
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic-responsive solid acid catalysts for esterification†
Dan Xue *ab,
Yun Jianga and
Fangxia Zhenga
*ab,
Yun Jianga and
Fangxia Zhenga
aSchool of Chemical Engineering, University of Science and Technology Liaoning, Anshan 114051, China
bKey Lab of Eco-restoration of Regional Contaminated Environment (Shenyang University), Ministry of Education, Shenyang 110036, China. E-mail: danxue@ustl.edu.cn
First published on 15th September 2023
Abstract
Two kinds of magnetic-responsive solid acid catalysts were designed and prepared via an in situ polymerization of poly(ionic liquid)s (PILs) on the surface of Fe3O4@SiO2 NPs for the catalyzed esterification of palmitic acid and methanol. They were characterized using XRD, TGA, VSM, NMR spectroscopy, FTIR spectroscopy, XPS, SEM, and GC techniques. The results confirmed the preparation of solid acid catalysts. Meanwhile, they possessed excellent catalytic activity and recyclability. The effect of the reaction conditions on the esterification was investigated through single-factor analyses, and the proposed catalytic mechanism of the prepared solid acid catalysts in the esterification are also discussed. Under the optimal reaction conditions (10 wt% catalyst, 6 h, 70 °C, and molar ratio (MR) of methanol to palmitic acid of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the conversion rate of palmitic acid could reach 94% and 79% with Fe3O4@SiO2-poly(1-vinyl-3-ethylimidazolium phosphotungstate) (Fe3O4@SiO2–P([VLIM]PW)) and Fe3O4@SiO2-poly(1-vinylimidazole-3-propyl sulfonate) (Fe3O4@SiO2–P([VLIM]SO3)) NPs serving as catalysts, respectively. Furthermore, the Fe3O4@SiO2–P([VLIM]PW) NPs could still maintain a high catalytic activity even after being reused 5 times without significant deactivation.
:1), the conversion rate of palmitic acid could reach 94% and 79% with Fe3O4@SiO2-poly(1-vinyl-3-ethylimidazolium phosphotungstate) (Fe3O4@SiO2–P([VLIM]PW)) and Fe3O4@SiO2-poly(1-vinylimidazole-3-propyl sulfonate) (Fe3O4@SiO2–P([VLIM]SO3)) NPs serving as catalysts, respectively. Furthermore, the Fe3O4@SiO2–P([VLIM]PW) NPs could still maintain a high catalytic activity even after being reused 5 times without significant deactivation.
1. Introduction
Esterification is one of the most important reactions in the pharmaceutical and chemical industries. However, the esterification of organic acids to esters is much slower than catalyzed esterification kinetically.1 Thus, it is critical to develop an appropriate catalyst for esterification. The catalyst could be an acid or base.2–4 Normally, a base catalyst is more effective than an acid catalyst in the esterification reaction due to its excellent catalytic activities.5 However, a base catalyst could directly react with an organic acid and the corresponding salt has to be removed from the product for the purification of the product.6 Meanwhile, homogeneous acid catalysts, such as sulfuric acid, hydrochloride acid, and nitric acid, suffer from problems related to equipment corrosion, environmental pollution, and non-recyclability.7–9 Based on the above analysis, solid acid catalysts could avoid the above problems. Meanwhile, the purification processes for the product could be greatly reduced.Biodiesel is a mixture of different fatty acid esters, which could be prepared by the esterification of fatty acids and short chain alcohols. Compared with traditional fossil fuels, biodiesel not only is renewable and biodegradable but also possesses similar functions in terms of performance and engine durability.10–12 Furthermore, it emits less CO and particles in combustion. Therefore, the employment of renewable biodiesel is a feasible solution to help alleviate the current energy and environmental crisis.13–15 In the recent years, researchers have paid more attention to developing solid acid catalysts for the preparation of biodiesel. Several kinds of solid acid catalysts, such as polymers,16,17 ionic liquids (ILs),18–20 poly (ionic liquids) (PILs),21–23 and carbon-derived solid acids and their composites,24–26 have been prepared and applied in various fields.27 Among the above-mentioned materials, acidic PILs are the most prominent catalysts owing to their remarkable properties of designability, environmental friendliness, strong acidity, high stability and so on.28–30 However, they are easily soluble in polar solvents, which brings difficulties in the recovery of the catalyst and purification of the product. Thus, it would be necessary to immobilize the acid PILs on the surface of recyclable solid materials.
A number of studies have reported that heteropoly acids with the Keggin structure, sulfonic acid, and their salts, including phosphotungstic acid (PTA), vanadium-substituted phosphomolybdic acid, phosphomolybdic acid, heterogeneous cesium hydrogen phosphotungstate, and sulfonated imidazolium ionic liquid, could serve as effective catalysts owing to their strong acid properties.9,31–34 Thus, the combination of organic cation and anion groups with strong acid properties is interesting as they would form a kind of novel ionic liquid (IL). These could act as effective solid acid catalysts. The immobilization of ILs through chemical bonds should be considered to avoid the leaching or deactivation. As is well known, magnetic materials can be easily separated and recycled by an external magnet. Some magnetic-responsive solid acid catalysts have been successfully prepared and applied in esterification.35–38 However, there are scant reports on the immobilization of PILs on the surface of magnetic-responsive materials through chemical bonding via a simple method. Therefore, in this work, we designed and prepared two kinds of recyclable and efficient solid acid catalysts via a series of simple reactions, such as solvothermal, sol–gel, polymerization, and ion exchange: phosphotungstic (PW)- and sulfonic (SO3)-based PILs-modified Fe3O4@SiO2 NPs, respectively (Fig. 1). These solid acid catalysts were applied in the preparation of biodiesel, which was achieved by the esterification of palmitic acid (model compound) and methanol to evaluate their catalytic activity. The prepared solid acid catalysts were characterized by XRD, VSM, TGA, NMR, XPS, FTIR, SEM, and GC. Meanwhile, the effects of the reaction conditions on the esterification reaction and conversion rate of palmitic acid were investigated through single-factor analysis. Furthermore, the proposed catalytic mechanism of the prepared solid acid in the esterification process and the recyclability of the magnetic-responsive solid acid catalysts were investigated and are discussed as well.
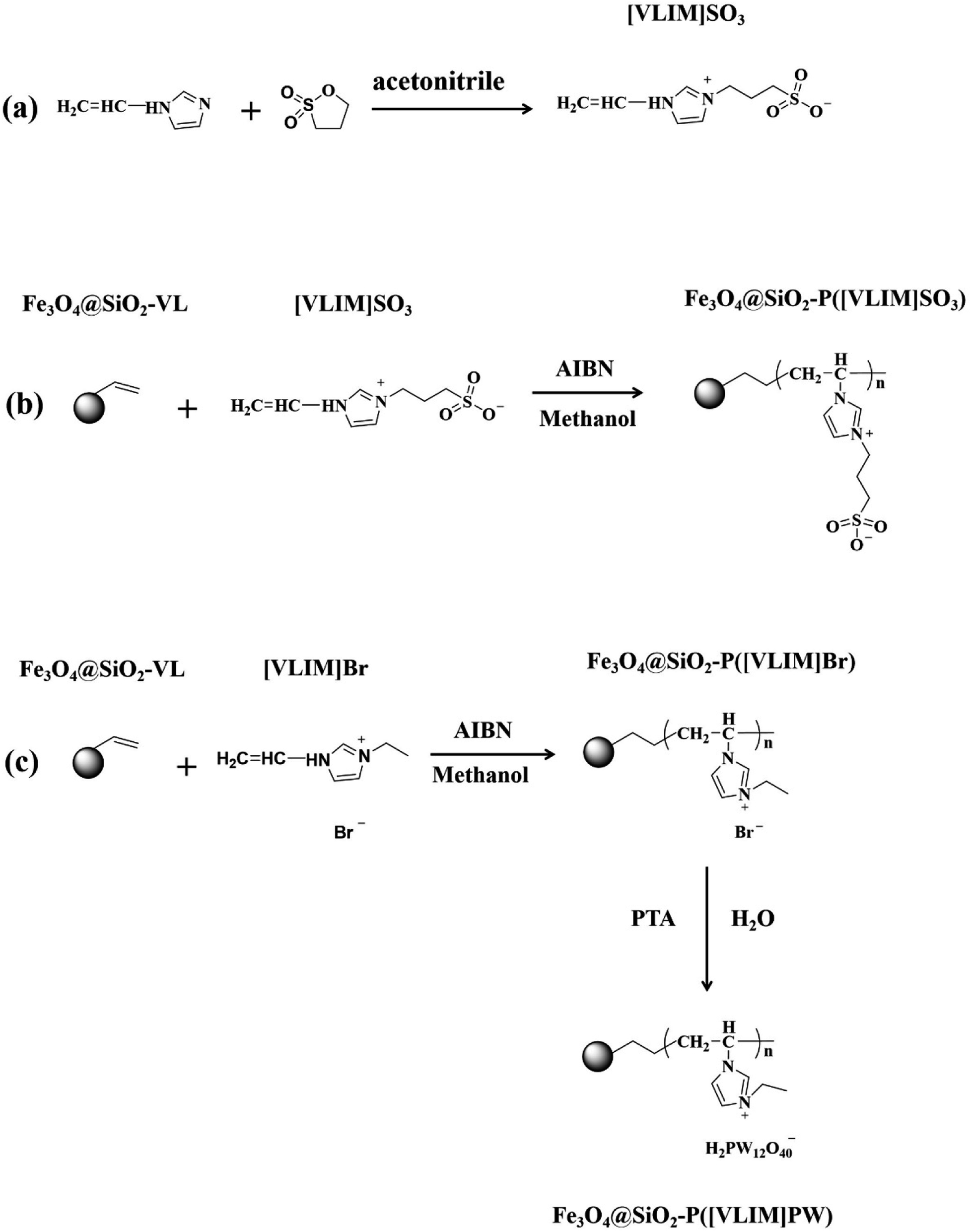 | ||
| Fig. 1 Schematic of the preparation of the magnetic-responsive solid acid catalysts. | ||
2. Experimental
2.1. Materials
Ethylene glycol, iron (III) chloride hexahydrate, sodium acetate trihydrate, tetraethyl orthosilicate (TEOS), ammonium hydroxide (NH3·H2O, 25 wt%), and potassium hydroxide (KOH) were purchased from Sinopharm Chemical Reagent Co., Ltd. 2,2-Azobis-(2-methylpropionitrile) (AIBN), vinyltrimethoxysilane (VL-TMC), 1-vinyl-3-ethylimidazolium bromide ([VLIM]Br), PTA, 1-vinylimidazole (VLIM), 1,3-propanesultone, ethanol, acetonitrile, methanol, and palmitic acid were purchased from Adamas Reagent Co., Ltd. All the reagents were analytical grade and used as received.2.2. Preparation of Fe3O4@SiO2-VL NPs
First, 0.1 g of Fe3O4@SiO2, which was first prepared as per our previous work,39,40 was first dispersed in a solution of 60 mL of ethanol under ultrasonication. Then, 200 μL of VL-TMC was added and stirred for 24 h at room temperature. Afterward, the mixture was heated to 80 °C for 5 h under continuous stirring. After the modification, the obtained black precipitate was collected with a magnet and washed with deionized water and ethanol three times to remove residual reactants. Finally, the product, named as Fe3O4@SiO2-VL NPs, was obtained after drying at 50 °C in a vacuum oven for 24 h.2.3. Preparation of [VLIM]SO3
First, 0.94 g of 1-vinylimidazole and 1.12 g of 1,3-propanesultone were slowly added into 60 mL of acetonitrile, placed in an ice bath, and continuously stirred for 0.5 h. After that, the mixture was heated to 60 °C and stirred for 24 h under a nitrogen atmosphere. The product was washed with acetonitrile to remove any unreacted reagents and dried in a vacuum oven at 50 °C for 24 h. The achieved white solid powder was named as [VLIM]SO3 (Fig. 1a).2.4. Preparation of Fe3O4@SiO2–P([VLIM]SO3) NPs
First, 0.1 g of Fe3O4@SiO2-VL NPs, 1 g of [VLIM]SO3, and 0.01 g of AIBN were dispersed in 60 mL of methanol under ultrasonication for 3 min to form a uniform suspension at room temperature. Then, a nitrogen atmosphere was flowed into the mixture for 0.5 h to remove dissolved oxygen. Afterward, the mixture was heated to 80 °C and continuously stirred for 24 h under a nitrogen atmosphere. The obtained black precipitate was collected with a magnet and washed with methanol to remove any residual reactants. Then it was dried in a vacuum oven at 50 °C for 24 h. The achieved black solid powder was named as Fe3O4@SiO2–P([VLIM]SO3) NPs (Fig. 1b).2.5. Preparation of Fe3O4@SiO2–P([VLIM]PW) NPs
Fe3O4@SiO2–P([VLIM]PW) NPs were prepared in a two-step method. The first step was the preparation of Fe3O4@SiO2–P([VLIM]Br) NPs, which followed a similar method to that used for Fe3O4@SiO2–P([VLIM]SO3), except that the monomer used for the polymerization was [VLIM]Br. In the second step, 0.1 g of Fe3O4@SiO2–P([VLIM]Br) NPs and 1.0 g of PTA were dispersed in 30 mL of deionized water, and then stirred at 80 °C for 24 h. The obtained black precipitate was collected with a magnet and washed with deionized water and ethanol several times to remove any residual PTA, and then dried at 50 °C in a vacuum oven for 24 h. Thus, Fe3O4@SiO2–P([VLIM]PW) NPs were obtained (Fig. 1c).2.6. Preparation of biodiesel
Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs were, respectively, used as the catalysts in the preparation of biodiesel. A stoichiometric amount of catalyst, methanol, and palmitic acid were successively added in to a round-bottom flask equipped with a reflux condenser and mechanical stirrer. The mixture was stirred at 70 °C for a certain time to obtain the biodiesel.For the recycling studies, the catalyst was washed and recovered with hot water after it was collected by a magnet. Afterward, the catalyst was further dried in a vacuum oven at 50 °C for 24 h for it to be reused. The recycled catalyst was reused under the same conditions.
As the amount of methanol was excessive and sufficient to drive the reaction forward toward the conversion of palmitic acid to palmitic methyl ester in the esterification, thus the conversion rate of palmitic acid was used to evaluate the catalytic activity, and it was calculated using the expression below.

3. Results and discussion
3.1. Characterization of [VLIM]SO3
The molecular structure of [VLIM]SO3 was characterized by 1H NMR and 13C NMR spectra, and their data are listed as follows. These spectra proved that [VLIM]SO3 was successfully synthesized.1H NMR (500 MHz, CD3OD) δ: 2.39 (m, 2H), 2.88 (t, J = 10 Hz, 2H), 4.51 (t, J = 10 Hz, 2H), 5.46 (dd, J = 10, 5 Hz, 1H), 5.96 (dd, J = 20, 10 Hz, 1H), 7.30 (dd, J = 20, 10, 1H), 7.84 (s, 1H), 8.03 (s, 1H), 9.36 (s, 1H) (Fig. S1†).
13C NMR (500 MHz, CD3OD) δ: 27.03, 109.94, 120.84, 124.60, 129.99, 136.94 (Fig. S2†).
3.2. Characterization of the solid acid catalysts
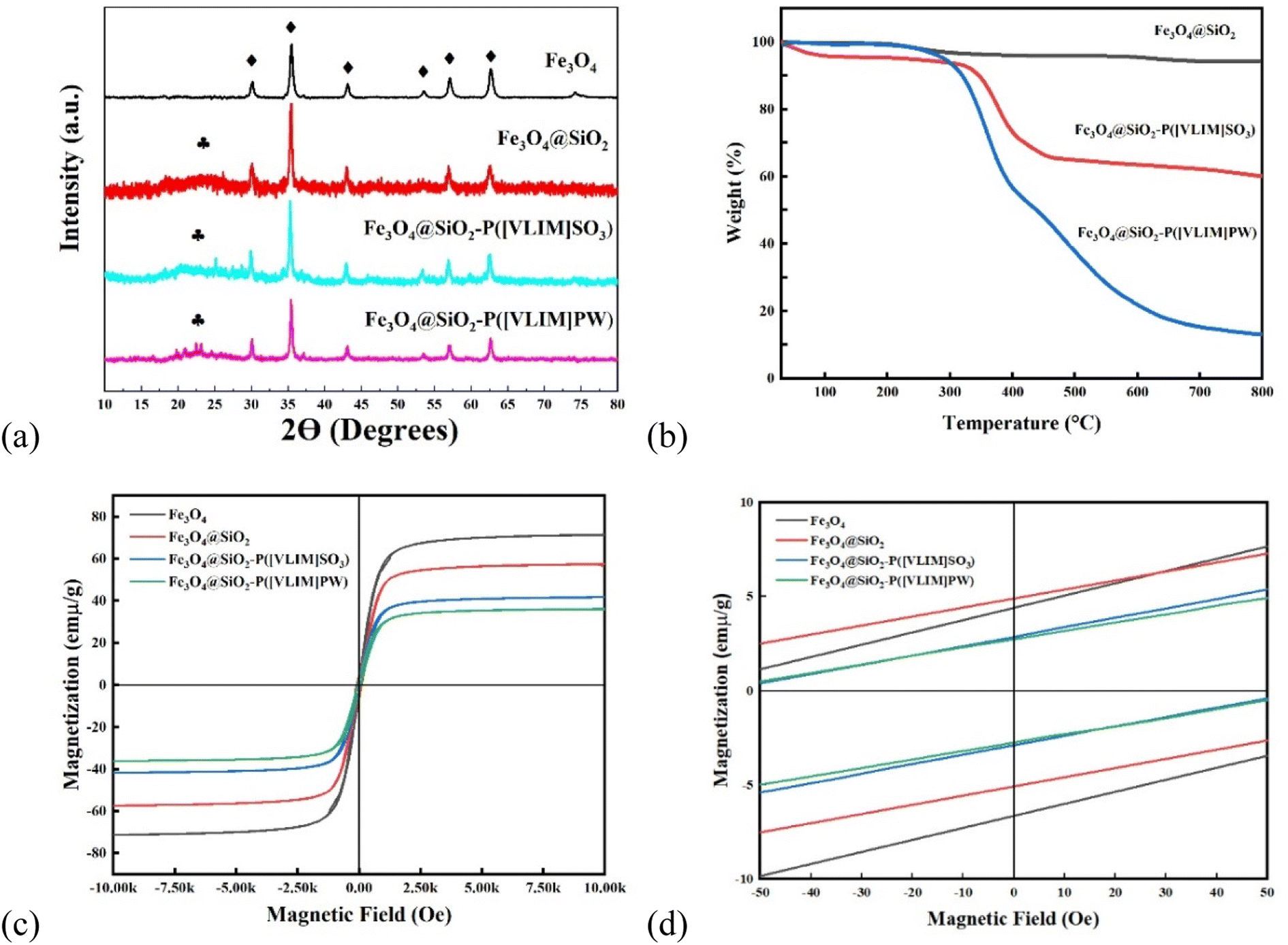 | ||
| Fig. 2 Characterization of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2–P([VLIM]SO3), and Fe3O4@SiO2–P([VLIM]PW) NPs: (a) XRD, (b) TGA, (c) VSM, and (d) enlarged VSM image. | ||
000 to +10000 Oe. In order to calculate the saturation magnetization (Ms), the magnetization (M) was plotted vs. 1/H. From the extrapolation of the curves, the Ms values of the samples were found to decrease from 71, 57, and 42 emμ g−1 to 36 emμ g−1 for Fe3O4, Fe3O4@SiO2 NPs, Fe3O4@SiO2–P([VLIM]SO3), and Fe3O4@SiO2–P([VLIM]PW), respectively (Fig. 2c). We inferred that this was caused by them being covered with a layer of non-magnetic SiO2, poly([VLIM]SO3), and poly([VLIM]PW) on the surface of the Fe3O4 NPs, respectively. The Ms value of the Fe3O4@SiO2–P([VLIM]PW) NPs was lower than that of Fe3O4@SiO2–P([VLIM]SO3) NPS, which might be caused by the layer of poly ([VLIM]PW) being thicker than that of poly ([VLIM]SO3) on the surface of the magnetic-responsive Fe3O4@SiO2 NPs. Furthermore, the TGA results showed the weight content of poly([VLIM]PW) was higher than that of poly([VLIM]SO3) on the surface of the Fe3O4@SiO2 NPs in the as-prepared solid acid catalysts. Thus, we inferred that the thickness of poly([VLIM]PW) was thicker than that of poly([VLIM]SO3) on the surface of the Fe3O4@SiO2 NPs. Also, the coercivity (Hc) of these samples was not zero (Oe), which confirmed that they had the properties for displaying superparamagnetic performances (Fig. 2d). Based on the above analysis, Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs had strong magnetic responsiveness and they could be easily recycled in an applied magnetic field.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds (Fig. 3c). Meanwhile, the peaks at 400.2 and 398.3 eV in the N 1s spectrum were assigned to CN and C–N bonds44 (Fig. 3d). These peaks were also both observed in the XPS spectra of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs, suggesting that the poly(vinyl imidazole) group had grafted on to the surface of the Fe3O4@SiO2 NPs.45 In addition, the characterized peaks at 36.4 and 38.1 eV in the W 4f spectrum were assigned to W 4f7/2 (WO3) and W 4f5/2 (WO3) of the PW group46 (Fig. 3e). The peaks at 164.8 and 167.6 eV in the S 2p spectrum were ascribed to S 2p3/2 and S 2p1/2 of SO3, while the peaks at 170.9 and 172.9 eV were attributed to S 2p3/2 and S 2p1/2 of SO4 of poly([VLIM]SO3)47 (Fig. 3f). All the spectral results confirmed that poly([VLIM]SO3) and poly([VLIM]PW) had respectively grafted on to the surface of the Fe3O4@SiO2 NPs, thus confirming Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs were achieved.
N bonds (Fig. 3c). Meanwhile, the peaks at 400.2 and 398.3 eV in the N 1s spectrum were assigned to CN and C–N bonds44 (Fig. 3d). These peaks were also both observed in the XPS spectra of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs, suggesting that the poly(vinyl imidazole) group had grafted on to the surface of the Fe3O4@SiO2 NPs.45 In addition, the characterized peaks at 36.4 and 38.1 eV in the W 4f spectrum were assigned to W 4f7/2 (WO3) and W 4f5/2 (WO3) of the PW group46 (Fig. 3e). The peaks at 164.8 and 167.6 eV in the S 2p spectrum were ascribed to S 2p3/2 and S 2p1/2 of SO3, while the peaks at 170.9 and 172.9 eV were attributed to S 2p3/2 and S 2p1/2 of SO4 of poly([VLIM]SO3)47 (Fig. 3f). All the spectral results confirmed that poly([VLIM]SO3) and poly([VLIM]PW) had respectively grafted on to the surface of the Fe3O4@SiO2 NPs, thus confirming Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs were achieved.
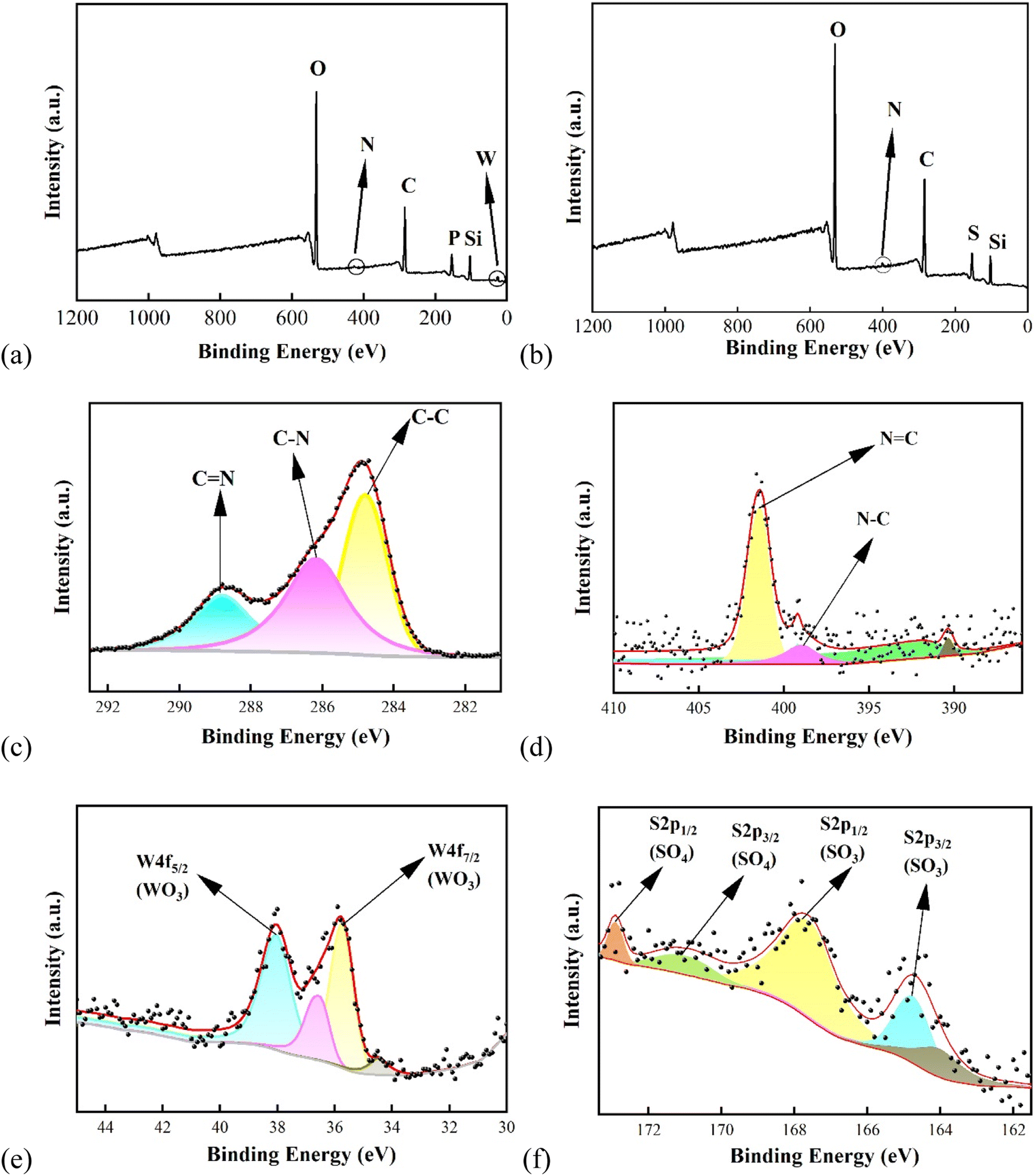 | ||
| Fig. 3 XPS spectra of the solid acid catalysts. Survey spectra of (a) Fe3O4@SiO2–P([VLIM]PW), (b) Fe3O4@SiO2–P([VLIM]SO3); and high-resolution spectra of (c) C 1s, (d) N 1s, (e) W 4f, and (f) S 2p. | ||
C, CN, and C–H bonds of the imidazole group, respectively.48 These above peaks could be observed in both curves. Furthermore, the characteristic peaks at 820, 890, 989, and 1080 cm−1, respectively, indicating O-b1-W, O-b2-W, WO, and P–O of the Keggin structure, were observed with the Fe3O4@SiO2–P([VLIM]PW) NPs;8 while the characterized peak of SO at 1175 cm−1 was observed with the Fe3O4@SiO2–P([VLIM]SO3) NPs.34,48 All the peaks indicated the as-designed solid acid catalysts were achieved.
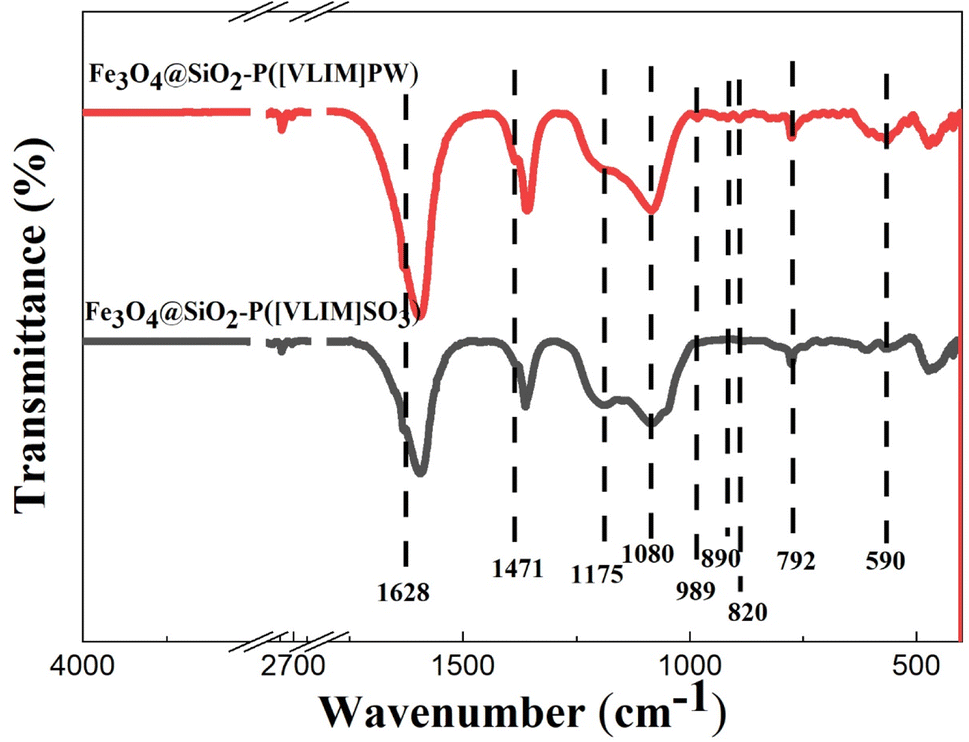 | ||
| Fig. 4 FTIR spectra of the solid acid catalysts. | ||
 | ||
| Fig. 5 SEM images of the samples: (a) Fe3O4, (b) Fe3O4@SiO2, (c) Fe3O4@SiO2–P([VLIM]SO3), and (d) Fe3O4@SiO2–P([VLIM]PW) NPs. | ||
where CH+ is the acid concentration of the catalyst (mmol g−1), and CKOH and mcat. are the molar concentration of the KOH–ethanol solution and the weight of the catalyst, respectively. The acidity values of the Fe3O4@SiO2–P([VLIM]SO3) NPs and Fe3O4@SiO2–P([VLIM]PW) NPs were 2.3 and 3.8 mmol g−1, respectively. It was obvious that the acidity of the Fe3O4@SiO2–P([VLIM]PW) NPs was higher than that of the Fe3O4@SiO2–P([VLIM]SO3) NPs, suggesting that the catalytic activity of the Fe3O4@SiO2–P([VLIM]PW) NPs would be better than that of the Fe3O4@SiO2–P([VLIM]SO3) NPs.
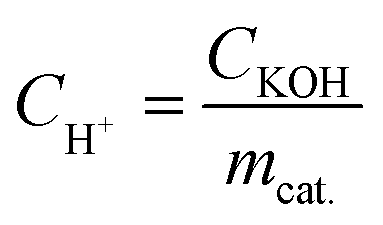
3.3. Catalytic properties of the solid acid catalysts
The catalytic properties of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs were investigated by the catalyzed esterification of palmitic acid and methanol. The esterification reaction was carried out at 70 °C for 6 h while the MR of methanol to palmitic acid was 18:1. As shown in Fig. 6, Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs both presented strong catalytic activity in the esterification. The conversion rate was significantly improved with the increase in the dosage of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs in the mixture, reaching up to 75% and 92%, respectively. Meanwhile, the results indicated that the catalytic activity of Fe3O4@SiO2–P([VLIM]PW) was higher than that of the Fe3O4@SiO2–P([VLIM]SO3) NPs in the esterification. We inferred that this could be attributed to the high loading content and monodispersion of poly([VLIM]PW) covered on the surface of the Fe3O4@SiO2 NPs. These results were consistent with those of the TGA and SEM images. When the dosage of Fe3O4@SiO2–P([VLIM]PW) NPs was 10 wt% (weight percentage based on palmitic acid), the conversion rate could reach up to 92%. However, the conversion rate gradually decreased when the dosage of solid acid catalysts was more than 10 wt% in the mixture, which was probably caused by aggregation of the Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs in the mixture, and thus their catalytic activity decreased.
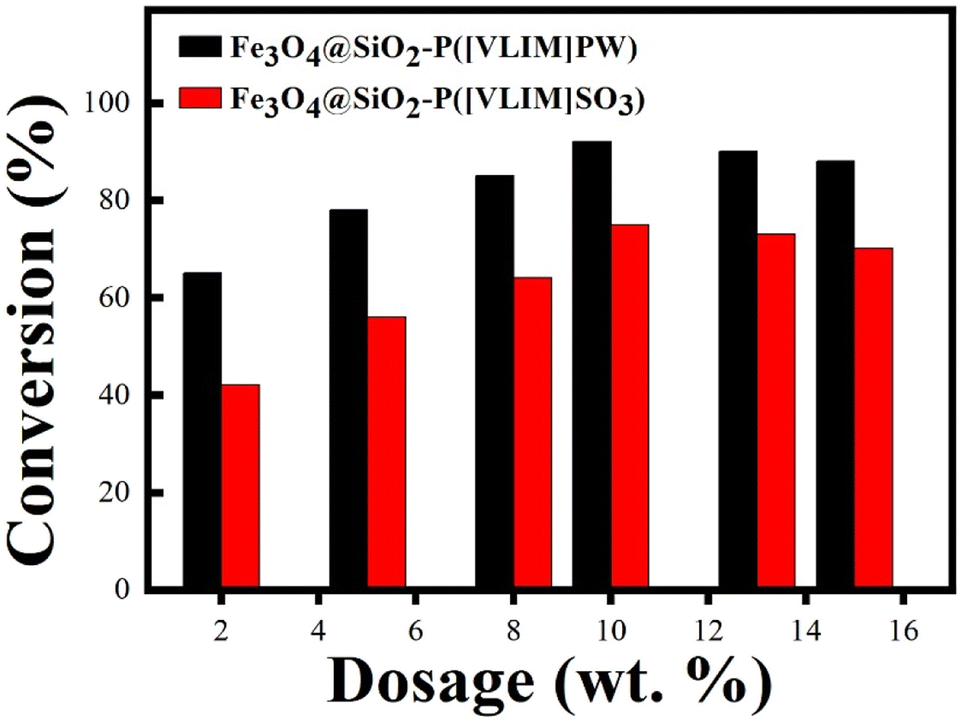 | ||
| Fig. 6 Conversion rate of palmitic acid in the esterification catalyzed by Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs with different dosages (70 °C, 6 h, MR of methanol to palmitic acid of 18:1). | ||
A series of experiments were carried out according to single-factor analyses to determine the optimal reaction conditions for the preparation of biodiesel by the esterification of palmitic acid and methanol. According to the results obtained from Fig. 6, the dosage of Fe3O4@SiO2–P([VLIM]PW) NPs was selected as 10 wt%. The effects of changing the reaction factors, such as reaction time, reaction temperature, and MR of methanol to palmitic acid, were investigated, and the corresponding results are shown in Fig. 7. The results indicated that the optimal reaction conditions were as following: reaction time, temperature, and MR of methanol to palmitic acid of 6 h, 70 °C, and 12:1, respectively. Under the above reaction conditions, the conversion rate could reach up to 94%.
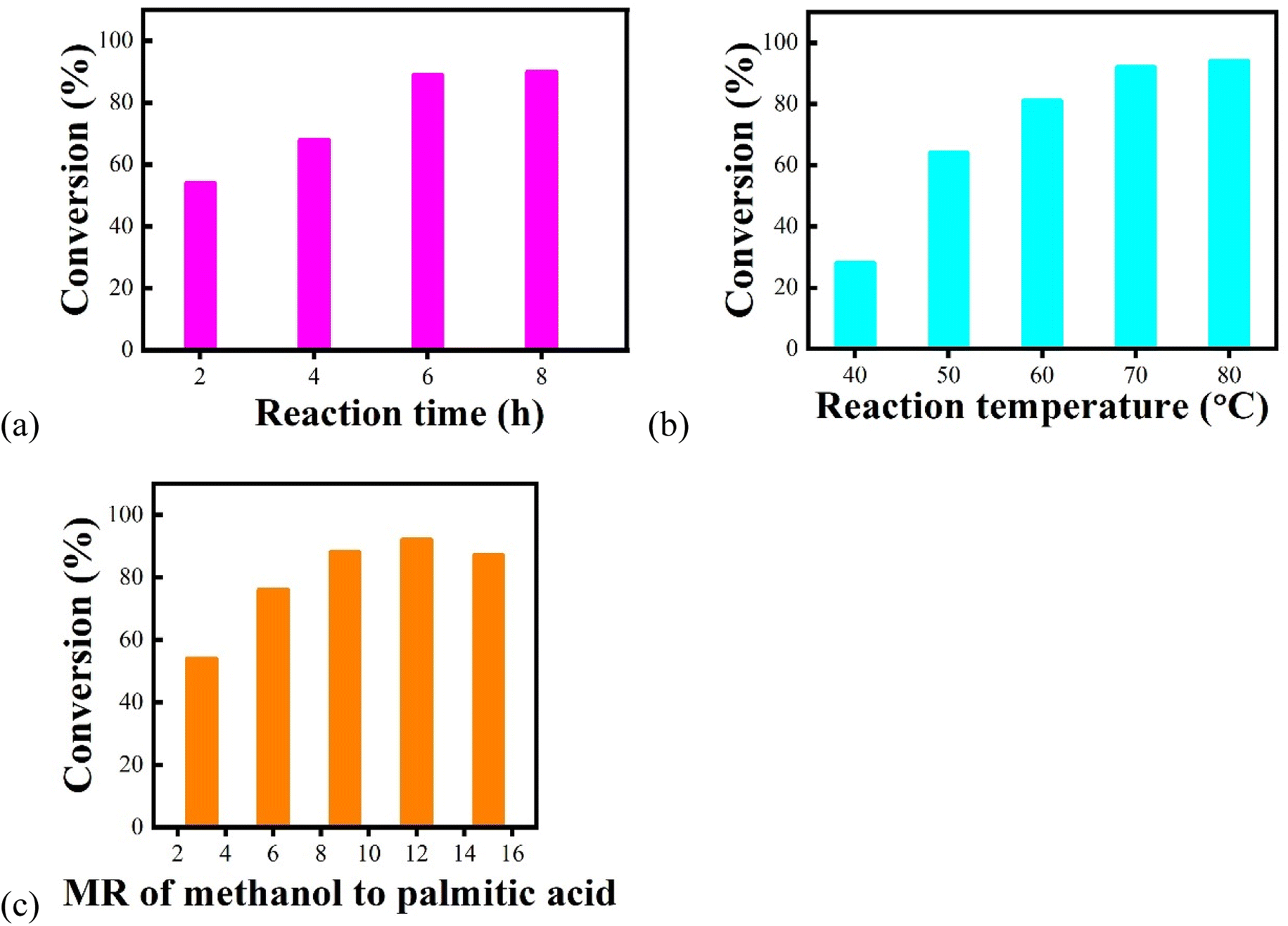 | ||
| Fig. 7 Effects of changing different reaction factors on the conversion rate with 10 wt% Fe3O4@SiO2–P([VLIM]PW) NPs as the catalyst: (a) reaction time (70 °C, MR of methanol to palmitic acid of 18:1), (b) reaction temperature (6 h, MR of methanol to palmitic acid of 18:1), and (c) MR of methanol to palmitic acid (70 °C, 6 h). | ||
3.4. Catalytic stability
From the viewpoints of economical consideration and environmental concerns, the stability properties of catalysts are greatly important. Thus, experiments to investigate the catalyst reuse potential were conducted in the esterification of palmitic acid and methanol with Fe3O4@SiO2–P([VLIM]PW) NPs as the catalyst under the optimal reaction conditions mentioned above. The results are shown in Fig. 8, where it can be seen that even after being reused for 5 times, the conversion rate was still as high as 84%. These results confirmed that the Fe3O4@SiO2–P([VLIM]PW) NPs possessed excellent reusability potential, and thus they could be suitable to serve as efficient solid acid catalysts in the esterification.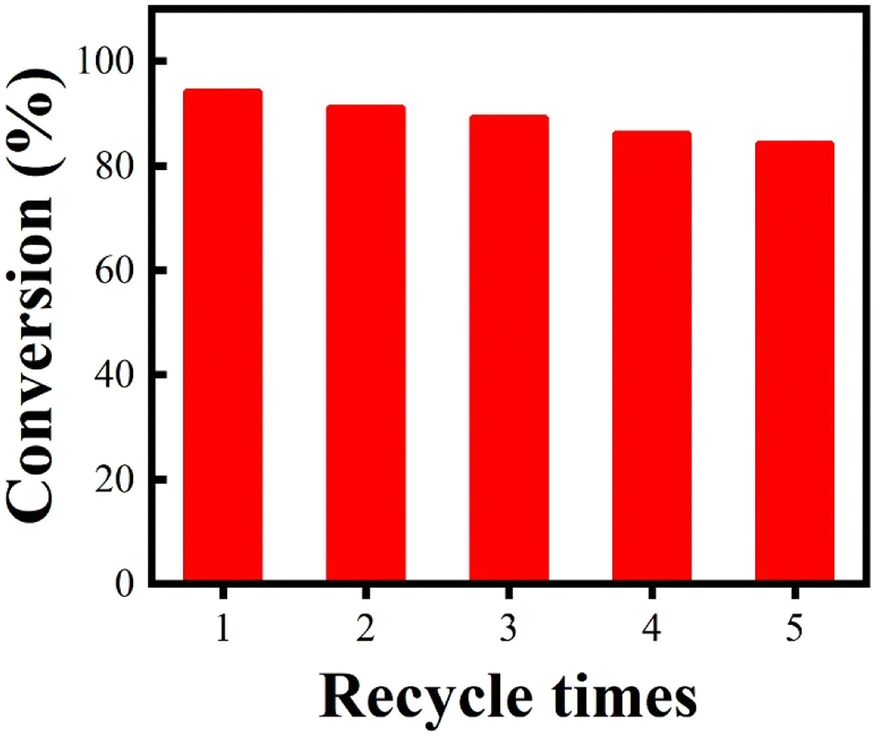 | ||
| Fig. 8 Reusability of Fe3O4@SiO2–P([VLIM]PW) NPs as catalysts for the esterification of palmitic acid and methanol. Reaction conditions: RM of methanol to palmitic acid of 15:1, 10 wt% catalyst, 70 °C, 6 h. | ||
The acidity of the reused Fe3O4@SiO2–P([VLIM]PW) NPs was also investigated, and the value was 3.2 mmol g−1 after recycling the catalyst 5 times. The Fe3O4@SiO2–P([VLIM]PW) NPs showed a slight decrease in acidity, whereas their SEM image and VSM value showed no change. However, in the FTIR spectra of the used Fe3O4@SiO2–P([VLIM]PW) NPs, there was a decrease in the intensity of the characteristic peak for phosphotungstate (Fig. S3†), indicating a slight deactivation of the reused catalyst.
3.5. Mechanism of the solid acid-catalyzed esterification
Based on the results from recent reports about catalyzed esterification kinetics,34,50,51 we think the catalyzed esterification reaction in this work was accomplished via the chemisorption of palmitic acid on the surface of the magnetic-responsive solid acid catalysts. The esterification was a nucleophilic reaction. The electron cloud density of poly([VLIM]PW) and poly([VLIM]SO3) was high, thus Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs could provide protons to palmitic acid. This resulted in palmitic acid being easily absorbed on the surface of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs. The COH+ was generated from the reaction of the active sites on the magnetic-responsive solid acid with the CO of palmitic acid. Then, the –COOH group of palmitic acid was protonated and a carbocation was formed. After that, nucleophilic attack from methanol to the above carbocation was performed, and then the tetrahedral intermediate was achieved. Finally, the tetrahedral intermediate was respectively moved away from the surface of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs via proton migration, and thus palmitic acid methyl ester was obtained. Meanwhile, the catalytic activities of Fe3O4@SiO2–P([VLIM]SO3) and Fe3O4@SiO2–P([VLIM]PW) NPs were regenerated after the esterification, respectively. Furthermore, we think the enhanced catalytic performance of the prepared solid acid catalysts in this work was caused by the PILs being uniformly dispersed on the Fe3O4@SiO2 NPs with a large specific surface area. Thus, a greater number of activated sites on the magnetic-responsive solid acid catalysts could be applied in the esterification and the catalytic performance was improved.
4. Conclusions
In summary, two kinds of magnetic-responsive solid acid catalysts were designed and prepared by an in situ polymerization on the surface of magnetic nanoparticles. They were characterized by XRD, VSM, NMR, FTIR, TGA, XPS, SEM, and GC. The results showed that the solid acid catalysts with magnetic responsiveness were about 350 nm in diameter, and their surface was rough due to the coverage of poly([VLIM]PW) and poly([VLIM]SO3) on the surface of the Fe3O4@SiO2 NPs, respectively. The weight contents of the poly([VLIM]PW) and poly([VLIM]SO3) coverings were about 87% and 40%, respectively. The catalytic activity of the solid acid catalysts was evaluated by the catalyzed esterification of palmitic acid and methanol. By comparison, the catalytic properties of Fe3O4@SiO2–P([VLIM]PW) NPs were better than that of the Fe3O4@SiO2–P([VLIM]SO3) NPs, which was due to the greater number of activated sites and high loading content of poly([VLIM]PW) covered on the surface of the Fe3O4@SiO2 NPs as presented in the esterification. Under the optimal reaction conditions (10 wt% catalyst, 6 h, 70 °C, and MR of methanol to palmitic acid of 12:1), the conversion rate of palmitic acid in the esterification could reach up to 94% when the Fe3O4@SiO2–P([VLIM]PW) NPs served as catalysts. Furthermore, the Fe3O4@SiO2–P([VLIM]PW) NPs could still maintain a high catalytic activity even after being reused 5 times without significant deactivation. More importantly, this work proposes a simple method to cover acidic PILs on the surface of magnetic particles. This method has great potential for the preparation of solid acid catalysts with magnetic responsiveness, which could be used in many domains, such as catalysis and waste treatment.
Author contributions
Dan Xue: conceptualization, writing – original draft & review & editing, investigation, visualization, methodology, validation, formal analysis, data curation, supervision, resources. Yun Jiang: investigation. Fang Xia Zheng: investigation.Conflicts of interest
The authors state that there are no conflicts to declare.Acknowledgements
This work is funded by National Natural Science Foundation of China (Project No. 52003137) and open fund from the Key Lab of Eco-restoration of Regional Contaminated Environment (Shenyang University), Ministry of Education (Grant No. KF-22-10). The authors would like to thank Gao Jilong from Shiyanjia Lab (https://www.shiyanjia.com/) for the XPS analysis.References
- D. D. Bala, M. Misra and D. Chidambaram, J. Cleaner Prod., 2017, 142, 4169–4177 CrossRef CAS.
- J. Gaidukevič, J. Barkauskas, A. Malaika, V. Jasulaitienė and M. Kozłowski, Appl. Surf. Sci., 2021, 554, 149588–149700 CrossRef.
- K.-S. Lin, N. V. Mdlovu, H.-Y. Chan, K. C.-W. Wu, J. C.-S. Wu and Y.-T. Huang, Catal. Today, 2022, 397–399, 145–154 CrossRef CAS.
- M. O. Abdelmigeed, E. G. Al-Sakkari, M. S. Hefney, F. M. Ismail, A. Abdelghany, T. S. Ahmed and I. M. Ismail, Renewable Energy, 2021, 165, 405–419 CrossRef CAS.
- A. Naeem, I. Wali Khan, M. Farooq, T. Mahmood, I. Ud Din, Z. Ali Ghazi and T. Saeed, Bioresour. Technol., 2021, 328, 124831–124838 CrossRef CAS PubMed.
- M. Racar, I. Šoljić Jerbić, Z. Glasovac and A. Jukić, Renewable Energy, 2023, 202, 1046–1053 CrossRef CAS.
- J. Li and X. Liang, Energy Convers. Manage., 2017, 141, 126–132 CrossRef CAS.
- R. Sree and S. Kuriakose, J. Energy Chem., 2015, 24, 87–92 CrossRef.
- H. Li, J. Wang, X. Ma, Y. Wang, G. Li, M. Guo, P. Cui, W. Lu, S. Zhou and M. Yu, Renewable Energy, 2021, 179, 1191–1203 CrossRef CAS.
- P. X. Pham, K. T. Nguyen, T. V. Pham and V. H. Nguyen, ACS Omega, 2020, 5, 20842–20853 CrossRef CAS PubMed.
- W. Xie, Y. Xiong and H. Wang, Renewable Energy, 2021, 174, 758–768 CrossRef CAS.
- G. Knothe and L. F. Razon, Prog. Energy Combust. Sci., 2017, 58, 36–59 CrossRef.
- A. K. Sodhi, S. Tripathi and K. Kundu, Clean Technol. Environ. Policy, 2017, 19, 1799–1807 CrossRef CAS.
- M. Hamza, M. Ayoub, R. B. Shamsuddin, A. Mukhtar, S. Saqib, I. Zahid, M. Ameen, S. Ullah, A. G. Al-Sehemi and M. Ibrahim, Environ. Technol. Innovation, 2021, 21, 101200–101220 CrossRef CAS.
- X. Zhang, N. Li, Z. Wei, B. Dai and S. Han, Renewable Energy, 2022, 196, 737–748 CrossRef CAS.
- Y. Wang, G. Yang, J. He, G. Sun, Z. Sun and Y. Sun, Process Saf. Environ. Prot., 2020, 141, 333–343 CrossRef CAS.
- N. A. Negm, M. A. Betiha, M. S. Alhumaimess, H. M. A. Hassan and A. M. Rabie, J. Cleaner Prod., 2019, 238, 117854–117869 CrossRef CAS.
- I. Tankov, Z. Mustafa, R. Nikolova, A. Veli and R. Yankova, Fuel, 2022, 307, 121876–121887 CrossRef CAS.
- F. Huang, Y. Su, Y. Tao, W. Sun and W. Wang, Fuel, 2018, 226, 417–422 CrossRef CAS.
- X. Lin, X. Ling, J. Chen, M. Li, T. Xu and T. Qiu, Green Chem., 2019, 21, 3182–3189 RSC.
- X.-X. Han, Y.-F. He, C.-T. Hung, L.-L. Liu, S.-J. Huang and S.-B. Liu, Chem. Eng. Sci., 2013, 104, 64–72 CrossRef CAS.
- Y. Bian, Q. Shan, C. Guo, C. Liu and J. Zhang, Catal. Lett., 2021, 151, 3523–3531 CrossRef CAS.
- Y. Leng, J. Wang, D. R. Zhu, X. Q. Ren, H. Q. Ge and L. Shen, Angew. Chem., Int. Ed., 2009, 48, 168–171 CrossRef CAS PubMed.
- Z. Yu, X. Chen, Y. Zhang, H. Tu, P. Pan, S. Li, Y. Han, M. Piao, J. Hu, F. Shi and X. Yang, Chem. Eng. J., 2022, 430, 133059–133069 CrossRef CAS.
- J. Yu, Y. Wang, L. Sun, Z. Xu, Y. Du, H. Sun, W. Li, S. Luo, C. Ma and S. Liu, ACS Omega, 2021, 6, 7896–7909 CrossRef CAS PubMed.
- M. Fan, Z. Si, W. Sun and P. Zhang, Fuel, 2019, 252, 254–261 CrossRef CAS.
- T. Parangi and M. K. Mishra, Comments Inorg. Chem., 2019, 39, 90–126 CrossRef CAS.
- F. F. Roman, A. E. Ribeiro, A. Queiroz, G. G. Lenzi, E. S. Chaves and P. Brito, Fuel, 2019, 239, 1231–1239 CrossRef CAS.
- M. A. Hanif, S. Nisar and U. Rashid, Catal. Rev., 2017, 59, 165–188 CrossRef CAS.
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef CAS PubMed.
- J. Zhang, X. Liu, M. N. Hedhili, Y. Zhu and Y. Han, ChemCatChem, 2011, 3, 1294–1298 CrossRef CAS.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- J. Zhang, M. Sun, X. Liu and Y. Han, Catal. Today, 2014, 233, 77–82 CrossRef CAS.
- P. Fan, S. Xing, J. Wang, J. Fu, L. Yang, G. Yang, C. Miao and P. Lv, Fuel, 2017, 188, 483–488 CrossRef CAS.
- Y.-T. Wang, X.-X. Yang, J. Xu, H.-L. Wang, Z.-B. Wang, L. Zhang, S.-L. Wang and J.-L. Liang, Renewable Energy, 2019, 139, 688–695 CrossRef CAS.
- P. Ke, D. Zeng, J. Wu, J. Cui, X. Li and G. Wang, ACS Omega, 2019, 4, 22119–22125 CrossRef CAS PubMed.
- H. Jing, X. Wang, Y. Liu and A. Wang, Chin. J. Catal., 2015, 36, 244–251 CrossRef CAS.
- J. Gardy, E. Nourafkan, A. Osatiashtiani, A. F. Lee, K. Wilson, A. Hassanpour and X. Lai, Appl. Catal., B, 2019, 259, 118093–118094 CrossRef CAS.
- D. Xue, Q. B. Meng and X. M. Song, ACS Appl. Mater. Interfaces, 2019, 11, 10967–10974 CrossRef CAS PubMed.
- D. Xue, X. Song and F. Liang, RSC Adv., 2017, 7, 25450–25454 RSC.
- C. Sun, D. Xu, C. Hou, H. Zhang, Y. Li, Q. Zhang, H. Wang and M. Zhu, J. Mech. Behav. Biomed. Mater., 2021, 121, 104593–104601 CrossRef CAS PubMed.
- Y. Yin, X. Huang, W. Wang and X. Liu, Solid State Sci., 2023, 136, 107109–107118 CrossRef CAS.
- S. Y. Lai, K. H. Ng, C. K. Cheng, H. Nur, M. Nurhadi and M. Arumugam, Chemosphere, 2021, 263, 128244–128266 CrossRef CAS PubMed.
- D. Yuan, J. Zhao, Q. Zhang, P. Lu, Y. Wang, Y. He, Z. Liu, Y. Liu, X. Zhao and C. Meng, J. Mater. Chem. A, 2022, 10, 12656–12668 RSC.
- C. Liu, S. Qiu, P. Du, H. Zhao and L. Wang, Nanoscale, 2018, 10, 8115–8124 RSC.
- S. Zou, H. Wang, S. Li, B. Lu, J. Zhao and Q. Cai, Appl. Surf. Sci., 2022, 574, 11 CrossRef.
- X. Li, Z. Zhou, Y. Zhao, D. Ramella and Y. Luan, Appl. Organomet. Chem., 2020, 34, 13 Search PubMed.
- Y. Wang, D. Zhao, L. Wang, X. Wang, L. Li, Z. Xing, N. Ji, S. Liu and H. Ding, Fuel, 2018, 216, 364–370 CrossRef CAS.
- Y. Wang, D. Wang, M. Tan, B. Jiang, J. Zheng, N. Tsubaki and M. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 26767–26775 CrossRef CAS PubMed.
- D. D. Bala, V. Vasquez and D. Chidambaram, Ind. Eng. Chem. Res., 2018, 57, 14948–14956 CrossRef CAS.
- Y. Wang, D. Zhao, G. Chen, S. Liu, N. Ji, H. Ding and J. Fu, Renewable Energy, 2019, 133, 317–324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05350d |
| This journal is © The Royal Society of Chemistry 2023 |