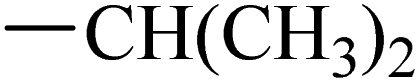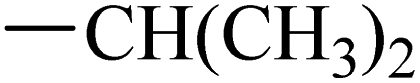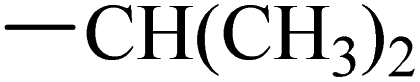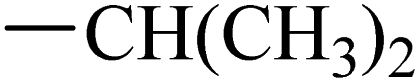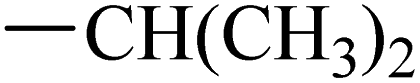DOI:
10.1039/D3RA05542F
(Paper)
RSC Adv., 2023,
13, 28462-28480
Design, synthesis and biological evaluation of VEGFR-2/HDAC dual inhibitors as multitargeted antitumor agents based on fruquintinib and vorinostat†
Received
15th August 2023
, Accepted 19th September 2023
First published on 27th September 2023
Abstract
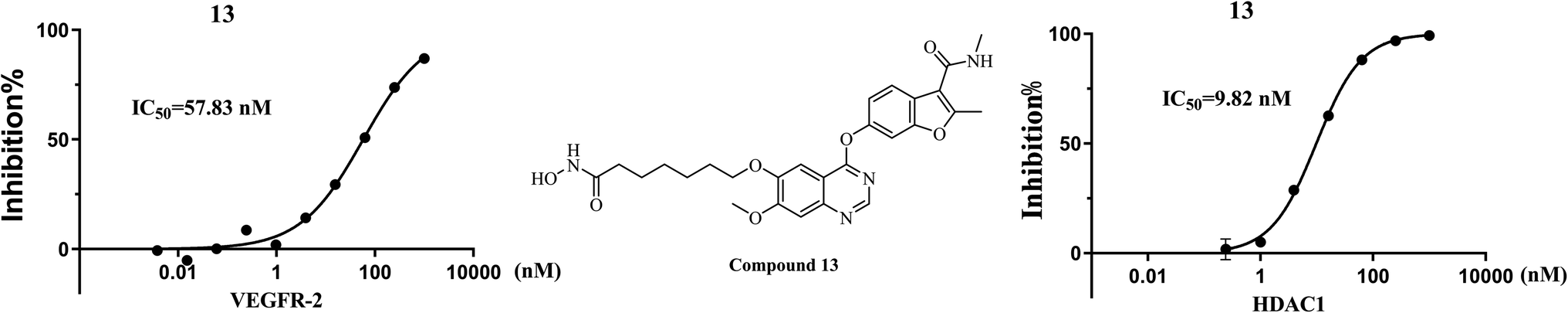
Herein, a series of 4-(benzofuran-6-yloxy)quinazoline derivatives as VEGFR-2/HDAC dual inhibitors were designed and synthesized based on fruquintinib and vorinostat. Among them, compound 13 exhibited potent inhibitory activity against VEGFR-2 and HDAC1 with IC50 values of 57.83 nM and 9.82 nM, and displayed moderate to significant antiproliferative activity against MCF-7, A549, HeLa and HUVEC. The cellular mechanism studies revealed that compound 13 arrested the cell cycle at the S and G2 phases, and induced significant apoptosis in HeLa cells. Tube formation assay in HUVECs demonstrated that 13 had a significant anti-angiogenic effect. Additionally, a molecular docking study supported the initial design strategy. These results highlighted that 13 was a valuable VEGFR-2/HDAC dual inhibitor and deserved further study for cancer therapy.
Introduction
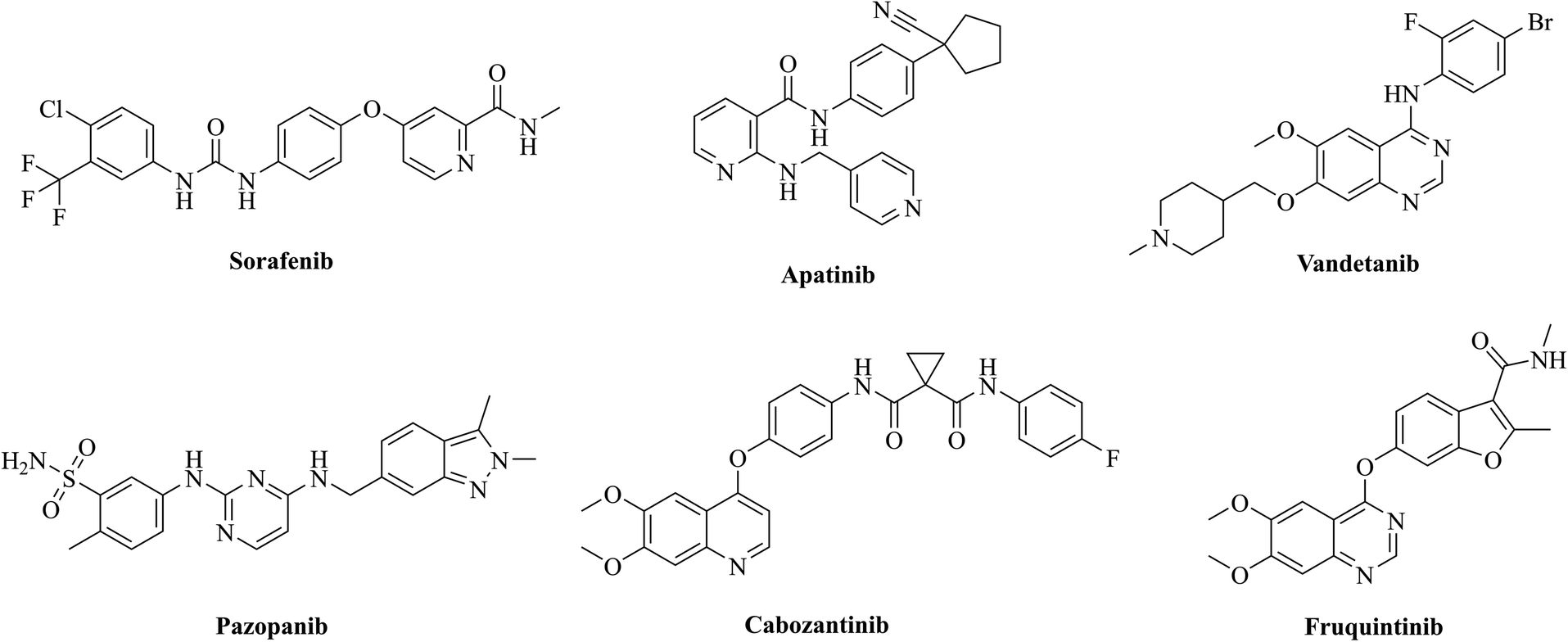
Tumour angiogenesis is an essential marker of tumor growth, infiltration and metastasis.1,2 Vascular endothelial growth factors (VEGFs) are some of the most active and specific proangiogenic factors known, and their kinase receptors include VEGFR-1, VEGFR-2 and VEGFR-3.3,4 Among them, VEGFR-2 is a key signaling receptor in the process of angiogenesis.5 The abnormal upregulation of VEGFR-2 could be used as an important marker to indicate the development of numerous diseases, especially in various malignant tumors, and inhibition of the VEGFR-2 pathway could lead to an effective antitumor angiogenic response.6,7 To date, a variety of monoclonal antibodies and small molecule inhibitors targeting VEGFR-2 have been approved for the treatment of solid tumors,8,9 such as ramucirumab, sorafenib, apatinib, vandetanib, pazopanib, cabozantinib, and fruquintinib, etc. (Fig. 1). However, drug resistance has occurred in most patients treated with VEGFR inhibitors,10–12 limiting the effectiveness of these inhibitors. To overcome the drug resistance, combination therapy is often required in clinical practice.13,14
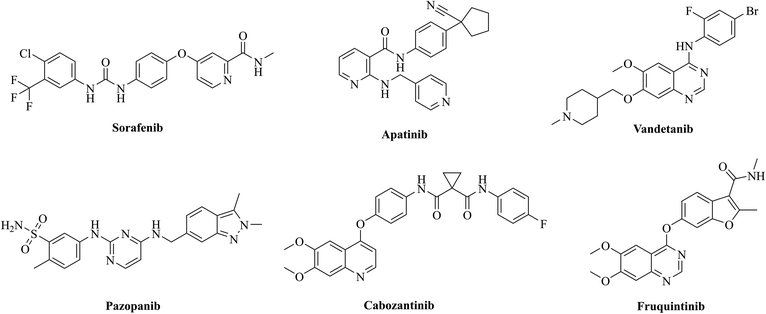 |
| | Fig. 1 Chemical structures of representative VEGFR-2 inhibitors. | |
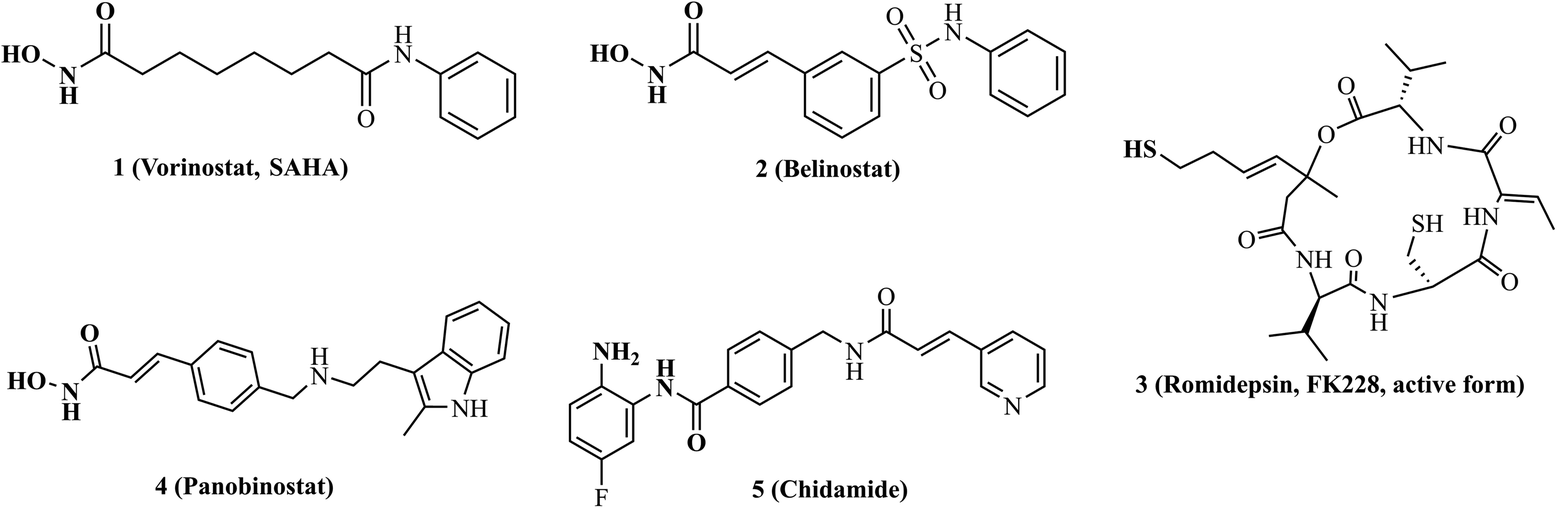
For the last decade, targeting epigenetic aberration has been considered as an important strategy for the treatment of tumors.15,16 As key mediators of epigenetic regulation, overexpression of histone deacetylases (HDACs) and their dysregulated functions have been observed in many tumor types.17 Several HDAC inhibitors (vorinostat, belinostat, romidepsin, panobinostat and chidamide, Fig. 2) have been available in the clinic for reversing aberrant epigenetic changes associated with tumor treatments.18 Notably, accumulating evidences demonstrate that combining HDAC inhibitors with VEGFR-2 inhibitors shows great potential for overcoming VEGFR-2 inhibitors resistance and improving antitumor efficacy.19–22 Considering that multi-drugs combination therapy is associated with significant side effects, poor patient compliance, complex pharmacokinetic properties and drug–drug interactions. Therefore, the development of individual drug targeting VEGFR-2 along with HDACs exhibits great promise.23 Until now, some VEGFR-2/HDAC dual inhibitors have been reported,24–27 such as pazopanib-based VEGFR-2/HDAC dual inhibitors designed by Zhang et al.28 and vandetanib-based VEGFR-2/HDAC dual inhibitors designed by Shi's group.29,30
 |
| | Fig. 2 Chemical structures of representative HDAC inhibitors. | |
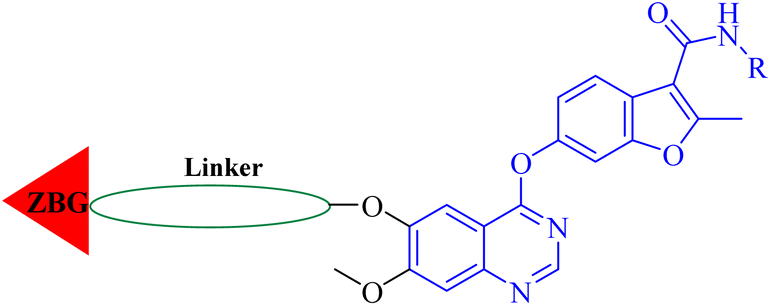
Herein, we designed and synthesized a series of 4-(benzofuran-6-yloxy)quinazoline-based derivatives based on the pharmacophore of the VEGFR inhibitor fruquintinib and the HDAC inhibitor, and screened for their inhibitory activity against VEGFR-2 and HDAC1 at the enzyme level. Furthermore, the compound with potent inhibitory activity against both VEGFR-2 and HDAC1 was selected for further in vitro antitumor activity evaluation and investigation of mechanism of action.
Results and discussion
Design strategy
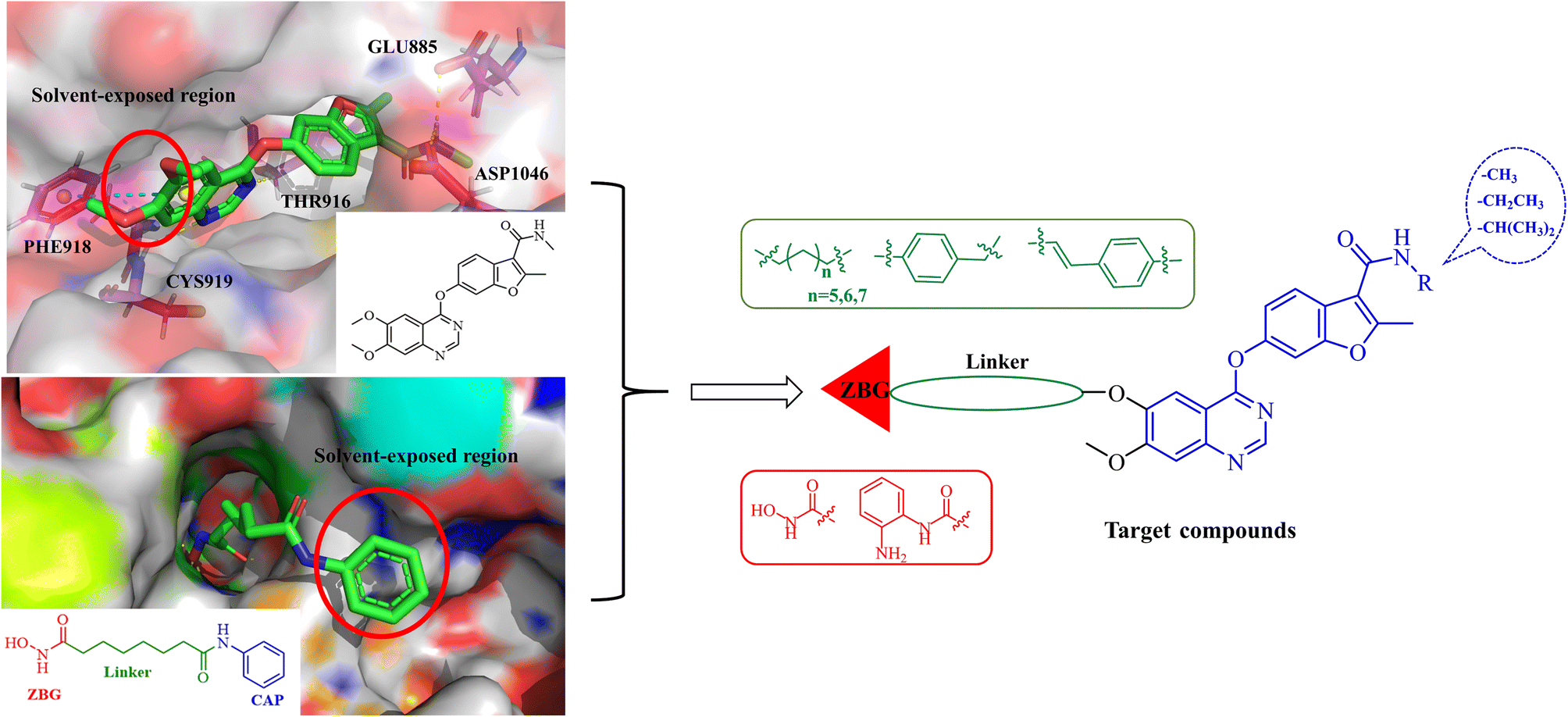
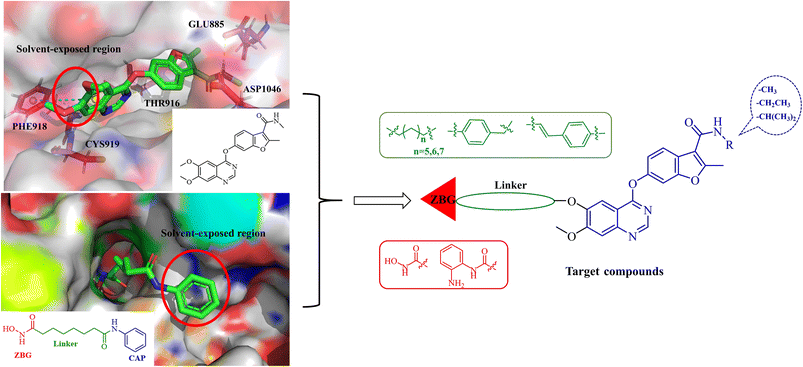
Fruquintinib is a novel and oral VEGFR small molecule tyrosine kinase inhibitor (TKI) developed by Hutchison MediPharma, with high affinity and selectivity for VEGFR-1, -2 and -3. In September 2018, fruquintinib was approved in China for treatment of metastatic colorectal cancer (CRC) patients who have failed at least two prior systemic anti-neoplastic therapies. As co-crystallization structure of fruquintinib and VEGFR-2 have not been reported, to better understand the binding of fruquintinib to VEGFR-2, a molecular docking study of fruquintinib and the ATP-binding pocket of VEGFR-2 was carried out. As showed in Fig. 3, the benzofuran moiety was accommodated in the mostly hydrophobic ATP-binding cleft and formed important hydrophobic interactions. The secondary amine group of amide backbone made two hydrogen bonds with Asp1046 and Glu885. The nitrogen atoms at 1 and 3-positions of the dimethoxyquinazoline ring created two hydrogen bonds with Cys919 of the hinge region and THR916, respectively. Importantly, the two methoxyl groups at 6 and 7-positions of the dimethoxyquinazoline ring extended into the solvent region and had no interaction with VEGFR-2 kinase. The pharmacophore of most HDAC inhibitors includes three parts, a CAP structure that interacts with the amino acid residues around the entrance of active pocket, a zinc ion binding group (ZBG) and a linker that is responsible for the connection of the CAP to ZBG and interacts with the hydrophobic channel of active pocket. Among them, the CAP is located around the entrance of active pocket, greater structural changes can be tolerated. Therefore, to optimally fuse the pharmacophore of HDAC inhibitor and fruquintinib, we retained the quinazoline and benzofuran moieties of fruquintinib, which formed key binding interactions with VEGFR-2, and introduced the linkers and ZBGs of HDAC inhibitors at the 6-position of quinazoline ring to design a series of 4-(benzofuran-6-yloxy)quinazoline-based derivatives (Fig. 3).
 |
| | Fig. 3 Design of fruquintinib-based VEGFR/HDAC dual inhibitors. | |
Chemistry
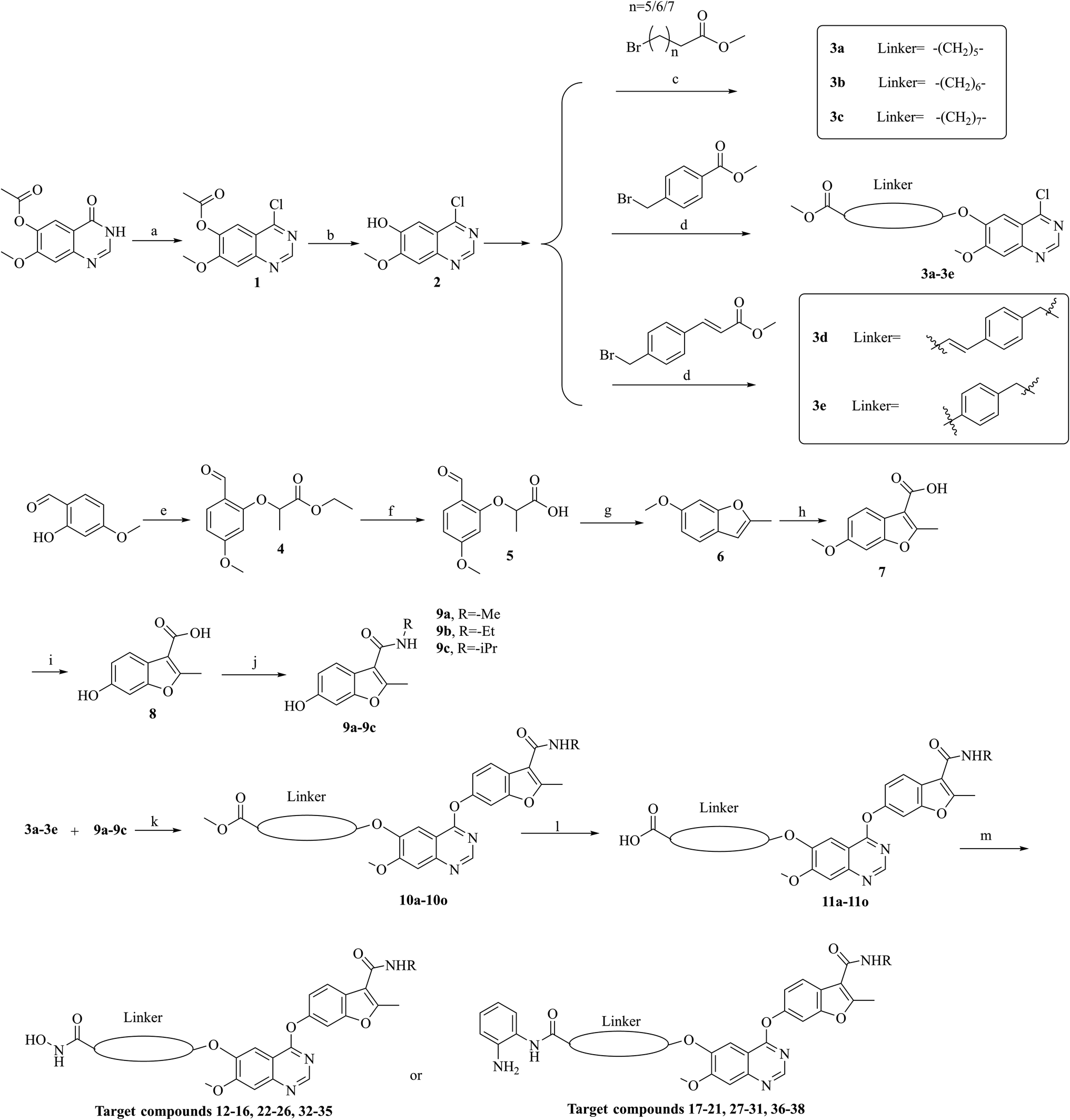
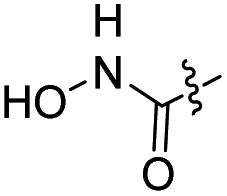
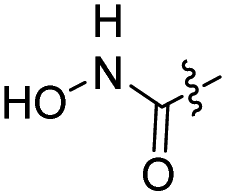
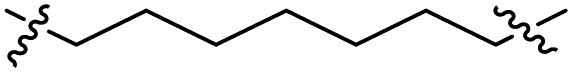
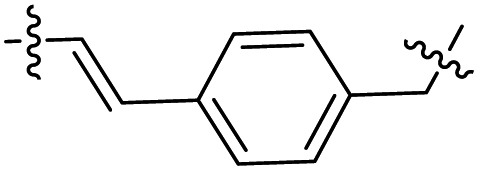
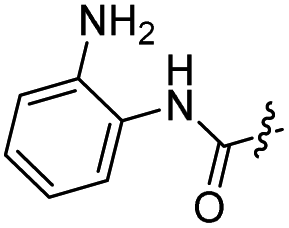
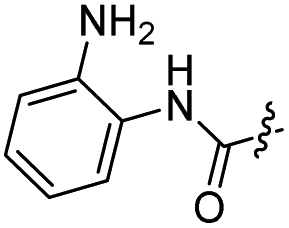
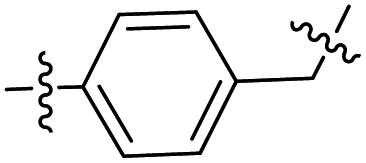
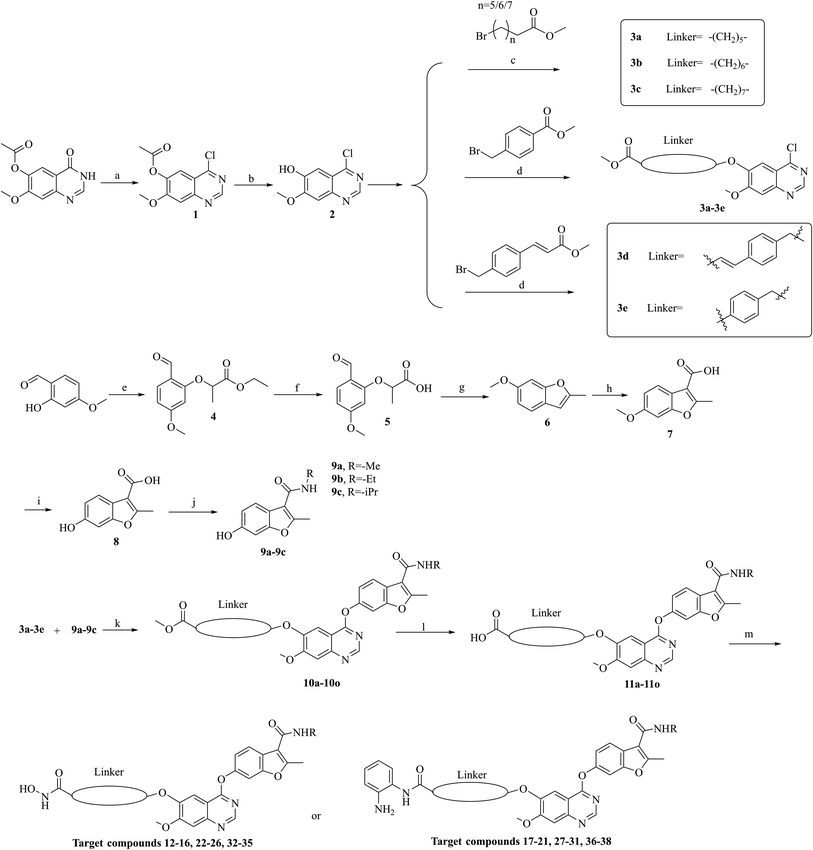
The synthesis of target compounds 12–38 is illustrated in Scheme 1. The commercially available starting material 3,4-dihydro-7-methoxy-4-oxoquinazolin-6-yl acetate was chlorinated by thionyl chloride in the presence of catalytic amount of N,N-dimethylformamide (DMF) to obtain intermediate 1, which was further hydrolysed by lithium hydroxide in the tetrahydrofuran and water at 35 °C to obtain intermediate 2. The intermediate 2 reacted with different linkers to yield key intermediates 3a–3e, respectively. In addition, 2-hydroxy-4-methoxybenzaldehyde reacted with ethyl 2-bromopropionate in N,N-dimethylformamide via a nucleophilic substitution reaction to produce intermediate 4. The 4 was further hydrolysed by sodium hydroxide in ethanol to obtain intermediate 5, which underwent an intramolecular cyclocondensation reaction to afford intermediate 6. Then, the intermediate 7 was prepared by the Friedel–Craft reaction of 6 and oxalyl chloride using the aluminium trichloride as a catalyst. The 7 was further demethylated in the presence of boron tribromide to give intermediate 8. The key intermediates 9a–9c were prepared by an amidation reaction of 8 with methylamine, ethylamine or isopropylamine, respectively. Next, 3a–3e reacted with 9a–9c in acetonitrile and potassium carbonate to yield intermediates 10a–10o, which were hydrolysed by lithium hydroxide to obtain intermediate 11a–11o. Finally, target compounds were afforded through an amidation reaction of 11a–11o and hydroxylamine hydrochloride or o-phenylenediamine in the presence of HATU and DIEA at room temperature.
 |
| | Scheme 1 Reagents and conditions: (a) SOCl2, DMF, 80 °C, 4 h. (b) LiOH, THF/H2O, 35 °C. (c) K2CO3, MeCN, 80 °C. (d) Cs2CO3, DMF, rt. (e) Ethyl 2-bromopropionate, K2CO3, DMF, rt. (f) NaOH, C2H5OH, rt. (g) NaOAc, Ac2O, reflux. (h) Oxalyl chloride, AlCl3, DCM, rt. (i) BBr3, DCM, rt. (j) HATU, DIEA, NH2R, DMF, rt. (k) K2CO3, MeCN, 80 °C. (l) LiOH, THF/H2O, 35 °C. (m) HATU, DIEA, NH2OH·HCl or o-phenylenediamine, DMF. | |
Biological activity evaluations
All the target compounds were evaluated for their inhibitory activities against VEGFR-2 and HDAC1 at a concentration of 100 nM with fruquintinib and SAHA as the positive controls. As shown in Table 1, we found that several compounds (12–14, 22 and 23) displayed outstanding inhibitory activity against both VEGFR-2 and HDAC1, suggesting that the design of VEGFR-2/HDAC dual inhibitors by introducing the linkers and ZBGs of HDAC inhibitor at the 6-position of quinazoline of fruquintinib is an effective strategy. Among them, compound 13 showed the strongest inhibitory activity with inhibition rates of 93.20% and 90.24%, which is more potent than fruquintinib and SAHA. Further, by comparison of the inhibitory activity of compounds 12–21, 22–31 and 32–38 against VEGFR-2, a clear structure–activity relationship (SAR) was observed that the inhibitory activity of target compounds showed a significant decrease as the size of R group increased, indicating that introduction of the bulky group at this position is not well tolerated. In contrast, changes of the ZBGs and linkers caused no regular change in VEGFR-2 inhibitory activity, the reason might be that the ZBGs and linkers were far from the ATP binding pocket of VEGFR-2 and had less effect on inhibitory activity. In addition, the inhibitory activity of target compounds against HDAC1 was investigated. We found that compounds (12–16, 22–26 and 32–35) bearing isohydroxamic acid exhibited more potent inhibition on the HDAC1 than these compounds bearing o-phenylenediamine (17–21, 27–31 and 36–38), suggesting that isohydroxamic acid as ZBG contributed more to the inhibitory potency against HDAC1 than o-phenylenediamine. Further investigations were performed to study the effects of different linkers on inhibitory activity, the results revealed that compounds bearing flexible aliphatic chains as linkers (12–14, 22–24, 32, 33) showed significantly stronger inhibitory effects on HDAC1 than those bearing rigid phenyl chains (15–16, 25–26, 34, 35), and the six carbons length aliphatic chain was the most favourable for HDAC1 inhibitory activity.
Table 1 Inhibitory activity of compounds 12–38 against VEGFR-2 and HDAC1
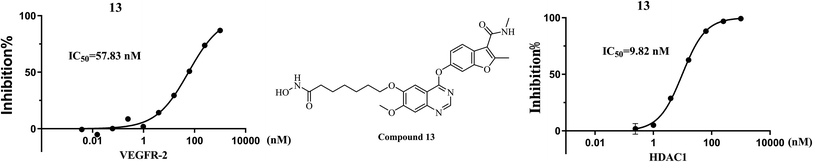
In view of the outstanding inhibitory activity of 13, which was further determined the half-maximal inhibitory concentrations (IC50) against VEGFR-2 and HDAC1. The final concentrations were from 1 μM with 4-fold dilution. The results (Fig. 4) showed that compound 13 exhibited potent inhibitory activity against both VEGFR-2 and HDAC1, with IC50 values of 57.83 nM and 9.82 nM, respectively.
 |
| | Fig. 4 Dose–response curves of compound 13 against VEGFR-2 and HDAC1. Assays were performed in replicates (n ≥ 2). | |
To further explore the antitumor effect, 13 was evaluated for its antiproliferative activity in vitro against HeLa (human cervical cancer cell line), MCF-7 (human breast cancer cell line) and A549 (human lung cancer cell line) by CCK-8 assay. As shown in Table 2, 13 exhibited moderate to good antiproliferative activity against MCF-7 and HeLa with IC50 values of 17.86 μM and 1.49 μM, which was comparable to combination of SAHA and fruquintinib, and displayed significantly stronger antiproliferative activity against A549 than combination of SAHA and fruquintinib.
Table 2 In vitro antiproliferative activity of 13 against HeLa, MCF-7 and A549
| Compound |
IC50 (μM) ± SDa |
| Hela |
MCF-7 |
A549 |
| IC50 values are displayed as averages from three independent experiments ± SD. |
| 13 |
1.49 ± 0.16 |
17.86 ± 0.54 |
4.56 ± 0.33 |
| SAHA |
1.03 ± 0.33 |
11.82 ± 0.14 |
3.54 ± 0.25 |
| Fruquintinib |
8.35 ± 0.47 |
>20 |
>20 |
Fruquintinib + SAHA (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
1.38 ± 0.36 |
18.50 ± 0.16 |
8.12 ± 0.23 |
Flow cytometry analysis
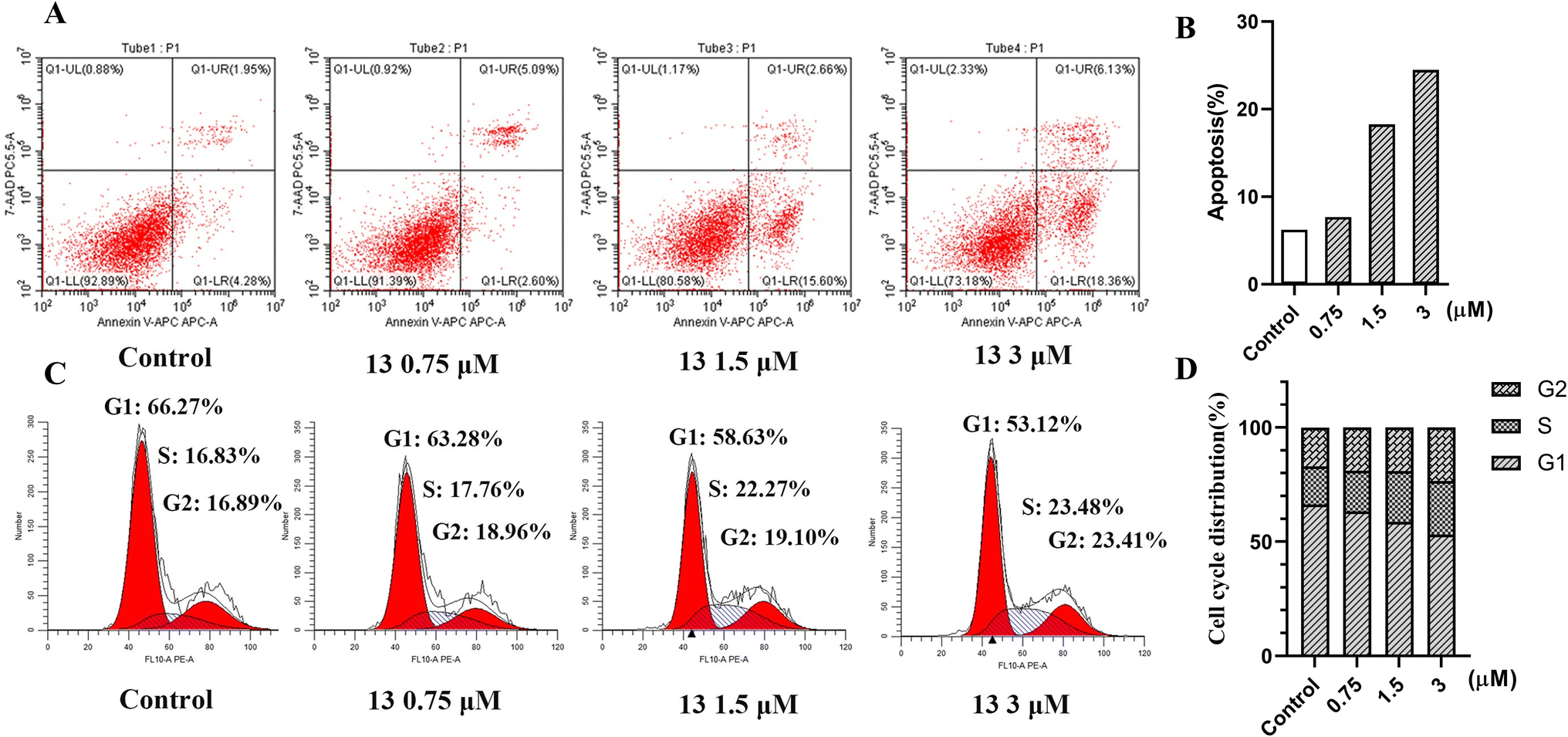
Inhibition of HDAC significantly induced apoptosis and blocked the cell cycle. To investigate the cellular mechanisms of compound 13, its effects on the apoptosis and cell cycle were tested in HeLa cells by the flow cytometry analysis. As shown in Fig. 5A and B, compared to the control, apoptosis analysis by Annexin-V APC/7-AAD revealed that the treatment of HeLa cells with different concentrations (0.75 μM, 1.5 μM and 3 μM) of 13 for 48 h caused a significant dose-dependent increase (7.69%, 18.26% and 24.49%) in the population of apoptotic cells, indicating that compound 13 could significantly induce apoptosis in a dose-dependent manner. In addition, as shown in Fig. 5C and D, cell cycle analysis by PI staining showed that the treatment with different concentrations (0.75 μM, 1.5 μM and 3 μM) of 13 led to the increase of S and G2 fraction from 16.83 to 23.48 and 16.89 to 23.41, respectively, suggesting that 13 could arrest the HeLa cell cycle at the S phase and G2 phase in a dose-dependent manner.
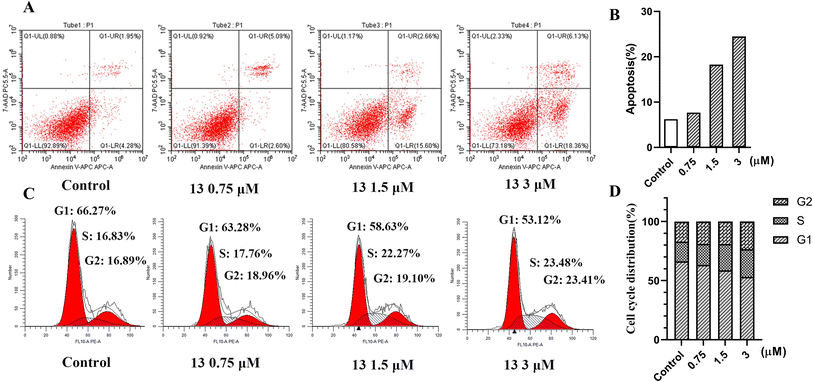 |
| | Fig. 5 (A) Apoptosis analysis of compound 13 in HeLa cells for 48 h by flow cytometry. (B) Ability of compound 13 to induce apoptosis in HeLa cells, apoptosis% = early apoptosis (Q1-LR) + late apoptosis (Q1-UR). (C) Cell cycle analysis of compound 13 in HeLa cells for 48 h by flow cytometry. (D) Percentages of the cell cycle for compound 13 with different concentrations in HeLa cells. | |
Cytotoxicity on HUVECs and tube formation assay
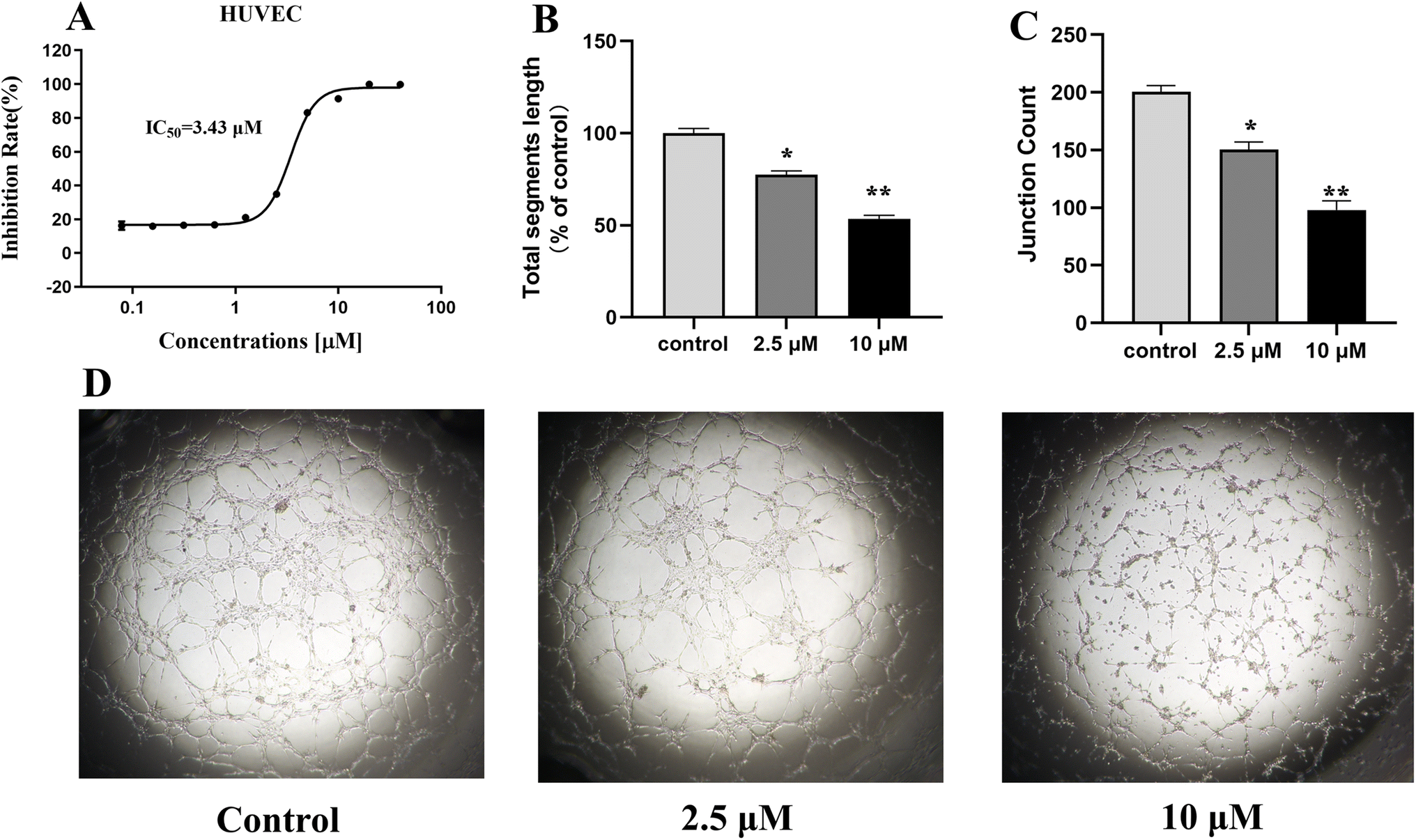
The formation of microvascular tube is one of the key functions in angiogenesis. Due to the significant VEGFR-2 inhibitory activity of compound 13 at the enzyme level, which was further selected to investigate the anti-angiogenic effect. Firstly, we carried out a cytotoxicity test of compound 13 for 72 h against HUVEC using CellTiter-Glo® (CTG) luminescent cell viability assay, and the result (Fig. 6A) showed that compound 13 displayed effective inhibitory activity on HUVEC with IC50 value of 3.44 μM, suggesting that 13 might have potential anti-angiogenic activity. Therefore, the effect of 13 on tube formation was further investigated. As showed in Fig. 6B–D, 13 suppressed the tube length in a dose-dependent manner. Compared to control, the tubule lengths of HUVECs were decreased to 77% and 53%, and the junctions were decreased from 200 to 150 and 98 after the treatment with 2.5 μM and 10 μM of 13 for 72 h, respectively, suggesting that 13 had significant anti-angiogenic effect.
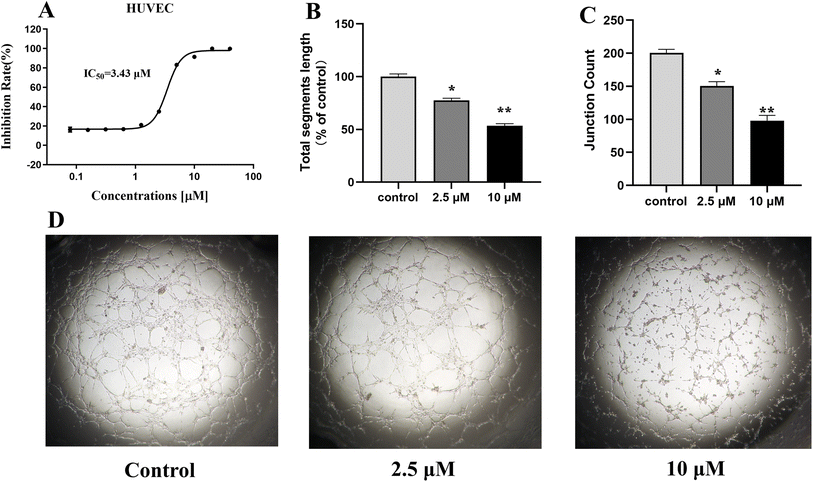 |
| | Fig. 6 Anti-angiogenic effect of 13 in vitro. (A) Cytotoxicity of 13 on HUVEC. (B and C) Quantitative analysis of 13 on the tubule length and junctions of HUVECs. (D) Representative images of the tubular network of HUVECs treated with DMSO or 13. Assays were performed in replicates (n ≥ 2). All data were expressed as the mean ± SEM. *P < 0.05, **P < 0.01, compared to the control group, ordinary one-way ANOVA multiple comparisons. | |
Molecular docking study
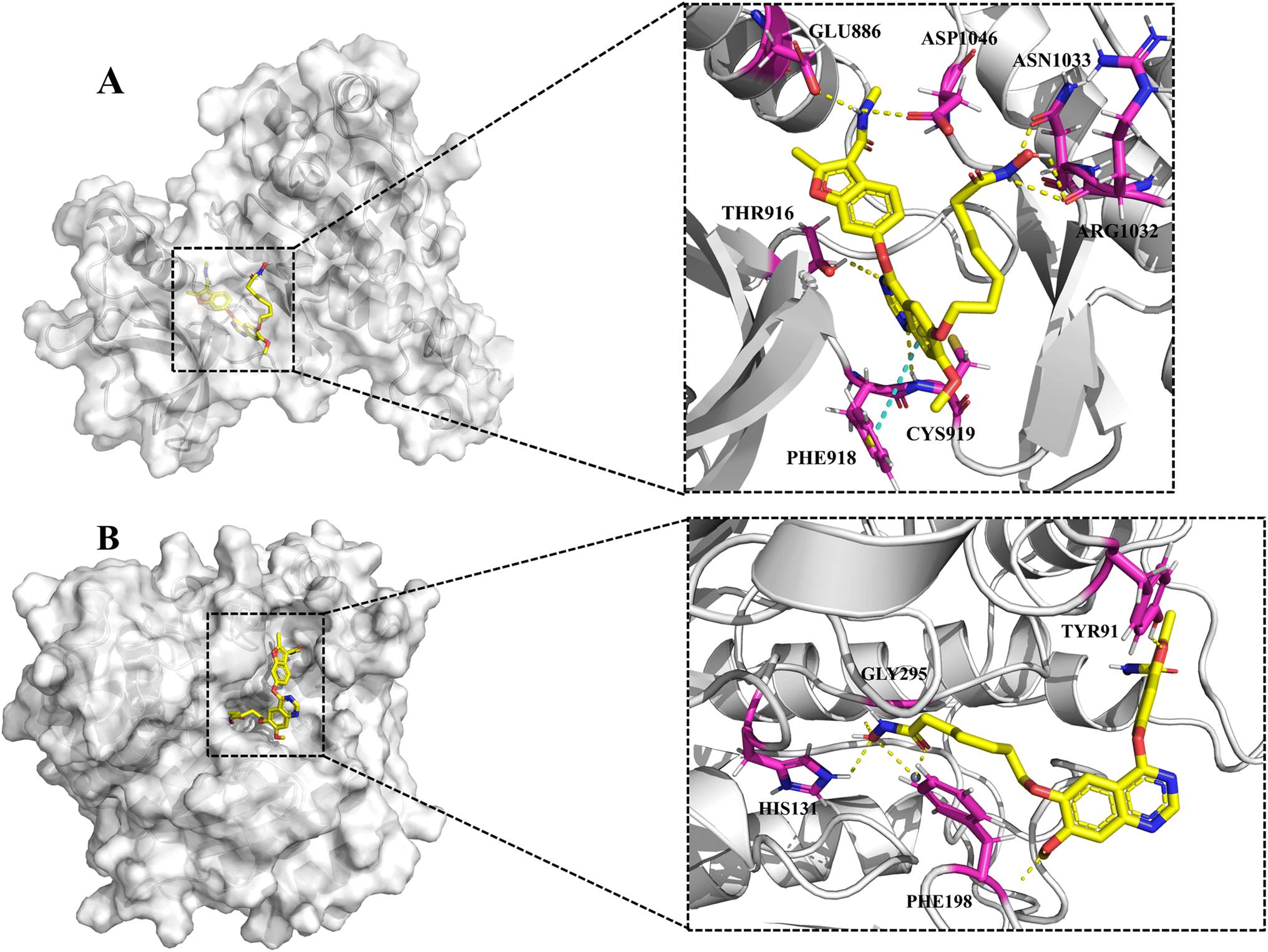
To explore the putative binding mode of 13 with HDAC1 and VEGFR-2, molecular docking studies of 13 were performed using the VEGFR-2/naphthamide inhibitor complex (PDB ID: 3B8R) and HDAC1/SAHA complex (PDB ID: 1C3S) as templates. As shown in Fig. 7A, the binding mode of 13 with VEGFR-2 revealed that the N,2-dimethyl-6-(quinazolin-4-yloxy)benzofuran-3-carboxamide moiety of 13 could occupy the ATP-binding pocket of VEGFR-2 in a similar way to the fruquintinib. The quinazoline ring formed two hydrogen bonds with Cys919 of the hinge region and THR916, and established a parallel π–π interaction with PHE918. The amide group on benzofuran formed two hydrogen bonds with Asp1046 and Glu885, respectively. Notably, compared with fruquintinib, the isohydroxamic acid tail extending into the solvent region, formed extra hydrogen bonds with ASN1033 and ARG1032, respectively. On the other hand, the binding mode of 13 with HDAC1 (Fig. 7B) revealed that the hydroxamic acid group could enter into the bottom of active site and formed multiple key interactions, including chelation with Zn2+, hydrogen bonds with HIS131 and Gly295. The aliphatic chain occupied the hydrophobic channel and created important hydrophobic interactions. The 4-(benzofuran-6-yloxy)quinazoline terminal backbone was located in the entrance of active site, the methoxy on the quinazoline ring and the oxygen atom of benzofuran formed two hydrogen bonds with PHE198 and TYR91, respectively. These finding provided strong support for the potent inhibitory activity of 13 against VEGFR-2 and HDAC1.
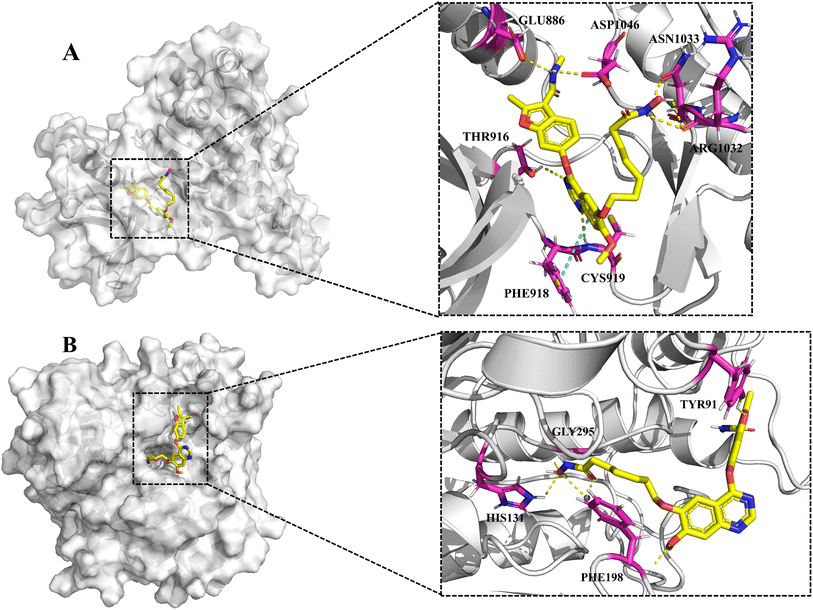 |
| | Fig. 7 (A) The proposed binding mode of 13 in the ATP-binding pocket of VEGFR-2 (PDB: 3B8R). (B) The proposed binding mode of 13 in the active pocket of HDAC1 (PDB: 1C3S). | |
Conclusion
In this paper, we designed and synthesized a series of 4-(benzofuran-6-yloxy)quinazoline-based derivatives as VEGFR-2/HDAC dual inhibitors. Biological activity evaluations led to the identification of compound 13, which exhibited the most potent inhibitory activity against VEGFR-2 and HDAC1 with IC50 values of 57.83 nM and 9.82 nM, and displayed moderate to significant antiproliferative activity against MCF-7, A549, HeLa and HUVEC with IC50 values of 17.86 μM, 4.59 μM, 1.49 μM and 3.43 μM. Further mechanism of action studies revealed that compound 13 blocked the cell cycle at the S and G2 phases, and induced significant apoptosis in HeLa cells, as well as exhibiting significant anti-angiogenic activity. Additionally, molecular docking studies showed that compound 13 could enter well into the ATP-binding pocket of VEGFR-2 and active pocket of HDAC1, and created multiple key interactions, which supported the potential inhibitory activity of 13 against VEGFR-2 and HDAC1. Overall, these above findings suggested that 13 could be as a promising lead targeting VEGFR-2 and HDAC1, and deserved further optimization and investigation for cancer therapy. Melting points were determined on a Melting Point XT5B apparatus (KeYi, Beijing). The purities of target compounds were determined by high-performance liquid chromatography (HPLC), performed on an Elite EClassical 3100 machine with a C18 reverse-phase column (Agilent 5 TC-C18, 4.6 mm × 250 mm) at room temperature. Mobile phase A (100% methanol) and mobile phase B (0.1% TFA in water) were used in a gradient elution program (0 min, phase A: 60%, phase B: 40%; 30 min, phase A: 100%, phase B: 0%) with a flow rate of 1.0 mL min−1. UV detection was conducted at 230 nm.
Experimental section
Chemistry
All reagents and solvents were obtained from commercially available sources and were used without further purification. All the reactions were monitored by TLC using silica gel GF/UV 254. Column chromatography was performed using silica gel (200–300 mesh). The 1H and 13C NMR spectroscopy were performed using Bruker AV-500/400 with tetramethylsilane (TMS) as an internal standard. High resolution mass spectrometry was performed on an Agilent Accurate-Mass UHPLC-QTOF 1290-6545 in ESI mode.
Synthesis of intermediate 1
3,4-Dihydro-7-methoxy-4-oxoquinazolin-6-yl acetate (5.60 g, 24 mmol) was added into 70 mL thionyl chloride, and then 10 mL N,N-dimethylformamide (DMF) was added. The mixture was heated at 80 °C for 4 h. TLC monitored completion of the reaction, then the mixture was cooled to room temperature and concentrated to afford a yellow solid as intermediate 1 (5.72 g, 95%).
Synthesis of intermediate 2
Intermediate 1 (5.70 g, 23 mmol) and LiOH (1.10 g, 46 mmol) were added to 60 mL THF–H2O (4:1, v/v) and stirred at 35 °C for 1 h. TLC monitored reaction completion, then the reaction mixture was concentrated under reduced pressure to remove THF and ice water was added. The solution was adjusted to pH 3–4 with 1.0 M hydrochloric acid. The filter cake was collected by filtration and dried to give a yellow solid as intermediate 2 (4.75 g, 86%).
Synthesis of intermediate 3a
Intermediate 2 (1.25 g, 4.9 mmol), K2CO3 (1.39 g, 10.1 mmol) and methyl 6-bromohexanoate (2.11 g, 10.1 mmol) were added to 3 mL acetonitrile and the mixture was stirred at 80 °C for 8 h. TLC monitored reaction completion. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated to give a residue, which was purified by column chromatography to obtain a yellow solid as intermediate 3a (1.22 g, 61%). 1H NMR (400 MHz, chloroform-d) δ 8.88 (s, 1H), 7.39 (s, 1H), 7.35 (s, 1H), 4.27–4.16 (m, 2H), 4.07 (s, 3H), 3.70 (s, 3H), 2.46–2.35 (m, 2H), 2.07–1.94 (m, 2H), 1.85–1.72 (m, 2H), 1.65–1.55 (m, 2H).
Synthesis of intermediate 3b
Intermediate 3b was prepared from intermediate 2 (0.61 g, 2.8 mmol) and methyl 7-bromoheptanoate (1.08 g, 4.8 mmol) in a similar manner as described for compound 3a (0.62 g, 63%). 1H NMR (400 MHz, chloroform-d) δ 8.88 (s, 1H), 7.39 (s, 1H), 7.35 (s, 1H), 4.26–4.16 (m, 2H), 4.06 (s, 3H), 3.68 (s, 3H), 2.43–2.30 (m, 2H), 2.02–1.93 (m, 2H), 1.78–1.68 (m, 2H), 1.62–1.53 (m, 2H), 1.52–1.41 (m, 2H).
Synthesis of intermediate 3c
Intermediate 3c was prepared from intermediate 2 (0.3 g, 1.4 mmol) and methyl 8-bromooctanoate (0.6 g, 2.5 mmol) in a similar manner as described for compound 3a (0.33 g, 65%). 1H NMR (500 MHz, DMSO-d6) δ 8.86 (s, 1H), 7.44 (s, 1H), 7.36 (s, 1H), 4.17 (t, J = 6.5 Hz, 2H), 4.01 (s, 3H), 3.58 (s, 3H), 2.30 (t, J = 7.4 Hz, 2H), 1.88–1.74 (m, 2H), 1.60–1.50 (m, 2H), 1.50–1.41 (m, 2H), 1.40–1.32 (m, 2H), 1.32–1.24 (m, 2H).
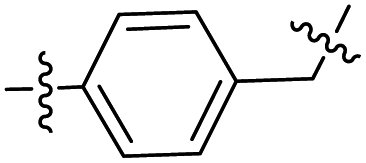
Synthesis of intermediate 3d
Intermediate 2 (0.35 g, 1.6 mmol), Cs2CO3 (1.12 g, 3.1 mmol) and methyl 3-(4-bromomethyl)cinnamate (1.82 g, 0.8 mmol) were added to 3 mL DMF and the mixture was stirred at room temperature for 2 h. TLC monitored completion of the reaction. 20 mL water was poured into the mixture and stirred for 30 min. The mixture solution was filtered to obtain a brown solid as intermediate 3d (0.45 g, 74%).
Synthesis of intermediate 3e
Intermediate 3e was prepared from intermediate 2 (0.51 g, 2.3 mmol) and methyl 4-bromomethylbenzoate (0.71 g, 3.1 mmol) in a similar manner as described for compound 3d (0.59 g, 72%).
Synthesis of intermediate 4
2-Hydroxy-4-methoxybenzaldehyde (10.12 g, 66 mmol), K2CO3 (14.11 g, 101 mmol) and ethyl 2-bromopropionate (9.4 mL, 72.6 mmol) were added to 40 mL DMF and the reaction was stirred at room temperature for 2 h. TLC monitored completion of the reaction. 40 mL ice water was added and then extracted with ethyl acetate (3 × 40 mL). The organic layer was washed with saturated salt water and dried with anhydrous Na2SO4. The organic layer was concentrated to yield colourless liquid as intermediate 4 (9.98 g, 60%). 1H NMR (500 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.68 (d, J = 8.7 Hz, 1H), 6.68 (dd, J = 8.7, 1.5 Hz, 1H), 6.61 (d, J = 2.2 Hz, 1H), 5.25 (q, J = 6.7 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 1.58 (d, J = 6.8 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H).
Synthesis of intermediate 5
Intermediate 4 (10.00 g, 40 mmol) and NaOH (2.00 g, 50 mmol) were added into 40 mL ethanol and stirred for 1 h. After complete reaction by TLC, the mixture was poured into 100 mL water and the pH was adjusted to 2–3 with 1.0 M hydrochloric acid. The solution was filtered and dried to give a white solid as intermediate 5 (7.91 g, 89%). 1H NMR (500 MHz, DMSO-d6) δ 10.27 (s, 1H), 7.68 (d, J = 8.7 Hz, 1H), 6.69 (dd, J = 8.7, 2.2 Hz, 1H), 6.59 (d, J = 2.2 Hz, 1H), 5.15 (q, J = 6.8 Hz, 1H), 3.83 (s, 3H), 1.57 (d, J = 6.7 Hz, 3H).
Synthesis of intermediate 6
Intermediate 5 (8.89 g, 40 mmol) and NaOAc (9.84 g, 120 mmol) were added to 30 mL acetic anhydride and stirred at reflux for 2 h. After complete reaction by TLC, the mixture was cooled to room temperature and poured into 100 mL ice water, and neutralized with 1.0 M sodium hydroxide. The solution was extracted with ethyl acetate (3 × 50 mL), the organic layer was dried over anhydrous Na2SO4 and concentrated to yield brown liquid as intermediate 6 (5.16 g, 80%). 1H NMR (500 MHz, DMSO-d6) δ 7.37 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 1.5 Hz, 1H), 6.81 (dd, J = 8.5, 2.3 Hz, 1H), 6.52–6.34 (m, 1H), 3.77 (s, 3H), 2.39 (s, 3H).
Synthesis of intermediate 7
AlCl3 (15.11 g, 110 mmol) was added to 50 mL dichloromethane at 0 °C, and then oxalyl chloride (9.6 mL, 110 mmol) was added dropwise and kept below 0 °C for stirring for 3 min. Intermediate 6 (7.62 g, 0.047 mol) was added and stirred at room temperature for 30 min. TLC monitored completion of the reaction. Dichloromethane was concentrated and ice water was added. The mixture was stirred for 30 min and filtered to afford green solid (7.86 g, 82%). 1H NMR (500 MHz, DMSO-d6) δ 7.74 (d, J = 8.6 Hz, 1H), 7.20 (d, J = 2.3 Hz, 1H), 6.93 (dd, J = 8.7, 2.3 Hz, 1H), 3.80 (s, 3H), 2.69 (s, 3H).
Synthesis of intermediate 8
Intermediate 7 (3.25 g, 15 mmol) was added to 20 mL dichloromethane and boron tribromide (7.58 mL, 75 mmol) was added dropwise at 0 °C. The mixture was stirred at room temperature for 1 h. TLC monitored completion of the reaction. The reaction solution was poured into 50 mL ice water and stirred until no air bubbles were released, and then the mixture was filtered to give a brown solid as intermediate 8 (2.51 g, 87%). 1H-NMR (400 MHz, DMSO-d6) δ 12.80 (s, 1H), 9.58 (s, 1H), 7.64 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.79 (dd, J = 8.5, 2.1 Hz, 1H), 2.66 (s, 3H).
Synthesis of intermediate 9a
A mixture of intermediate 8 (0.54 g, 2.8 mmol), HATU (1.8 g, 4.2 mmol), DIEA (1.16 mL, 5.6 mmol) and methylamine solution (0.42 mL, 5.6 mmol) was stirred for 1 h. TLC monitored completion of the reaction. 20 mL ice water was added and the mixture was extracted with dichloromethane (100 mL × 3). The organic layer was concentrated to give a residue, which was purified by column chromatography to obtain a yellow solid as intermediate 9a (0.43 g, 70%). 1H NMR (500 MHz, DMSO-d6) δ 9.63 (s, 1H), 7.79 (d, J = 4.7 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 6.77 (dd, J = 8.5, 2.1 Hz, 1H), 2.79 (d, J = 4.6 Hz, 3H), 2.55 (s, 3H).
Synthesis of intermediate 9b
Intermediate 9b was prepared from intermediate 8 (0.3 g, 1.5 mmol) and ethylamine (0.08 g, 1.9 mmol) in a similar manner as described for compound 9a (0.26 g, 77%). 1H NMR (400 MHz, chloroform-d) δ 7.50–7.38 (m, 1H), 6.98 (d, J = 3.7 Hz, 1H), 6.89–6.81 (m, 1H), 5.86 (s, 1H), 5.40 (s, 1H), 3.62–3.48 (m, 2H), 2.70 (s, 3H), 1.35–1.30 (m, 3H).
Synthesis of intermediate 9c
Intermediate 9c was prepared from intermediate 8 (1 g, 5.2 mmol) and isopropylamine (0.94 mL, 10.4 mmol) in a similar manner as described for compound 9a (0.91 g, 75%). 1H NMR (500 MHz, chloroform-d) δ 7.39 (d, J = 8.5 Hz, 1H), 6.99 (d, J = 2.2 Hz, 1H), 6.88 (dd, J = 8.5, 2.2 Hz, 1H), 5.77 (d, J = 8.0 Hz, 1H), 4.42–4.29 (m, 1H), 2.68 (s, 3H), 1.32 (d, J = 6.5 Hz, 6H).
Synthesis of intermediate 10a
A mixture of intermediate 3a (0.12 g, 0.35 mmol), K2CO3 (0.071 g, 0.51 mmol) and 9a (0.09 g, 0.44 mmol) in 3 mL acetonitrile was stirred at 80 °C for 8 h. TLC monitored completion of the reaction. The mixture was hot filtered and the filtrate is concentrated to obtain a residue, which was purified by column chromatography to afford a yellow solid as intermediate 10a (0.12 g, 67%). HRMS (ESI) for C27H29N3O7 [M–H]−. Calcd: 506.1927, found: 506.1954.
Synthesis of intermediate 10b
Intermediate 10b was prepared from intermediate 3b (2.1 g, 5.8 mmol) and 9a (1.2 g, 5.8 mmol) in a similar manner as described for compound 10a (2.18 g, 67%). HRMS (ESI) for C28H31N3O7 [M–H]−. Calcd: 520.2084, found: 520.2034.
Synthesis of intermediate 10c
Intermediate 10c was prepared from intermediate 3c (2.42 g, 6.6 mmol) and 9a (1.36 g, 6.6 mmol) in a similar manner as described for compound 10a (2.40 g, 68%). HRMS (ESI) for C29H33N3O7 [M–H]−. Calcd: 534.2240, found: 534.2196.
Synthesis of intermediate 10d
Intermediate 10c was prepared from intermediate 3d (1.78 g, 4.6 mmol) and 9a (0.95 g, 4.6 mmol) in a similar manner as described for compound 10a (1.81 g, 75%). 1H NMR (400 MHz, DMSO-d6) δ 8.55 (s, 1H), 8.04 (d, J = 7.9 Hz, 2H), 7.98 (s, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.77 (s, 1H), 7.69 (d, J = 7.8 Hz, 2H), 7.63 (s, 1H), 7.45 (s, 1H), 7.25 (d, J = 8.5 Hz, 1H), 5.45 (s, 2H), 4.03 (s, 3H), 3.88 (s, 3H), 2.84 (s, 3H), 2.65 (s, 3H).
Synthesis of intermediate 10e
Intermediate 10e was prepared from intermediate 3e (1.22 g, 3.4 mmol) and 9a (0.69 g, 3.4 mmol) in a similar manner as described for compound 10a (1.11 g, 62%). 1H NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H), 7.98 (s, 1H), 7.79 (d, J = 8.2 Hz, 3H), 7.75 (s, 1H), 7.70 (d, J = 16.1 Hz, 1H), 7.63 (s, 1H), 7.58 (d, J = 7.8 Hz, 2H), 7.43 (s, 1H), 7.25 (d, J = 8.3 Hz, 1H), 6.68 (d, J = 13.5 Hz, 1H), 5.37 (s, 2H), 4.02 (s, 3H), 3.74 (s, 3H), 2.84 (s, 3H), 2.65 (s, 3H).
Synthesis of intermediate 10f
Intermediate 10f was prepared from intermediate 3a (0.56 g, 1.7 mmol) and 9b (0.36 g, 1.7 mmol) in a similar manner as described for compound 10a (0.62 g, 72%). HRMS (ESI) for C28H31N3O7 [M–H]−. Calcd: 520.2084, found: 520.2100.
Synthesis of intermediate 10g
Intermediate 10g was prepared from intermediate 3b (0.25 g, 0.71 mmol) and 9b (0.16 g, 0.71 mmol) in a similar manner as described for compound 10a (0.24 g, 63%). HRMS (ESI) for C29H33N3O7 [M–H]−. Calcd: 534.2240, found: 520.2206.
Synthesis of intermediate 10h
Intermediate 10h was prepared from intermediate 3c (1.24 g, 0.34 mmol) and 9b (0.74 g, 3.4 mmol) in a similar manner as described for compound 10a (1.26 g, 68%). HRMS (ESI) for C30H35N3O7 [M–H]−. Calcd: 548.2397, found: 548.2410.
Synthesis of intermediate 10i
Intermediate 10i was prepared from intermediate 3d (1.38 g, 3.5 mmol) and 9b (0.78 g, 3.5 mmol) in a similar manner as described for compound 10a (1.22 g, 60%).
Synthesis of intermediate 10j
Intermediate 10j was prepared from intermediate 3e (1.25 g, 3.5 mmol) and 9b (0.76 g, 3.5 mmol) in a similar manner as described for compound 10a (1.24 g, 66%). HRMS (ESI) for C32H29N3O7 [M–H]−. Calcd: 566.1927, found: 566.1887.
Synthesis of intermediate 10k
Intermediate 10k was prepared from intermediate 3a (0.78 g, 2.3 mmol) and 9c (0.53 g, 2.3 mmol) in a similar manner as described for compound 10a (0.96 g, 78%). HRMS (ESI) for C29H33N3O7 [M–H]−. Calcd: 534.2240, found: 534.2223.
Synthesis of intermediate 10l
Intermediate 10l was prepared from intermediate 3b (1.51 g, 4.3 mmol) and 9c (0.99 g, 4.3 mmol) in a similar manner as described for compound 10a (1.63 g, 69%). HRMS (ESI) for C30H35N3O7 [M–H]−. Calcd: 548.2397, found: 548.2410.
Synthesis of intermediate 10m
Intermediate 10m was prepared from intermediate 3c (1.49 g, 4.1 mmol) and 9c (0.95 g, 4.1 mmol) in a similar manner as described for compound 10a (1.44 g, 63%). HRMS (ESI) for C31H37N3O7 [M–H]−. Calcd: 562.2553, found: 562.2589.
Synthesis of intermediate 10n
Intermediate 10m was prepared from intermediate 3d (1.15 g, 2.99 mmol) and 9c (0.71 g, 3.04 mmol) in a similar manner as described for compound 10a (1.21 g, 69%).
Synthesis of intermediate 10o
Intermediate 10o was prepared from intermediate 3e (1.07 g, 2.99 mmol) and 9c (0.68 g, 2.92 mmol) in a similar manner as described for compound 10a (1.11 g, 69%).
Synthesis of intermediate 11a
Intermediate 10a (1.72 g, 3.4 mmol) was added in 20 mL THF/H2O (4:1), and LiOH (0.33 g, 13.61 mmol) was added. The mixture was stirred at 35 °C for 8 h. TLC monitored completion of the reaction. THF was removed under reduced pressure and ice water was added. The solution was adjusted to pH 3–4 with 1.0 M hydrochloric acid. The filter cake was collected by filtration and dried to afford a yellow solid as intermediate 11a (1.32 g, 79%), which is directly followed by the next step without purification.
Synthesis of intermediate 11b
Intermediate 11b was prepared from intermediate 10b (1.79 g, 3.2 mmol) and LiOH (0.31 g, 12.9 mmol) in a similar manner as described for compound 11a (1.39 g, 80%).
Synthesis of intermediate 11c
Intermediate 11c was prepared from intermediate 10c (0.88 g, 1.64 mmol) and LiOH (0.16 g, 6.51 mmol) in a similar manner as described for compound 11a (0.73 g, 86%).
Synthesis of intermediate 11d
Intermediate 11d was prepared from intermediate 10d (1.29 g, 2.33 mol) in a similar manner as described for compound 11a (1.11 g, 88%).
Synthesis of intermediate 11e
Intermediate 11e was prepared from intermediate 10e (1.36 g, 2.5 mmol) in a similar manner as described for compound 11a (1.17 g, 89%).
Synthesis of intermediate 11f
Intermediate 11f was prepared from intermediate 10f (1.18 g, 2.26 mmol) and LiOH (0.21 g, 8.75 mmol) in a similar manner as described for compound 11a (0.88 g, 77%).
Synthesis of intermediate 11g
Intermediate 11g was prepared from intermediate 10g (1.21 g, 2.26 mol) and LiOH (0.22 g, 9.04 mmol) in a similar manner as described for compound 11a (0.99 g, 84%).
Synthesis of intermediate 11h
Intermediate 11h was prepared from intermediate 10h (1.62 g, 2.95 mmol) and LiOH (0.28 g, 11.8 mmol) in a similar manner as described for compound 11a (1.33 g, 84%).
Synthesis of intermediate 11i
Intermediate 11i was prepared from intermediate 10i (0.86 g, 1.52 mmol) and LiOH (0.5 g, 6.07 mmol) in a similar manner as described for compound 11a (0.73 g, 87%).
Synthesis of intermediate 11j
Intermediate 11j was prepared from intermediate 10j (1.80 g, 3.33 mmol) and LiOH (0.32 g, 13.32 mmol) in a similar manner as described for compound 11a (1.38 g, 79%).
Synthesis of intermediate 11k
Intermediate 11k was prepared from intermediate 10k (1.28 g, 2.39 mol) and LiOH (0.23 g, 9.57 mmol) in a similar manner as described for compound 11a (0.97 g, 78%).
Synthesis of intermediate 11l
Intermediate 11l was prepared from intermediate 10l (0.92 g, 1.68 mmol) and LiOH (0.16 g, 6.51 mmol) in a similar manner as described for compound 11a (0.75 g, 84%).
Synthesis of intermediate 11m
Intermediate 11m was prepared from intermediate 10m (1.38 g, 2.45 mol) and LiOH (0.23 g, 9.81 mmol) in a similar manner as described for compound 11a (1.17 g, 87%).
Synthesis of intermediate 11n
Intermediate 11n was prepared from intermediate 10n (1.66 g, 2.86 mol) and LiOH (0.27 g, 11.41 mmol) in a similar manner as described for compound 11a (1.42 g, 88%).
Synthesis of intermediate 11o
Intermediate 11o was prepared from intermediate 10o (1.64 g, 2.95 mmol) and LiOH (0.28 g, 11.81 mmol) in a similar manner as described for compound 11a (1.34 g, 84%).
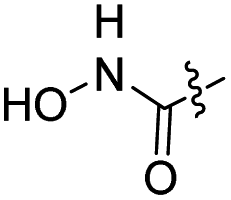
Synthesis of target compound 12
Hydroxylamine hydrochloride (24 mg, 0.36 mmol) and DIEA (58 μL, 0.36 mmol) were added to 2 mL DMF and stirred at 35 °C for 1 h to prepare solution A. 11a (0.12 g, 0.23 mmol) was added to 2 mL DMF, followed by HATU (0.13 g, 0.36 mmol) and DIEA (77 μL, 0.48 mmol), stirred for 5 min, and then which was added dropwise to solution A. The mixture was stirred at room temperature for 1 h. TLC monitored completion of the reaction. 10 mL ice water was added and stirred for 30 min. The filter cake was collected by filtration and dried to afford a white solid, which was purified by column chromatography as target compound 12 (80 mg, 65%). HPLC: tR 9.110 min, purity: 94.1%. Mp: 192.3–194.3. HRMS (ESI) for C26H28N4O7 [M–H]−. Calcd: 507.1880, found: 507.1889. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.70 (s, 1H), 8.53 (s, 1H), 7.99 (q, J = 4.5 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.59 (s, 1H), 7.39 (s, 1H), 7.25 (dd, J = 8.5, 2.1 Hz, 1H), 4.17 (t, J = 6.4 Hz, 2H), 4.00 (s, 3H), 2.84 (d, J = 4.5 Hz, 3H), 2.65 (s, 3H), 1.99 (t, J = 7.3 Hz, 2H), 1.86–1.77 (m, 2H), 1.64–1.55 (m, 2H), 1.48–1.39 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.51, 165.52, 163.71, 158.73, 156.35, 153.36, 152.62, 149.96, 149.90, 149.24, 124.06, 121.13, 118.46, 113.11, 110.22, 107.25, 106.00, 101.85, 69.05, 56.65, 32.70, 28.75, 26.54, 25.64, 25.34, 14.14.
Synthesis of target compound 13
Compound 13 was prepared from intermediate 11b (25 mg, 0.049 mmol) in a similar manner as described for compound 12 (15 mg, 60%). HPLC: tR 11.430 min, purity: 94.4%. Mp: 163.0–165.0. HRMS (ESI) for C27H30N4O7 [M–H]−. Calcd: 521.2036, found: 521.2047. 1H NMR (500 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.73 (s, 1H), 8.50 (s, 1H), 8.02 (d, J = 4.7 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 2.1 Hz, 1H), 7.54 (s, 1H), 7.35 (s, 1H), 7.23 (dd, J = 8.5, 2.2 Hz, 1H), 4.13 (t, J = 6.5 Hz, 2H), 3.98 (s, 3H), 2.83 (d, J = 4.4 Hz, 3H), 2.63 (s, 3H), 1.96 (t, J = 7.3 Hz, 2H), 1.85–1.72 (m, 2H), 1.56–1.48 (m, 2H), 1.47–1.40 (m, 2H), 1.35–1.29 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.72, 165.49, 163.80, 158.75, 156.31, 153.32, 152.58, 149.92, 149.84, 149.14, 124.06, 121.12, 118.45, 113.06, 110.17, 107.12, 105.97, 101.73, 69.05, 56.63, 32.67, 28.85, 28.76, 26.55, 25.71, 25.54, 14.13.
Synthesis of target compound 14
Compound 14 was prepared from intermediate 11c (56 mg, 0107 mmol) in a similar manner as described for compound 12 (35 mg, 60%). HPLC: tR 13.928 min, purity: 95.9%. Mp: 187.7–189.7. HRMS (ESI) for C28H32N4O7 [M–H]−. Calcd: 535.2193, found: 535.2199. 1H NMR (500 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.67 (s, 1H), 8.52 (s, 1H), 7.99 (d, J = 4.7 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.58 (s, 1H), 7.39 (s, 1H), 7.25 (dd, J = 8.5, 2.1 Hz, 1H), 4.17 (t, J = 6.5 Hz, 2H), 4.00 (s, 3H), 2.84 (d, J = 4.5 Hz, 3H), 2.65 (s, 3H), 1.95 (t, J = 7.3 Hz, 2H), 1.83–1.77 (m, 2H), 1.54–1.48 (m, 2H), 1.47–1.42 (m, 2H), 1.38–1.33 (m, 2H), 1.30–1.24 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.59, 165.52, 163.71, 158.73, 156.36, 153.35, 152.61, 149.98, 149.89, 149.22, 124.06, 121.14, 118.47, 113.11, 110.22, 107.23, 106.01, 101.81, 69.12, 56.66, 32.72, 28.99, 28.94, 28.92, 26.55, 25.88, 25.54, 14.14.
Synthesis of target compound 15
Compound 15 was prepared from intermediate 11d (44 mg, 0.081 mmol) in a similar manner as described for compound 12 (32 mg, 73%). HPLC: tR 9.438 min, purity: 92.5%. Mp: 203.2–205.2. HRMS (ESI) for C28H24N4O7 [M–H]−. Calcd: 527.1567, found: 527.1574. 1H NMR (400 MHz, DMSO-d6) δ 11.27 (s, 1H), 9.09 (s, 1H), 8.55 (s, 1H), 8.00 (s, 1H), 7.86–7.78 (m, 3H), 7.76 (s, 1H), 7.66–7.53 (m, 2H), 7.44 (s, 1H), 7.26 (s, 1H), 5.39 (s, 2H), 4.02 (s, 3H), 2.84 (s, 3H), 2.65 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.62, 164.46, 163.71, 158.75, 156.39, 153.35, 152.85, 149.85, 149.45, 139.92, 128.22(2C), 127.57(2C), 124.11, 121.19, 120.56, 118.43, 116.80, 113.11, 110.12, 107.42, 106.00, 102.66, 70.19, 56.76, 26.55, 14.14.
Synthesis of target compound 16
Compound 16 was prepared from intermediate 11e (44 mg, 0.086 mmol) in a similar manner as described for compound 12 (28 mg, 62%). HPLC: tR 12.207 min, purity: 93.3%. Mp: 225.1–227.1. HRMS (ESI) for C30H26N4O7 [M–H]−. Calcd: 553.1723, found: 553.1738. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.05 (s, 1H), 8.56 (s, 1H), 7.99 (s, 1H), 7.82 (s, 1H), 7.75 (s, 1H), 7.63 (s, 3H), 7.57 (s, 2H), 7.49 (d, J = 15.0 Hz, 1H), 7.43 (s, 1H), 7.25 (s, 1H), 6.50 (d, J = 15.0 Hz, 1H), 5.35 (s, 2H), 4.01 (s, 3H), 2.84 (s, 3H), 2.65 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.61, 163.70, 163.62, 158.75, 156.40, 153.36, 152.84, 149.86, 149.49, 149.42, 138.44, 138.11, 135.08, 128.94(2C), 128.12(2C), 124.10, 121.19, 119.78, 118.44, 113.11, 110.14, 107.43, 106.01, 102.62, 70.34, 56.74, 26.56, 14.15.
Synthesis of target compound 17
11a (0.15 g, 0.30 mmol) was added into 4 mL DMF at room temperature. Then the HATU (0.16 g, 0.42 μL), DIEA (140 mg, 0.84 mmol) and o-phenylenediamine (46 mg, 0.42 mmol) were added and the mixture was stirred for 1 h. After complete reaction by TLC, 15 mL ice water was added and the mixture was filtered to give a white solid, which was further dried and purified by column chromatography to obtain a white powder as compound 17 (0.13 g, 73%). HPLC: tR 12.708 min, purity: 91.0%. Mp: 207.8–209.8. HRMS (ESI) for C32H36N5O6 [M–H]−. Calcd: 582.2353, found: 582.2331. 1H NMR (500 MHz, DMSO-d6) δ 9.11 (s, 1H), 8.52 (s, 1H), 7.97 (d, J = 4.6 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.61 (d, J = 2.1 Hz, 1H), 7.58 (s, 1H), 7.38 (s, 1H), 7.25 (dd, J = 8.5, 2.2 Hz, 1H), 7.15 (d, J = 6.1 Hz, 1H), 6.89 (t, J = 6.8 Hz, 1H), 6.72 (d, J = 6.5 Hz, 1H), 6.53 (t, J = 6.8 Hz, 1H), 4.17 (t, J = 6.5 Hz, 2H), 4.00 (s, 3H), 2.84 (d, J = 4.4 Hz, 3H), 2.65 (s, 3H), 2.33 (t, J = 7.4 Hz, 2H), 1.88–1.78 (m, 2H), 1.67–1.57 (m, 2H), 1.52–1.44 (m, 2H), 1.41–1.37 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 171.65, 165.51, 163.71, 158.73, 156.37, 153.36, 152.60, 149.99, 149.90, 149.23, 142.19, 126.13, 125.71, 124.06, 121.14, 118.45, 116.75, 116.43, 113.11, 110.23, 107.22, 105.99, 101.82, 69.14, 56.65, 36.25, 29.12, 29.02, 26.54, 25.91, 25.74, 14.14.
Synthesis of target compound 18
Compound 18 was prepared from intermediate 11b (32 mg, 0.063 mmol) in a similar manner as described for compound 17 (28 mg, 74%). HPLC: tR 10.570 min, purity: 92.5%. Mp: 201.7–203.7. HRMS (ESI) for C33H35N5O6 [M–H]−. Calcd: 596.2509, found: 596.2498. 1H NMR (500 MHz, DMSO-d6) δ 9.11 (s, 1H), 8.52 (s, 1H), 7.97 (d, J = 4.6 Hz, 1H), 7.81 (d, J = 8.6 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.58 (s, 1H), 7.38 (s, 1H), 7.25 (dd, J = 8.5, 2.1 Hz, 1H), 7.16 (dd, J = 7.8, 1.6 Hz, 1H), 6.88 (td, J = 7.6, 1.6 Hz, 1H), 6.72 (dd, J = 8.0, 1.5 Hz, 1H), 6.52 (td, J = 7.5, 1.5 Hz, 1H), 4.18 (t, J = 6.5 Hz, 2H), 4.00 (s, 3H), 2.84 (d, J = 4.4 Hz, 3H), 2.65 (s, 3H), 2.34 (t, J = 7.4 Hz, 2H), 1.89–1.78 (m, 2H), 1.71–1.59 (m, 2H), 1.58–1.47 (m, 2H), 1.47–1.37 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 171.64, 165.52, 163.71, 158.73, 156.37, 153.36, 152.61, 149.99, 149.90, 149.24, 142.24, 126.12, 125.73, 124.11, 124.07, 121.14, 118.46, 116.69, 116.39, 113.12, 110.23, 107.24, 105.99, 101.82, 69.12, 56.65, 36.21, 28.89, 26.54, 26.41, 25.82, 25.74, 14.14.
Synthesis of target compound 19
Compound 19 was prepared from intermediate 11c (30 mg, 0.058 mmol) in a similar manner as described for compound 17 (27 mg, 77%). HPLC: tR 14.940 min, purity: 93.5%. Mp: 164.7–166.7. HRMS (ESI) for C34H37N5O6 [M–H]−. Calcd: 610.2666, found: 610.2678. 1H NMR (500 MHz, DMSO-d6) δ 9.13 (s, 1H), 8.52 (s, 1H), 7.98 (q, J = 4.6 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.60 (s, 1H), 7.39 (s, 1H), 7.25 (dd, J = 8.5, 2.1 Hz, 1H), 7.16 (dd, J = 7.8, 1.6 Hz, 1H), 6.89 (td, J = 7.6, 1.6 Hz, 1H), 6.72 (dd, J = 8.0, 1.5 Hz, 1H), 6.53 (td, J = 7.5, 1.5 Hz, 1H), 4.20 (t, J = 6.5 Hz, 2H), 4.00 (s, 3H), 2.84 (d, J = 4.5 Hz, 3H), 2.65 (s, 3H), 2.37 (t, J = 7.4 Hz, 2H), 1.92–1.82 (m, 2H), 1.75–1.66 (m, 2H), 1.58–1.48 (m, 4H). 13C NMR (126 MHz, DMSO) δ 171.65, 165.51, 163.71, 158.73, 156.37, 153.36, 152.60, 149.99, 149.90, 149.23, 142.19, 126.13, 125.71, 124.14, 124.06, 121.14, 118.45, 116.75, 116.43, 113.11, 110.23, 107.22, 105.99, 101.82, 69.14, 56.65, 36.24, 29.12, 29.02, 28.98, 26.54, 25.91, 25.74, 14.14.
Synthesis of target compound 20
Compound 20 was prepared from intermediate 11d (29 mg, 0.053 mmol) in a similar manner as described for compound 17 (25 mg, 74%). HPLC: tR 11.348 min, purity: 95.5%. Mp: 230.2–232.2. HRMS (ESI) for C34H29N5O6 [M–H]−. Calcd: 602.2040, found: 602.2055. 1H NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.56 (s, 1H), 8.05 (d, J = 7.9 Hz, 2H), 7.99 (s, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.79 (s, 1H), 7.68 (s, 1H), 7.65 (d, J = 6.4 Hz, 2H), 7.46 (s, 1H), 7.26 (d, J = 8.5 Hz, 1H), 7.19 (d, J = 7.9 Hz, 1H), 6.99 (t, J = 7.7 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.61 (t, J = 7.7 Hz, 1H), 5.44 (s, 2H), 4.93 (s, 2H), 4.04 (s, 3H), 2.84 (s, 3H), 2.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.63, 165.53, 163.70, 158.75, 156.43, 153.36, 152.88, 149.86, 149.45, 143.57, 140.18, 134.79, 130.12, 128.47(2C), 128.05(2C), 127.19, 127.01, 124.12, 123.71, 121.20, 118.45, 116.75, 116.59, 113.12, 110.15, 107.48, 106.02, 102.73, 70.20, 56.79, 26.55, 14.15.
Synthesis of target compound 21
Compound 21 was prepared from intermediate 11e (41 mg, 0.084 mmol) in a similar manner as described for compound 17 (42 mg, 84%). HPLC: tR 14.393 min, purity: 93.7%. Mp: 211.5–213.5. HRMS (ESI) for C36H31N5O6 [M–H]−. Calcd: 628.2196, found: 628.2204. 1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.55 (s, 1H), 8.00 (s, 1H), 7.79 (d, J = 13.5 Hz, 2H), 7.87–7.74 (m, 2H), 7.66–7.54 (m, 4H), 7.45 (s, 1H), 7.35 (s, 1H), 7.27 (s, 1H), 6.94 (d, J = 13.6 Hz, 2H), 6.77 (s, 1H), 6.59 (s, 1H), 5.37 (s, 2H), 4.97 (s, 2H), 4.02 (s, 3H), 2.84 (s, 3H), 2.65 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.62, 163.93, 163.71, 158.76, 156.40, 153.36, 152.85, 149.87, 149.52, 149.43, 142.05, 139.66, 138.28, 135.15, 129.01(2C), 128.28(2C), 126.29, 125.24, 124.11, 123.97, 123.02, 121.20, 118.45, 116.80, 116.51, 113.12, 110.15, 107.44, 106.02, 102.63, 70.38, 56.75, 26.56, 14.15.
Synthesis of target compound 22
Compound 22 was prepared from intermediate 11f (0.12 g, 0.24 mmol) in a similar manner as described for compound 12 (0.09 g, 73%). HPLC: tR 10.463 min, purity: 97.7%. Mp: 170.2–172.2. HRMS (ESI) for C27H30N4O7 [M–H]−. Calcd: 521.2036, found: 521.2047. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.69 (s, 1H), 8.52 (s, 1H), 8.09 (s, 1H), 7.79 (d, J = 5.1 Hz, 1H), 7.61 (dd, J = 13.1, 3.8 Hz, 2H), 7.39 (s, 1H), 7.26 (d, J = 7.4 Hz, 1H), 4.17 (s, 2H), 4.00 (s, 3H), 3.38–3.32 (m, 2H), 2.65 (s, 3H), 2.08–1.92 (m, 2H), 1.87–1.74 (m, 2H), 1.66–1.53 (m, 2H), 1.51–1.39 (m, 2H), 1.22–1.13 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 169.50, 165.54, 162.98, 158.57, 156.31, 153.35, 152.62, 149.94, 149.87, 149.22, 124.14, 121.14, 118.47, 113.31, 110.20, 107.23, 106.01, 101.80, 69.03, 56.65, 34.24, 32.69, 28.75, 25.64, 15.39, 15.37, 14.12.
Synthesis of target compound 23
Compound 23 was prepared from intermediate 11g (22 mg, 0.042 mmol) in a similar manner as described for compound 12 (18 mg, 70%). HPLC: tR 12.845 min, purity: 92.3%. Mp: 146.5–148.7. HRMS (ESI) for C28H32N4O7 [M–H]−. Calcd: 535.2193, found: 535.2182. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.68 (s, 1H), 8.52 (s, 1H), 8.10 (s, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.59 (s, 1H), 7.39 (s, 1H), 7.26 (d, J = 8.5 Hz, 1H), 4.27–4.11 (m, 2H), 4.00 (s, 3H), 2.64 (s, 3H), 2.04–1.88 (m, 2H), 1.87–1.74 (m, 2H), 1.60–1.47 (m, 3H), 1.50–1.40 (m, 2H), 1.40–1.28 (m, 2H), 1.17 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.54, 162.98, 162.90, 158.57, 156.34, 153.35, 152.62, 149.97, 149.87, 149.21, 124.14, 121.14, 118.47, 113.31, 110.21, 107.23, 106.01, 101.79, 69.08, 56.66, 34.24, 32.69, 28.89, 28.79, 25.74, 25.55, 15.39, 14.12.
Synthesis of target compound 24
Compound 24 was prepared from intermediate 11h (42 mg, 0.078 mmol) in a similar manner as described for compound 12 (29 mg, 67%). HPLC: tR 10.480 min, purity: 91.8%. Mp: 160.7–162.7. HRMS (ESI) for C29H34N4O7 [M–H]−. Calcd: 549.2349, found: 549.2371. 1H NMR (400 MHz, chloroform-d) δ 10.34 (s, 1H), 8.67 (s, 1H), 8.51 (s, 1H), 8.09 (t, J = 5.5 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.62 (s, 1H), 7.58 (s, 1H), 7.38 (s, 1H), 7.25 (d, J = 8.5 Hz, 1H), 4.17 (t, J = 6.5 Hz, 2H), 3.99 (s, 3H), 3.31 (s, 2H), 2.63 (s, 3H), 1.94 (t, J = 7.4 Hz, 2H), 1.86–1.74 (m, 2H), 1.56–1.47 (m, 2H), 1.46–1.40 (m, 2H), 1.37–1.30 (m, 2H), 1.26–1.21 (m, 2H), 1.17 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.05, 165.52, 162.99, 158.58, 156.32, 153.35, 152.60, 149.96, 149.86, 149.20, 128.29, 124.14, 118.46, 113.30, 110.19, 107.19, 105.99, 101.74, 69.09, 56.63, 34.24, 33.90, 32.72, 29.00, 28.94, 25.88, 25.55, 15.38, 14.11.
Synthesis of target compound 25
Compound 25 was prepared from intermediate 11i (22 mg, 0.040 mmol) in a similar manner as described for compound 12 (16 mg, 70%). HPLC: tR 16.720 min, purity: 92.0%. Mp: 235.6–237.6. HRMS (ESI) for C29H26N4O7 [M–H]−. Calcd: 541.1723, found: 541.1731. 1H NMR (500 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.10 (t, J = 5.7 Hz, 1H), 8.02 (s, 1H), 8.00 (s, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.76 (s, 1H), 7.66 (s, 1H), 7.64 (s, 1H), 7.62 (d, J = 2.2 Hz, 1H), 7.44 (s, 1H), 7.25 (dd, J = 8.5, 2.2 Hz, 1H), 5.42 (s, 2H), 4.02 (s, 3H), 3.34–3.32 (m, 2H), 2.64 (s, 3H), 1.23 (d, J = 6.2 Hz, 1H), 1.17 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.18, 165.20, 162.53, 158.14, 155.94, 152.90, 152.43, 149.39, 149.02, 148.94, 141.27, 130.61, 129.54(2C), 127.67(2C), 123.73, 120.74, 117.98, 112.87, 109.67, 107.02, 105.55, 102.29, 69.67, 56.33, 33.80, 14.94, 13.67.
Synthesis of target compound 26
Compound 26 was prepared from intermediate 11j (26 mg, 0.049 mmol) in a similar manner as described for compound 12 (18 mg, 67%). HRMS (ESI) for C31H28N4O7 [M–H]−. Calcd: 567.1880, found: 567.1791. 1H NMR (500 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.10 (t, J = 5.6 Hz, 1H), 7.85 (s, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.75 (dd, J = 17.2, 8.1 Hz, 1H), 7.66 (d, J = 2.1 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 8.1 Hz, 2H), 7.50 (s, 1H), 7.47 (d, J = 2.7 Hz, 1H), 7.29 (dd, J = 8.5, 2.1 Hz, 1H), 6.51 (d, J = 15.8 Hz, 1H), 5.39 (s, 2H), 4.05 (s, 3H), 3.38–3.29 (m, 2H), 2.65 (s, 3H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 166.64, 163.08, 162.91, 162.78, 158.79, 153.26, 151.86, 150.19, 149.50, 138.33, 137.80, 137.80, 135.20, 128.97, 128.81(2C), 128.14(2C), 124.60, 121.33, 119.86, 118.15, 113.35, 110.17, 105.84, 103.17, 70.60, 57.09, 34.25, 15.37, 14.14.
Synthesis of target compound 27
Compound 27 was prepared from intermediate 11f (28 mg, 0.055 mmol) in a similar manner as described for compound 17 (25 mg, 76%). HPLC: tR 11.988 min, purity: 98.1%. Mp: 146.1–148.1. HRMS (ESI) for C33H35N5O6 [M–H]−. Calcd: 596.2509, found: 596.2466. 1H NMR (500 MHz, DMSO-d6) δ 9.13 (s, 1H), 8.52 (s, 1H), 8.10 (t, J = 5.6 Hz, 1H), 7.78 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.60 (s, 1H), 7.39 (s, 1H), 7.26 (dd, J = 8.5, 2.1 Hz, 1H), 7.16 (dd, J = 7.7, 1.6 Hz, 1H), 6.92–6.85 (m, 1H), 6.71 (d, J = 6.6 Hz, 1H), 6.53 (t, J = 7.3 Hz, 1H), 4.84 (s, 1H), 4.20 (t, J = 6.5 Hz, 2H), 4.00 (s, 3H), 3.34 (d, J = 5.0 Hz, 2H), 2.64 (s, 3H), 2.37 (t, J = 7.2 Hz, 2H), 1.97–1.81 (m, 2H), 1.80–1.63 (m, 2H), 1.63–1.47 (m, 2H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 171.57, 165.55, 162.99, 158.57, 156.34, 153.35, 152.62, 149.97, 149.87, 149.23, 142.35, 126.16, 125.75, 124.14, 124.01, 121.14, 118.48, 116.62, 116.32, 113.31, 110.21, 107.23, 106.01, 101.81, 69.03, 56.65, 36.19, 34.25, 28.83, 25.70, 25.53, 15.39, 14.12.
Synthesis of target compound 28
Compound 28 was prepared from intermediate 11g (21 mg, 0.040 mmol) in a similar manner as described for compound 17 (19 mg, 77%). HRMS (ESI) for C34H37N5O6 [M–H]−. Calcd: 610.2666, found: 610.2672. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.53 (s, 1H), 8.09 (t, J = 5.4 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.63 (s, 1H), 7.60 (s, 1H), 7.40 (s, 1H), 7.26 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 7.8 Hz, 1H), 6.88 (t, J = 7.5 Hz, 1H), 6.71 (d, J = 7.9 Hz, 1H), 6.52 (t, J = 7.5 Hz, 1H), 4.82 (s, 2H), 4.19 (t, J = 6.2 Hz, 2H), 4.01 (s, 3H), 2.65 (s, 3H), 2.34 (t, J = 7.3 Hz, 2H), 1.92–1.77 (m, 2H), 1.71–1.58 (m, 2H), 1.58–1.47 (m, 2H), 1.47–1.37 (m, 2H), 1.18 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.15, 165.08, 162.51, 158.11, 155.89, 152.89, 152.16, 149.52, 149.41, 148.77, 141.89, 125.68, 125.29, 123.68, 123.57, 120.68, 118.01, 116.15, 115.88, 112.85, 109.76, 106.79, 105.55, 101.34, 68.64, 56.20, 35.74, 33.78, 28.43, 25.36, 25.27, 14.92, 13.66.
Synthesis of target compound 29
Compound 29 was prepared from intermediate 11h (24 mg, 0.045 mmol) in a similar manner as described for compound 17 (19 mg, 68%). HPLC: tR 13.960 min, purity: 99.2%. Mp: 142.2–144.2. HRMS (ESI) for C35H39N5O6 [M–H]−. Calcd: 624.2822, found: 624.2805. 1H NMR (500 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.51 (s, 1H), 8.15 (t, J = 5.6 Hz, 1H), 7.78 (d, J = 8.5 Hz, 1H), 7.64–7.58 (m, 2H), 7.57 (d, J = 4.4 Hz, 1H), 7.37 (s, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.24 (d, J = 10.3 Hz, 1H), 7.18 (d, J = 9.1 Hz, 1H), 6.86 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 6.52 (t, J = 7.6 Hz, 1H), 4.19–4.13 (m, 2H), 3.99 (d, J = 5.1 Hz, 3H), 3.35–3.31 (m, 2H), 2.63 (s, 3H), 2.42–2.25 (m, 2H), 1.85–1.78 (m, 2H), 1.49–1.43 (m, 2H), 1.43–1.37 (m, 2H), 1.37–1.32 (m, 2H), 1.32–1.24 (m, 2H), 1.17 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 175.16, 165.52, 163.70, 158.72, 156.33, 153.34, 152.62, 149.95, 149.87, 149.21, 124.07, 121.16, 118.46, 113.11, 110.20, 107.22, 106.00, 101.78, 69.06, 56.65, 56.27, 34.21, 29.46, 28.85, 28.75, 26.55, 25.74, 24.96, 14.17.
Synthesis of target compound 30
Compound 30 was prepared from intermediate 11i (29 mg, 0.052 mmol) in a similar manner as described for compound 17 (26 mg, 77%). HPLC: tR 12.810 min, purity: 92.2%. Mp: 188.7–190.8. HRMS (ESI) for C35H31N5O6 [M–H]−. Calcd: 616.2196, found: 616.2206. 1H NMR (400 MHz, DMSO-d6) δ 9.71 (s, 1H), 8.55 (s, 1H), 8.10 (t, J = 5.7 Hz, 1H), 8.04 (d, J = 7.9 Hz, 2H), 7.84–7.75 (m, 2H), 7.67 (d, J = 7.9 Hz, 2H), 7.45 (s, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 6.99 (t, J = 7.6 Hz, 1H), 6.80 (d, J = 7.9 Hz, 1H), 6.62 (t, J = 7.5 Hz, 1H), 5.44 (s, 2H), 4.92 (s, 2H), 4.03 (s, 3H), 3.36–3.31 (m, 3H), 2.65 (s, 3H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.64, 165.55, 163.00, 158.59, 156.43, 153.35, 152.87, 149.84, 149.44, 143.63, 140.18, 134.79, 128.47(2C), 128.06(2C), 127.19, 127.02, 124.20, 123.68, 121.19, 118.44, 116.72, 116.58, 113.31, 110.14, 107.46, 106.00, 102.74, 70.20, 56.78, 34.25, 15.38, 14.12.
Synthesis of target compound 31
Compound 31 was prepared from intermediate 11j (19 mg, 0.036 mmol) in a similar manner as described for compound 17 (17 mg, 76%). HPLC: tR 15.352 min, purity: 95.0%. Mp: 184.1–186.4. HRMS (ESI) for C37H33N5O6 [M–H]−. Calcd: 642.2353, found: 642.2368. 1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.55 (d, J = 1.4 Hz, 1H), 8.10 (t, J = 5.7 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.78 (s, 1H), 7.70 (d, J = 7.9 Hz, 2H), 7.65–7.55 (m, 4H), 7.45 (s, 1H), 7.35 (d, J = 7.9 Hz, 1H), 7.26 (d, J = 8.5 Hz, 1H), 6.99–6.89 (m, 2H), 6.77 (d, J = 7.9 Hz, 1H), 6.59 (t, J = 7.6 Hz, 1H), 5.37 (s, 2H), 5.03–4.92 (m, 2H), 4.02 (s, 3H), 2.65 (s, 3H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.64, 163.92, 162.98, 158.59, 156.41, 153.36, 152.85, 149.86, 149.52, 149.44, 142.10, 139.66, 138.29, 135.15, 129.02(2C), 128.28(2C), 126.29, 125.24, 125.15, 124.19, 123.02, 121.20, 118.44, 116.77, 116.48, 113.32, 110.15, 107.45, 106.00, 102.63, 70.38, 56.75, 34.25, 15.39, 14.13.
Synthesis of target compound 32
Compound 32 was prepared from intermediate 11k (24 mg, 0.046 mmol) in a similar manner as described for compound 12 (17 mg, 69%). HPLC: tR 16.600 min, purity: 88.4%. Mp: 153.4–155.4. HRMS (ESI) for C28H32N4O7 [M–H]−. Calcd: 535.2193, found: 535.2199. 1H NMR (500 MHz, DMSO-d6) δ 8.50 (s, 1H), 8.00 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 2.0 Hz, 1H), 7.56 (d, J = 4.3 Hz, 1H), 7.36 (d, J = 3.1 Hz, 1H), 7.24 (dd, J = 8.5, 2.0 Hz, 1H), 4.23–4.09 (m, 3H), 3.99 (s, 3H), 2.61 (s, 3H), 2.25 (t, J = 7.3 Hz, 2H), 1.87–1.76 (m, 2H), 1.64–1.54 (m, 2H), 1.50–1.42 (m, 2H), 1.21 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.98, 165.53, 162.38, 158.13, 156.30, 153.32, 152.58, 149.92, 149.83, 149.19, 124.33, 121.12, 118.42, 113.62, 110.18, 107.16, 105.92, 101.77, 69.01, 56.62, 41.22, 34.13, 28.74, 25.63, 24.72, 22.81(2C), 14.05.
Synthesis of target compound 33
Compound 32 was prepared from intermediate 11m (46 mg, 0.083 mmol) in a similar manner as described for compound 12 (29 mg, 61%). HRMS (ESI) for C30H36N4O7 [M–H]−. Calcd: 563.2506, found: 563.2511. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.50 (d, J = 5.3 Hz, 1H), 7.98 (d, J = 7.9 Hz, 1H), 7.71 (dd, J = 8.6, 3.6 Hz, 1H), 7.59 (dd, J = 5.4, 2.2 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 9.7 Hz, 1H), 7.24 (dt, J = 8.0, 2.2 Hz, 1H), 4.20–4.08 (m, 3H), 3.99 (s, 3H), 2.61 (s, 3H), 1.95 (t, J = 7.3 Hz, 2H), 1.86–1.75 (m, 2H), 1.55–1.45 (m, 2H), 1.48–1.40 (m, 2H), 1.38–1.31 (m, 2H), 1.29–1.24 (m, 2H), 1.21 (d, J = 6.6 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 169.63, 165.54, 162.39, 158.14, 156.32, 153.33, 152.58, 149.96, 149.83, 149.19, 124.33, 121.12, 118.43, 113.62, 110.19, 107.18, 105.93, 101.75, 69.10, 56.63, 53.97, 41.23, 32.71, 28.99, 28.93, 25.88, 25.54, 22.81(2C), 14.04.
Synthesis of target compound 34
Compound 34 was prepared from intermediate 11n (48 mg, 0.089 mmol) in a similar manner as described for compound 12 (34 mg, 69%). HPLC: tR 17.733 min, purity: 91.4%. Mp: 237.1–239.9. HRMS (ESI) for C30H28N4O7 [M–H]−. Calcd: 555.1880, found: 555.1877. 1H NMR (500 MHz, DMSO-d6) δ 10.61 (s, 1H), 8.72 (s, 1H), 8.16 (d, J = 6.6 Hz, 2H), 7.99 (d, J = 7.8 Hz, 1H), 7.84 (s, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.65 (s, 1H), 7.49 (s, 1H), 7.28 (d, J = 8.1 Hz, 1H), 5.48 (s, 2H), 4.17–4.12 (m, 1H), 4.06 (s, 3H), 2.63 (s, 3H), 1.22 (d, J = 6.6 Hz, 6H). 13C NMR (126 MHz, DMSO) δ 166.10, 166.03, 162.33, 162.25, 158.28, 153.29, 149.61, 140.86, 133.68, 128.81(2C), 128.08(2C), 127.61, 127.33, 124.64, 121.27, 118.27, 118.25, 113.67, 113.64, 110.16, 105.86, 103.07, 70.28, 57.00, 41.25, 22.83(2C), 14.07.
Synthesis of target compound 35
Compound 35 was prepared from intermediate 11o (31 mg, 0.054 mmol) in a similar manner as described for compound 12 (22 mg, 69%). HRMS (ESI) for C32H30N4O7 [M–H]−. Calcd: 581.2036, found: 581.2061. 1H NMR (500 MHz, DMSO-d6) δ 8.52 (s, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.74 (s, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.60 (s, 1H), 7.54 (d, J = 7.9 Hz, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.41 (s, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.16 (d, J = 15.9 Hz, 1H), 6.42 (d, J = 15.9 Hz, 1H), 5.30 (s, 2H), 4.17–4.10 (m, 1H), 4.00 (s, 3H), 2.89 (s, 3H), 1.21 (d, J = 6.6 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 168.05, 165.64, 162.45, 158.16, 153.32, 152.78, 149.79, 149.44, 144.00, 138.90, 134.44, 128.82(2C), 128.80(2C), 128.53, 124.38, 121.15, 119.86, 118.39, 113.59, 110.11, 108.45, 107.26, 105.91, 102.64, 70.25, 56.73, 41.26, 22.79(2C), 14.03.
Synthesis of target compound 36
Compound 36 was prepared from intermediate 11k (32 mg, 0.061 mmol) in a similar manner as described for compound 17 (31 mg, 83%). HRMS (ESI) for C34H37N5O6 [M–H]−. Calcd: 610.2666, found: 610.2672. 1H NMR (500 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.51 (s, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.57 (s, 1H), 7.37 (s, 1H), 7.24 (dd, J = 8.5, 2.1 Hz, 1H), 7.18 (dd, J = 7.8, 1.5 Hz, 1H), 6.88 (t, J = 8.3 Hz, 1H), 6.71 (d, J = 9.4 Hz, 1H), 6.52 (t, J = 8.3 Hz, 1H), 4.89 (s, 2H), 4.19–4.15 (m, 2H), 4.16–4.11 (m, 1H), 3.99 (s, 3H), 2.62 (s, 3H), 2.38 (t, J = 7.4 Hz, 2H), 1.92–1.82 (m, 2H), 1.75–1.65 (m, 2H), 1.58–1.46 (m, 2H), 1.21 (d, J = 6.4 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 171.63, 165.54, 162.40, 158.14, 156.31, 153.33, 152.58, 149.94, 149.83, 149.19, 142.29, 126.08, 125.70, 124.33, 124.05, 121.12, 118.43, 116.56, 116.33, 113.62, 110.19, 107.17, 105.94, 101.76, 69.02, 56.62, 41.23, 36.18, 28.84, 25.70, 25.56, 22.81(2C), 14.05.
Synthesis of target compound 37
Compound 37 was prepared from intermediate 11n (21 mg, 0.039 mmol) in a similar manner as described for compound 17 (19 mg, 78%). HPLC: tR 14.002 min, purity: 90.2%. Mp: 188.7–190.7. HRMS (ESI) for C36H33N5O6 [M–H]−. Calcd: 630.2353, found: 630.2369. 1H NMR (500 MHz, DMSO-d6) δ 9.78 (s, 1H), 8.54 (s, 1H), 8.06 (d, J = 7.9 Hz, 2H), 8.01 (d, J = 7.8 Hz, 1H), 7.76 (s, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 2.1 Hz, 1H), 7.43 (s, 1H), 7.25 (dd, J = 8.5, 2.1 Hz, 1H), 7.20 (d, J = 7.8 Hz, 1H), 7.03–6.94 (m, 1H), 6.81 (dd, J = 8.0, 1.5 Hz, 1H), 6.61 (t, J = 7.6 Hz, 1H), 5.42 (s, 2H), 4.43 (s, 2H), 4.17–4.09 (m, 1H), 4.02 (s, 3H), 2.62 (s, 3H), 1.22 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 165.64, 162.40, 158.17, 156.40, 153.34, 152.84, 149.81, 149.42, 143.53, 140.17, 134.76, 128.51(2C), 128.03(2C), 127.21, 124.39, 123.79, 121.18, 118.40, 116.79, 116.67, 113.63, 110.13, 107.41, 105.94, 102.69, 70.19, 56.50, 41.24, 22.81(2C), 14.06.
Synthesis of target compound 38
Compound 38 was prepared from intermediate 11o (24 mg, 0.042 mmol) in a similar manner as described for compound 17 (22 mg, 79%). HPLC: tR 13.910 min, purity: 88.1%. Mp: 209.1–211.6. HRMS (ESI) for C38H35N5O6 [M–H]−. Calcd: 656.2509, found: 656.2487. 1H NMR (500 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.68 (s, 1H), 7.99 (d, J = 7.9 Hz, 1H), 7.83 (s, 1H), 7.75 (d, J = 3.3 Hz, 1H), 7.75–7.72 (m, 2H), 7.65–7.62 (m, 3H), 7.51–7.48 (m, 1H), 7.47 (s, 1H), 7.42–7.37 (m, 2H), 7.37–7.33 (m, 1H), 7.27 (dd, J = 8.5, 2.1 Hz, 1H), 6.96 (d, J = 15.7 Hz, 1H), 5.41 (s, 2H), 4.17–4.12 (m, 1H), 4.04 (s, 3H), 2.63 (s, 3H), 1.22 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, DMSO) δ 166.12, 164.89, 162.33, 162.25, 158.25, 156.92, 153.31, 152.43, 149.82, 149.68, 147.62, 141.29, 138.75, 134.70, 129.12(2C), 128.53(2C), 127.92, 127.08, 125.99, 125.91, 124.58, 124.01, 121.69, 121.26, 118.28, 113.67, 110.17, 106.18, 105.88, 102.91, 70.46, 56.93, 41.24, 22.83, 22.80, 14.07.
HDAC1 inhibition assay
Prepare 1× assay buffer (modified Tris buffer). Transfer compounds to assay plate by Echo in 100% DMSO. The final fraction of DMSO is 1%. Prepare enzyme solution (BPS, Cat. No. 50051) in 1× assay buffer. Add trypsin and Ac-peptide substrate in 1× assay buffer to make the substrate solution. Transfer 15 μL of enzyme solution to assay plate or for low control transfer 15 μL of 1× assay buffer. Incubate at room temperature for 15 minutes. Add 10 μL of substrate solution to each well to start reaction. Read the plate on Envision with excitation at 355 nm and emission at 460 nm. Fit the data in Excel to obtain inhibition values using the equation: inhibition% = (max − signal)/(max − min) × 100. IC50 values were calculated by Graphpad prism using the equation: log(inhibitor) vs. response − variable slope (four parameters).
VEGFR-2 inhibition assay
Transfer different concentrations of compounds to a 384-well assay plate in duplicates. Add 10 μL of kinase solution (Carna, Cat. No. 08-191, Lot. No. 07CBS-0540) to each well of the assay plate, except for control wells without enzyme (add 5 μL of 1× kinase buffer instead). Add 10 μL of substrate solution of fluorescein-poly GT (Invitrogen, Cat. No. PV3611, Lot. No. 374293B) and ATP to each well of the assay plate to start reaction. Add 20 μL of detection solution (Invitrogen, Cat. No. PV3552, Lot. No. 1508164B) to each well of the assay plate to stop the reaction. Mix briefly with Centrifuge and incubate at room temperature for 60 minutes before reading on a plate reader for fluorescence. Collect data on Envision with excitation at 340 nm and emission at 520 nm and 495 nm. Calculate ratio of RFU 520 nm/RFU 495 nm. Convert ratio values to percent inhibition values. Percent inhibition = (max − sample ratio)/(max − min) × 100. IC50 values were calculated by Graphpad prism using the equation: log(inhibitor) vs. response − variable slope (four parameters).
Cell cycle analysis
HeLa cells were treated with different concentrations (0.75 μM, 1.5 μM and 3 μM) of 13 for 48 h and were harvested by digestion with 0.25% trypsin and centrifugation (1000 rpm for 15 min) with PBS. The cell pellets were suspended with 70% ethanol at 4 °C overnight. Afterward, cells were washed with phosphate buffered saline (PBS) twice and incubated with 100 μL RNase at 37 °C for 30 min and followed by incubation with 400 μL PI solution for 30 min in the dark at 4 °C. Cells were analyzed by flow cytometry system (BECKMAN COULTER CytoFLEX) and red fluorescence was recorded at 488 nm.
Apoptosis study
Briefly, cells were treated with different concentrations (0.75 μM, 1.5 μM and 3 μM) of compound 13 for 48 h, and were harvested and washed twice with PBS (1000 rpm, 5 min). Then cells were resuspended in 500 μL binding buffer. 5 μL of annexin V-APC and 5 μL 7-AAD were added to each tube and the cells were gently vortexed and incubated for 15 min at room temperature in the dark. The stained cells were analyzed by a flow cytometer (BECKMAN COULTER CytoFLEX).
CCK-8 assay
Stock solutions of tested compounds were prepared in DMSO at a concentration of 1 mmol L−1. Before use, the stock solutions were further diluted to the corresponding concentration by culture medium. The HeLa, MCF-7 and A549 cell lines were purchased from Cell Bank of Chinese Academy of Sciences and were cultured in the RPMI 1640 medium (Gibco) containing 10% FBS (Gibco) and 1% penicillin/streptomycin (Solarbio) and incubated at 37 °C in a humidified atmosphere containing 5% CO2. Cells were harvested by the 0.25% trypsin–EDTA solution (Beyotime) from the cell-culture flask and seeded in a 96-well plate at a density of 5 × 104 cells per well and incubated for 24 h. The tested compounds were added to the culture medium such that the final concentration of DMSO was less than 0.1% in the reaction. After 48 h, the CCK-8 solution (10 μL, Beyotime) was added to each well and incubated at 37 °C for 2 h. Absorbance at 450 nm was determined with a molecular device microplate reader. Cell viabilities were calculated using Excel. IC50 values were obtain by SPSS software. All experiments were performed in three times.
CellTiter-Glo® luminescent cell viability assay
Make 500× cpd solution in DMSO. Dilute compound with growth medium to 10× final concentration. Add 2 μL 1000× cpd to 98 μL growth medium. The HUVEC cell line was from PreceDo Biotechnology Co., Ltd and was cultured in the BEGM cell culture medium containing 10% FBS (Gibco) and 1% penicillin/streptomycin (Corning). Cells were seeded in a 96-well plate at a desired density and incubated for 24 h. Add 10 μL 10× cpds to the into 96-well plate according to the plate map. Final DMSO concentration in each well is 0.2%. Incubate at 37 °C, 5% CO2 for 72 h. Equilibrate the assay plate to room temperature before measurement. Add 50 μL of CellTiter-Glo® reagent (Promega) into each well. Mix contents for 2 minutes on an orbital shaker to induce cell lysis. Incubate at room temperature for 10 minutes to stabilize luminescent signal. Record luminescence on Paradigm. Inhibition rate (Inh%) was calculated relative to vehicle (DMSO) treated control wells using following formula: Inhibition rate (Inh%) = 100 − (RLU compound − RLU blank)/(RLU control − RLU blank) × 100%. Data were analyzed using Graphpad 7.0, fitting to a 4-parameter equation to generate concentration response curves.
Tube formation assay
After treated with different final concentrations (0 μM, 2.5 μM, 10 μM) of compound 13 for 72 h, the HUVECs were collected. 2 × 104 cells per well HUVECs were seeded on Matrigel's surface, then cultured for 4 h. The tube formation was then observed and photographed with a microscope. Use image pro plus software to calculate the total length of tubes.
Docking study
The crystal structures of VEGFR-2 (PDB code 3B8R) and HDAC1 (PDB code 1C3S) were obtained from Protein Data Bank. Water molecules and cocrystallized ligands were removed from the proteins using Pymol software. Compound 13 was created by ChemDraw 2D and saved as PDB format by ChemDraw 3D. Proteins and compound were prepared using the Autodock tool 1.5.6, such as adding hydrogen atoms, computing Gasteiger and so on. The grid-enclosing box was placed on the centroid of the co-crystallized ligand. Docking was carried out by the Autodock vina to obtain the molecular docking results. Molecular graphic manipulations and visualizations were performed using Pymol software.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Fujian Provincial Health Technology Project (2020QNA060, China), the Startup Fund for scientific research, Fujian Medical University (2019QH1123, China) and the Fujian Province Natural Science Foundation (2021J01309, China). Thank the Instrumental Analysis Center of Huaqiao University.
References
- J. Folkman, JNCI, J. Natl. Cancer Inst., 1990, 82, 4–7 CrossRef CAS PubMed.
- P. Carmeliet, Nat. Med., 2003, 9, 653–660 CrossRef CAS PubMed.
- X. Yang, Y. Zhang, Y. Yang, S. Lim, Z. Cao, J. Rak and Y. Cao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13932–13937 CrossRef CAS PubMed.
- A.-K. Olsson, A. Dimberg, J. Kreuger and L. Claesson-Welsh, Nat. Rev. Mol. Cell Biol., 2006, 7, 359–371 CrossRef CAS PubMed.
- X. Wang, A. M. Bove, G. Simone and B. Ma, Front. Cell Dev. Biol., 2020, 8, 599281 CrossRef PubMed.
- A. A. Shah, A. M. Kamal and S. Akhtar, Curr. Drug Metab., 2021, 22, 50–59 CAS.
- K. Holmes, O. L. Roberts, A. M. Thomas and M. J. Cross, Cell. Signalling, 2007, 19, 2003–2012 CrossRef CAS PubMed.
- K. Cheng, C. F. Liu and G. W. Rao, Curr. Med. Chem., 2021, 28, 2540–2564 CAS.
- F. W. Peng, D.-K. Liu, Q. W. Zhang, Y. G. Xu and L. Shi, Expert Opin. Ther. Pat., 2017, 27, 987–1004 CrossRef CAS PubMed.
- G. Bergers and D. Hanahan, Nat. Rev. Cancer, 2008, 8, 592–603 CrossRef CAS PubMed.
- S. Vyse, F. Mccarthy, M. Broncel, A. Paul, J. P. Wong, A. Bhamra and P. H. Huang, J. Proteomics, 2018, 170, 130–140 CrossRef CAS PubMed.
- R. N. Gacche and R. J. Meshram, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1846, 161–179 CrossRef CAS PubMed.
- J. A. Hill and L. E. Cowen, Future Microbiol., 2015, 10, 1719–1726 CrossRef CAS PubMed.
- G. R. Zimmermann, J. Lehár and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS PubMed.
- S. E. Bates, N. Engl. J. Med., 2020, 383, 650–663 CrossRef CAS PubMed.
- J. E. Lee and M. Y. Kim, Semin. Cancer Biol., 2022, 83, 4–14 CrossRef CAS PubMed.
- B. Barneda-Zahonero and M. Parra, Mol. Oncol., 2012, 6, 579–589 CrossRef CAS PubMed.
- M. Manal, M. J. N. Chandrasekar, J. Gomathi Priya and M. J. Nanjan, Bioorg. Chem., 2016, 67, 18–42 CrossRef CAS PubMed.
- R. Aggarwal, S. Thomas, N. Pawlowska, I. Bartelink, J. Grabowsky, T. Jahan, A. Cripps, A. Harb, J. Leng, A. Reinert, I. Mastroserio, T.-G. Truong, C. J. Ryan and P. N. Munster, J. Clin. Oncol., 2017, 35, 1231–1239 CrossRef CAS PubMed.
- S. Tavallai, H. A. Hamed, S. Grant, A. Poklepovic and P. Dent, Cancer Biol. Ther., 2014, 15, 578–585 CrossRef PubMed.
- L. Booth, J. L. Roberts, C. Sander, J. Lee, J. M. Kirkwood, A. Poklepovic and P. Dent, Oncotarget, 2017, 8, 16367–16386 CrossRef PubMed.
- J. J. Mcclure, X. Li and C. J. Chou, Adv. Cancer Res., 2018, 138, 183–211 CrossRef CAS PubMed.
- R.-G. Fu, Y. Sun, W.-B. Sheng and D. F. Liao, Eur. J. Med. Chem., 2017, 136, 195–211 CrossRef CAS PubMed.
- N. Upadhyay, K. Tilekar, S. Safuan, A. P. Kumar, M. Schweipert, F. J. Meyer-Almes and C. S. Ramaa, RSC Med. Chem., 2021, 12, 1540–1554 RSC.
- X. J. Liu, H.-C. Zhao, S. J. Hou, H.-J. Zhang, L. Cheng, S. Yuan, L. R. Zhang, J. Song, S. Y. Zhang and S.-W. Chen, Bioorg. Chem., 2023, 133, 106425 CrossRef CAS PubMed.
- X. Xue, Y. Zhang, Y. Liao, D. Sun, L. Li, Y. Liu, Y. Wang, W. Jiang, J. Zhang, Y. Luan and X. Zhao, Invest. New Drugs, 2022, 40, 10–20 CrossRef CAS PubMed.
- Y. Luan, J. Li, J. A. Bernatchez and R. Li, J. Med. Chem., 2019, 62, 3171–3183 CrossRef CAS PubMed.
- J. Zang, X. Liang, Y. Huang, Y. Jia, X. Li, W. Xu, C. J. Chou and Y. Zhang, J. Med. Chem., 2018, 61, 5304–5322 CrossRef CAS PubMed.
- F. W. Peng, T.-T. Wu, Z.-W. Ren, J. Y. Xue and L. Shi, Bioorg. Med. Chem. Lett., 2015, 25, 5137–5141 CrossRef CAS PubMed.
- F. W. Peng, J. Xuan, T. T. Wu, J. Y. Xue, Z. W. Ren, D. K. Liu, X. Q. Wang, X. H. Chen, J. W. Zhang, Y. G. Xu and L. Shi, Eur. J. Med. Chem., 2016, 109, 1–12 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Junjie Ma
*b and
Junjie Ma