
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modified selective optical sensor for selenium determination based on incorporating xylenol orange in a poly(vinyl chloride) membrane
Abeer M. E. Hassana,
Reem F. Alshehrib,
Salah M. El-Bahyc,
Alaa S. Amin *d and
Mai Aishe
*d and
Mai Aishe
aChemistry Department, Faculty of Pharmacy, October 6th University, October, Egypt
bChemistry Department, Faculty of Science, Taibah University, Kingdom of Saudi Arabia
cChemistry Department, Turabah University College, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
dChemistry Department, Faculty of Science, Benha University, Benha, Egypt. E-mail: asamin2005@hotmail.com
eChemistry Department, Faculty of Science, Port Said University, Port Said, Egypt
First published on 27th November 2023
Abstract
A novel optical sensor has been developed to measure selenium ions. The sensor membrane was created by mixing xylenol orange (XO) and sodium tetraphenylborate (NaTPB) with a plasticized poly(vinyl chloride) membrane that contained o-nitrophenyl octyl ether (o-NPOE) as a plasticizer. XO was previously established for use in a colorimeter to measure selenium in water and other media. At pH 6.6, the color of the detecting membrane changed from orange to pink when in contact with Se4+ ions. Various variables affecting the uptake efficiency were evaluated and optimized. Under optimum conditions (i.e., 30% PVC, 60% o-NPOE, and 5.0% of both XO and NaTPB for 5.0 min as the response time), the proposed sensor displayed a linear range 10–175 ng mL−1 with the detection and quantification limits of 3.0 and 10 ng mL−1, respectively. Also, the precision (RSD%) was better than 2.2% for six replicate determinations of 100 ng mL−1 Se4+ in various membranes. For the detection of Se4+, the selectivity of the sensor membrane was investigated for a number of possible interfering inorganic cations, but no appreciable interference was found. With the use of a 0.3 M HCl solution, the sensor was successfully restored, and the response that may have been reversible and reproducible exhibited an RSD% of less than 2.0%. The sensor has been successfully used to analyze Se4+ ions in environmental and biological materials.
Introduction
Selenium (Se) is a significant microelement that has a variety of beneficial impacts on diseases and human health. As the levels of Se are incredibly variable across different populations and areas, it is crucial to regulate the status and intake of Se for specific populations. Selenium is one of the trace minerals needed for proper functioning of the body. To maintain homeostasis, human body cells require the right nutrients. Micronutrients, including vitamins, antioxidants, and trace minerals, are essential for numerous regenerative processes, controlling oxidative stress, and establishing immunity against infections.1Selenium is widely distributed in nature in relatively small concentrations in rocks, plants, coal, and other fossil fuels. It exists in environmental samples and living organisms because of the catalytic effects of its compounds in metabolism.2 Owing to its antioxidant properties and the presence of selenoproteins, selenium is a vital trace element for all living organisms.3 Selenium has to be present in relatively small amounts for maintaining human and animal health, but in excessive concentrations, it could be hazardous. The organic and inorganic forms of selenium as well as its oxidation configuration are poisonous in nature. In natural waters, the inorganic forms of selenium are more toxic than organic forms, and selenite Se(IV) is more toxic than selenate Se(VI).4,5
The World Health Organization (WHO) has set a limit for the selenium concentration in drinking water at 40 ng mL−1.6,7 While the European Union (EU) and the U.S. Environmental Protection Agency (EPA) have set the limit levels of 50 ng mL−1 and 10 ng mL−1, respectively.8,9 The Se speciation analysis in water becomes difficult due to the need for very sensitive and precise analytical techniques.
Selenium(IV) fractionation issues in soils with plenty of organic matter have received much attention.10 One of the many physiological tasks performed by selenium(IV) is the protection of cell membranes against oxidative damage. Severe exposure to elemental selenium and selenium oxides can cause a variety of ailments, including bronchitis, stomach aches, coughing, and irritation of the respiratory tract. When selenium levels in the body are above optimal levels, the neurological system is affected. Se(IV) has been discovered to possess anticancer properties. There is growing evidence that selenium is crucial for both human and animal reproduction and growth.11
Selenium is considered to be an economically critical mineral in some countries. A low-cost, portable sensor could be useful during the processing of Se (for example, for semiconductors) and for downstream process monitoring. Se speciation in food is crucial for a better understanding of the detection of this metalloid, mainly because selenium absorption has been found to be higher in organic molecules. The low concentration of each species to be identified and the intricacy of the matrix are factors in many selenium speciation analysis issues.12,13
Various instrumental analytical techniques such as atomic absorption spectroscopy (AAS),14–17 atomic fluorescence spectroscopy (AFS),18 hydride generation atomic fluorescence spectrometry (HGAFS),19 electrothermal atomic absorption spectrometry (ETAAS),20 inductively coupled plasma optical emission spectrometry (ICP-OES),21 hydride generation system and atomic fluorescence spectrometer (HG-AFS),22 hydride generation atomic fluorescence spectrometer (HG-AFS),23 integrated coupled plasma mass spectroscopy,24 inductively coupled plasma mass spectrometry (ICP-MS),25,26 spectrofluorimetry,27,28 and stripping voltammetry29 were used for the determination of selenium in different water, food, and environmental samples. A comprehensive evaluation of these advanced instrumental approaches includes disadvantages related to these techniques, such as expensive investment costs, significant electricity consumption during continuous analysis, the requirement for frequent maintenance of these instruments, and laborious analytical methodology. However, the majority of these methods are complex, relatively costly, time-consuming, and difficult to implement. This prompted the development of a novel and easily deployable optical sensor for the detection of selenate Se(VI). The optical technique has the ability to detect specific oxidation states compared to AAS, and ICP-MS is included.
Spectrophotometry is more accessible, quicker, and less expensive. There are numerous reagents available for selenium spectrophotometric measurement. The spectrophotometric methods for the determination of selenium used J-acid,30 Leuco crystal violet,31 resazurin,32 sodium salt of hexamethyleneimine carbodithioate,33 1-naphthylamine-7-sulphonic acid34 and variamine blue,35 which have several disadvantages. However, these procedures can only yield the total Se level or are not accurately reliable for detecting ultra-trace Se4+. As a result, preliminary species preconcentration is needed before detection using the above techniques. Additionally, they can offer the required sensitivity and selectivity for environmental checking. However, it lacks selectivity and is hampered by interferences from mutual anions and cations. Consequently, establishing rapid, simple, and effective methods for selenium monitoring at trace levels in water, food, environmental, and biological samples is crucial.
Recently, there has been a focus on the development of chemical methods that reduce the use of compounds that could endanger human health and cause environmental contamination. An alternative strategy is recommended in order to reduce the amount of reagent needed. In the past three decades, interest in the development of optical chemical sensors—also known as optodes or optrodes—as viable substitutes for electrochemical sensors has increased.36 These sensors have lower detection limits and high sensitivity.37 Additionally, they may benefit from spectrum characteristics related to the analyte or analyte-specific indication.38 Furthermore, they are not impacted by electrical noise and do not require internal or external reference devices and extended preconditioning times.39 The advantages of optical sensor methodology have attracted the attention of the scientific community, as they allow the development of low-cost or cost-competitive systems with faster response times and a wider nano-concentration range and low detection and quantification limits compared with conventional technologies,14–29,36–39 in addition to the ability to detect specific oxidation states compared to the AAS and ICP-MS methods.14–26
The membrane optode matrix is chosen based on analyte permeability, good mechanical properties, plasticizability, affordability, ease of miniaturization and remote sensing, suitability for immobilizing the chromophore and extractant, and minimal sample manipulation.40–42 Polyvinyl chlorides are the most often utilized polymers in optical sensors. For many applications, they compare favorably with sol–gel matrices and have several advantageous characteristics. The polymers are most frequently utilized in sensors with visible spectrophotometric detections due to their transparency.43 Numerous sensors have been described in trace analyses of different analytes, including metal ions.44–49
Yellow amorphous xylenol orange is easily soluble in conventional organic solvents. Our research team previously employed this reagent for colorimetric tests to examine how selenium interacts with it.50 The ultimate goal of the current work is to successfully incorporate XO into plasticized PVC film and develop a novel optical sensor with excellent sensitivity and selectivity for the quick measurement of Se4+ ions in environmental and biological samples.
Experimental
Reagents
All the chemicals were of analytical grade. Xylenol orange was used without further purification from Sigma (St. Louis, MO, USA). High molecular weight poly(vinyl chloride) (PVC), dioctyl adipate (DOA), dioctyl sebacate (DOS), tributylphosphate (TBP), dibutyl phthalate (DBP), ortho-nitrophenyl octyl ether (o-NPOE), sodium tetraphenylborate (NaTPB), and tetrahydrofurane (THF), Na2EDTA were from Fluka (Buchs, Switzerland) or Merck (Darmstadt, Germany) Chemical Companies. Universal buffer solutions of pH 2.75–10.63 were prepared as described earlier.51Selenium stock solutions of 5 × 10−3 M were prepared by dissolving an appropriate weight of sodium selenite obtained from BDH Chemical Company (Poole, UK) in the least amount of bidistilled water and completing the volume in a 50 mL measuring flask. Working solutions were obtained by suitable dilution of the stock solution with bidistilled water.
Instrumentation
An Orion research model 601 A/digital ionalyzer pH meter was accomplished for examining the pH of solutions. Electrothermal atomic absorption spectrometry (PerkinElmer Analysis 700 model, Norwalk, CT, USA) was used in this investigation. Selenium EDL lamp was used at 200 mA current, 196.0 nm wavelength, and 2.0 nm spectral band pass. Selenium measurements were carried out as peak area. Operating conditions of selenium in a graphite furnace were adjusted according to manufacturers. Drying, ashing, and atomization temperatures were optimized before analysis. Argon was used at 250 mL min−1 flow rate. A Hamilton syringe (10 μL) was performed to transport minor Se4+ ion volumes into the cell. The sensor thickness was acquired by a digital microscope (Ray Vision Y 103) that was coupled with a video camera (JVC TK-C 751 EG) and a digital micrometer (Mitutoy, Japan) with an accuracy of ±0.001 mm. UV-vis spectrophotometer model V 53 from JASCO (Tokyo, Japan) was attained for recording the spectra and the absorbance assessments. The absorbance assessments were achieved inside a quartz cuvette by mounting the samples of optical membrane sensors (3.0 cm × 1.0 cm). The absorbance assessments of the optical membrane sensor samples were established with respect to air and a blank sensor sample.Membrane preparation
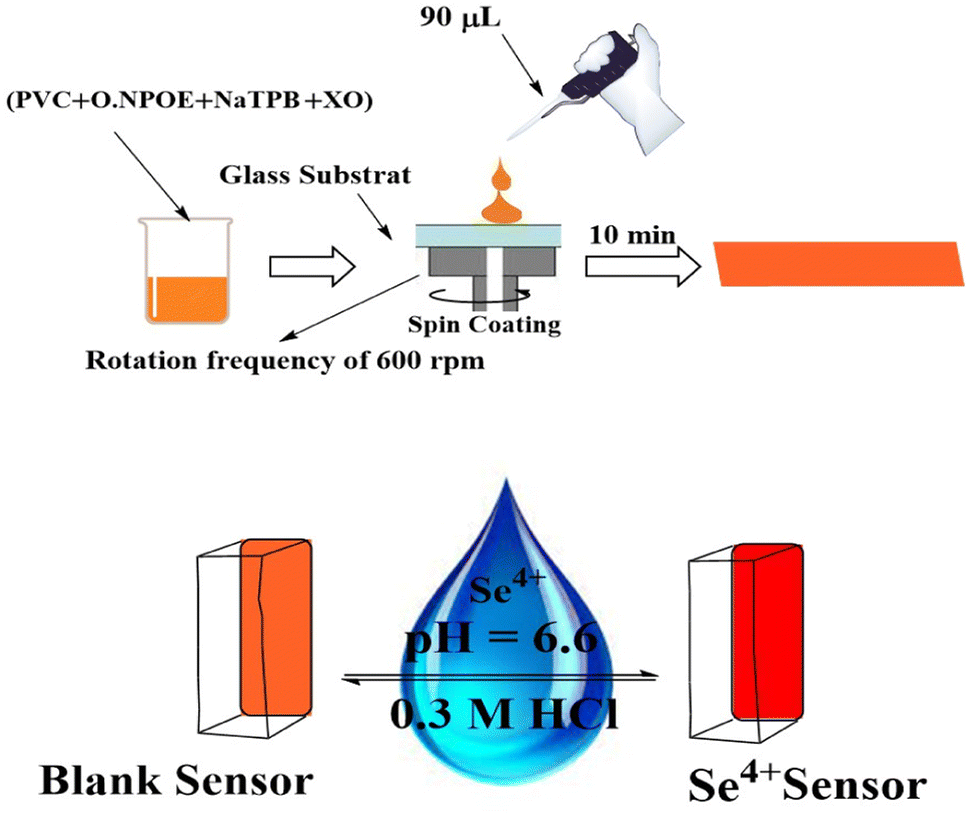
The membrane has the right quantities of active ingredients. In a glass vial, 3.0 mL THF was used to dissolve 24 mg of PVC (30%), 48 mg of o-NPOE (60.0%), 4.0 mg of XO (5.0%), and 4.0 mg of NaTPB (5.0%). Instantaneously, the mixture was vigorously mixed to attain total homogeneity. The spin-on apparatus was used to spin a glass plate (1 mm × 9 mm × 50 mm), which had been thoroughly cleaned with pure THF to remove any organic contaminants. 90 μL of the aforementioned solution was injected into the glass plate. The membrane was placed in ambient air and spun for 30 s at 600 revolutions per minute before being left to dry for 10 min. Furthermore, the thickness of the PIM was assessed using a digital microscope (Ray Vision Y 103) that was connected to a video camera (JVC TK-C 751 EG). Control membranes were prepared in the same manner. Nevertheless, D2EHPA and CPAHPD were exceptional from the membrane solution. Control membranes were prepared in the same manner. Nevertheless, XO was not presented in the membrane solution. In order to construct the control optical sensor, which is made up of PVC and o-NPOE, 24 mg of PVC and 48 mg of o-NPOE were dissolved in 3.0 mL of THF. The mixture was stirred for 10 min, poured into a 9.0 cm diameter flat-bottomed Petri dish, and left to evaporate for two days.Colorimetric procedure
The sensing membrane was placed in a 1.0 cm quartz cell mounted in the spectrophotometer that was already filled with 2.5 mL of the buffer solution of pH 6.6 containing various quantities of Se4+, and it was vigorously stirred for 5.0 min. At 585 nm, the net absorbance of the sensor was determined in comparison to a blank membrane. The blank membrane was defined as a glass-supported membrane that was put in the test solution without Se4+ ions. A freshly made sensor membrane was used for every measurement. The absorbance of the samples were examined at 585 nm.Determination of total Se and Se(VI) species
The determination of total inorganic Se needed a pre-reduction step since the absorption band was produced by a selective radical cation reaction between Se(IV) species and XO. A volume of 3.0 mL of concentrated HCl was added to 10 mL of the sample, and the mixture was heated on a hot plate at 100 °C for at least 30 min to completely reduce Se(VI) to Se(IV). The process for determining total inorganic selenium was then carried out using the reduced samples after they had been allowed to cool at a temperature of 25 ± 2 °C. Finally, the concentration of Se(VI) was calculated by subtracting Se(IV) from total Se.Interference studies
Standard solutions including 100 ng mL of Fe(III) and 2.0 mg mL−1 of one of the following base metal ions frequently found in water samples, Ca(II), Mg(II), Al(III), Cu(II), Pb(II), Co(II), Ni(II), Cd(II), Fe(II), Pd(II), Cr(III), Te(IV), Cr(VI) and Zn(II), were prepared. The solutions were examined by the newly established membrane for this study.Water sample collection
All sampling materials were washed overnight with a 10% (v/v) HNO3 solution, rinsed with ultrapure water, and finally, three times with the sample prior to collection. For tap water samples, domestic water was left to run for 20 min, after which a volume of approximately 500 mL was collected in an HDPE bottle. Clean HDPE bottles were used to collect samples of sea, rain, and subsurface water; 500 mL of each type of sample was taken at a depth of 5.0 cm. Sample aliquots were immediately filtered through 0.22 μm pore size PTFE membrane filters (Millipore Corporation) and stored at 4 °C. Sand residues present in seawater were separated by centrifugation prior to filtration. In the Egyptian city of Port Said, seawater was collected. In Benha city (Egypt), samples of rain were taken during a rainstorm, while in El-Madina city (Saudi Arabia), samples of underground water were taken from a well. After neutralization with NH4OH and completion with buffer pH 6.6, the cooled samples were transferred to a 10 mL measuring flask, mixed thoroughly, and then subjected to the indicated sensor and FAAS procedures for analysis.Application to food and beverage samples
CS-M-3 mushroom (Boletus edulis) and NIST SRM 1946 fish tissue (100 mg), canned fish, cultivated mushroom, black tea, green tea, coffee, egg, honey, onion, garlic, cow meat, chicken meat, salami, cheese, cabbage, potato, boiled wheat, canned tomato (500 mg), mixed fruit juice and ice tea (1.0 mL) were digested with 6.0 mL HNO3 (65%) and 2.0 mL H2O2 (30%) in a Milestone Ethos D model closed vessel microwave digestion system according to a reported procedure52 and diluted to 50 mL with deionized water. In the same manner, and omitting the sample, a blank digestion was also created. On samples that had been digested, the optimized procedure previously described was used. Additionally, ETAAS was used to determine the selenium content.Determination of selenium in soil sample
A known weight of selenium was mixed with 20 g of the soil sample, fused with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sodium carbonate and potassium nitrate mixture in a nickel crucible and extracted with water. The filtrate of the extract was treated with 20 mL of 10 M hydrochloric acid and then heated to expel chlorine and oxides of nitrogen. For the solution to have an appropriate concentration of selenium, the solution was further diluted with water. To remove the iron present in the soil, a portion of the stock solution was run through the cation exchange resin. Utilizing the sensor mentioned above and using the conventional addition method, the selenium contents were determined.
:1 sodium carbonate and potassium nitrate mixture in a nickel crucible and extracted with water. The filtrate of the extract was treated with 20 mL of 10 M hydrochloric acid and then heated to expel chlorine and oxides of nitrogen. For the solution to have an appropriate concentration of selenium, the solution was further diluted with water. To remove the iron present in the soil, a portion of the stock solution was run through the cation exchange resin. Utilizing the sensor mentioned above and using the conventional addition method, the selenium contents were determined.
Determination of Se in human hair
A known amount of human hair was digested with HCl–HNO3 (3 + 2 v/v; 10 mL) for 10 min. According to the above-described methodology, the solution was cooled, neutralized with diluted NaOH solution, and then analyzed for Se concentration.Determination of Se in a cosmetic sample
A known weight of a cosmetic sample (lipstick) was dissolved in alcohol to extract any organic material. The residue was gently heated with concentrated HNO3 (10 mL) for 10 min, and the contents were cooled and then boiled with HCl (10 mL) for 10 min to convert Se(VI) to Se(IV). The sample residue was cooled and then treated with 0.5 M H2SO4, 5.0 mL of diluted NaOH solution, neutralization, and bidistilled water dilution to a specified volume (50 mL). The sensor technique for Se was used to analyze the resultant solution.Results and discussion
Preliminary investigations
According to the previous study,50 at a pH 6.6, the main species of XO was presented in the solution as L2− that forms a colored radical cation with Se4+ and Te4+. It was found that selenium may be determined colorimetrically by optimizing the fabrication of a sensor by including XO and NaTPB in a plasticized PVC membrane containing o-NPOE, whereas Te4+ radical cation is not created. When Se4+ ions diffused into the membrane, it formed radical cations with XO, so the membrane color changed from orange to pink. Fig. 1 shows the absorption spectra of the blank and XO sensors in various concentrations of Se4+.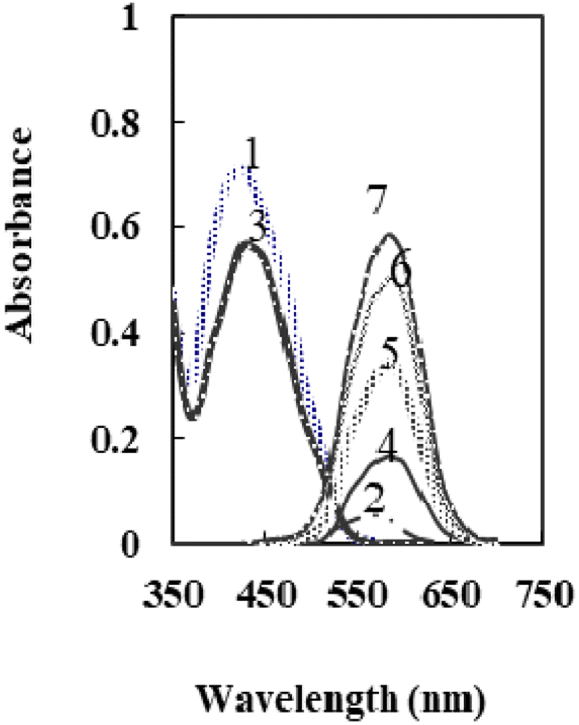 | ||
| Fig. 1 The absorption spectra of 1: XO in solution, 2: XO-Se in solution, 3: XO sensor, and 4–7: XO-Se sensor at 50, 100, 150, and 175 ng mL−1. | ||
A schematic showing the sensing moiety and its expected interaction with Se(IV) is shown in Scheme 1.
 | ||
| Scheme 1 The sensing moiety of the membrane sensor and its reactivity in the presence of different [Se4+]. | ||
As can be seen, the membrane spectrum without Se4+ ions exhibits a maximum absorbance at λ = 428 nm, whereas the membrane spectrum with Se4+ ions exhibits a maximum absorbance at 585 nm. Comparing the blank and the radical cation spectra of the sensor (Fig. 1) with those in solution,50 it was observed that the absorbance maxima of both immobilized reagent and radical cation had a bathochromic shift. These results imply that the immobilized reagent's structural conformation is more planar than that of its solution equivalent.53 The maximal absorbance wavelengths of the membrane are shown at 585 nm in the current work, where Se4+ reacts with XO in the sensor membrane. Because of its high selectivity and sensitivity at this maximum, this wavelength was chosen for future studies. It was essential to elucidate the influence of all factors that can possibly affect the prepared sensor.
Optimization of the method
To take full advantages of the sensor, amounts of the sensor ingredients and reaction conditions should be optimized. In the one-at-a-time optimization procedure, the absorbance of the membrane in the presence of 100 ng mL−1 Se4+ at 585 nm was used as the analytical signal.Effect of membrane composition
The different components utilized in each sensor, such as the base matrix, solvent mediator, ionophore, and the essential additive used in the membrane construction, have a major impact on the response characteristics and working concentration range. As a result, choosing the sensor matrix is crucial. It was observed that high molecular weight PVC could be used as the membrane base. This selection was due to several parameters, such as appropriate transmittance, suitable immobilization of XO as the reagent without any leakage, good mechanical stability, and reliable permeability to Se4+ ions.Solvent mediators (plasticizers) must be physically compatible with the polymer utilized to prepare the sensor membrane to produce a homogeneous organic phase. As potential plasticizers, several solvents, including DOA, DOS, TBP, DBP, and o-NPOE, were evaluated in this study. As seen in Fig. 2, the membranes containing DOA or DOS had improper physical properties, indicating that these membrane solvents did not cause a suitable signal for the proposed membrane sensor. Also, as shown in Fig. 2, TBP and DBP plasticizers showed good sensitivity, but membranes containing these plasticizers showed reagent leakage at short times. The membrane containing o-NPOE was the appropriate selection with respect to high sensitivity and minimum leakage of XO from the membrane. As shown in Table 1, the membrane sensors with a weight ratio of o-NPOE to PVC of 2.0 provided better absorbances. Thus, 48 mg o-NPOE (60.0%) was selected as an optimum value.
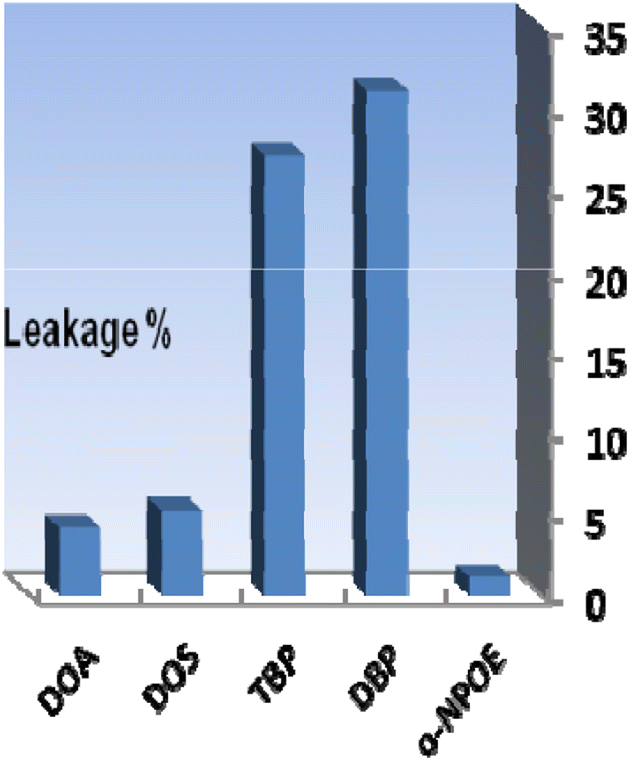 | ||
| Fig. 2 The effect of plasticizer nature on the membrane leakage% after 30 min. Conditions: [Se4+] = 100 ng mL−1, T = 25 °C; the membrane layer contained 24 mg of PVC, 48 mg of each plasticizer, and 4.0 mg of XO. | ||
| Sensor | PVC (mg) | o-NPOE (mg) | XO (mg) | NaTPB (mg) | Response time (min) | Absorbancea (585 nm) |
|---|---|---|---|---|---|---|
| a Mean absorbance (n = 4) of every parameter is recorded from four solutions of 100 ng mL−1 Se4+ (pH 6.6). | ||||||
| 1 | 24 | 40 | 4 | 4 | 5 | 0.336 |
| 2 | 24 | 44 | 4 | 4 | 5 | 0.421 |
| 3 | 24 | 48 | 4 | 4 | 5 | 0.501 |
| 4 | 24 | 52 | 4 | 4 | 5 | 0.442 |
| 5 | 24 | 56 | 4 | 4 | 5 | 0.271 |
| 6 | 24 | 48 | 1 | 4 | 5 | 0.231 |
| 7 | 24 | 48 | 2 | 4 | 5 | 0.384 |
| 8 | 24 | 48 | 3 | 4 | 5 | 0.453 |
| 9 | 24 | 48 | 4 | 4 | 5 | 0.502 |
| 10 | 24 | 48 | 5 | 4 | 5 | 0.453 |
| 11 | 24 | 48 | 4 | 1 | 5 | 0.237 |
| 12 | 24 | 48 | 4 | 2 | 5 | 0.308 |
| 13 | 24 | 48 | 4 | 3 | 5 | 0.436 |
| 14 | 24 | 48 | 4 | 4 | 5 | 0.500 |
| 15 | 24 | 48 | 4 | 5 | 5 | 0.417 |
| 16 | 24 | 48 | 4 | 4 | 1 | 0.269 |
| 17 | 24 | 48 | 4 | 4 | 3 | 0.462 |
| 18 | 24 | 48 | 4 | 4 | 5 | 0.500 |
| 19 | 24 | 48 | 4 | 4 | 7 | 0.499 |
| 20 | 24 | 48 | 4 | 4 | 10 | 0.498 |
XO performs two different functions in the proposed sensor membrane: chromoionophore and ionophore. Optimizing its concentration in the membrane composition is, therefore, necessary. The effect of different amounts of XO on the membrane response is shown in Table 1. As can be seen, the absorbance rose with increasing XO concentrations up to 4.0 mg and fell with higher concentrations caused by membrane leakages. Therefore, the optimal value of 4.0 mg XO (5.0%) was chosen.
The addition of an anionic additive, such as NaTPB, promotes the ion–exchange equilibrium by allowing for the entire mass transfer of Se4+ ions into the membrane and by reducing response time.54 Thus, in the subsequent studies, the effect of NaTPB (4.0 mg) as an anionic additive was tested on the membrane properties with different plasticizers. As can be seen in Fig. 3, the presence of NaTPB caused an increase in the sensor responses and reagent leakages from all membranes. It is clear that utilizing a sensor with o-NPOE allowed for the greatest achievable response and the least amount of XO leaking from the membrane. The amount of NaTPB was investigated in the range of 1.0–5.0 mg. The results are given in Table 1. It is shown that the highest absorbance is recorded by using 4.0 mg of NaTPB. Lower concentrations saw a drop in associated absorbances due to less mass transfer of Se4+, while higher concentrations saw a decrease due to XO leakage. Therefore, 4.0 mg NaTPB (5.0%) was selected as the optimal amount in the membrane composition.
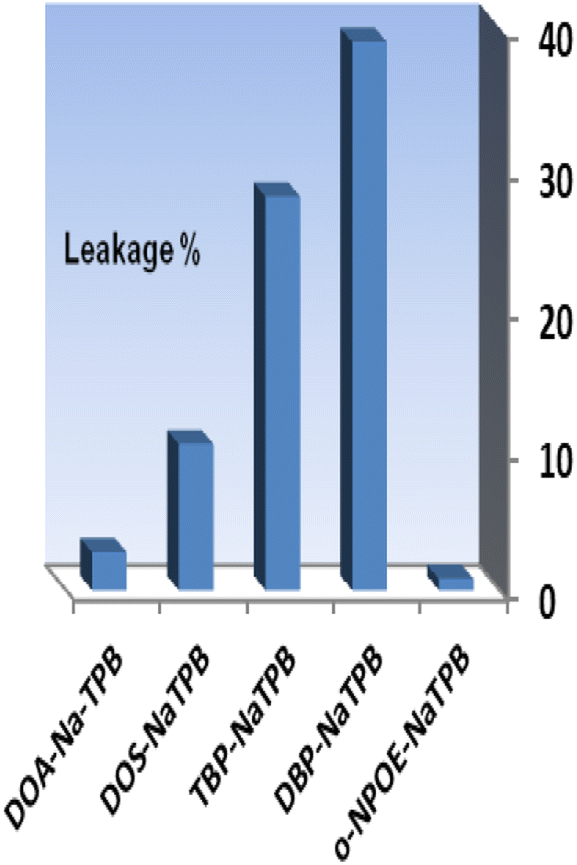 | ||
| Fig. 3 The effect of the anionic additive with different plasticizers on the membrane leakage% after 5.0 min. Conditions: [Se4+] = 100 ng mL−1 and T = 25 °C; the membrane layer contained 24 mg of PVC, 48 mg of each plasticizer, and 4.0 mg of XO. | ||
Effect of pH
The effect of pH variation on the sensor response was investigated in the range of 2.65–10.50. At pH 6.6, optode absorbance reaches its peak (Fig. 4). The low concentration of chromoionophore in the membrane, which serves as a potential binding site and prevents Se4+ ions from completely permeating, accounts for a drop in the metal uptake efficiency at pH < 6.6. At pH > 7.0, hydrolysis of Se4+ ions occurs, and hydroxide species probably caused the incomplete diffusion of Se4+ ions into the membrane. Therefore, a buffer of pH 6.6 was chosen in all experiments.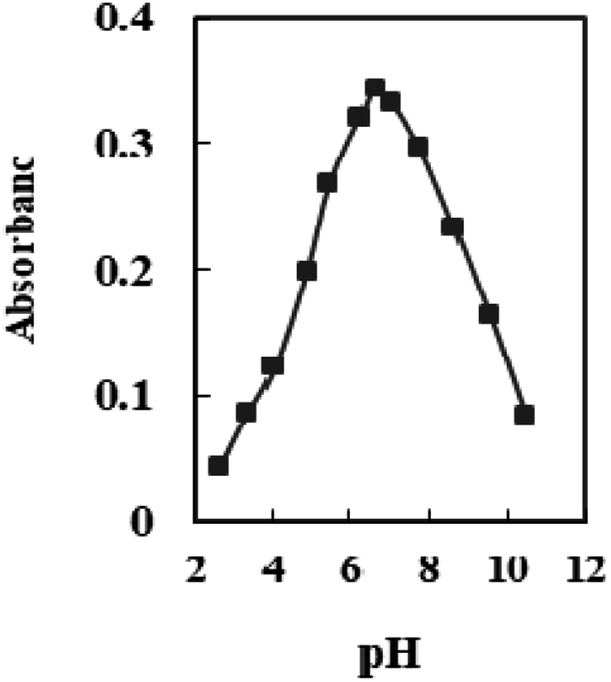 | ||
| Fig. 4 The absorbance of the sensor in solutions containing 100 ng mL−1 Se4+ at different pH values. | ||
Effect of response time
The definition of response time for sensors is the time it takes for metal ions to diffuse from the solution into the membrane (the slowest phase in the radical cation process). The effect of this parameter on the sensor response was investigated in the range of 1.0–10 min. As seen in Table 1, a time interval of at least 5.0 min is required for quantitative uptake at 25 ± 2.0 °C (Fig. 5). It was found that response time has a reverse relation toward initial Se4+ ion concentrations and the response time significantly increased from 3.0 min to 5.0 min by raising the Se4+ ion concentration from 50–150 ng mL−1. In general, the response time is lower in concentrated solutions than in dilute solutions (Table 2).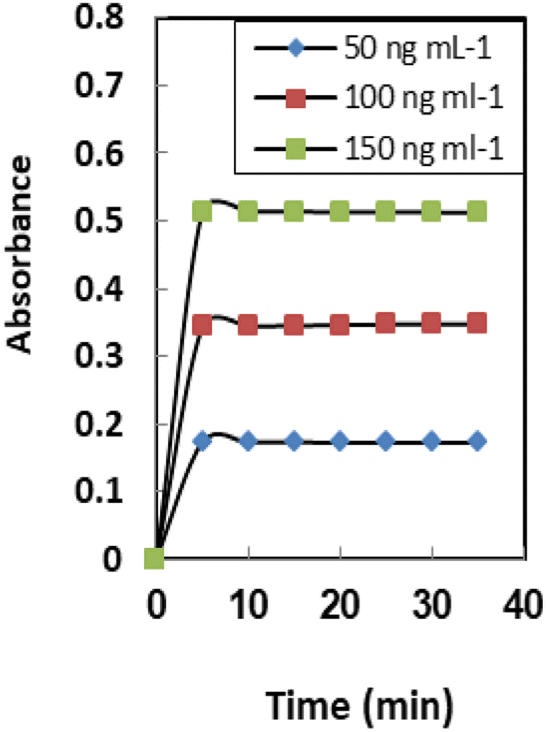 | ||
| Fig. 5 The response time of the sensor membranes for different concentrations of Se4+. Conditions: pH 6.6 and T = 25 °C; the membrane layer contained 24 mg of PVC, 48 mg o-NPOE, and 4.0 mg of both XO and NaTPB. | ||
| Ion | TR | Ion | TR |
|---|---|---|---|
| Na+, K+, Li+ | 12500 |
Al3+, Fe3+, CO32− | 4000 |
| Ca2+, Mg2+, acetate | 11000 |
Fe2+, NH2OH | 3500 |
| Ag+, Cu2+, NO3− | 1000 | Cd2+, Ni2+, SO42− | 3000 |
| Sr2+, Ba2+, S2O32− | 8500 | Hg2+, Co2+, oxalate | 2750 |
| Ge4+, Ti4+, citrate | 7000 | Cu2+, Sn2+, HCO32− | 2500 |
| Bi2+, Sn2+, Mn2+ | 6000 | Pb2+, Pd2+, SCN− | 2000 |
| Zr4+, Cr6+, succinate | 5500 | Au3+, La3+, Cr3+ | 1750 |
| Mo6+, W6+, Br− | 5000 | Se6+, Te6+, NH4+ | 1500 |
| Th4+, UO22+, B4O72− | 4500 | Te4+ | 120 |
Effect of stirring
Stirring the Se4+ solution has a large influence on the response of the formed sensor. About tenfold enhancement was achieved when the Se4+ solution was stirred compared to the non-stirred solution. This observation can be explained by the movement of Se4+ions towards the immobilized XO. The Se4+ ion diffusion through the membrane to the XO has been sped up by the stirring procedure, which has also sped up the Se4+ ion-XO reaction. Regarding the non-stirring procedure, the concentration gradient is the only factor that influences the diffusion of Se4+ ions across the membrane.55The average thickness of the prepared sensor membrane was established to be 31 ± 2 mm. The thickness of the membrane was assessed during the experiments by a digital microscope (Ray Vision Y 103) connected to a video camera (JVC TK-C 751EG). This thickness of the membrane achieved is appropriate for ion mobility for the reaction of Se4+ with XO. This is because the membrane is not too thick (>100 mm) and not too thin (<5 mm) and is reasonable to be applied as a transducer for sensor membrane depending on the co-extraction principle.56
Membrane properties
The properties of the sensor membrane were measured by recording absorbance changes at 585 nm from individual solutions of 50, 100, and 150 ng mL−1 Se4+. It is obvious from Fig. 5 that, in all three cases, the sensors reached 95% response after 5.0 min of stirring. The stability of membranes was tested for 3.0 h, and during this period, the mean difference of absorbances for the mentioned solutions was ±0.011. The membrane responses were stable for 15 days in the air.One of the key features of sensor membranes is their ability for regeneration, which enables repeated use of the tested sensor and much lower reagent use. Given that the membrane is not entirely reversible on its own, it can be regenerated using an appropriate stripping reagent. The time it takes for the sensor to return to its steady state baseline after being loaded with the regenerating solution is how the regeneration time is measured. As stripping reagents, a number of substances, including HCl, H2SO4, HNO3, KBr, KI, and KSCN, were studied. Complete regeneration with HNO3 and H2SO4 solutions was relatively long, and the sensors in contact with these solutions only recovered 80% of their initial absorbance after 10 min. Although all the other reagents were efficient for regenerating the sensor membranes, the shortest regeneration time was achieved with the HCl solution. The effect of the concentration of the stripping reagent was also illustrated. The best results were observed with 0.3 M HCl solution, although lower concentrations of down to 0.1 M can also be applied with increased regeneration times. The mechanism of regeneration is represented as SeCl4 is rapidly formed while using 0.3 M HCl rather than 0.1 M HCl due to the slow formation of SeCl2. The proposed mechanism for the interaction between the XO sensor and the Se4+ ions and its complexation and regeneration is depicted in Scheme 2.
 | ||
| Scheme 2 Schematic representation for the preparation, complexation, and regeneration of the formed optical sensor. | ||
The regeneration times of the sensors after contact with the 50, 100, 150 ng mL−1 Se4+ solutions were 40, 40, and 50 s, respectively, when 0.3 M HCl was used as the stripping reagent (Fig. 6). It should be mentioned that there is no requirement for the regeneration step if the measurement is brought from lower to higher Se4+ ion levels.
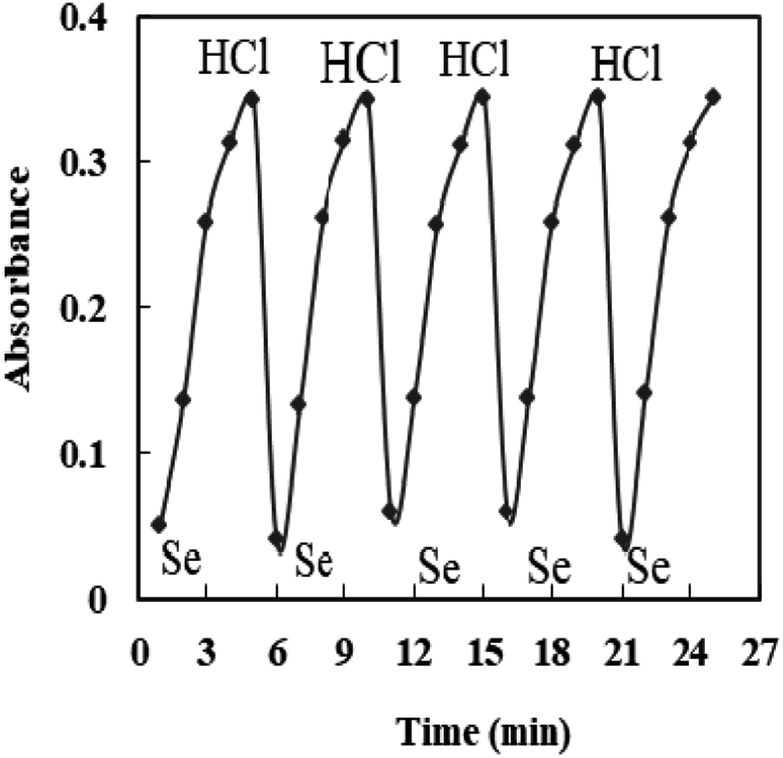 | ||
| Fig. 6 The reversibility of the sensor exposed to 100 ng mL−1 Se4+ and 0.3 M HCl. | ||
Interference
The relative sensitivity of the sensor membrane to the principal ion over concurrent ions in the solution is one of its most significant characteristics.57 The cations and anions that could interact with the optode's ionophore or species that might react with Se4+ ions and reduce the diffusion and migration efficiency are the interferences. The concentration that results in an error of more than ±5.0% in the absorbance, known as the tolerance limit for Se4+ ions, was chosen as the tolerance limit.58,59 In this regard, the absorbance of the sensor before and after the addition of a fixed amount of interference ions into 100 ng mL−1 of Se4+ ion solution was recorded, as shown in Fig. 7, and results are given in Table 3. The results confirmed that the sensor membrane exhibited excellent selectivity toward Se4+ ions with respect to the other coexisting interference ions at λmax = 585 nm except for Te4+ ions, which at concentrations of >120 fold mass ratios do not interfere. Taking into account that the maximum acceptable concentration of those species in water, food, and biological samples was lower than the tolerance limit found, the procedure could be useful for Se determination.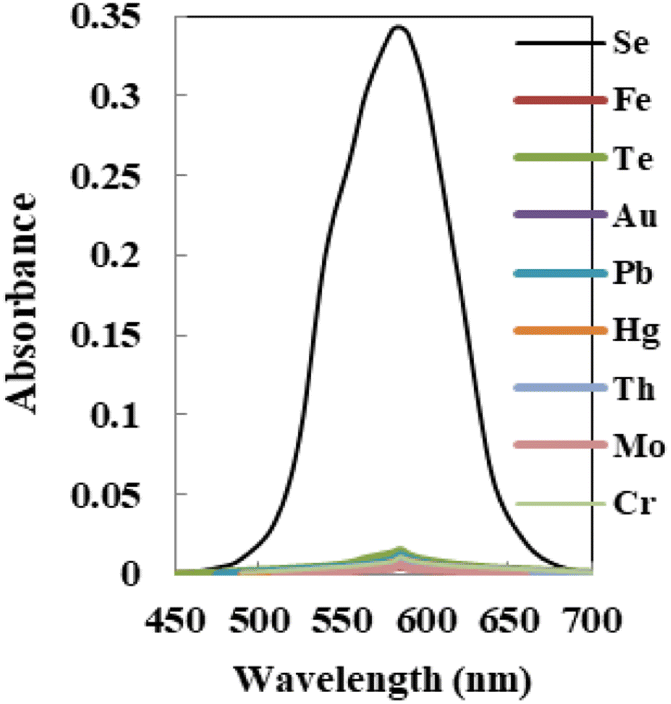 | ||
| Fig. 7 The absorption spectra of the sensor with each potential interfering metal tested on 100 ng mL−1 of Se4+ using the membrane. | ||
| Sample | [Se4+] and [Se6+]; ng mL−1 | Found by the sensor; ng mL−1 | Total Se, ETAAS | |
|---|---|---|---|---|
| [Se4+] | Total SE | |||
| 1 | [Se4+] = 50; [Se6+] = 0.0 | 49.6 ± 0.07 | 49.6 ± 0.51 | 50.7 ± 0.25 |
| 2 | [Se4+] = 50; [Se6+] = 25 | 49.5 ± 0.12 | 75.4 ± 0.68 | 74.4 ± 0.41 |
| 3 | [Se4+] = 50; [Se6+] = 50 | 50.7 ± 0.11 | 99.2 ± 0.74 | 101.2 ± 0.36 |
| 4 | [Se4+] = 70; [Se6+] = 70 | 70.5 ± 0.08 | 140.8 ± 0.91 | 138.6 ± 0.29 |
| 5 | [Se4+] = 80; [Se6+] = 40 | 80.5 ± 0.09 | 119.6 ± 0.77 | 121.2 ± 0.26 |
| 6 | [Se4+] = 80; [Se6+] = 80 | 79.4 ± 0.14 | 158.6 ± 0.83 | 161.8 ± 0.37 |
| 7 | [Se4+] = 40; [Se6+] = 80 | 39.5 ± 0.10 | 121.4 ± 0.69 | 118.9 ± 0.28 |
| 8 | [Se4+] = 60; [Se6+] = 100 | 60.8 ± 0.15 | 158.6 ± 0.58 | 162.4 ± 0.43 |
Speciation studies
The suggested optical sensor was examined as a promising candidate for the Se4+/Se6+ speciation due to its excellent selectivity for Se4+. In solutions including various amounts of Se4+ and Se6+ ions, the concentration of Se4+ ion can be assessed under optimal experimental circumstances with the offered optode. Then, the total concentration of selenium can also be detected in the same way after the reduction of Se6+ to Se4+. Finally, the Se6+ ion concentration is assessed by subtracting the amount of Se4+ from the total amount of selenium.To make a comparison, ETAAS also evaluated the overall amount of selenium. Table 3 shows the outcomes for the selenium speciation at different Se4+/Se6+ ratios. Table 3 shows that, within the experimental errors given, the total amounts of selenium measured by the suggested sensor membrane and those identified by the ETAAS technique are in good agreement. Furthermore, in all Se4+/Se6+ mixtures checked, the amount of both Se4+ and Se6+ ions present in the initial solutions can be assessed precisely.
Analytical characteristics
Table 4 lists the analytical features of the improved sensor membrane, including the regression equation, linear range (Fig. 8), detection and quantification limits, and selenium determination reproducibility. The limits of detection and quantification, defined as CDL = 3SB/m and CQL = 10SB/m (where CDL, CQL, SB, and m are the limit of detection, the limit of quantification, standard deviation of the blank, and slope of the calibration equation, respectively), were 3.0 and 10 ng mL−1. Additionally, 2.2% was the relative standard deviation (RSD%) for six replicate analyses of 100 ng mL−1 Se4+ in different membranes. This shows that the produced sensors' responses are repeatable and that the individual measurements did not significantly deviate from one another.| Parameters | Proposed sensor |
|---|---|
| a For six replicate determinations of 100 ng mL−1 Se4+. | |
| pH | 6.6 |
| λmax (nm) | 585 |
| Beer's range (ng mL−1) | 10–175 |
| Ringbom range (ng mL−1) | 25–160 |
| Molar absorptivity (L mol−1 cm−1) | 7.17 × 107 |
| Detection limit (ng mL−1) | 3.0 |
| Quantification limit (ng mL−1) | 10 |
| Reproducibility (RSD%)a | 2.2 |
|
|
| Regression equation | |
| Slope (ng mL−1) | 16.4 |
| Intercept | −0.04 |
| Correlation coefficient (r) | 0.9980 |
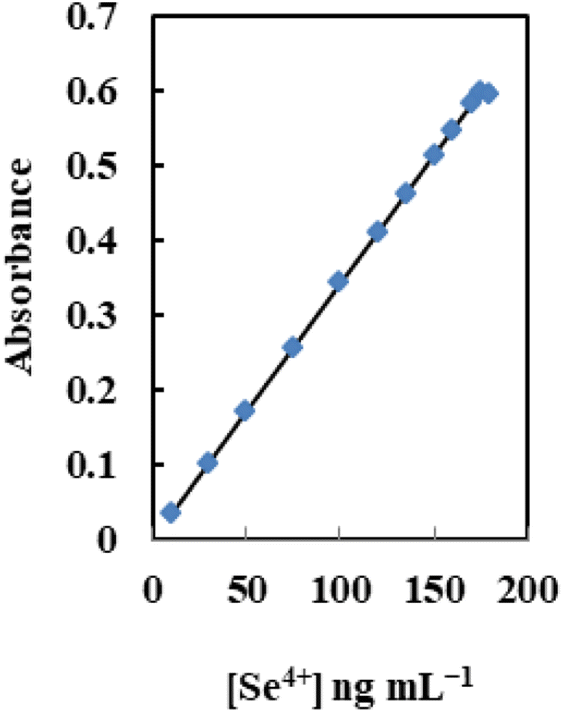 | ||
| Fig. 8 The dynamic range of the sensor at the optimum conditions for different [Se4+]. | ||
Table 5 presents a comparison between the proposed optode and the other reported techniques recommended in the literature19,20,22,27,60–69 for the determination of selenium. It is clear that the suggested approach has many benefits, including simplicity, low cost, rapidity, minimal toxicity, and relatively high selectivity. Comparing the given sensor to the reported sophisticated approaches, the linear range and detection limit are excellent; however, a review of the literature reveals that there have been no reports on a sensor membrane with chip reagents for assessing Se4+ ions in solutions. Furthermore, the offered method is simple and rapid compared with the reported procedures. Although the results obtained in the proposed sensor were primarily focused on Se4+ detection, the proposed sensor may be readily applied for the determination of selenium in real samples without interference from other metal ions.
| Mediaa | Technique | LOD (ng L−1) | RSD (%) | Ref. |
|---|---|---|---|---|
| a MSPME: Magnetic solid phase micro extraction, SPE: solid phase micro extraction, LOD: limit of detection RSD: relative standard deviation, PF: preconcentration factor, CHLLME: continuous homogeneous liquid–liquid microextraction, cxMWCNTs: oxidized multiwall carbon nanotubes. | ||||
| SPE (Chemically modified mesoporous silica) | ICP-OES | 2560 | 3.84 | 22 |
| SPE (CTAB modified alkyl silica) | ICP-OES | 100 | 3.6 | 60 |
| SPE (Modified multi-wall carbon nanotubes) | ICP-MS | 16 | 6.2 | 61 |
| SPE (GO-TiO2) | GFAAS | 40 | 9.4 | 62 |
| SPE (2,6-Diamino-4-phenil-1,3,5-triazine bonded silica gel) | GFAAS | 15 | <8 | 63 |
| SPE (Magnetic multi-walled carbon nanotubes) | HGAFS | 13 | 2.3 | 64 |
| Vortex assisted based liquid–liquid micro extraction (VA-LLME) method | ETAAS | 70 | 4.6 | 65 |
| oxMWCNTs | ETAAS | 30 | 4 | 20 |
| MSPME (Polystyrene-g-polyoleic acid-g-polyethylene glycol graft copolymer) | ETAAS | 6.60 | 3.2 | 66 |
| SPE (Polyvinyl chloride) | HG-ICP-OES | 30 | 5.7 | 67 |
| Au-coated W-coil atom trap | HG-AAS | 21 | 3.2 | 68 |
| SPE (Mg–FeCO3 layered double hydroxides loaded cellulose fibre) | HGAFS | 11 | 3.3 | 69 |
| CHLLME | HGAFS | 10 | 3.8 | 19 |
| On-line flow injection analysis | Spectrofluorometry | 270 | 2.0 | 27 |
| 2,3-Dichloro-6-(2,7-dihydroxy-naphthylazo)quinoxaline | Spectrophotometry | 60 | 1.2 | 70 |
| Optical sensor membrane | Colorimetrry | 3.0 | 2.2 | This work |
Validation and application
The proposed sensor was used to measure selenium in actual water samples in order to assess the analytical applicability of the proposed sensor. Six different types of water samples were therefore chosen, including tap, mineral, subterranean, rain, sea, and river water. The results of the proposed methodology are recorded in Table 6. As seen, the mean recoveries for the addition of different concentrations of selenium to water samples are in the range of 98.66–101.98%. Therefore, the proposed sensor membrane can be successfully applied for the determination of Se4+ in various water samples.| Samples | Added [ng mL−1] | Founda [ng mL−1] | Recovery (%) | t-testb | F-valueb | |
|---|---|---|---|---|---|---|
| Sensor | ETAAS | |||||
| a Mean ± SD.b Theoretical values for t and F values at 95% confidence level for five degrees of freedom are 2.57 and 5.05, respectively.c μg kg−1. | ||||||
| Tap water | — | 0.40 ± 0.06 | 0.45 ± 0.85 | |||
| 40 | 40.75 ± 0.08 | 41.00 ± 0.77 | 100.87 | 1.86 | ||
| 80 | 79.60 ± 0.13 | 81.00 ± 1.05 | 99.00 | 3.77 | ||
| Mineral water | — | 0.30 ± 0.07 | 0.27 ± 1.15 | |||
| 50 | 50.50 ± 0.11 | 49.80 ± 1.20 | 100.40 | 1.67 | ||
| 100 | 99.90 ± 0.18 | 101.0 ± 0.95 | 99.60 | 3.93 | ||
| Underground water | — | 0.65 ± 0.14 | 0.60 ± 0.85 | |||
| 60 | 60.35 ± 0.09 | 61.0 ± 1.05 | 99.50 | 2.11 | ||
| 120 | 121.0 ± 0.10 | 120.2 ± 1.30 | 100.29 | 4.05 | ||
| Rain water | — | 0.25 ± 0.15 | 0.25 ± 0.90 | |||
| 70 | 69.90 ± 0.07 | 70.70 ± 0.80 | 99.50 | 1.83 | ||
| 140 | 140.6 ± 0.12 | 139.5 ± 1.00 | 100.25 | 3.93 | ||
| River water | — | 0.55 ± 0.06 | 0.50 ± 1.10 | |||
| 80 | 80.05 ± 0.08 | 81.00 ± 1.25 | 99.94 | 1.47 | ||
| 160 | 161.3 ± 0.13 | 159.6 ± 0.75 | 100.47 | 3.73 | ||
| Ice tea | — | 1.00 ± 0.08 | 1.00 ± 1.15 | — | ||
| 55 | 55.25 ± 0.16 | 57.2 ± 0.95 | 98.66 | 4.04 | ||
| 110 | 113.2 ± 0.20 | 108.5 ± 0.85 | 101.98 | 1.97 | ||
| Mixed fruit juice | — | 75.5 ± 0.25 | 75.0 ± 1.20 | |||
| 50 | 126.5 ± 0.21 | 124.2 ± 1.45 | 100.80 | 3.78 | ||
| 100 | 173.7 ± 0.13 | 177.0 ± 0.95 | 98.97 | 2.03 | ||
| Fresh cabbagec | — | 1.12 ± 0.07 | 1.15 ± 1.05 | |||
| 75 | 77.00 ± 0.11 | 74.65 ± 1.30 | 101.16 | 1.88 | ||
| 150 | 149.7 ± 0.17 | 153.0 ± 1.55 | 99.06 | 3.75 | ||
| Soilc | — | 0.25 ± 0.09 | 0.25 ± 0.75 | |||
| 65 | 65.75 ± 0.14 | 64.30 ± 1.00 | 100.77 | 2.14 | ||
| 130 | 128.8 ± 0.19 | 131.8 ± 1.35 | 98.88 | 4.16 | ||
| Cosmetic preparation (5.0 g lipstick) | — | 253.5 ± 0.04 | 254.0 ± 1.55 | |||
| 100 | 365.7 ± 0.11 | 335.8 ± 2.05 | 103.45 | 2.04 | ||
| 175 | 415.4 ± 0.13 | 435.5 ± 1.85 | 96.90 | 3.89 | ||
| — | 225.8 ± 0.15 | 220.8 ± 1.65 | ||||
| Human hair (0.5 g) | 65 | 280.5 ± 0.18 | 300.5 ± 0.19 | 98.15 | 1.83 | |
| 130 | 360.7 ± 0.20 | 330.4 ± 0.19 | 101.38 | 3.65 | ||
The developed method was applied to the quantitative determination of traces of selenium in real matrices such as soil and human hair samples, viz., cosmetic preparations. The results of an analysis of the above samples (Table 6) compare favorably with those from the ETAAS method.20
The performance of the proposed sensor was assessed using the t-value (for accuracy) and F-test (for precision) compared with the ETAAS method.20 The mean values were obtained by a Student's t-test and F-test at 95% confidence limits for five degrees of freedom.71 The outcomes demonstrated that the calculated values (Table 6) did not surpass the theoretical values. The advantage of the suggested method over prior ones is that it has a larger range of determination, greater precision, greater stability, and requires less time.
Various microwave-digested food samples, such as canned tomato, potato, coffee, black tea, green tea, honey, egg, garlic, salami, onion, cultured mushroom, canned fish, cow meat, boiled wheat, cheese, and chicken meat, were also effectively tested using the current method. The results are given in Table 7. These samples contained different matrix media. Selenium levels were determined at μg kg−1 levels in analyzed food samples. The selenium ions found in the analyzed food, beverage, and water samples had levels that were suitable for human consumption. Some food and drink samples can significantly increase selenium consumption.
| Samples | Selenium (μg kg−1)a ± SD | t-testb | F-valuec | |
|---|---|---|---|---|
| Sensor | ETAAS | |||
| a Mean ± SD.b Theoretical values for t-test at 95% confidence level for five degrees of freedom is 2.57.c Theoretical values for F-value at 95% confidence level for five degrees of freedom is 5.05. | ||||
| Canned tomato | 187 ± 0.11 | 185 ± 1.07 | 1.27 | 2.63 |
| Potato | 99 ± 0.09 | 100 ± 0.84 | 1.53 | 2.96 |
| Coffee | 188 ± 0.16 | 185 ± 0.88 | 1.07 | 2.35 |
| Black tea | 284 ± 0.19 | 280 ± 1.32 | 1.33 | 2.75 |
| Green tea | 143 ± 0.11 | 145 ± 1.03 | 1.48 | 2.62 |
| Honey | 197 ± 0.18 | 200 ± 0.96 | 1.22 | 2.48 |
| Garlic | 344 ± 0.20 | 345 ± 1.24 | 1.36 | 2.84 |
| Egg | 167 ± 0.14 | 165 ± 1.13 | 1.74 | 3.17 |
| Salami | 160 ± 0.10 | 160 ± 0.79 | 1.11 | 2.39 |
| Onion | 273 ± 0.24 | 270 ± 0.92 | 1.29 | 2.67 |
| Cultivated mushroom | 215 ± 0.12 | 217 ± 0.69 | 1.45 | 2.72 |
| Canned fish | 430 ± 0.35 | 427 ± 0.85 | 1.67 | 2.98 |
| Cows meat | 226 ± 0.17 | 230 ± 1.17 | 1.41 | 2.56 |
| Boiled wheat | 135 ± 0.12 | 140 ± 0.95 | 1.63 | 3.04 |
| Cheese | 263 ± 0.22 | 260 ± 1.28 | 1.16 | 2.53 |
| Chicken meat | 158 ± 0.13 | 155 ± 1.05 | 1.44 | 2.69 |
Conclusions
The suggested sensor membrane, which is based on an optical sensor membrane linked with spectrophotometry, is a low-cost, precise, sensitive, and highly selective method for determining selenium. The suggested method offers a broad dynamic range, trustworthy reproducibility, and good limits of detection and quantification, in addition to being quick and easy. In addition to being quick and simple to use, the proposed sensor also offers a detection limit comparable to previous techniques, according to a comparison of the proposed sensor with previously reported techniques for selenium determination (Table 5). It is crucial to note that this method is being used for the first time and has not previously been mentioned in the literature as being used for selenium speciation and determination. Finally, the selenium in actual environmental and biological samples may be monitored with success using the developed optical sensor membrane.Author contributions
Abeer Hassan and Reem Alshehri: conceptualization, data curation, investigation, methodology, visualization, validation, writing – original draft, writing – review & editing. Salah El-Bahy and Alaa Amin: conceptualization, methodology, data curation, investigation, supervision, validation, writing – original draft, writing – review & editing. Mai Aish: conceptualization, investigation, methodology, validation, writing - original draft, writing – review & editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The researchers acknowledge the Deanship of Scientific research, Taif University, for funding this work. The authors would like to acknowledge financial support from the Department of Chemistry, Faculty of Science, Taibah, Benha, and Port Said Universities for providing instrumental facilities.References
- P. T. Bhattacharya, S. R. Misra and M. Hussain, Scientifica, 2016, 5464373 Search PubMed.
- H. Asiabi, Y. Yamini, S. Seidi, M. Shamsayei, M. Safari and F. Rezaei, Anal. Chim. Acta, 2016, 922, 37–47 CrossRef CAS PubMed.
- K. Kocot, R. Leardi, B. Walczak and R. Sitko, Talanta, 2015, 134, 360–365 CrossRef CAS PubMed.
- L. C. Herrero, G. J. Barciela, M. S. García and C. R. M. Peña, Anal. Chim. Acta, 2013, 804, 37–49 CrossRef PubMed.
- M. Tuzen and O. Z. Pekiner, Food Chem., 2015, 188, 619–624 CrossRef CAS PubMed.
- WHO, Selenium in drinking-water. Background document for preparation of WHO Guidelines for drinking-water quality, World Health Organization (WHO/SDE/WSH/03.04/13), Geneva, 2003 Search PubMed.
- WHO, Guidelines for Drinking-Water Quality, World Health Organization, 2011 Search PubMed.
- EU, Directive of the European Parliament and of the Council on the Quality of Water Intended for Human Consumption, European Commission, 2017 Search PubMed.
- EPA, National Primary Drinking Water Regulation Table, U.S. Environmental Protection Agency, U.S.A., 2009 Search PubMed.
- M. Bujdos, J. Kubove and V. Stresko, Anal. Chim. Acta, 2000, 408, 103–109 CrossRef CAS.
- H. D. Mistry, P. F. Broughton, L. Redman and L. Poston, Am. J. Obstet. Gynecol., 2012, 206, 21–30 CrossRef CAS PubMed.
- S. Otles, Methods of Analysis of Food Components and Additives, CRC Press, 2005 Search PubMed.
- M. B. Melwanki and J. Seetharamappa, Turk. J. Chem., 2000, 24, 287–291 CAS.
- B. V. Samlafo, P. O. Yeboah and Y. J. Serfor-Armah, Appl. Sci. Technol., 2011, 16, 1–2 Search PubMed.
- N. Altunay, A. Elik and K. Katin, J. Food Compos. Anal., 2021, 99, 103871 CrossRef CAS.
- L. Chirita, E. Covaci, A. Mot, M. Ponta, A. Gandeac and T. Frentiu, J. Anal. At. Spectrom., 2021, 36, 267–272 RSC.
- B. G. Nieto, J. Gismera, T. Sevilla and J. R. Procopio, Anal. Chim. Acta, 2022, 1202, 339637 CrossRef PubMed.
- M. Llaver, A. L. Chapana and R. G. Wuilloud, Talanta, 2021, 222, 121460 CrossRef CAS PubMed.
- A. Shishov, M. Wieczorek, P. Kościelniak, D. Dudek-Adamska, A. Telk, L. Moskvin and A. Bulatov, Talanta, 2018, 181, 359–365 CrossRef CAS PubMed.
- A. M. Martínez, S. Vázquez, R. Lara, L. D. Martínez and P. Pacheco, Spectrochim. Acta, Part B, 2018, 140, 22–28 CrossRef.
- M. Murillo, N. Carrio, M. Quintana, G. Sanabria, R. Manuel, L. Duarte and F. Ablan, J. Trace Elem. Med. Biol., 2005, 19, 23–27 CrossRef CAS PubMed.
- H. Sereshti, Y. E. Heravi, S. Samadi, A. Badiei and N. H. Roodbari, Food Anal. Methods, 2013, 6, 548–558 CrossRef.
- D. Mazej, I. Falnoga, M. Veber and V. Stibilj, Talanta, 2006, 68, 558–568 CrossRef CAS PubMed.
- J. Pinho, J. Canario, R. Cesario and C. Vale, Anal. Chim. Acta, 2005, 551, 207–212 CrossRef CAS.
- Y. Yuan, Y. Shao, F. Yang, H. Yu, Y. Zhang and M. Wen, Spectrochim. Acta, Part B, 2023, 203, 106664 CrossRef CAS.
- Y. Chen, N. Guo, L. Zhang, K. Hu, J. Yang and K. Shi, J. Radioanal. Nucl. Chem., 2023, 332, 1071–1081 CrossRef CAS.
- D. G. Santarossa and L. P. Fernández, Talanta, 2017, 172, 31–36 CrossRef CAS PubMed.
- M. Acosta, L. P. Fernandez and M. C. Talio, J. Fluoresc., 2023, 17, 1–10 Search PubMed.
- P. Devi, R. Jain, A. Thakur, M. Kumar, N. K. Labhsetwar, M. Nayak and P. Kumar, Trends Anal. Chem., 2017, 95, 69–85 CrossRef CAS.
- R. Manish, K. N. Ramachandran and V. K. Gupta, Talanta, 1994, 41, 1623–1626 CrossRef CAS PubMed.
- D. Agarwal, G. Sunitha and V. K. Gupta, J. Indian Chem. Soc., 1998, 73, 151–154 Search PubMed.
- A. Safavi and A. Afkhami, Anal. Lett., 1995, 28, 1095–1101 CrossRef CAS.
- M. N. Pathare and A. D. Sawant, Anal. Lett., 1995, 28, 317–334 CrossRef CAS.
- K. Pyrzynska, Anal. Sci., 1997, 13, 629–632 CrossRef CAS.
- H. D. Revanasiddappa and T. N. Kiran Kumar, Anal. Sci., 2001, 17, 1309–1312 CrossRef CAS PubMed.
- M. R. Ganjali, M. Hosseini, M. Hariri, F. Faridbod and P. Norouzi, Sens. Actuators, B, 2009, 142, 90–96 CrossRef CAS.
- W. R. Seitz, in Optical Ion Sensing Fiber Optic Chemical Sensors Biosensors II, ed. O. S. Wolfbeis, CRC Press, Bocaraton, FL, 1991, pp. 1–19 Search PubMed.
- M. Shamsipur, M. Sadeghi, K. Alizadeh, H. Sharghi and R. Khalifeh, Anal. Chim. Acta, 2008, 630, 57–66 CrossRef CAS PubMed.
- G. Absalan, M. Asadi, S. Kamran, S. Torabi and L. Sheikhian, Sens. Actuators, B, 2010, 147, 31–36 CrossRef CAS.
- Y. Kalyan, A. K. Pandey, P. R. Bhagat, R. Acharya, V. Natarajan, G. R. K. Naidu and A. V. R. Reddy, J. Hazard. Mater., 2009, 166, 377–382 CrossRef CAS PubMed.
- M. R. Ganjali, R. Zare-Dorabei and P. Norouzi, Sens. Actuators, B, 2009, 143, 233–238 CrossRef.
- Y. Kalyan, A. K. Pandey, G. R. K. Naidu and A. V. R. Reddy, Spectrochim. Acta, Part A, 2009, 74, 1235–1241 CrossRef CAS PubMed.
- S. Rastegarzadeh, N. Pourreza and I. Saeedi, J. Hazard. Mater., 2010, 173, 110–114 CrossRef CAS PubMed.
- H. H. El-Feky, A. S. Amin and E. M. I. Moustafa, RSC Adv., 2022, 12, 18431–18440 RSC.
- E. M. I. Moustafa, A. S. Amin and M. A. El-Attar, Anal. Biochem., 2022, 654, 114835 CrossRef CAS PubMed.
- H. H. El-Feky, S. El-Bahy and A. S. Amin, Anal. Biochem., 2022, 651, 114720 CrossRef CAS PubMed.
- H. H. El-Feky, A. M. Askar and A. S. Amin, RSC Adv., 2021, 11, 35300–35310 RSC.
- A. S. Amin, S. El-Bahy and H. H. El-Feky, Anal. Biochem., 2022, 643, 114579 CrossRef CAS PubMed.
- L. dlC. Coo and I. S. Martinez, Talanta, 2004, 64, 1317–1322 CrossRef CAS PubMed.
- A. S. Amin and M. M. Zareh, Anal. Lett., 1996, 29, 2177–2189 CrossRef CAS.
- H. T. S. Britton, Hydrogen Ions, Chapman and Hall, London, UK, 4th edn, 1952, p. 1168 Search PubMed.
- A. Naeemullah, M. Tuzen and T. G. Kazi, J. Ind. Eng. Chem., 2018, 57, 188–192 CrossRef.
- A. Safavi and M. Bagheri, Sens. Actuators, B, 2005, 107, 53–58 CrossRef CAS.
- M. Shamsipur, T. Poursaberi, A. R. Karami, M. Hosseini, A. Momeni, N. Alizadeh, M. Yousefi and M. R. Ganjali, Anal. Chim. Acta, 2004, 501, 55–60 CrossRef CAS.
- M. Ahmad and R. Narayanaswamy, Sens. Actuators, B, 2002, 81, 259–266 CrossRef CAS.
- C. C. Li and M. S. Kuo, Anal. Sci., 2002, 18, 607–609 CrossRef CAS PubMed.
- M. R. Baezzat and M. Karimi, Int. J. ChemTech Res., 2013, 5, 2503–2507 CAS.
- H. Tavallali and M. Yazdandoust, Eurasian J. Anal. Chem., 2008, 3, 284–287 Search PubMed.
- C. Sanchez-Pedreno, J. A. Ortuno, M. I. Albero, M. S. Garcia and M. V. Valero, Anal. Chim. Acta, 2000, 414, 195–203 CrossRef CAS.
- C. Xiong, M. He and B. Hu, Talanta, 2008, 76, 772–779 CrossRef CAS PubMed.
- H. Peng, N. Zhang, M. He, B. Chen and B. Hu, Talanta, 2015, 131, 266–272 CrossRef CAS PubMed.
- Y. Zhang, B. Chen, S. Wu, M. He and B. Hu, Talanta, 2016, 154, 474–480 CrossRef CAS PubMed.
- D. Mendil, Z. Demirci, O. D. Uluozlu, M. Tuzen and M. Soylak, Food Chem., 2017, 221, 1394–1399 CrossRef CAS PubMed.
- Y. Wang, J. Xie, Y. Wu, X. Hu, C. Yang and Q. Xu, Talanta, 2013, 112, 123–128 CrossRef CAS PubMed.
- J. Ali, M. Tuzen, X. Feng and T. G. Kazi, Food Chem., 2021, 344, 128706 CrossRef CAS PubMed.
- A. N. Acikkapia, M. Tuzena and H. Baki, Food Chem., 2019, 284, 1–7 CrossRef PubMed.
- L. A. Escudero, P. H. Pacheco, J. A. Gasquez and J. A. Salonia, Food Chem., 2015, 169, 73–79 CrossRef CAS PubMed.
- M. Atasoy and I. Kula, Food Chem., 2022, 369, 130938 CrossRef CAS PubMed.
- M. L. Chen and M. I. An, Talanta, 2012, 95, 31–35 CrossRef CAS PubMed.
- A. S. Amin, J. AOAC Int., 2014, 97, 586–592 CrossRef CAS PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson/Prentice Hall, Harlow, UK, 5th edn, 2005, pp. 58–67 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |