
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe relationship between structure and excited-state properties in polyanilines from geminal-based methods†
Seyedehdelaram Jahani ,
Katharina Boguslawski and
Paweł Tecmer*
,
Katharina Boguslawski and
Paweł Tecmer*
Institute of Physics, Faculty of Physics, Astronomy, and Informatics, Nicolaus Copernicus University in Toruń, Grudziadzka 5, 87-100 Toruń, Poland. E-mail: ptecmer@fizyka.umk.pl
First published on 20th September 2023
Abstract
We employ state-of-the-art quantum chemistry methods to study the structure-to-property relationship in polyanilines (PANIs) of different lengths and oxidation states. Specifically, we focus on leucoemeraldine, emeraldine, and pernigraniline in their tetramer and octamer forms. We scrutinize their structural properties, HOMO and LUMO energies, HOMO–LUMO gaps, and vibrational and electronic spectroscopy using various Density Functional Approximations (DFAs). Furthermore, the accuracy of DFAs is assessed by comparing them to experimental and wavefunction-based reference data. We perform large-scale orbital-optimized pair-Coupled Cluster Doubles (oo-pCCD) calculations for ground and electronically excited states and conventional Configuration Interaction Singles (CIS) calculations for electronically excited states in all investigated systems. The EOM-pCCD+S approach with pCCD-optimized orbitals allows us to unambiguously identify charge transfer and local transitions across the investigated PANI systems—an analysis not possible within a delocalized canonical molecular orbital basis obtained, for instance, by DFAs. We show that the low-lying part of the emeraldine and pernigraniline spectrum is dominated by charge transfer excitations and that polymer elongation changes the character of the leading transitions. Furthermore, we augment our study with a quantum informational analysis of orbital correlations in various forms of PANIs.
1 Introduction
Organic-based semiconductors are essential building blocks of organic electronic devices, such as field-effect transistors, light-emitting diodes, memory cells, solar cells, and sensors.1 The research progress in organic electronics has been greatly accelerated by the discovery of conducting polymers in 1977.2 The importance of this scientific discovery led to the 2000 Nobel prize in chemistry “for the discovery and development of conductive polymers”.3 Among the conducting polymers, the most studied are polyanilines (PANIs). Due to their environmental stability,4,5 cost-effectiveness, ease of synthesis,6 and controllable electrical conductivity,7,8 PANIs became a very popular conducting polymer. PANIs find applications in catalysis,9,10 energy storage,11 battery electrode materials,12 sensors,13 and solar cells.14,15 PANIs usually act as a donor and the fullerene containing-unit as an acceptor in the latter. Thus, the PANIs' Highest Occupied Molecular Orbital (HOMO) energy level dictates the electron-donating properties.What distinguishes PANIs from other conducting polymers is their existence at different oxidation states with specific conducting properties by electronic or protonic doping.8 Different forms are obtained by varying the average oxidation state and the degree of protonation16 according to the general formula17
{[–(C6H6)–NH–(C6H6)–NH–]1−x[–(C6H6)–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (C6H4)N–]x}n. (C6H4)N–]x}n.
| (1) |
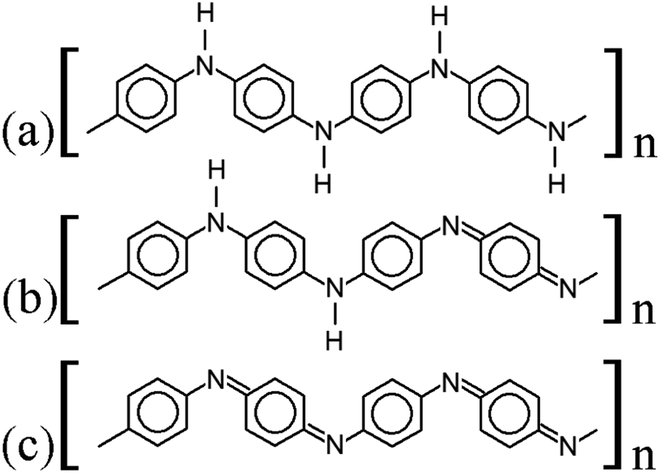 | ||
| Fig. 1 Lewis structures of PANIs. (a) leucoemeraldine, (b) emeraldine, and (c) pernigraniline drawn with ChemPlot.20 | ||
Experimental studies related to PANIs and their derivatives are the primary source of information on their structural, physical, and chemical characteristics.17,21,22 This includes, among other things, chemical, electrochemical and gas-phase preparations, redox and polymerization mechanisms, and examinations of chemical, physical, electrochemical properties, and molecular structures.21,23,24 Further modifications and tuning of PANI-based materials with desired properties could greatly benefit from reliable quantum chemical predictions. Unfortunately, such studies are limited due to computational difficulties. It is well-known that such systems bear a non-negligible amount of multi-reference character, but their molecular size prohibits standard multi-configurational methods. Despite that, several attempts have been made to model the electronic structures of PANIs using quantum chemistry. One of the earliest applications is a quantum-chemical prediction of optical absorption spectra of some model PANI compounds using the intermediate neglect of differential overlap (INDO) model combined with the configuration interaction (CI) approach.25 The authors were among the first to notice the importance of the torsion angle between the quinoid rings and the C–N–C backbone. Semi-empirical methods were also used to study the hydration, stacking, and solvent effects of PANIs.26,27 Moreover, simplified model systems of PANI were studied using Density Functional Approximations (DFAs).28,29 The Hartree–Fock (HF) and DFA optimized structures of PANIs at different oxidation states and unit lengths were investigated by Lim et al.,19 Mishra et al.,30 and Romanova et al.31 The aforementioned studies point to an HF failure, incorrectly distributing conjugation along the polymer chain and contradicting the X-ray experimental findings.23 However, the resulting properties strongly depend on the choice of the exchange–correlation (xc) functional. Mishra and Tandon30 used DFAs to investigate the infrared (IR) and Raman spectra of LB and its oligomers. Zhang et al.32 studied electronically excited states of model PANI complexes with water using time-dependent DFT (TD-DFT).
In this work, we reexamine the electronic structures and properties of PANIs using various approximations to the xc functional and unconventional electron correlation methods based on the pair Coupled Cluster Doubles (pCCD) model,33–36 initially introduced as the Antisymmetric Product of 1-reference orbital Geminal (AP1roG) ansatz.33 An additional advantage of pCCD-based methods is the possibility to optimize all orbitals at the correlated level and a quantitative description of orbital-based correlations using concepts from quantum information theory.37,38 The pCCD model combined with an orbital optimization protocol39–41 proved to be a reliable tool for modeling complex electronic structures and potential energy surfaces featuring strong correlation.40,42–44 Extensions to excited states within the Equation of Motion (EOM) formalism45,46 allow us to model double electron excitations,47–49 a known struggle for standard EOM-CCSD-based approaches.50 Furthermore, pCCD-based models allow us to gain qualitative insights into electronic structures and scrutinize them using localized orbitals, reflecting the intuitive picture of Lewis structures as pairs of electrons present from the early days of quantum chemistry. Specifically, working in a localized basis, we will be able to unambiguously dissect electronic excitations into different types, for instance, local or charge-transfer ones. All these features are desired in quantum chemical descriptions of electronic structures and properties of conducting polymers. Thus, pCCD-based quantum chemistry methods are promising alternatives to DFAs which might significantly speed up the structure-to-properties search in organic electronics and guide the experimental synthesis of new conductive polymers.
This work is organized as follows. Section 2 summarizes the computational methodology. Section 3 scrutinizes the ground- and electronic excited-states properties of selected PANIs combined with a quantum information analysis of orbital correlations. We conclude in Section 4.
2 Computational details
2.1 DFT calculations
All structure optimizations and vibrational frequency calculations were performed with the Turbomole6.6 (ref. 51 and 52) software package using the BP86 (ref. 53 and 54) xc functional and the def2-TZVP basis set.55,56 The optimized xyz structures are provided in Tables S1–S7 of the ESI.† These structures were later used for the calculation of electronic excitation energies within the TD-DFT57,58 framework using the Amsterdam Density Functional (v.2018) program package,59,60 the BP86,53,54 PBE,61 PBE0,62 and CAM-B3LYP63 xc functionals, and the triple-ζ polarization (TZ2P) basis set.642.2 pCCD-based methods
All pCCD33,34,36,39 calculations were carried out in a developer version of the PyBEST software package65,66 using the cc-pVDZ basis set67 and the DFT optimized structures. For the ground-state pCCD calculations, we employed the variational orbital optimization protocol.39–41 The Pipek–Mezey localized orbitals68 were used as a starting point for orbital optimization. Our numerical experience showed that using localized orbitals accelerates the orbital optimization process as the final pCCD natural orbitals are typically localized and bear some resemblance with split-localized orbitals.69
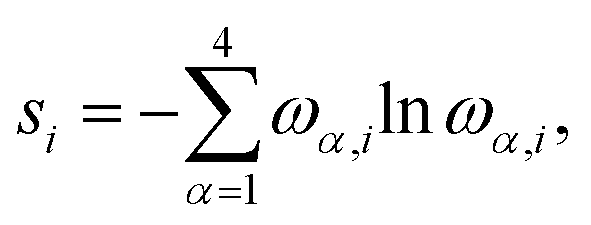 | (2) |
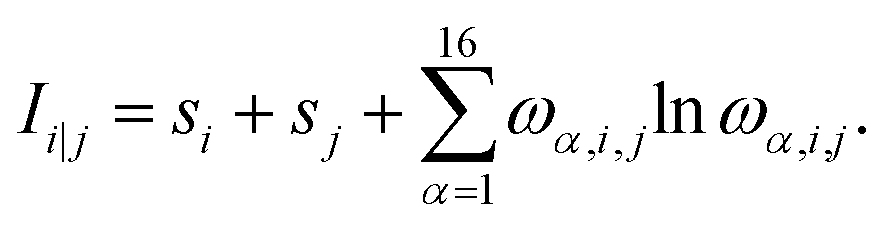 | (3) |
3 Results and discussion
In the following, we discuss the structural, vibrational, and electronically excited-state parameters of the aniline binary compound and selected PANIs in their tetramer and octamer structural arrangements. Since we aim at elucidating the structure-to-property relationship in polyanilines, we require chemically reasonable structures. In the following, we show that DFT indeed allows us to obtain reliable molecular structures, which are then used to model electronically excited states and selected properties. The results are compared to experiments and other theoretical predictions. Furthermore, the TD-DFT excitation energies obtained from different xc functionals are compared to wave-function calculations. Finally, we use an orbital entanglement and correlation analysis of orbital interactions for assessing the electronic structures and changes in electron correlation effects in PANIs of various oxidation states and lengths.3.1 Ground-state optimized electronic structures
The optimized structure of aniline, a small building block of PANIs, is shown in Fig. 2. All optimized bond lengths between N–H are roughly equal to 1.016 Å, while the C–H bond length equals 1.09 Å. The optimized structures of leucoemeraldine (1t), emeraldine (2t), and pernigraniline (3t) in the tetramer form and in their corresponding octamer forms (1o, 2o, and 3o) are visualized in Fig. S1 and S2 of the ESI,† respectively. Fig. 3 shows the corresponding Lewis structures, highlighting that aniline is a building block of PANIs. Our DFT calculations predict C–C and C–N bond distances between 1.3 and 1.4 Å (see Table 1). The bond angle between two phenyl rings in 1t and 1o and 2t and 2o is almost the same and averages to 125°. In contrast, the dihedral angles between the rings feature an average value of around 26.33°. For 3t and 3o, the bond angle between the two phenyl rings is pretty similar except for the angle N58–C59–C60, which increases to 126.4°. The dihedral angles of 3t and 3o significantly grow to 48.1° and 47.4°, respectively, compared to 1t and 1o. The total torsion angle between the phenyl rings is one of the main factors that govern the band gaps, conjugation length, and electrical conductivity, all of which are important factors in determining the electronic properties of PANIs. For this purpose, we collected the tilt angles (as indicated in Fig. 4 and collected in Table 2) for all the tetramer (t) and octamer (o) forms of the investigated PANIs. Our data suggest that the tilt angle for 3t and 3o significantly decreases compared to the remaining PANI compounds. That coincides with the theoretically best conductive properties of 3t and 3o over the remaining PANIs at lower oxidation states.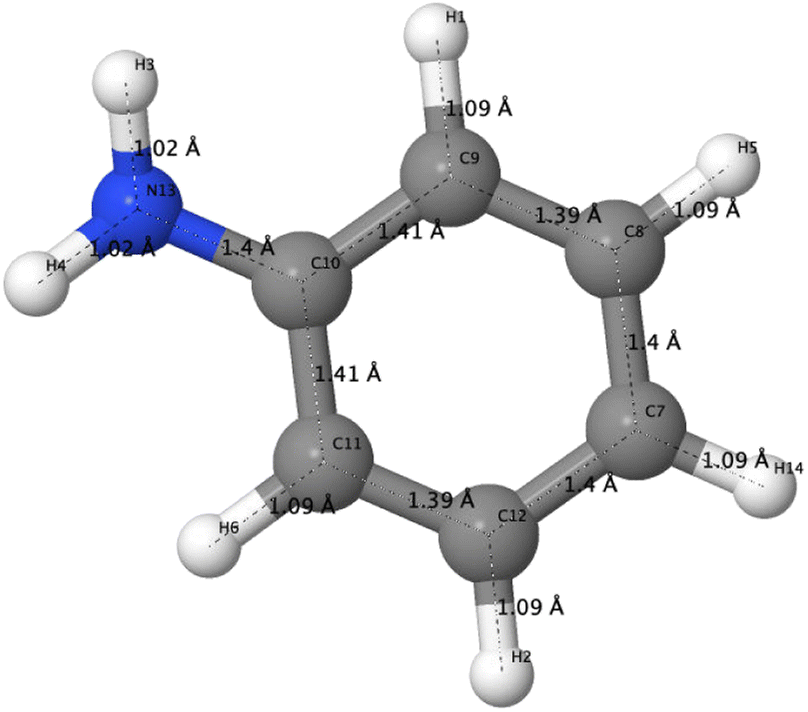 | ||
| Fig. 2 DFT-optimized structure of aniline including bond lengths in Å. | ||
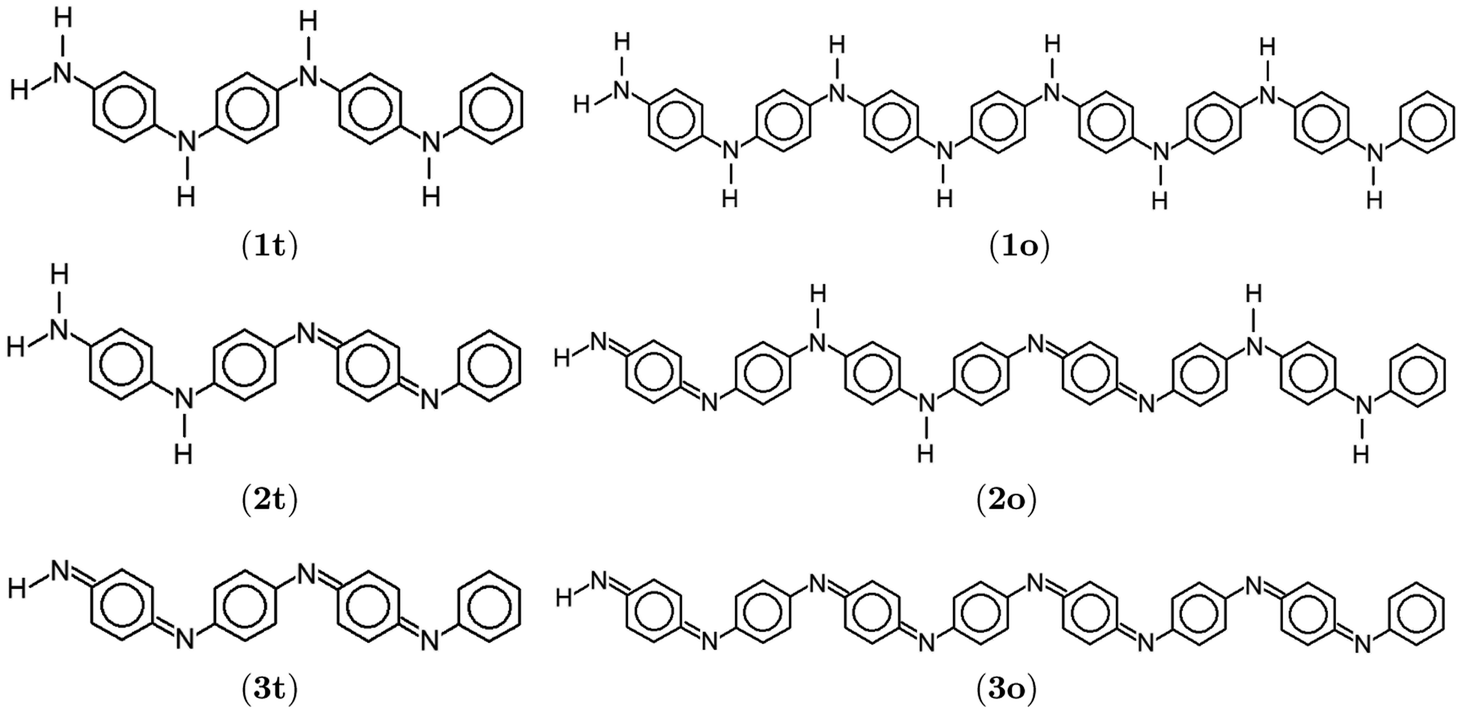 | ||
| Fig. 3 Lewis structures of polyanilies drawn with ChemPlot.20 Subfigures display leucoemeraldine (1), emeraldine (2), and pernigraniline (3) in the tetramer (t) and octamer (o) forms. | ||
| 1t | 1o | ||
|---|---|---|---|
| Geometrical parameters | Bond length [Å] | Geometrical parameters | Bond length [Å] |
| N7–C4, N7–C8 | 1.393, 1.406 | N58–C55, N58–C59 | 1.393, 1.404 |
| C4–C3, C8–C9 | 1.409, 1.406 | C55–C54, C59–C60 | 1.409, 1.406 |
| C3–C2, C9–C10 | 1.395, 1.390 | C54–C53, C60–C61 | 1.395, 1.391 |
| C2–C1, C10–C11 | 1.397, 1.409 | C53–C52, C61–C62 | 1.397, 1.408 |
| C1–C6, C11–C12 | 1.399, 1.407 | C52–C57, C62–C63 | 1.399, 1.408 |
| C6–C5, C12–C13 | 1.392, 1.393 | C57–C56, C63–C64 | 1.392, 1.392 |
| C5–C4, C13–C8 | 1.411, 1.405 | C56–C55, C64–C59 | 1.411, 1.406 |
| 1t | 1o | ||
|---|---|---|---|
| Geometrical parameters | Bond angle [°] | Geometrical parameters | Bond angle [°] |
| C4–N7–C8 | 129.1 | C55–N58–C59 | 129.4 |
| N7–C4–C3 | 123.1 | N58–C55–C54 | 123.0 |
| N7–C8–C9 | 122.8 | N58–C59–C60 | 123.1 |
| 1t | 1o | ||
|---|---|---|---|
| Geometrical parameters | Dihedral angle [°] | Geometrical parameters | Dihedral angle [°] |
| C8–N7–C4–C3 | 15.1 | C59–N58–C55–C54 | 22.6 |
| C4–N7–C8–C9 | 36.3 | C55–N58–C59–C60 | 28.1 |
| 2t | 2o | ||
|---|---|---|---|
| Geometrical parameters | Bond length [Å] | Geometrical parameters | Bond length [Å] |
| N7–C4, N7–C8 | 1.398, 1.394 | N58–C55, N58–C59 | 1.397, 1.399 |
| C4–C3, C8–C9 | 1.408, 1.411 | C55–C54, C59–C60 | 1.408, 1.407 |
| C3–C2, C9–C10 | 1.395, 1.386 | C54–C53, C60–C61 | 1.395, 1.391 |
| C2–C1, C10–C11 | 1.397, 1.416 | C53–C52, C61–C62 | 1.397, 1.407 |
| C1–C6, C11–C12 | 1.398, 1.419 | C52–C57, C62–C63 | 1.398, 1.406 |
| C6–C5, C12–C13 | 1.392, 1.387 | C57–C56, C63–C64 | 1.392, 1.391 |
| C5–C4, C13–C8 | 1.409, 1.411 | C56–C55, C64–C59 | 1.410, 1.408 |
| 2t | 2o | ||
|---|---|---|---|
| Geometrical parameters | Bond angle [°] | Geometrical parameters | Bond angle [°] |
| C4–N7–C8 | 129.9 | C55–N58–C59 | 129.6 |
| N7–C4–C3 | 122.9 | N58–C55–C54 | 123.1 |
| N7–C8–C9 | 123.3 | N58–C59–C60 | 123.1 |
| 2t | 2o | ||
|---|---|---|---|
| Geometrical parameters | Dihedral angle [°] | Geometrical parameters | Dihedral angle [°] |
| C8–N7–C4–C3 | 25.5 | C59–N58–C55–C54 | 19.5 |
| C4–N7–C8–C9 | 22.2 | C55–N58–C59–C60 | 29.7 |
| 3t | 3o | ||
|---|---|---|---|
| Geometrical parameters | Bond length [Å] | Geometrical parameters | Bond length [Å] |
| N7–C4, N7–C8 | 1.389, 1.313 | N58–C55, N58–C59 | 1.388, 1.314 |
| C4–C3, C8–C9 | 1.414, 1.457 | C55–C54, C59–C60 | 1.415, 1.457 |
| C3–C2, C9–C10 | 1.394, 1.357 | C54–C53, C60–C61 | 1.394, 1.358 |
| C2–C1, C10–C11 | 1.398, 1.454 | C53–C52, C61–C62 | 1.398, 1.453 |
| C1–C6, C11–C12 | 1.400, 1.456 | C52–C57, C62–C63 | 1.400, 1.455 |
| C6–C5, C12–C13 | 1.391, 1.357 | C57–C56, C63–C64 | 1.391, 1.358 |
| C5–C4, C13–C8 | 1.413, 1.455 | C56–C55, C64–C59 | 1.413, 1.455 |
| 3t | 3o | ||
|---|---|---|---|
| Geometrical parameters | Bond angle [°] | Geometrical parameters | Bond angle [°] |
| C4–N7–C8 | 123.4 | C55–N58–C59 | 123.4 |
| N7–C4–C3 | 123.4 | N58–C55–C54 | 123.4 |
| N7–C8–C9 | 123.4 | N58–C59–C60 | 126.4 |
| 3t | 3o | ||
|---|---|---|---|
| Geometrical parameters | Dihedral angle [°] | Geometrical parameters | Dihedral angle [°] |
| C8–N7–C4–C3 | 48.1 | C59–N58–C55–C54 | 47.4 |
| C4–N7–C8–C9 | 10.7 | C55–N58–C59–C60 | 11.1 |
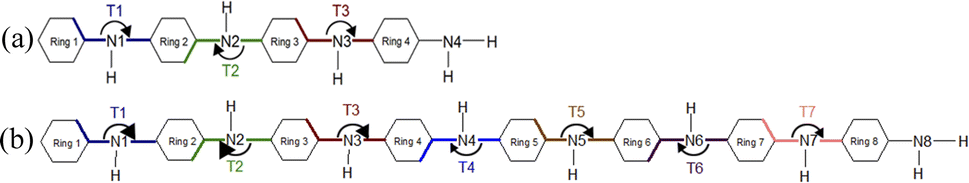 | ||
| Fig. 4 Schematic representation of the T1–T7 ring tilt angles in the tetramer (a) and octamer (b) models of PANIs. | ||
| 1t | 1o | 2t | 2o | 3t | 3o | |
|---|---|---|---|---|---|---|
| T1 | 166.6 | 159.7 | 157.2 | 162.8 | 137.6 | 138.4 |
| T2 | 194.9 | 206.3 | 212.9 | 204.7 | 191.7 | 192.7 |
| T3 | 165.0 | 159.0 | 168.3 | 148.7 | 141.9 | 146.1 |
| T4 | 201.9 | 192.4 | 194.3 | |||
| T5 | 156.3 | 158.0 | 146.9 | |||
| T6 | 205.2 | 206.4 | 193.5 | |||
| T7 | 158.8 | 149.2 | 142.6 |
We should also stress that the effect of various approximate xc functionals on the torsional angle of PANI is discussed in ref. 31. The studies conclude that different xc functionals provide qualitatively the same torsional angles.
3.2 Vibrational spectra
Aniline and PANIs have been a significant target of structural and electronic studies, experimentally and theoretically, for many years.7,17,19,30,80,81 Table 3 presents a complete vibrational assignment of all fundamental vibrations and a comparison to experimental data.17 Most importantly, all theoretical data agrees with experimental results for aniline and PANIs. The vibrational spectra of all investigated PANIs are reconstructed in Fig. S3 of the ESI† using the Gabedit software package. In the spectrum of aniline, two peaks appear at 1612 and 1580 cm−1. The former is assigned to the –NH2 bending and the latter to the C–C ring-stretching vibration of the phenyl group. The remaining leading vibrations of the Raman and IR spectra are located at 1276 cm−1 and correspond to the ring-stretching mode mainly attributed to the C–N stretching. The band at 1147 cm−1 results from the C–H bending mode. All the characteristic features of the aniline vibrational spectrum are present in all investigated PANIs, except for the –NH2 peak that is absent in 3t and 3o.| Molecule | Exp.17 | This work | ||
|---|---|---|---|---|
| Freq. [cm−1] | Intensity [km mol−1] | Assignment | ||
| Aniline | 1620 | 1612 | 153.261 | N–H2 bending |
| 1603 | 1580 | 4.343 | C–C ring stretching | |
| 1276 | 1276 | 53.756 | C–N stretching | |
| 1176, 1155 | 1147 | 1.333 | C–H bending | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Tetramer (t) | ||||
| 1t | 1618 | 1621 | 13.936 | C–C ring stretching |
| 1616 | 18.881 | C–C ring stretching | ||
| 1615 | 26.274 | C–C ring stretching | ||
| 1599 | 206.853 | C–C ring stretching | ||
| 1602 | 62.197 | N–H2 bending | ||
| 1219 | 1221 | 15.378 | C–N stretching | |
| 1219 | 6.463 | C–N stretching | ||
| 1181 | 1165 | 4.293 | C–H bending | |
| 1163 | 0.161 | C–H bending | ||
| 2t | 1617 | 1619 | 91.950 | C–C ring stretching |
| 1606 | 506.207 | N–H2 bending | ||
| 1519 | 1519 | 254.253 | CN stretching |
|
| 1220, 1219 | 1227 | 6.510 | C–N stretching | |
| 1222 | 3.694 | C–N stretching | ||
| 1182 | 1168 | 2.785 | C–H bending | |
| 1155 | 132.108 | C–H bending | ||
| 1153 | 150.552 | C–H bending | ||
| 1144 | 1.584 | C–H bending | ||
| 3t | 1612, 1553 | 1556 | 49.940 | C–C ring stretching |
| 1582, 1579 | 1588 | 2.021 | CC stretching |
|
| 1581 | 5.799 | CC stretching |
||
| 1480 | 1496 | 77.580 | CN stretching |
|
| 1219 | 1235 | 0.642 | C–N stretching | |
| 1229 | 7.934 | C–N stretching | ||
| 1218 | 22.962 | C–N stretching | ||
| 1157 | 1157 | 2.481 | C–H bending | |
|
||||
| Octamer (o) | ||||
| 1o | 1618 | 1622 | 5.474 | C–C ring stretching |
| 1617 | 0.271 | C–C ring stretching | ||
| 1616 | 5.865 | C–C ring stretching | ||
| 1615 | 16.793 | C–C ring stretching | ||
| 1599 | 206.640 | C–C ring stretching | ||
| 1602 | 70.742 | N–H2 bending | ||
| 1219 | 1221 | 1.003 | C–N stretching | |
| 1220 | 7.350 | C–N stretching | ||
| 1219 | 41.414 | C–N stretching | ||
| 1181 | 1165 | 6.165 | C–H bending | |
| 1164 | 0.405 | C–H bending | ||
| 1163 | 1.103 | C–H bending | ||
| 2o | 1617 | 1616 | 23.481 | C–C ring stretching |
| 1519 | 1515 | 169.019 | CN stretching |
|
| 1220, 1219 | 1224 | 45.260 | C–N stretching | |
| 1223 | 21.564 | C–N stretching | ||
| 1221 | 4.281 | C–N stretching | ||
| 1182 | 1169 | 1.030 | C–H bending | |
| 1166 | 3.486 | C–H bending | ||
| 1165 | 76.322 | C–H bending | ||
| 1157 | 113.823 | C–H bending | ||
| 1155 | 455.013 | C–H bending | ||
| 1149 | 0.902 | C–H bending | ||
| 1143 | 21.476 | C–H bending | ||
| 3o | 1612, 1553 | 1586 | 8.260 | C–C ring stretching |
| 1581 | 5.022 | C–C ring stretching | ||
| 1582, 1579 | 1589 | 5.578 | CC stretching |
|
| 1574 | 7.336 | CC stretching |
||
| 1480 | 1490 | 158.490 | CN stretching |
|
| 1472 | 0.771 | CN stretching |
||
| 1219 | 1218 | 53.816 | C–N stretching | |
| 1157 | 1157 | 8.286 | C–H bending | |
| 1148 | 269.192 | C–H bending | ||
For 1t we observe several characteristic vibrations of the benzene ring, such as those peaked at 1599, 1615, 1616, and 1621 cm−1, which correspond to a C–C stretching vibrational mode for ring 1, 2, 3, and 4, respectively, (cf. Fig. 4 for ring labels) and two C–H bending vibrational modes at 1165 and 1163 cm−1. The bands at 1221, and 1219 cm−1 correspond to the C–N stretching vibrational mode for N1, N2, and N3 respectively, while the –NH2 bending mode is positioned at 1602 cm−1 (the atomic labels are indicated in Fig. S1 of the ESI†).
For 2t, the C–C ring-stretching is located at 1619 cm−1, and the CN stretching mode at 1519 cm−1. The two peaks at 1222 (N1) and 1227 (N2 and N3) cm−1 are due to a C–N stretching mode (see also Fig. S1 of the ESI† for atomic labels). The C–H bending vibrational mode of the benzene ring can be characterized by a Raman band at 1168, 1155, 1153, and 1144 cm−1, respectively. The –NH2 bending mode is positioned at 1606 cm−1.
3t features the fundamental bands of CC stretching modes at 1581 and 1588 cm−1 and a C–C ring-stretching mode at 1556 cm−1. The Raman band at 1496 cm−1 corresponds to a CN stretching vibrational mode, while the C–N stretching mode is positioned at 1217, 1228, and 1234 cm−1. The C–H bending mode is predicted at 1157 cm−1.
Comparing the characteristic vibrational features of 1t, 2t, and 3t, we note a redshift of the C–C ring stretching and C–H bending frequencies. Moreover, we observe a blueshift of the N–H2 bending vibrations from 1t to 2t. Essentially the same vibrational features as for 1t, 2t, and 3t are observed for 1o, 2o, and 3o, respectively. The only difference is the larger number of peaks and a negligible increase in characteristic vibrational frequencies by about 1–2 cm−1 for longer polymer chains (cf. Table 3).
3.3 HOMO–LUMO gaps from DFAs
The HOMO and LUMO molecular orbitals of 1t, 2t, 3t, 1o, 2o, and 3o obtained from different xc functionals (BP86, PBE, PBE0, and CAM-B3LYP) are depicted in Fig. S5–S8 of the ESI.† All xc functionals predict similar HOMO and LUMO π- and π*-type molecular orbitals delocalized over the whole molecular structures. The HOMO and LUMO energies and the HOMO–LUMO gaps are summarized in Table S8 and visualized in Fig. S4 of the ESI.† Both generalized gradient approximations to the xc functional (BP86 and PBE) predict identical HOMO–LUMO gaps for aniline and almost identical for all PANIs. The PBE0 xc functional with an admixture of 25% of HF exchange roughly doubles the HOMO–LUMO gaps. The range-separated CAM-B3LYP xc functional further widens the HOMO–LUMO gaps by about 20–25%. Specifically, CAM-B3LYP predicts the HOMO–LUMO gap of 0.29 eV for aniline, and 0.196 eV for 1t, 0.155 eV for 2t, and 0.162 eV for 3t, respectively. The HOMO–LUMO gap is only slightly affected (lowered by around 0.01 eV) in the longer PANIs (1o, 2o, and 3o). Finally, we should note that our DFA calculations do not show any clear trend of the HOMO–LUMO gap with respect to the formal oxidation state of PANIs.3.4 Electronic excitation energies
A significant feature of conjugated polymers often studied theoretically and experimentally is the electronic structure of their valence band. The desired donor properties feature high-intensity electronic transitions with a dominant HOMO → LUMO character in the specific range of the spectrum.82 Therefore, we will scrutinize the lowest-lying electronic excitation energies obtained from different quantum chemistry methods to assess the structure-to-property relationship. Table 4 summarizes low-lying electronic transition energies and associated characteristics obtained from various xc functionals (BP86, PBE, PBE0, and CAM-B3LYP), CIS, and EOM-pCCD+S. The EOM-pCCD and EOM-pCCD-CCS excitation energies are reported in Table S9 of the ESI† for comparison.| Molecule | No. | Character | BP86 | PBE | PBE0 | CAM-B3LYP | EOM-pCCD+S | CIS |
|---|---|---|---|---|---|---|---|---|
| a The CAM-B3LYP ground-state calculations for 1o, 2o, and 3o did not converge due to numerical difficulties. | ||||||||
| Aniline | 1 | Energy | 4.422 | 4.410 | 4.820 | 4.912 | 6.005 | 5.821 |
| Weight | 0.900 | 0.900 | 0.880 | 0.860 | 0.485 | 0.618 | ||
| Character | H → L | H → L | H → L | H → L | H-1 → L | H → L | ||
| Intensity | 0.029 | 0.029 | 0.038 | 0.040 | — | — | ||
| 2 | Energy | 4.903 | 4.660 | 5.180 | 5.253 | 6.880 | 6.174 | |
| Weight | 0.850 | 0.810 | 0.550 | 0.950 | 0.445 | 0.551 | ||
| Character | H → L+2 | H → L+2 | H → L+2 | H → L+1 | H-1 → L+1 | H → L+1 | ||
| Intensity | 0.013 | 0.008 | 0.013 | 0.012 | — | — | ||
| 3 | Energy | 5.373 | 5.250 | 5.670 | 5.737 | 8.002 | 7.304 | |
| Weight | 0.660 | 0.820 | 0.430 | 0.810 | 0.309 | 0.578 | ||
| Character | H → L+1 | H → L+3 | H → L+1 | H → L+2 | H-11 → L+3 | H → L+2 | ||
| Intensity | 0.131 | 0.024 | 0.130 | 0.128 | — | — | ||
| 1t | 1 | Energy | 2.791 | 2.780 | 3.547 | 3.916 | 5.525 | 4.913 |
| Weight | 0.570 | 0.490 | 0.720 | 0.600 | 0.192 | 0.552 | ||
| Character | H → L | H → L | H → L | H → L | H-38 → L+3 | H → L | ||
| Intensity | 0.027 | 0.025 | 0.562 | 0.839 | — | — | ||
| 2 | Energy | 2.834 | 2.823 | 3.632 | 4.000 | 5.645 | 5.116 | |
| Weight | 0.720 | 0.770 | 0.690 | 0.530 | 0.231 | 0.534 | ||
| Character | H → L+2 | H → L+2 | H → L+1 | H → L+1 | H-32 → L+3 | H → L+1 | ||
| Intensity | 0.036 | 0.035 | 0.724 | 0.558 | — | |||
| 3 | Energy | 2.918 | 2.894 | 3.700 | 4.092 | 5.692 | 5.220 | |
| Weight | 0.440 | 0.430 | 0.880 | 0.600 | 0.293 | 0.484 | ||
| Character | H → L+1 | H → L+1 | H → L+2 | H → L+2 | H-33 → L+1 | H → L+2 | ||
| Intensity | 0.647 | 0.671 | 0.033 | 0.032 | — | — | ||
| 2t | 1 | Energy | 1.734 | 1.729 | 2.078 | 2.376 | 4.381 | 3.173 |
| Weight | 0.880 | 0.880 | 0.940 | 0.890 | 0.212 | 0.628 | ||
| Character | H → L | H → L | H → L | H → L | H-43 → L+1 | H → L | ||
| Intensity | 0.912 | 0.909 | 1.177 | 1.301 | — | — | ||
| 2 | Energy | 2.012 | 2.001 | 2.429 | 2.814 | 4.987 | 3.872 | |
| Weight | 0.890 | 0.890 | 0.890 | 0.740 | 0.332 | 0.509 | ||
| Character | H-1 → L | H-1 → L | H-1 → L | H-1 → L | H-42 → L | H-1 → L | ||
| Intensity | 0.091 | 0.087 | 0.022 | 0.001 | — | — | ||
| 3 | Energy | 2.583 | 2.573 | 3.304 | 3.896 | 5.821 | 4.972 | |
| Weight | 0.890 | 0.900 | 0.610 | 0.190 | 0.312 | 0.429 | ||
| Character | H-2 → L | H-2 → L | H-2 → L | H-4 → L | H-28 → L+6 | H-9 → L | ||
| Intensity | 0.001 | 0.001 | 0.021 | 0.674 | — | — | ||
| 3t | 1 | Energy | 1.676 | 1.671 | 2.087 | 2.413 | 4.468 | 3.216 |
| Weight | 0.580 | 0.580 | 0.940 | 0.870 | 0.255 | 0.617 | ||
| Character | H → L | H → L | H → L | H → L | H-45 → L+2 | H → L | ||
| Intensity | 0.566 | 0.564 | 1.198 | 1.327 | — | — | ||
| 2 | Energy | 1.770 | 1.763 | 2.450 | 2.916 | 5.123 | 3.923 | |
| Weight | 0.380 | 0.370 | 0.500 | 0.440 | 0.384 | 0.405 | ||
| Character | H → L | H → L | H → L+1 | H-1 → L | H-46 → L+1 | H-1 → L | ||
| Intensity | 0.378 | 0.367 | 0.010 | 0.025 | — | — | ||
| 3 | Energy | 2.012 | 2.002 | 2.539 | 2.942 | 5.284 | 4.049 | |
| Weight | 0.540 | 0.560 | 0.440 | 0.430 | 0.382 | 0.343 | ||
| Character | H → L+1 | H → L+1 | H → L+1 | H → L+1 | H-45 → L+2 | H → L+1 | ||
| Intensity | 0.005 | 0.005 | 0.011 | 0.021 | — | — | ||
| 1o | 1 | Energy | 2.381 | 2.364 | 3.266 | a— | 5.349 | 4.683 |
| Weight | 0.900 | 0.900 | 0.670 | a— | 0.111 | 0.408 | ||
| Character | H → L | H → L | H → L | a— | H-78 → L+9 | H → L | ||
| Intensity | 0.375 | 0.397 | 2.618 | a— | — | — | ||
| Energy | 2.473 | 2.456 | 3.470 | a— | 5.516 | 4.899 | ||
| Weight | 0.870 | 0.900 | 0.280 | a— | 0.165 | 0.342 | ||
| 2 | Character | H → L+1 | H → L+1 | H → L+4 | a— | H-53 → L+6 | H-1 → L | |
| Intensity | 0.085 | 0.061 | 0.111 | a— | — | — | ||
| 3 | Energy | 2.535 | 2.523 | 3.486 | a— | 5.548 | 5.043 | |
| Weight | 0.570 | 0.530 | 0.370 | a— | 0.179 | 0.267 | ||
| Character | H → L+2 | H → L+2 | H → L+3 | a— | H-52 → L+3 | H → L+2 | ||
| Intensity | 0.007 | 0.009 | 0.253 | a— | — | — | ||
| 2o | 1 | Energy | 0.970 | 0.970 | 1.783 | a— | 4.249 | 3.042 |
| Weight | 0.780 | 0.770 | 0.940 | a— | 0.204 | 0.559 | ||
| Character | H → L | H → L | H → L | a— | H-85 → L+1 | H → L | ||
| Intensity | 0.150 | 0.152 | 2.217 | a— | — | — | ||
| 2 | Energy | 1.272 | 1.268 | 1.996 | a— | 4.675 | 3.548 | |
| Weight | 0.410 | 0.400 | 0.950 | a— | 0.329 | 0.374 | ||
| Character | H → L+1 | H → L+1 | H → L+1 | a— | H-84 → L+2 | H-1 → L+1 | ||
| Intensity | 1.379 | 1.386 | 0.002 | a— | — | — | ||
| 3 | Energy | 1.347 | 1.345 | 2.088 | a— | 4.896 | 3.792 | |
| Weight | 0.290 | 0.290 | 0.910 | a— | 0.310 | 0.326 | ||
| Character | H-1 → L | H-1 → L | H-1 → L | a— | H-86 → L+3 | H-3 → L | ||
| Intensity | 0.013 | 0.010 | 0.004 | a— | — | — | ||
| 3o | 1 | Energy | 1.093 | 1.090 | 1.524 | a— | 3.959 | 2.598 |
| Weight | 0.480 | 0.480 | 0.930 | a— | 0.159 | 0.558 | ||
| Character | H → L+1 | H → L+1 | H → L | a— | H-3 → L+3 | H → L | ||
| Intensity | 0.090 | 0.069 | 3.561 | a— | — | — | ||
| 2 | Energy | 1.117 | 1.114 | 1.893 | a— | 4.370 | 3.135 | |
| Weight | 0.810 | 0.810 | 0.480 | a— | 0.174 | 0.408 | ||
| Character | H → L | H → L | H-1 → L | a— | H-81 → L+4 | H-1 → L | ||
| Intensity | 1.970 | 1.976 | 0.000 | a— | — | — | ||
| 3 | Energy | 1.349 | 1.344 | 2.028 | a— | 4.679 | 3.524 | |
| Weight | 0.550 | 0.560 | 0.490 | a— | 0.216 | 0.382 | ||
| Character | H-2 → L | H-2 → L | H → L+1 | a— | H-84 → L+3 | H-2 → L | ||
| Intensity | 0.003 | 0.009 | 0.000 | a— | — | — | ||
PANIs significantly lower the electronic transitions observed in the aniline model system. Specifically, they fall in an energetic descending order 1t < 1o < 3t < 3o < 2t < 2o, indicating that emeraldine has the lowest-lying electronic transitions among them all.21 That contradicts the common experimental knowledge about the absorption spectra of PANI, which is expected to be in the range of 2–3.2 eV for leucoemeraldine, 1.6–3.1 eV for emeraldine, and 1.0–1.8 eV for pernigraniline, respectively.21,25 We should stress that the electrical conductivity of PANIs does not directly depend on the position of their excited states but on the type of dopant, the extent of doping, and the polymer length.83 Such features combined with the given range of excitation energies and their type can, in turn, affect the conjugation properties.
Moving from structures t to o, we observe a lowering of excitations by about 0.3–0.4 eV. The absolute values of excitation energies and, to some extent, their characteristics strongly depend on the applied xc functional.
Based on previous TD-DFT benchmarks and analysis of excitation energies, we do not expect any outstanding performance from semi-local xc functionals like BP86 and PBE, as they tend to underestimate electron excitations.84–86 Addition of HF exchange introduces some non-local effects in the xc kernel and improves the overall performance of TD-DFT. We expect further enhancement of the description of charge-transfer states with range-separated hybrids.87–91 Thus, we anticipate the PBE0 and CAM-B3LYP results to be more reliable, although limited to model single electronic transitions and electronic structures well-described by a single Slater determinant. A significant difference between the PBE0 and CAM-B3LYP excitation energies can be used to identify partial charge-transfer states.90 Based on that, we anticipate that all investigated PANI structures have some partial charge-transfer character, with aniline being the exception. The nature of PBE0 and CAM-B3LYP transitions is very similar, except for structure 1t, where the order of the 2-nd and 3-rd excited state changes. The PBE0 and CAM-B3LYP excitation energies are comparable in magnitude to the CIS data: electronic transitions' ordering and main character are virtually the same. They differ, however, in the absolute values of excitation energies (cf. Table 4), where CIS predicts much higher excitation energies. The most considerable discrepancies are observed for the aniline molecule (up to 1.5 eV) and are reduced to approximately 1 eV in PANIs.
3.4.2.1 Collective contributions to excitation energies. Table 5 summarizes the collective contributions to each excited state, where all the excitation contributions are grouped according to their character. We can see the qualitative differences in the low-lying transitions between the leucoemeraldine (1t and 1o) and the remaining structures; while the lower part of the leucoemeraldine spectrum is dominated by the LPN → B (N lone pair to benzenoid ring) and B → B electronic excitations, in the remaining systems, the electrons are mainly transferred to the quinoid ring (Q). Specifically, the electronic spectrum of emeraldine (2t and 2o) and pernigraniline (3t and 3o) are best described by the LPN → Q, Q → Q, and B → Q electronic transitions. Their collective contributions increase for higher-lying states. Thus, the local nature of pCCD-optimized orbitals allows us to dissect the character of each transition in PANIs and their structure-to-property relationship. Specifically, the LPN → Q transitions in leucoemeraldine can be classified as charge-transfer (CT) type and the B → B as local (L) in nature. All B → Q excitations in emeraldine and pernigraniline are of CT type, and Q → Q are L type. The LPN → Q electronic transitions in emeraldine have dominantly CT character, but in pernigraniline, they have mixed CT/L nature with a diminishing CT character in the longer polymer structure (3o).
| Molecule | no. | LPN → B | LPN → Q | σN → Q | B → B | B → Q | Q → B | Q → Q |
|---|---|---|---|---|---|---|---|---|
| 1t | 1 | 12.4% | 38.8% | |||||
| 2 | 13.7% | 40.3% | ||||||
| 3 | 16.5% | 51% | ||||||
| 2t | 1 | 12.5% | 1.2% | 1.5% | 25.8% | 24.2% | ||
| 2 | 29.3% | 4.6% | 1.6% | 26.5% | 2.9% | |||
| 3 | 0.64% | 5.6% | 62.7% | 1.3% | 1.9% | |||
| 3t | 1 | 10.3% | 0.9% | 8.1% | 19.2% | 21.7% | ||
| 2 | 19.8% | 3.5% | 4.3% | 27.3% | 16.4% | |||
| 3 | 28.3% | 4.5% | 21.7% | 15.3% | ||||
| 1o | 1 | 9.3% | 14.1% | |||||
| 2 | 9.3% | 32.2% | ||||||
| 3 | 11.2% | 36.8% | ||||||
| 2o | 1 | 10.3% | 1.1% | 1.4% | 20.9% | 25.3% | ||
| 2 | 0.63% | 16.7% | 3.8% | 7.2% | 25.9% | 12.5% | ||
| 3 | 25.3% | 5.1% | 1.5% | 23.3% | 3.2% | |||
| 3o | 1 | 7.5% | 1.1% | 13.9% | 19.3% | |||
| 2 | 11.08% | 1.7% | 14.9% | 9% | ||||
| 3 | 17.7% | 0.6% | 1% | 15% | 0.5% | 4.3% |
An additional feature of the leucoemeraldine electron spectrum (not shown in Table 5) is the partial contribution of double excitations in the B → B transitions. Such excitations are also partially present in the 3-rd excited state of 2t but somehow disappear in 2o. The “pure” double electronic transitions in all the investigated systems are presented in the upper part of the spectrum, as shown in Table S9 of the ESI.†
3.4.2.2 Analysis of the leading contributions. For 1t, all three lowest excitations have leading contributions from the
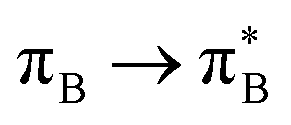 , where B indicates the benzenoid rings. They differ between themselves in the admixture of transitions from the nitrogen lone pair (LPN) orbital to the
, where B indicates the benzenoid rings. They differ between themselves in the admixture of transitions from the nitrogen lone pair (LPN) orbital to the 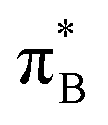 and σ* orbitals. In the second and third excited state of 1t, transitions of the πB → σ* character appear additionally. Upon polymer elongation (1o), the excitations become almost solely dominated by the
and σ* orbitals. In the second and third excited state of 1t, transitions of the πB → σ* character appear additionally. Upon polymer elongation (1o), the excitations become almost solely dominated by the 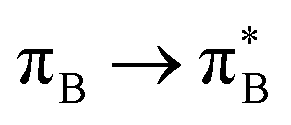 transitions.
transitions.The electronic spectrum of 2t is very complex and involves transitions of many characters. The leading contributions for the first excited state come from the nitrogen lone-pairs (LPN), πN, and πB orbitals to the 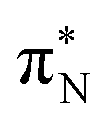 orbital (where the subscript N underlines that the orbital is centered at the nitrogen atom). Additionally, we find smaller but non-negligible contributions of type
orbital (where the subscript N underlines that the orbital is centered at the nitrogen atom). Additionally, we find smaller but non-negligible contributions of type 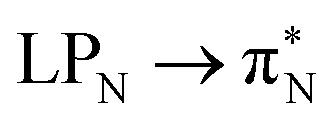 ,
, 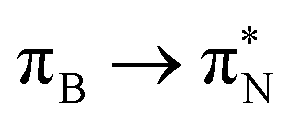 , and
, and 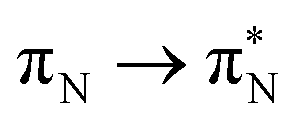 , where the index Q indicates the quinoid ring.
, where the index Q indicates the quinoid ring. 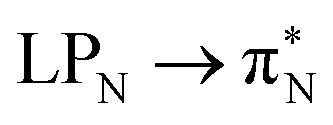 and
and 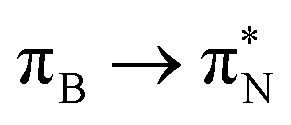 electronic transitions dominate the second excited state and
electronic transitions dominate the second excited state and 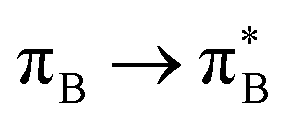 the third excited state of 2t, respectively. Moving to 2o, we observe a more organized spectrum composed of less diverse transitions. Specifically, these are
the third excited state of 2t, respectively. Moving to 2o, we observe a more organized spectrum composed of less diverse transitions. Specifically, these are 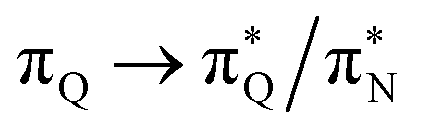 and
and 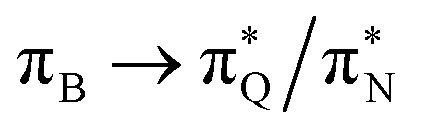 transitions for the first,
transitions for the first, 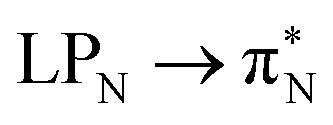 ,
, 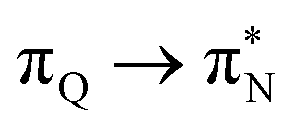 , and
, and 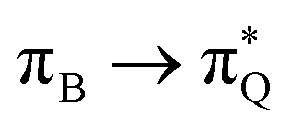 transitions for the second, and the
transitions for the second, and the 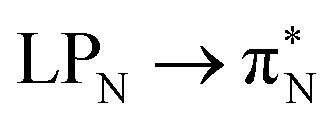 and
and 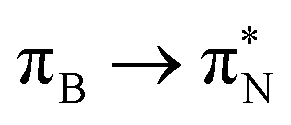 transitions for the third excited state, respectively. Thus, the elongation of emeraldine (2) profoundly affects its low-lying electronic transitions, revealing the involvement of quinoid rings only in the octamer configuration.
transitions for the third excited state, respectively. Thus, the elongation of emeraldine (2) profoundly affects its low-lying electronic transitions, revealing the involvement of quinoid rings only in the octamer configuration.
The electronic spectrum of 3t is as complex as 2t, differing mainly in the increased involvement of quinoid orbitals and the presence of σN orbitals (see the corresponding orbitals in Fig. S10–S18 of the ESI†). The first excited state of 3t is dominated by 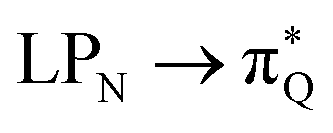 ,
, 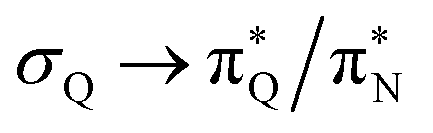 , and
, and 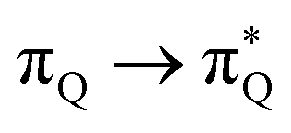 transitions, the second one by
transitions, the second one by 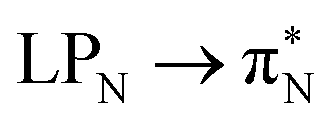 ,
, 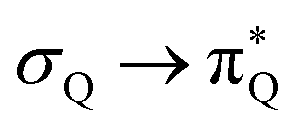 , and
, and 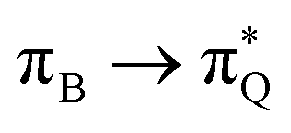 transitions, and the third one by
transitions, and the third one by 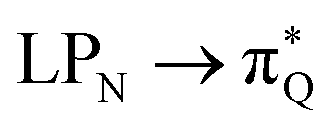 ,
, 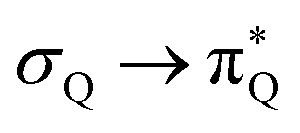 , and
, and 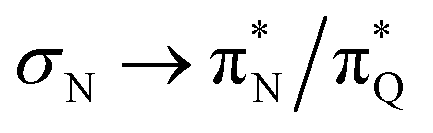 transitions. The electronic spectrum of the corresponding structure 3o is less complex, dominated by three main types of transitions. Specifically, these are
transitions. The electronic spectrum of the corresponding structure 3o is less complex, dominated by three main types of transitions. Specifically, these are 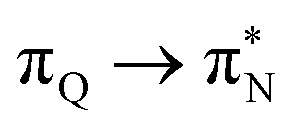 and
and 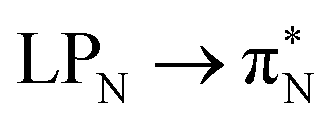 transitions for the first excited state,
transitions for the first excited state, 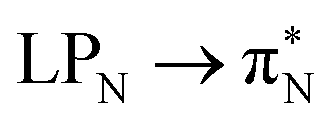 ,
, 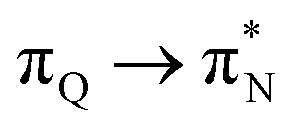 , and
, and 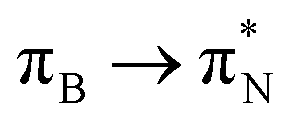 transitions for the second one, and
transitions for the second one, and 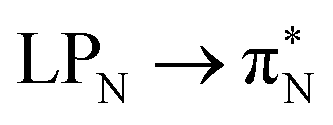 and
and 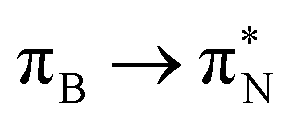 transitions for the third one, respectively.
transitions for the third one, respectively.
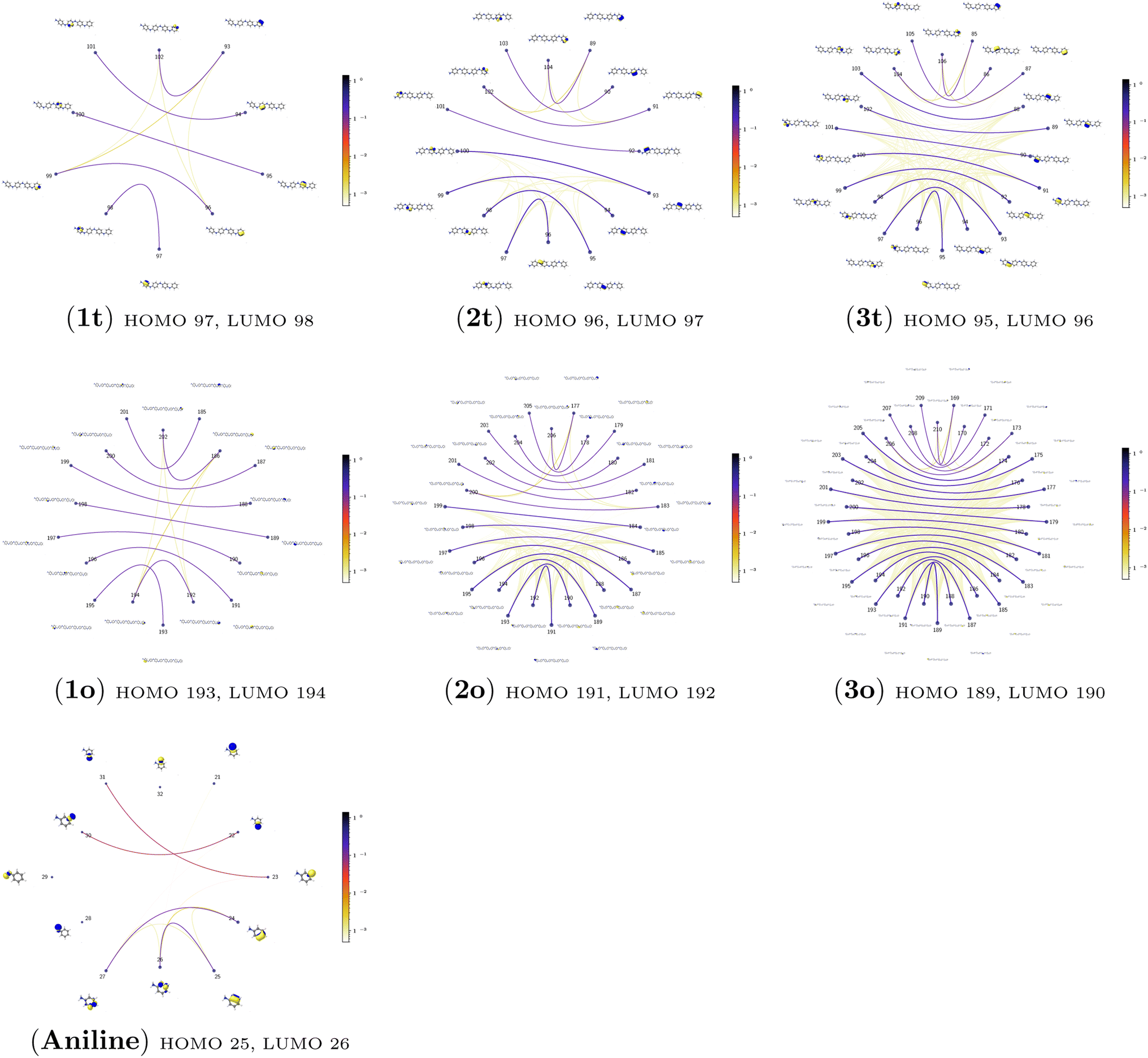 | ||
| Fig. 5 The orbital-pair mutual information for aniline and all investigated PANIs in their tetramer (t) and octamer (o) forms calculated from the pCCD ground-state wavefunctions within the PyBEST software package. | ||
The quantum information analysis of PANI structures points to an increased multi-reference character in longer polymer chains. That is highlighted by the growing number of strongly-correlated orbitals in Fig. 5. Thus, we anticipate that for such structures, pCCD-based methods should be superior to DFAs.
4 Conclusions
In this article, we employed modern quantum chemistry methods to investigate the electronic structures and properties, such as vibrational and electronic spectra, of the aniline molecule and PANIs at different oxidation states and lengths. We analyzed their structure-to-property relationship for the first time.The BP86-optimized electronic structures and vibrational frequencies of aniline and PANIs are in excellent agreement with the available experimental data indicating the right choice of the xc functional. The characteristic structural and vibrational features of PANIs in the tetramer form (1t, 2t, and 3t) are almost indistinguishable from their octamer counterparts (1o, 2o and 3o). Thus, the tetramer forms of PANIs are adequate models for longer polymer chains when considering structural and vibrational features, regardless of their oxidation states. However, the length of the PANI chain profoundly affects the electronic spectra and the overall electronic structure. Moving from aniline to polymeric structures, the mutual information analysis indicates the increased multi-reference character of the systems. Such observation calls into question the reliability of DFAs in predicting ground and excited-state properties in a balanced way. A numerical indication is already observed for the CAM-B3LYP xc functional having convergence issues for octamer structures (1o, 2o, and 3o). As an alternative, we propose to use pCCD-based methods that utilize the complete set of variationally optimized orbitals at the correlated level and can cope with such complex electronic structures. An additional advantage of pCCD-based methods is the optimization of all orbitals on an equal footing (up to a thousand basis functions in this work). The final pCCD orbitals are localized. The excitation energies are composed of many small components for such localized orbitals. We showed that working with localized orbitals (like pCCD-optimized ones) allows us to dissect the collective CT and L character of electronic transitions in each PANI for the first time. Specifically, we demonstrated that EOM-pCCD+S electronic spectra of emeraldine and pernigraniline have a dominant CT character and that polymer elongation changes the character of the leading transitions. Such an analysis is not possible using the delocalized canonical DFT orbitals. Our results highlight the strong structure-to-property relationship for electronic excitations, where the character of the excited states changes upon polymer elongation of the oxidized forms of PANIs. For instance, elongating the polymer 2 delocalizes the leading transitions of the first excited state over the whole quinoid ring (2o), while 2t features leading transitions to the quinoid 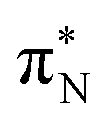 orbital. Similarly, the first excited state in 3 changes its character upon polymer elongation. While 3t features more delocalized leading transitions from the LPN to the quinoid rings, the dominant transitions in 3o are centered on the quinoid ring.
orbital. Similarly, the first excited state in 3 changes its character upon polymer elongation. While 3t features more delocalized leading transitions from the LPN to the quinoid rings, the dominant transitions in 3o are centered on the quinoid ring.
Finally, our work underlines the potential of pCCD-based methods in modeling organic electronics and motivates their further development. Based on previous studies,47,49 EOM-pCCD-based models provide reliable excited states' characters, while the overall excitation energies might be too high compared to experimental results. We should stress that adding dynamical correlation did not significantly improve excitation energies in polyenes.49 This observation suggests that other effects like basis set size or environmental effects have to be considered. One possibility to account for environmental effects is to use embedding methods.95 Such investigations are currently in progress in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. J. and P. T. acknowledge financial support from the SONATA BIS research grant from the National Science Centre, Poland (Grant No. 2021/42/E/ST4/00302). P. T. acknowledges the scholarship for outstanding young scientists from the Ministry of Science and Higher Education. The research leading to these results has received funding from the Norway Grants 2014–2021 via the National Centre for Research and Development. The authors thank Julia Romanova for providing us with the initial xyz structures of polyanilines. Calculations have been carried out using resources provided by the Wroclaw Centre for Networking and Supercomputing (http://wcss.pl), grant no. 411.Notes and references
- R. B. Salikhov, Y. N. Biglova and A. G. Mustafin, New organic polymers for solar cells, in Emerging Solar Energy Materials, ed. S. Ameen, IntechOpen Limited, London, 2018, pp. 83–104 Search PubMed.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098–1101 CrossRef CAS.
- H. Shirakawa, Rev. Mod. Phys., 2001, 73, 713–718 CrossRef CAS.
- V. G. Kulkarni, L. D. Campbell and W. R. Mathew, Synth. Met., 1989, 30, 321–325 CrossRef CAS.
- V. G. Kulkarni, W. R. Mathew, B. Wessling, H. Merkle and S. Blaettner, Synth. Met., 1991, 41, 1009–1012 CrossRef CAS.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2009, 42, 135–145 CrossRef CAS PubMed.
- A. K. Mishra, J. Comput. Sci., 2015, 10, 195–208 CrossRef.
- A. Ray, G. Asturias, D. Kershner, A. Richter, A. MacDiarmid and A. Epstein, Synth. Met., 1989, 29, 141–150 CrossRef.
- S. Chen, Z. Wei, X. Qi, L. Dong, Y.-G. Guo, L. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
- G. Wu, N. H. Mack, W. Gao, S. Ma, R. Zhong, J. Han, J. K. Baldwin and P. Zelenay, ACS Nano, 2012, 6, 9764–9776 CrossRef CAS PubMed.
- M. Silakhori, M. S. Naghavi, H. S. C. Metselaar, T. M. I. Mahlia, H. Fauzi and M. Mehrali, Materials, 2013, 6, 1608–1620 CrossRef PubMed.
- M. Liu, Y.-E. Miao, C. Zhang, W. W. Tjiu, Z. Yang, H. Peng and T. Liu, Nanoscale, 2013, 5, 7312–7320 RSC.
- M. Ates, Mater. Sci. Eng., C, 2013, 33, 1853–1859 CrossRef CAS PubMed.
- S. Ameen, M. S. Akhtar, Y. S. Kim, O.-B. Yang and H.-S. Shin, J. Phys. Chem. C, 2010, 114, 4760–4764 CrossRef CAS.
- Y. Zou, J. Pisciotta, R. B. Billmyre and I. V. Baskakov, Biotechnol. Bioeng., 2009, 104, 939–946 CrossRef CAS PubMed.
- H. S. A. Khalil, PhD thesis, Polytechnic University, 2001 Search PubMed.
- S. Quillard, G. Louarn, S. Lefrant and A. MacDiarmid, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 12496 CrossRef PubMed.
- A. F. R. A. G. Macdiarmid and J. C. Chiang, Synth. Met., 1987, 18, 285–290 CrossRef CAS.
- S. Lim, K. Tan, E. Kang and W. Chin, J. Chem. Phys., 2000, 112, 10648–10658 CrossRef CAS.
- M. Cihan Sorkun, D. Mullaj, J. V. A. Koelman and S. Er, Chem.: Methods, 2022, e202200005 CAS.
- E. Genies, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 1990, 36, 139–182 CrossRef CAS.
- S. Bhadra and D. Khastgir, Polym. Test., 2008, 27, 851–857 CrossRef CAS.
- L. Shacklette, J. Wolf, S. Gould and R. Baughman, J. Chem. Phys., 1988, 88, 3955–3961 CrossRef CAS.
- H.-S. Moon and J.-K. Park, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1431–1439 CrossRef CAS.
- B. Sjögren and S. Stafström, J. Chem. Phys., 1988, 88, 3840–3847 CrossRef.
- Z. T. de Oliveira Jr and M. Dos Santos, Chem. Phys., 2000, 260, 95–103 CrossRef.
- H. Zhekova, A. Tadjer, A. Ivanova, J. Petrova and N. Gospodinova, Int. J. Quantum Chem., 2007, 107, 1688–1706 CrossRef CAS.
- S. A. Jansen, T. Duong, A. Major, Y. Wei and L. T. Sein Jr, Synth. Met., 1999, 105, 107–113 CrossRef CAS.
- J. P. Foreman and A. P. Monkman, J. Phys. Chem. A, 2003, 107, 7604–7610 CrossRef CAS.
- A. K. Mishra and P. Tandon, J. Phys. Chem. B, 2009, 113, 14629–14639 CrossRef CAS PubMed.
- J. Romanova, J. Petrova, A. Ivanova, A. Tadjer and N. Gospodinova, J. Mol. Struct., 2010, 954, 36–44 CrossRef CAS.
- Y. Zhang, Y. Duan and J. Liu, Spectrochim. Acta, Part A, 2017, 171, 305–310 CrossRef CAS PubMed.
- P. A. Limacher, P. W. Ayers, P. A. Johnson, S. De Baerdemacker, D. Van Neck and P. Bultinck, J. Chem. Theory Comput., 2013, 9, 1394–1401 CrossRef CAS PubMed.
- T. Stein, T. M. Henderson and G. E. Scuseria, J. Chem. Phys., 2014, 140, 214113 CrossRef PubMed.
- P. Tecmer and K. Boguslawski, Phys. Chem. Chem. Phys., 2022, 24, 23026–23048 RSC.
- P. Tecmer and K. Boguslawski, Phys. Chem. Chem. Phys., 2022, 24, 23026–23048 RSC.
- K. Boguslawski and P. Tecmer, Int. J. Quantum Chem., 2017, 117, e25455 CrossRef.
- K. Boguslawski and P. Tecmer, Int. J. Quantum Chem., 2015, 115, 1289–1295 CrossRef CAS.
- K. Boguslawski, P. Tecmer, P. W. Ayers, P. Bultinck, S. De Baerdemacker and D. Van Neck, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 201106 CrossRef.
- K. Boguslawski, P. Tecmer, P. A. Limacher, P. A. Johnson, P. W. Ayers, P. Bultinck, S. De Baerdemacker and D. Van Neck, J. Chem. Phys., 2014, 140, 214114 CrossRef PubMed.
- K. Boguslawski, P. Tecmer, P. Bultinck, S. De Baerdemacker, D. Van Neck and P. W. Ayers, J. Chem. Theory Comput., 2014, 10, 4873–4882 CrossRef CAS PubMed.
- P. Tecmer, K. Boguslawski, P. A. Limacher, P. A. Johnson, M. Chan, T. Verstraelen and P. W. Ayers, J. Phys. Chem. A, 2014, 118, 9058–9068 CrossRef CAS PubMed.
- P. Tecmer, K. Boguslawski and P. W. Ayers, Phys. Chem. Chem. Phys., 2015, 17, 14427–14436 RSC.
- P. Tecmer, K. Boguslawski, M. Borkowski, P. S. Żuchowski and D. Kędziera, Int. J. Quantum Chem., 2019, 119, e25983 CrossRef.
- D. J. Rowe, Rev. Mod. Phys., 1968, 40, 153–166 CrossRef.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS.
- K. Boguslawski, J. Chem. Phys., 2016, 145, 234105 CrossRef PubMed.
- K. Boguslawski, J. Chem. Phys., 2017, 147, 139901 CrossRef PubMed.
- K. Boguslawski, J. Chem. Theory Comput., 2019, 15, 18–24 CrossRef CAS PubMed.
- J. D. Watts and R. J. Bartlett, J. Chem. Phys., 1994, 101, 3073–3078 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- TURBOMOLE V6.6, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2022, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com Search PubMed.
- J. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- A. Becke, Phys. Rev. A, 1988, 38, 3098–4000 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- S. Van Gisbergen, J. Snijders and E. Baerends, Comput. Phys. Commun., 1999, 118, 119–138 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2018, ADF2018.01, http://www.scm.com Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- K. Boguslawski, A. Leszczyk, A. Nowak, F. Brzęk, P. S. Żuchowski, D. Kędziera and P. Tecmer, Comput. Phys. Commun., 2021, 264, 107933 CrossRef CAS.
- K. Boguslawski, A. Leszczyk, A. Nowak, E. Sujkowski, F. Brzęk, P. S. Źuchowski, D. Kędziera and P. Tecmer, PyBESTv.1.2.0 Search PubMed.
- T. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS.
- L. Bytautas, J. Ivanic and K. Ruedenberg, J. Chem. Phys., 2003, 119, 8217–8224 CrossRef CAS.
- K. Boguslawski and P. Tecmer, Int. J. Quantum Chem., 2015, 115, 1289–1295 CrossRef CAS.
- K. Boguslawski, P. Tecmer, G. Barcza, O. Legeza and M. Reiher, J. Chem. Theory Comput., 2013, 9, 2959–2973 CrossRef CAS PubMed.
- A. Nowak, O. Legeza and K. Boguslawski, J. Chem. Phys., 2021, 154, 084111 CrossRef CAS PubMed.
- J. Rissler, R. M. Noack and S. R. White, Chem. Phys., 2006, 323, 519–531 CrossRef CAS.
- S. Szalay, M. Pfeffer, V. Murg, G. Barcza, F. Verstraete, R. Schneider and Ö. Legeza, Int. J. Quantum Chem., 2015, 115, 1342–1391 CrossRef CAS.
- G. Barcza, O. Legeza, K. H. Marti and M. Reiher, Phys. Rev. A, 2011, 83, 012508 CrossRef.
- L. Ding and C. Schilling, J. Chem. Theory Comput., 2020, 16, 4159–4175 CrossRef CAS PubMed.
- L. Ding, S. Mardazad, S. Das, S. Szalay, U. Schollwöck, Z. Zimborás and C. Schilling, J. Chem. Theory Comput., 2021, 17, 79–95 CrossRef CAS PubMed.
- K. Boguslawski, P. Tecmer and Ö. Legeza, Phys. Rev. B, 2016, 94, 155126 CrossRef.
- A. Nowak and K. Boguslawski, Phys. Chem. Chem. Phys., 2023, 25, 7289–7301 RSC.
- H. Piest, G. von Helden and G. Meijer, J. Chem. Phys., 1999, 110, 2010–2015 CrossRef CAS.
- P. M. Wojciechowski, W. Zierkiewicz, D. Michalska and P. Hobza, J. Chem. Phys., 2003, 118, 10900–10911 CrossRef CAS.
- Y. Cui, P. Zhu, X. Liao and Y. Chen, J. Mater. Chem. C, 2020, 8, 15920–15939 RSC.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS.
- T. Körzdörfer and J.-L. Brédas, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- M. Dreuw, J. L. Weisman and M. Head-Gordon, J. Chem. Phys., 2003, 119, 2943 CrossRef.
- M. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef PubMed.
- P. Tecmer, A. S. P. Gomes, U. Ekström and L. Visscher, Phys. Chem. Chem. Phys., 2011, 13, 6249–6259 RSC.
- P. Tecmer, R. Bast, K. Ruud and L. Visscher, J. Phys. Chem. A, 2012, 116, 7397–7404 CrossRef CAS PubMed.
- P. Tecmer, N. Govind, K. Kowalski, W. A. de Jong and L. Visscher, J. Chem. Phys., 2013, 139, 034301 CrossRef PubMed.
- A. Nowak, P. Tecmer and K. Boguslawski, Phys. Chem. Chem. Phys., 2019, 21, 19039–19053 RSC.
- N. Ben Amor, S. Evangelisti, T. Leininger and D. Andrae, in Basis Sets in Computational Chemistry, Springer, 2021, pp. 41–101 Search PubMed.
- J. J. Stewart, J. Mol. Model., 2019, 25, 7 CrossRef PubMed.
- R. Chakraborty, K. Boguslawski and P. Tecmer, Phys. Chem. Chem. Phys., 2023 10.1039/D3CP02502K.
Footnote |
| † Electronic supplementary information (ESI) available: BP86 optimized structures of aniline and polyanilines (xyz coordinates and 3-D structures with atom labels), HOMO and LUMO DFT molecular orbitals, HOMO and LUMO energies, and HOMO–LUMO gaps (BP86, PBE, PBE0, and CAM-B3LYP), oo-pCCD molecular orbitals involved in the electronic transitions, EOM-pCCD, EOM-pCCD-CCS excitation energies. See DOI: https://doi.org/10.1039/d3ra05621j |
| This journal is © The Royal Society of Chemistry 2023 |