de Assunção, C. S. Meireles, D. A. Cerqueira, G. Rodrigues Filho, C. A. Ribeiro, Y. Messaddeq and S. J. L. Ribeiro, Thermochim. Acta, 2008, 471, 61–69 CrossRef CAS.
- K.-Y. Lee and A. Bismarck, Cellulose, 2012, 19, 891–900 CrossRef CAS.
- J. Lease, T. Kawano and Y. Andou, Polym., 2021, 13, 4397 CAS.
- L. Szcześniak, A. Rachocki and J. Tritt-Goc, Cellulose, 2008, 15, 445–451 CrossRef.
- E. Vittadini, L. C. Dickinson and P. Chinachoti, Carbohydr. Polym., 2001, 46, 49–57 CrossRef CAS.
- S. Saraiva, P. Pereira, C. T. Paula, R. C. Rebelo, J. F. J. Coelho, A. C. Serra and A. C. Fonseca, Mater. Sci. Eng., C, 2021, 131, 112498 CrossRef CAS PubMed.
- M. Patowary, K. Pathak and R. Ananthakrishnan, RSC Adv., 2015, 5, 79852–79859 RSC.
- E. Hosono, S. Fujihara, I. Honma and H. Zhou, J. Am. Chem. Soc., 2005, 127, 13458–13459 CrossRef CAS.
- Y. Cheng, X. Zhang, C. Yin, J. Zhang, J. Yu and J. Zhang, Macromol. Rapid Commun., 2021, 42, 1–7 CrossRef.
- S. Guzman-Puyol, G. Tedeschi, L. Goldoni, J. J. Benítez, L. Ceseracciu, A. Koschella, T. Heinze, A. Athanassiou and J. A. Heredia-Guerrero, Food Hydrocolloids, 2022, 128, 107562 CrossRef CAS.
- N. B. Erdal and M. Hakkarainen, Biomacromolecules, 2022, 23, 2713–2729 CrossRef CAS PubMed.
- G. Singh, C. Chandoha-Lee, W. Zhang, S. Renneckar, P. J. Vikesland and A. Pruden, Water Res., 2016, 104, 137–146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04715f |
| This journal is © The Royal Society of Chemistry 2023 |