
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled synthesis of Mo2C micron flowers via vapor–liquid–solid method as enhanced electrocatalyst for hydrogen evolution reaction
Yuwei Wanga,
Jian Heb,
Yipeng Zangc,
Changbao Zhaoc,
Miaomiao Dib and
Bin Wang *bc
*bc
aCollege of Physical Science and Technology, Bohai University, Jinzhou, 121013, China
bCollege of Chemistry and Materials Engineering, Bohai University, Jinzhou, 121013, China. E-mail: wangbinlhx@163.com
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China
First published on 4th September 2023
Abstract
Mo2C demonstrates excellent performance in catalysis, and it has been found to possess excellent hydrogen evolution reaction (HER) catalytic activity and highly efficient nitrogen fixation. The catalytic activity of Mo2C is greatly influenced and restricted by the preparation method. Sintering and carbon deposition, which affect the catalytic activity of Mo2C, are inevitable in the traditional vapor–solid–solid (VSS) process. In this study, we report the controllable synthesis of α-Mo2C micron flowers by adjusting the growth temperature via a vapor–liquid–solid (VLS) process. The density of the Mo2C micron flowers is closely related to the concentration of Na2MoO4 aqueous solution. The as-grown Mo2C micron flowers dispersed with Pt are validated to be an enhanced collaborative electrocatalyst for HER against Pt/VSS-Mo2C.
Introduction
In recent years, transition metal carbides (TMCs) have been intensively researched for their specific chemical properties.1–3 TMCs are considered to be similar to precious metals in the aspect of electrochemistry and catalysis. Mo2C belonging to the TMC family is called quasi platinum catalyst, and it plays an important role in highly efficient nitrogen fixation4 and hydrogen evolution reaction (HER).5–9 Especially, as some metal atoms are dispersed on the surface of Mo2C crystals for collaborative catalysis, they exhibit excellent selectivity and superior activity for many catalytic reactions.10–13 However, many collaborative catalysis suffer from low mass-specific activity owing to the low metal loading.14 In order to optimize metal loading, the support Mo2C crystals should have a high specific surface area which can provide abundant surface sites to enhance the collaborative catalysis.As a catalytic material with excellent performance, the catalytic activity of Mo2C is greatly influenced and restricted by its preparation method.15–18 In earlier studies, the sintering of the as-grown Mo2C crystals was inevitable,19,20 influencing the structure and morphology of Mo2C crystals, which results in the reduction of the specific surface area and catalytic activity. Therefore, it is important to improve the preparation methods to reduce the sintering and thus increase the specific surface area of the Mo2C crystals.
Herein, we report the synthesis of α-Mo2C crystals via an atmospheric pressure vapor–liquid–solid (VLS) method with Na2MoO4 as the Mo precursor. The morphology of the Mo2C crystals could be controlled by adjusting the growth temperature. Mo2C micron flowers were obtained when the growth temperature was 780 °C. Compared with the vapor–solid–solid (VSS) mode, VLS mode has the advantages of good wettability and superior mobility, which can promote the lateral migration of Mo precursors and prevent the reactive materials from accumulating.11–25 Thus, the as-grown Mo2C crystals can form sheet morphology at an appropriate temperature comparing with the block morphology formation at higher temperatures or via the VSS mode. The advantage of VLS over VSS mode can be further demonstrated by comparing the HER catalytic activity of the as-grown Mo2C dispersed with Pt. Pt/VLS-Mo2C has a lower overpotential than Pt/VSS-Mo2C at a current density of 10 mA cm−2. Mo2C crystals grown using the VLS method is of great significance to improve their catalytic activity and expand their application fields.
Results and discussion
The CVD growth process of Mo2C on Au substrate is illustrated in Fig. 1a. The upper panel shows a typical VSS mode for the growth where (NH4)6Mo7O24 aqueous solution is used as the Mo precursor. As the growth temperature reaches 780 °C, (NH4)6Mo7O24 decomposed to form the solid state of MoO3 particles, which were then carbonized to produce Mo2C when C2H4 was introduced into the reaction chamber. Fig. 1b shows the SEM image of the as-grown Mo2C with 150 mg per mL (NH4)6Mo7O24 as the Mo precursor. The Mo2C demonstrated block morphology with size inconsistency.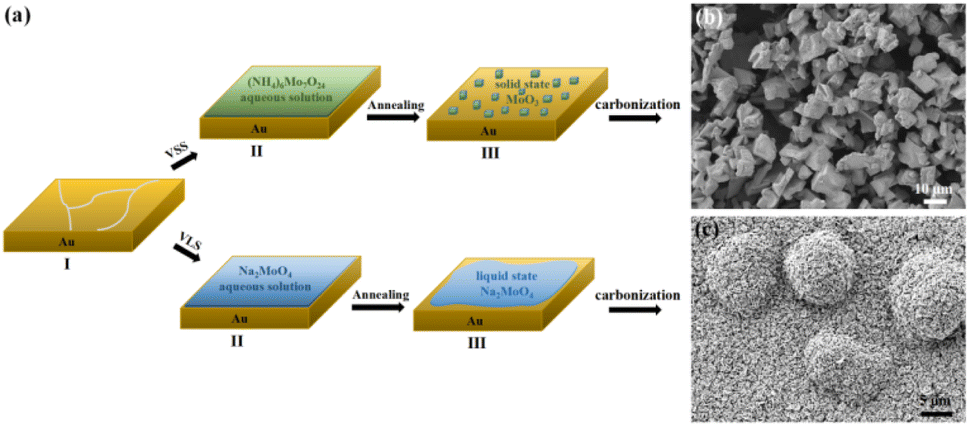 | ||
| Fig. 1 (a) Schematic illustration of the VSS and VLS growth process of Mo2C. (b) and (c) Typical SEM images of the Mo2C crystals grown with VSS and VLS mode, respectively. | ||
Mo2C micron flowers with high specific surface area were synthesized via the VLS mode, and the schemes are shown in the bottom panel of Fig. 1a. 150 mg per mL Na2MoO4 aqueous solution replaces (NH4)6Mo7O24 aqueous solution as the Mo precursor. It is worth noting that the melting point of Na2MoO4 is 687 °C, it melts into liquid state and forms a liquid–solid interface with Au substrate at the growth temperature (780 °C). Importantly, liquid has the advantage of a lower migration barrier, which is more beneficial to the unrestricted diffusion and homogeneous distribution of the precursors on the Au substrate. Thus, uniform Mo2C micron sheets can be synthesized via the VLS mode. Moreover, the liquid–solid interface is conducive to the lateral growth of Mo2C micron sheets. As the size and density increases, the Mo2C micron sheets gradually form the Mo2C micron flower morphology, as shown in Fig. 1c.
X-ray photoelectron spectroscopy (XPS) was conducted to evaluate the chemical composition and valence state of the Mo2C crystals. Fig. 2a shows the binding energies of Mo 3d peaks at 231.4 eV and 228.1 eV, which are attributed to the Mo 3d3/2 and Mo 3d5/2, respectively.26–30 In addition, two weak peaks were observed at 233.4 and 229.8 eV, representing the intermediate oxidation states of Mo (MoOx).28,29 The MoOx may have resulted either from the exposure of Mo2C to air or from the oxidization of Mo2C during the XPS measurement process. Fig. 2b shows the C 1s XPS spectrum, whereby the peak located at the lower binding energy of 283.3 eV was assigned to C–Mo,26,27,30 and those peaks at higher binding energies of 284.8, 286.3, and 288.1 eV can be ascribed to the carbons in the non-oxygenated C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO, respectively.28,29 The XPS signals confirmed the identity of the Mo2C crystals, as expected. Raman spectroscopy and XRD were conducted to evaluate the structure of the Mo2C crystals (Fig. 2c and d). Raman spectrum showed a well-defined characteristic peak at 652 cm−1, corresponding to the Ag mode of α-Mo2C crystal.31,32 The diffraction peaks of Mo2C in the X-ray diffraction (XRD) spectra were consistent with the standard XRD pattern of Mo2C (PDF#31-0871), demonstrating that the as-grown Mo2C crystals were α-Mo2C.
O, and O–CO, respectively.28,29 The XPS signals confirmed the identity of the Mo2C crystals, as expected. Raman spectroscopy and XRD were conducted to evaluate the structure of the Mo2C crystals (Fig. 2c and d). Raman spectrum showed a well-defined characteristic peak at 652 cm−1, corresponding to the Ag mode of α-Mo2C crystal.31,32 The diffraction peaks of Mo2C in the X-ray diffraction (XRD) spectra were consistent with the standard XRD pattern of Mo2C (PDF#31-0871), demonstrating that the as-grown Mo2C crystals were α-Mo2C.
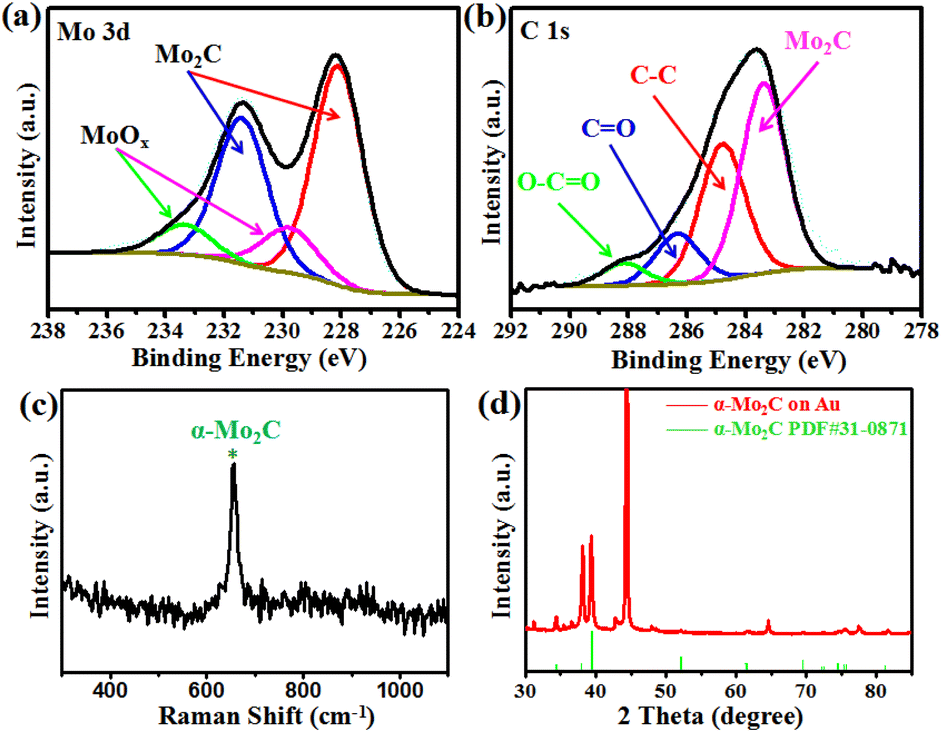 | ||
| Fig. 2 (a) and (b) XPS spectra acquired at the Mo 3d and C 1s regions. Raman spectrum (c) and XRD pattern (d) of the as-grown Mo2C nanocrystals. | ||
The morphology and density of Mo2C crystals can be tuned remarkably by changing the growth temperature and the concentration of Na2MoO4 aqueous solution. Fig. 3a and b present the SEM images of the Mo2C micron sheets grown with 30 and 75 mg per mL Na2MoO4 aqueous solutions at 780 °C, respectively. When the Na2MoO4 aqueous concentration was 30 mg mL−1, it provided a low concentration of Mo species, resulting in few nucleation sites, and thus, only a low quantity of Mo2C micron sheets appeared, as shown in Fig. 3a. By increasing the Na2MoO4 aqueous concentration to 75 mg mL−1, the shape of the Mo2C micron sheets became more evident, whereby some micron sheets have begun to form flower-like shapes. The inset in Fig. 3b is the SEM image of an individual Mo2C micron flower. Energy dispersive X-ray spectroscopy (EDS) mapping were recorded for the spatial distribution of the Mo and C elements (Fig. 3c and d), and both of them were found to be distributed uniformly in the micron flowers with sharp edges, exhibiting the uniformity of the Mo2C crystals. Subsequently, the influence of growth temperature was investigated, and the 75 mg per mL Na2MoO4 aqueous solution was used as the Mo precursor. As the growth temperature increased from 850 to 900 °C, the as-grown Mo2C crystals gradually adopt block morphology, as shown in Fig. 3e and f. The inset in Fig. 3f is the SEM image of an individual block Mo2C crystal.
 | ||
| Fig. 3 SEM images of the Mo2C crystals grown with different concentrations of Na2MoO4 aqueous solution: (a) 30 mg mL−1 and (b) 75 mg mL−1. (c) and (d) EDS elemental mapping of Mo and C of the Mo2C micron flower, respectively. SEM images of the Mo2C crystals grown with different temperatures: (e) 850 °C and (f) 900 °C. Insets in (b) and (f) are SEM images of the individual Mo2C crystal grown at 780 and 900 °C with high magnification, respectively. | ||
In order to further demonstrate the advantage of VLS in synthesizing Mo2C, the HER catalytic activities of VLS-Mo2C and VSS-Mo2C were compared. The samples of VLS-Mo2C (150 mg per mL Na2MoO4) and VSS-Mo2C [150 mg per mL (NH4)6Mo7O24] were synthesized on Au substrates, and both the two kinds of Mo2C were loaded with 2 nm Pt for the electrochemical test. The HER catalysis was evaluated in 1.0 M KOH solution using a typical three-electrode system with the studied materials as the working electrodes, Hg/HgO as the reference electrode, and the Pt foil as the counter electrode.
Fig. 4a shows the linear sweep voltammetry (LSV) curves of Pt/VLS-Mo2C, Pt/VSS-Mo2C, and Pt/Au with a scan rate of 5 mV s−1. Compared with the Pt/VSS-Mo2C and Pt/Au, the Pt/VLS-Mo2C has a lower overpotential of 52 mV versus the reversible hydrogen electrode (RHE) at a current density of 10 mA cm−2, indicating that the VLS mode can substantially improve the collaborative catalytic performance of Pt/Mo2C toward HER in alkaline condition. The derived Tafel slope of Pt/VLS-Mo2C and Pt/VSS-Mo2C is around 166 and 222 mV dec−1, respectively (Fig. 4b), indicating that the hydrogen evolution on both of them undergoes the Volmer mechanism, and water dissociation is the rate-determining step. Critically, a substantially decreased Tafel slope of Pt/VLS-Mo2C revealed that the sluggish water dissociation behavior had improved significantly. In addition, electrochemical impedance spectroscopy (Fig. 4c) showed that Pt/VLS-Mo2C possessed a lower charge transfer resistance than Pt/VSS-Mo2C. The significantly reduced impedance further suggest that Pt/VLS-Mo2C can substantially boost the interfacial electron-transfer kinetics between the Mo2C and Au foil, which promotes the HER dynamic process. The electrochemical surface areas of Pt/Mo2C crystals were further estimated by deriving the electrochemical double layer capacitance (Cdl) from the cyclic voltammetry studies, as shown in Fig. 4d–f. The Pt/VLS-Mo2C was found to have a larger Cdl of 13.2 mF cm−2 than Pt/VSS-Mo2C (11.1 mF cm−2), indicating that the VLS mode can increase the electrochemical surface areas of the as-grown Mo2C crystals.
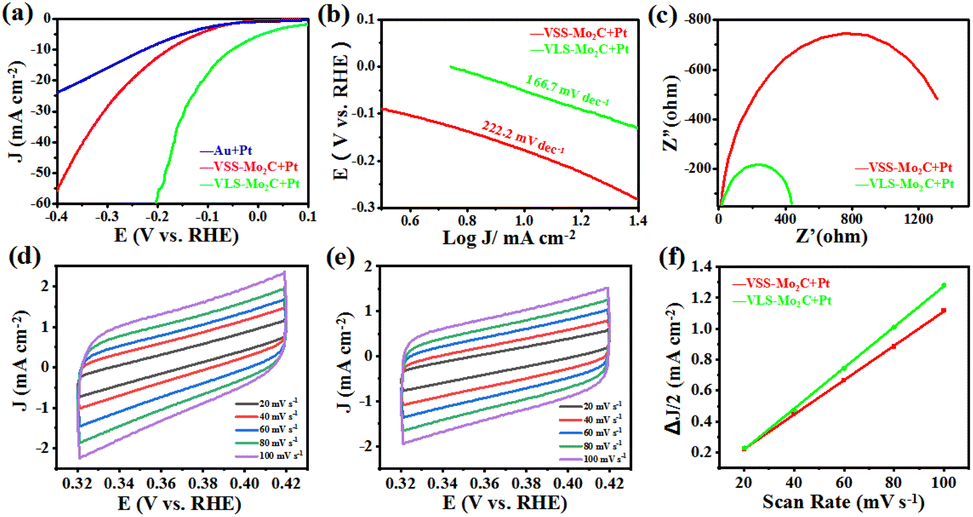 | ||
| Fig. 4 (a) The LSV curves of Pt/VLS-Mo2C, Pt/VSS-Mo2C, and Pt/Au with IR correction. (b) The corresponding Tafel slopes. (c) Nyquist plots of Pt/VLS-Mo2C and Pt/VSS-Mo2C collected at the open-circuit voltage. CV curves at different scan rates from 20 to 100 mV s−1 of (d) Pt/VLS-Mo2C and (e) Pt/VSS-Mo2C. (f) The plots of ΔJ versus scan rates for the Pt/VLS-Mo2C and Pt/VSS-Mo2C, respectively. | ||
We compared the Pt/VLS-Mo2C over the state-of-the-art of electrocatalysts for HER, as shown in Table 1. We believe that the VLS method could offer new insights into the synthetic approaches for Mo2C and provide new strategies for constructing metal-loading catalysts with high HER catalytic activity.
Conclusion
In summary, we demonstrated the VLS growth of α-Mo2C micron flowers, which were realized by using liquid precursor for the first time. The morphology and density of the Mo2C crystals could be controlled by tuning the growth temperature and concentration of Na2MoO4 aqueous solution. The unique flower-like structure produces a high specific surface area and abundant surface sites on the surface, increasing the Pt loading and enhancing the collaborative catalysis. The comparison between Pt/VLS-Mo2C and Pt/VSS-Mo2C in terms of HER catalytic activities further demonstrated the advantage of VLS in synthesizing Mo2C crystals. Our study not only offers new insights into the synthetic approaches for Mo2C but also provides a new strategy for constructing metal-loading catalysts with high catalytic activity.Author contributions
Bin Wang designed and conducted the VLS growth and analyzed the data. Yipeng Zang performed the HER of the materials. All the authors discussed and commented on the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21688102 and No. 21825203), the National Key R&D Program of China (No. 2016YFA0200200), and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB17020000).Notes and references
- A. W. H. Stefan, W. G. Robert, S. E. Daan and H. B. Johannes, ACS Catal., 2013, 3, 2837–2844 CrossRef.
- M. Q. Zeng, Y. X. Chen, J. X. Li, H. F. Xue, R. G. Mendes, J. X. Liu, T. Zhang, M. H. Rümmeli and L. Fu, Nano Energy, 2017, 33, 356–362 CrossRef CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- K. Ba, G. L. Wang, T. Ye, X. R. Wang, Y. Y. Sun, H. Q. Liu, A. Q. Hu, Z. Y. Li and Z. Z. Sun, ACS Catal., 2020, 10, 7864–7870 CrossRef CAS.
- W. Y. Sun, X. Q. Wang, J. Q. Feng, T. Li, Y. H. Huan, J. B. Qiao, L. He and D. L. Ma, Nanotechnol, 2019, 30, 385601–385608 CrossRef CAS PubMed.
- J. S. Li, Y. Wang, C. H. Liu, S. L. Li, Y. G. Wang, L. Z. Dong, Z. H. Dai, Y. F. Li and Y. Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- D. Geng, X. Zhao, Z. Chen, W. Sun, W. Fu, J. Chen, W. Liu, W. Zhou and K. P. Loh, Adv. Mater., 2017, 29, 1700072 CrossRef PubMed.
- Y. C. Liu, B. B. Huang, X. Hu and Z. L. Xie, Int. J. Hydrogen Energy, 2019, 44(7), 3702–3710 CrossRef CAS.
- X. F. Zhang, T. Lei, M. Xia, Q. H. Wei and Z. L. Xie, Dalton Trans., 2023, 52, 6267–6272 RSC.
- Y. Z. Ge, X. T. Qin, A. W. Li, Y. C. Deng, L. L. Lin, M. T. Zhang, Q. L. Yu, S. W. Li, M. Peng, Y. Xu, X. Y. Zhao, M. Q. Xu, W. Zhou, S. Y. Yao and D. Ma, J. Am. Chem. Soc., 2021, 143(2), 628–633 CrossRef CAS PubMed.
- L. L. Lin, Q. L. Yu, M. Peng, A. W. Li, S. Y. Yao, S. H. Tian, X. Liu, A. Li, Z. Jiang, R. Gao, X. D. Han, Y. W. Li, X. D. Wen, W. Zhou and D. Ma, J. Am. Chem. Soc., 2021, 143, 309–317 CrossRef CAS PubMed.
- X. Zhang, M. T. Zhang, Y. C. Deng, M. Q. Xu, L. C. Artiglia, W. Wen, R. Gao, B. B. Chen, S. Y. Yao, X. C. Zhang, M. Peng, J. Yan, A. W. Li, Z. Jiang, X. Y. Gao, S. F. Cao, C. Yang, A. J. Krop, J. N. Shi, J. L. Xie, M. S. Bi, J. A. Bokhoven, Y. W. Li, X. D. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589, 396–401 CrossRef CAS PubMed.
- S. Posada-Pérez, R. A. Gutiérrez, Z. J. Zuo, P. J. Ramírez, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, Catal. Sci. Technol., 2017, 7, 5332–5342 RSC.
- J. B. Wu, L. K. Xiong, B. T. Zhao, M. L. Liu and L. Huang, Small Methods, 2020, 4, 1900540 CrossRef CAS.
- X. Y. Li, D. Ma, L. M. Chen and X. H. Bao, Catal. Lett., 2007, 116, 63–69 CrossRef CAS.
- T. C. Xiao, A. P. E. York, H. Al-Megren, C. V. Williams, H. T. Wang and M. L. H. Green, J. Catal., 2001, 202(1), 100–109 CrossRef CAS.
- T. C. Xiao, A. P. E. York, V. C. Williams, H. Al-Megren, A. Hanif, X. Y. Zhou and M. L. H. Green, Chem. Mater., 2000, 12(12), 3896–3905 CrossRef CAS.
- S. R. Vallance, S. Kingman and D. H. Gregory, Chem. Commun., 2007, 7, 742–744 RSC.
- M. Saito and R. B. Anderson, J. Catal., 1980, 63, 438–446 CrossRef CAS.
- L. Volpe and M. Boudart, J. Solid State Chem., 1985, 59, 348–356 CrossRef CAS.
- H. Liu, G. P. Qi, C. S. Tang, M. L. Chen, Y. Chen, Z. W. Shu, H. Y. Xiang, Y. Y. Jin, S. S. Wang, H. M. Li, M. Ouzounian, T. S. Hu, H. G. Duan, S. S. Li, Z. Han and S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 13174–13181 CrossRef CAS PubMed.
- S. M. Feng, J. Y. Tan, S. L. Zhao, S. Q. Zhang, U. Khan, L. Tang, X. L. Zou, J. H. Lin, H. M. Cheng and B. L. Liu, Small, 2020, 2003357, 1–9 Search PubMed.
- S. S. Li, Y. C. Li, X. Y. Liu, Z. H. Hu, J. Wu, H. Nakajima, S. Liu, T. Okazaki, W. Chen, T. Minari, Y. Sakuma, K. Tsukagoshi, K. Suenaga, T. Taniguchi and M. Osada, Nanoscale, 2019, 11, 16122–16129 RSC.
- S. S. Li, Y. C. Lin, W. Zhao, J. Wu, Z. Wang, Z. H. Hu, Y. D. Shen, D. M. Tang, J. Y. Wang, Q. Zhang, H. Zhu, L. Q. Chu, W. J. Zhao, C. Liu, Z. P. Sun, T. Taniguchi, M. Osada, W. Chen, Q. H. Xu, A. T. S. Wee, K. Suenaga, F. Ding and G. Eda, Nat. Mater., 2018, 17, 535–542 CrossRef CAS PubMed.
- M. Zeng and L. Fu, Acc. Chem. Res., 2018, 51, 2839–2847 CrossRef CAS PubMed.
- H. Cheng, L. X. Ding, G. F. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
- J. Halim, S. Kota, M. R. Lukatskaya, M. Naguib, M. Q. Zhao, E. J. Moon, J. Pitock, J. Nanda, S. J. May, Y. Gogotsi and M. W. Barsoum, Adv. Funct. Mater., 2016, 26, 3118–3127 CrossRef CAS.
- Q. Gao, X. Y. Zhao, Y. Xiao, D. Zhao and M. H. Cao, Nanoscale, 2014, 6, 6151–6157 RSC.
- R. R. Li, S. G. Wang, W. Wang and M. H. Cao, Phys. Chem. Chem. Phys., 2015, 17, 24803–24809 RSC.
- D. C. Geng, X. X. Zhao, L. J. Li, P. Song, B. B. Tian, W. Liu, J. Y. Chen, D. Shi, M. Lin, W. Zhou and K. P. Loh, 2D Mater., 2017, 4, 011012 CrossRef.
- T. Li, W. Luo, H. Kitadai, X. Wang and X. Ling, Adv. Mater., 2019, 31, 1807160 CrossRef PubMed.
- T. C. Xiao, A. P. E. York, H. Al-Megren, C. V. Williams, H. T. Wang and M. L. H. Green, J. Catal., 2001, 202, 100–109 CrossRef CAS.
- T. Ouyang, A. N. Chen, Z. Z. He, Z. Q. Liu and Y. Tong, Chem. Commun., 2018, 54, 9901–9904 RSC.
- L. Ai, J. Su, M. Wang and J. Jiang, ACS Sustainable Chem. Eng., 2018, 6, 9912–9920 CrossRef CAS.
- W. Liu, X. T. Wang, F. Wang, K. F. Du, Z. F. Zhang, Y. Z. Guo, H. Y. Yin and D. H. Wang, Nat. Commun., 2021, 12, 6776 CrossRef CAS PubMed.
- T. T. Liu, H. Liu, X. J. Wu, Y. L. Niu, B. M. Feng, W. Li, W. H. Hu and C. M. Li, Electrochim. Acta, 2018, 281, 710–716 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |