
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy on the chemical reactivity difference of primary hydroxyl groups in iridoid glycosides†
Yuanfang Kong *a,
Jindan Xuabc,
Pengbo Zhuabc,
Shaopei Wangabc,
Bin Yangabc,
Jingyu Zhang*a and
Chunhong Dong*abc
*a,
Jindan Xuabc,
Pengbo Zhuabc,
Shaopei Wangabc,
Bin Yangabc,
Jingyu Zhang*a and
Chunhong Dong*abc
aHenan University of Chinese Medicine, Zhengzhou 450046, Henan, China. E-mail: kongyf@hactcm.edu.cn; jyzhang2004@126.com; chunhong_dong@hactcm.edu.cn
bHenan Polysaccharide Research Center, Zhengzhou 450046, Henan, China
cHenan Key Laboratory of Chinese Medicine for Polysaccharides and Drugs Research, Zhengzhou 450046, Henan, China
First published on 10th November 2023
Abstract
Iridoid glycoside, which belongs to the polyhydroxy compound, is a kind of active ingredient of traditional Chinese medicine with a wide range of sources, and has many pharmacological effects such as anti-cancer, anti-inflammatory, anti-virus, hypoglycemic and so on. Its structure contains many hydroxyl groups, including two primary hydroxyl groups. The chemical reactivity of primary hydroxyl groups has very little difference, so it is very important to control the selectivity of hydroxyl groups under certain conditions. In this paper, the difference between the two primary hydroxyl groups in iridoid glycoside was calculated based on computer simulation and verified this result through designed experiments. This study will provide an important way for site-directed modification of hydroxyl in iridoid glycoside in the future.
1 Introduction
The selective protection of hydroxyl groups on polyols, polyphenols and sugars has always been an important research topic in organic chemistry, especially sugar chemistry.1 When the specific sugar building blocks were synthesized, kinds of oligosaccharides with various functions and biological activities can also be synthesized.2 Through selective protection reactions on hydroxyl groups, complex organic chemical synthesis steps can be avoided. The ideal protecting groups are usually cheap, easily available, non-toxic, and easy to separate from the product after the protecting groups are removed.3 The hydroxyl protecting groups include ester protecting groups, ether protecting groups, acetal or ketal protecting groups and so on.4–7 Therefore, the chemical reactivity of different hydroxyl groups in polyhydroxy compounds deserves further study.Iridoid glycosides are a typical class of compounds with multiple hydroxyl groups. Most iridoid glycosides contain two primary hydroxyl groups, which were located on the aglycone and the glucose ring (C10-position and C6′-position), respectively, such as catalpol,8 geniposide9 and aucubin,10 as shown in Fig. 1. For the iridoid glycosides containing two primary hydroxyl groups, the position and number of modifications are worthy of attention when structural modification is carried out, and it is also difficult to modify the structure. Therefore, it is very important to control the selectivity of hydroxyl modification under certain reaction conditions.
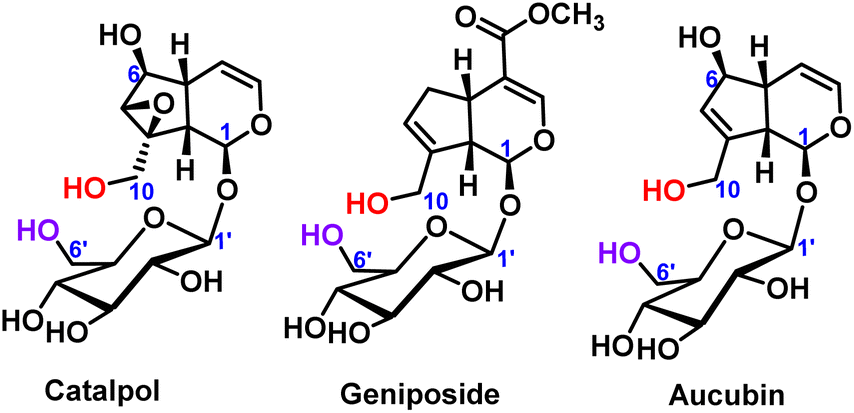 | ||
| Fig. 1 Chemical structures of catalpol, geniposide and aucubin. | ||
Many researchers mainly focused on the exploration of partial silicoetherification of catalpol hydroxyl groups, for example, first silicoetherification then esterification for catalpol structure, or full esterification for catalpol structure only. 2004, Pungitore et al.11 and García et al.12,13 conducted partial silico-etherification of catalpol then Tonn et al.14,15 explored silico-etherification followed by esterification and full esterification of catalpol, in addition, these silyl ether protective compounds have a DNA polymerase inhibitory effect. In 2019, Zhang et al.16 modified the C6-position esterified catalpol derivatives and synthesized a series of 8-hydroxyguanine DNA glycosylase 1 (OGG1) with significant effects on inhibits activity. These hydroxyl protection of catalpol were shown in Fig. 2.
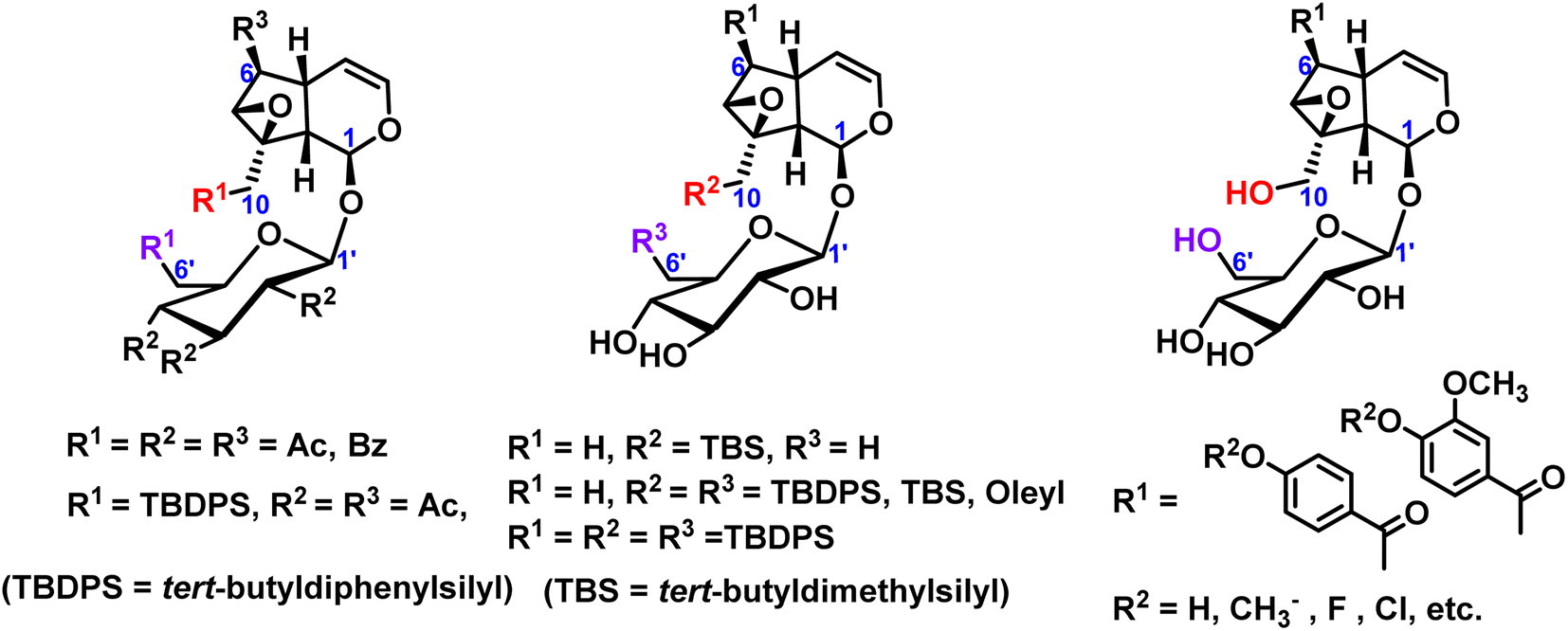 | ||
| Fig. 2 Modification of hydroxyl groups of catalpol. | ||
In iridoid glycosides, in addition to the protection of the hydroxyl groups of catalpol, the hydroxyl groups of geniposide and aucubin have also been reported. Lei et al.17 protected all the hydroxyl groups of geniposide with benzyl, and then further structurally modified to obtain a series of geniposide derivatives. Wang et al.18 reported geniposide was first converted to intermediate pentaacetyl-geniposide acid by hydrolyzing the ester bond with sodium hydroxide and acetylating the hydroxy compound with acetic anhydride, then reaction with different substituted amines and a series of geniposide derivatives were obtained. The use of tert-butyldimethylsilyl chloride permits the selective protection of aucubin at C6′-position of the sugar moiety and C10-position, or C6-position and C10-position, of the aglycone.19 These hydroxyl protection of geniposide and aucubin were shown in Fig. 3.
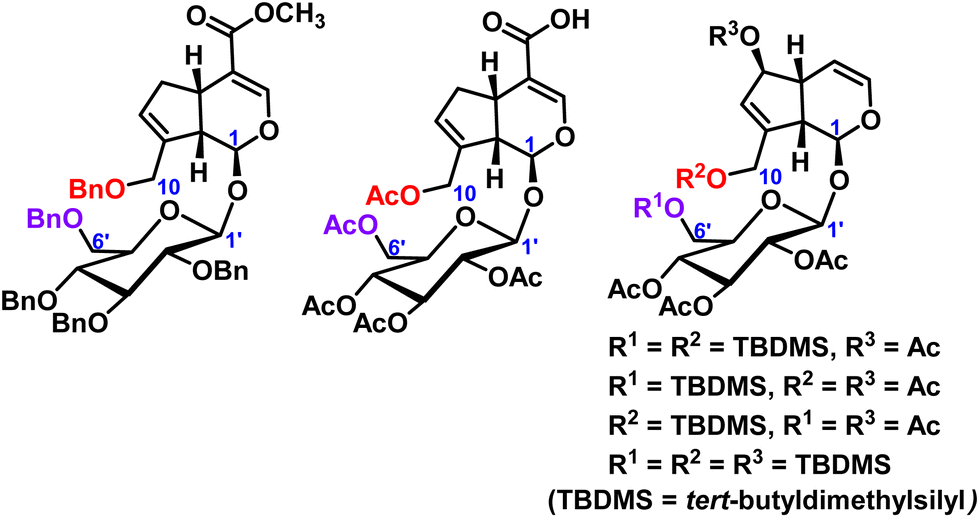 | ||
| Fig. 3 Modification of hydroxyl groups of geniposide and aucubin. | ||
However, there is no report on the chemical reactivity of two primary hydroxyl groups in iridoid glycosides. In order to modify the hydroxyl groups in iridoid glycosides, it is necessary for researchers to study this. According to the researches on iridoid glycosides previously reported by our research group20–22, computer simulation was used to explore the difference between the C10-position and C6′-position primary hydroxyl groups, and experiments were used to verify the difference and control the reaction conditions to generate a single product in order to obtain the higher yield in this paper.
2 Results and discussion
2.1 Computer simulation
Catalpol is a polyhydroxy compound containing six hydroxyl groups, including two primary hydroxyl groups and four secondary hydroxyl groups. Hydroxyl groups in catalpol structure were calculated by Gaussian software, and the structure of catalpol was optimized at the theoretical level of B3LYP/6-311++G(d,p) (Fig. 4).
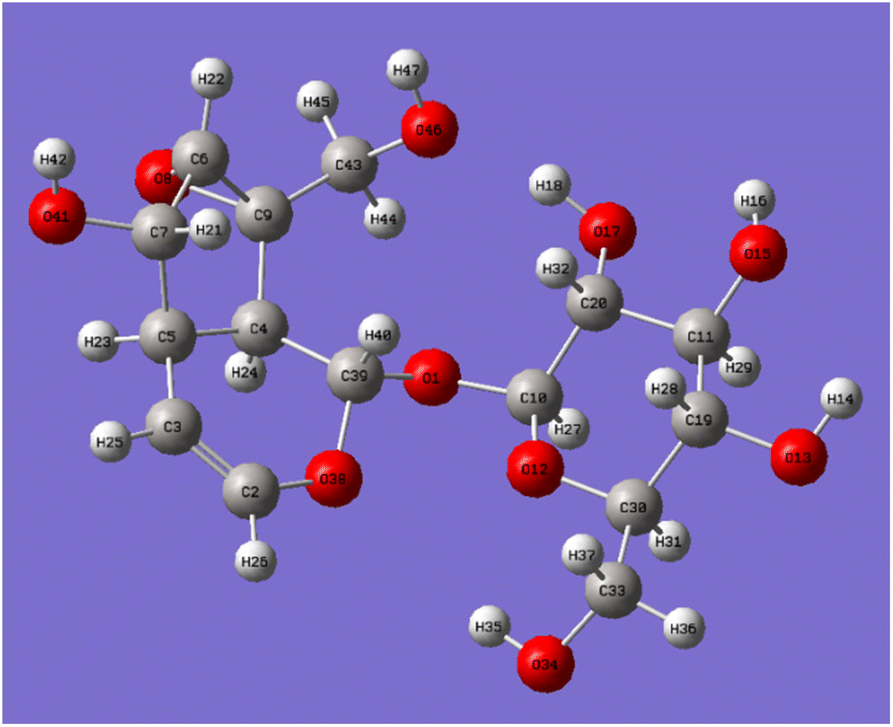 | ||
| Fig. 4 Optimized geometry of catalpol molecule. | ||
The optimized six C–O bond lengths connected to –OH were shown in Table 1. The numbers of hydroxyl groups (Fig. 4) were automatically numbered according to Gauss software, while the numbers in chemical structure were numbered according to the rules of IUPAC (International Union of Pure and Applied Chemistry), but in fact, they were the same hydroxyl groups. It can be seen from Table 1, the bond lengths of C43–O46 are the longest, 1.437. Scanning iridoid glycosides showed that the bond length of its hydroxyl group was 1.428. To scan the dihedral angle O17–H18⋯O46 of the hydroxyl group can be obtained an angle of 172.43°. It can be seen that it is affected by the hydrogen bond. When the angle of forming the hydrogen bond is larger, and the hydrogen bond is stronger, and the electron cloud density is more inclined to O atoms, so that the longer the bond length of C43–O46, the more unstable. The reaction is more likely to occur at the bond length of C43–O46, that is, the C10-position hydroxyl group is more likely to react than the C6′-position hydroxyl group.
| Geometrical parameters | Bond lengths (Å) | |
|---|---|---|
| Gauss software number | Chemical structure number | |
| a Note: according to the Gaussian software and the chemical structure of the carbon atom number is different, but the same row of carbon atoms is the same carbon atom in the catalpol structure. | ||
| C43–O46 | C10–O | 1.437 |
| C11–O15 | C3′–O | 1.425 |
| C7–O41 | C6–O | 1.422 |
| C19–O13 | C4′–O | 1.421 |
| C33–O34 | C6′–O | 1.418 |
| C20–O17 | C2′–O | 1.416 |
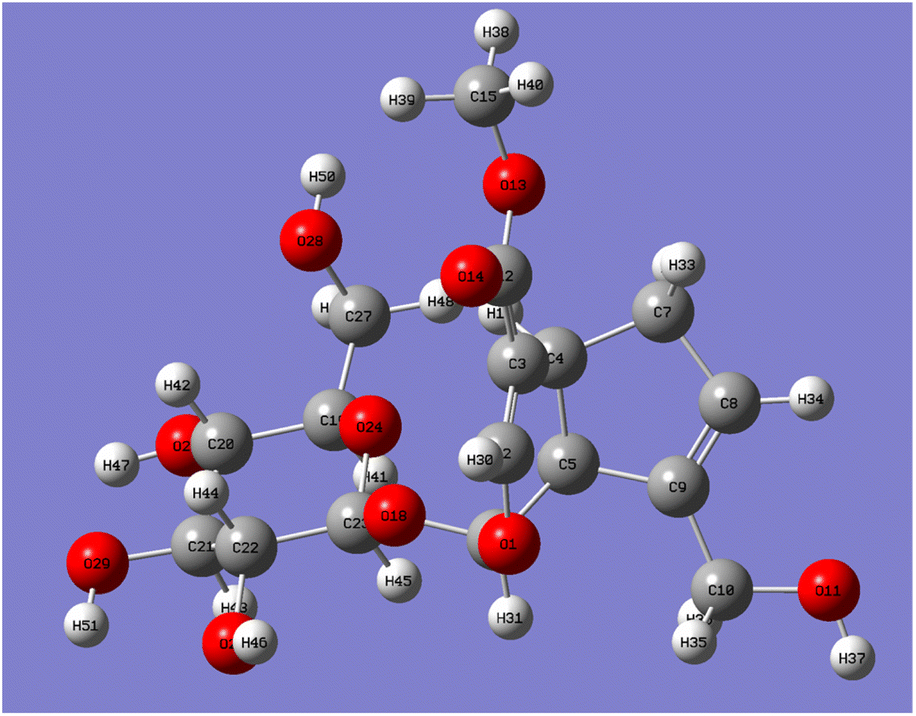 | ||
| Fig. 5 Optimized geometry of geniposide molecular. | ||
The five C–O bond lengths connected to the hydroxyl group after optimization were shown in Table 2. It can be seen from Table 2 that the bond lengths of C10–O11 and C27–O28 are longer, which are 1.4216 Å and 1.4133 Å, respectively, which are the hydroxyl groups at the C10-position on the five-membered ring and the hydroxyl groups at the C6′-position on the sugar ring. Compared with C27–O28, the bond length of C10–O11 is longer and more unstable, that is, compared with the C6′-position, the reaction is more likely to occur at the C10-position.
| Geometrical parameters | Bond lengths (Å) | |
|---|---|---|
| Gauss software number | Chemical structure number | |
| a Note: according to the Gaussian software and the chemical structure of the carbon atom number is different, but the same row of carbon atoms is the same carbon atom in the geniposide structure. | ||
| C10–O11 | C10–O | 1.422 |
| C27–O28 | C6′–O | 1.413 |
| C20–O26 | C4′–O | 1.406 |
| C22–O25 | C2′–O | 1.405 |
| C21–O29 | C3′–O | 1.405 |
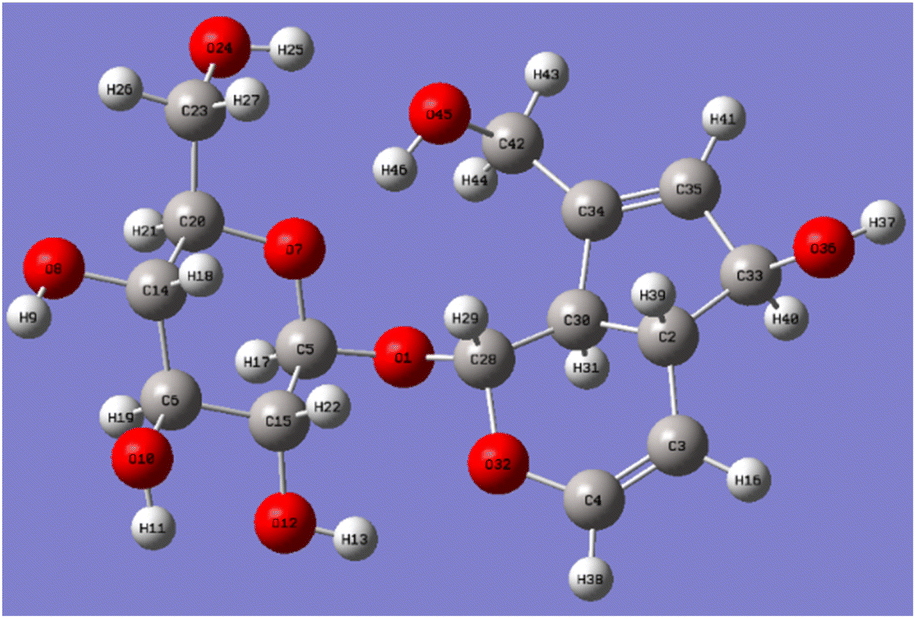 | ||
| Fig. 6 Optimized geometry of aucubin molecular. | ||
After optimizing the structure of aucubin, the six C–O bond lengths connected to the hydroxyl group are shown in Table 3. Aucubin is a polyhydroxy compound containing six hydroxyl groups, including two primary hydroxyl groups and four secondary hydroxyl groups. The chemical reactivity differences of different hydroxyl groups were calculated by Gaussian theory, and the aucubin was optimized at the theoretical level of B3LYP/6-311++G(d,p). The six C–O bond lengths connected to the hydroxyl group after optimization were shown in the Table 3, and the C42–O45 bond length at the C10-position is the longest, which is 1.42943 Å. Due to the influence of hydrogen bonds, the electron cloud density is more inclined to oxygen atoms, which makes the bond length of C42–O45 longer and more unstable. That is, compared with the C6′-position, the reaction is more likely to occur at the C10-position, which proves that the primary hydroxyl chemical reactivity on aucubin is better than that on glucose.
| Geometrical parameters | Bond lengths (Å) | |
|---|---|---|
| Gauss software number | Chemical structure number | |
| a Note: according to the Gaussian software and the chemical structure of the carbon atom number is different, but the same row of carbon atoms is the same carbon atom in the aucubin structure. | ||
| C42–O45 | C10–O | 1.429 |
| C33–O36 | C6–O | 1.427 |
| C6–O10 | C3′—O | 1.422 |
| C14–O8 | C4′—O | 1.418 |
| C15–O12 | C2′—O | 1.416 |
| C23–O24 | C6′—O | 1.415 |
Computer simulation was used to theoretically analyze the structure of catalpol, geniposide, and aucubin. By comparing their bond lengths, it was found that the primary hydroxyl chemical reactivity at the C10-position was higher than that at the C6′-position. In order to prove this result, the experimental data were further verified.
2.2 Experiments
2.2.1.1 Iodination reaction of iridoid glycosides. The iodination reaction of catalpol was carried out to explore the chemical reactivity of the two primary hydroxyl groups in catalpol.24 The temperature was 0 °C, the iodine element was 6 equivalents, and the reaction was carried out for 18 hours to obtain the compound 4 with only C10-position substitution, and the yield was up to 70%. It was further verified that the chemical reactivity of the hydroxyl group at the C10-position was higher than that of the hydroxyl group at the C6′-position (Scheme 1). After selective hydroxy iodination of catalpol at the C10-position, subsequent reactions were performed on the C10-position iodocatalpol to further verify the hydroxyl selectivity.
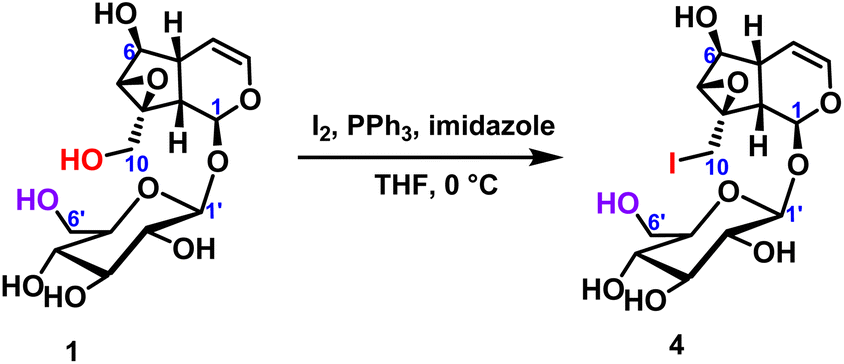 | ||
| Scheme 1 Iodine reaction of C10-hydroxyl of catalpol. | ||
2.2.1.2 Oxidation reaction of iridoid glycosides. The oxidation reaction of geniposide was occurred under the condition of IBX (2-iodoxybenzoic), and only the hydroxyl group at the C10-position was oxidized to form an aldehyde group, and 78% yield was obtained (Scheme 2).
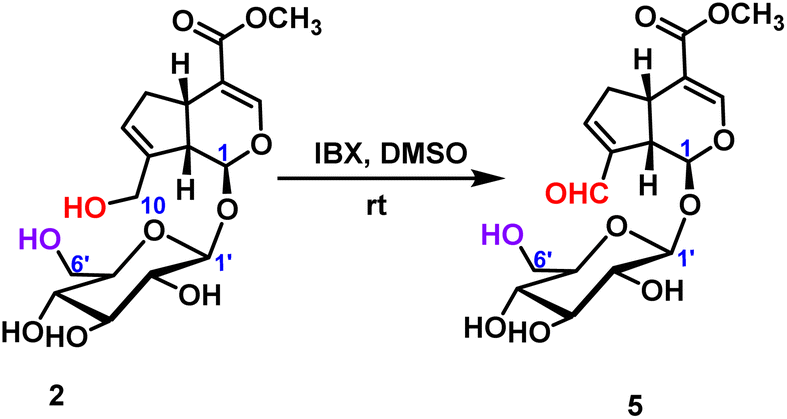 | ||
| Scheme 2 Oxidative reaction of C10-hydroxyl of geniposide. | ||
The oxidation of aucubin was carried out under the same conditions as geniposide to obtain the oxidation product with a C10-position aldehyde group in a yield of 67% (Scheme 3).
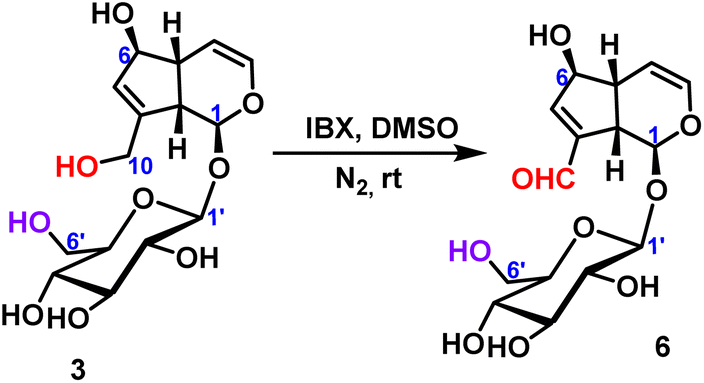 | ||
| Scheme 3 Oxidative reaction of C10-hydroxyl of aucubin. | ||
2.2.1.3 Reduction reaction of iridoid glycosides. For the reduction reaction, palladium chloride was used as the catalyst to obtain the target product 7 with the reduction of C10-position hydroxyl group, and the yield was 40% (Scheme 4). It was further verified that the chemical reactivity of C10-position hydroxyl group was higher than that of C6′-position hydroxyl group.
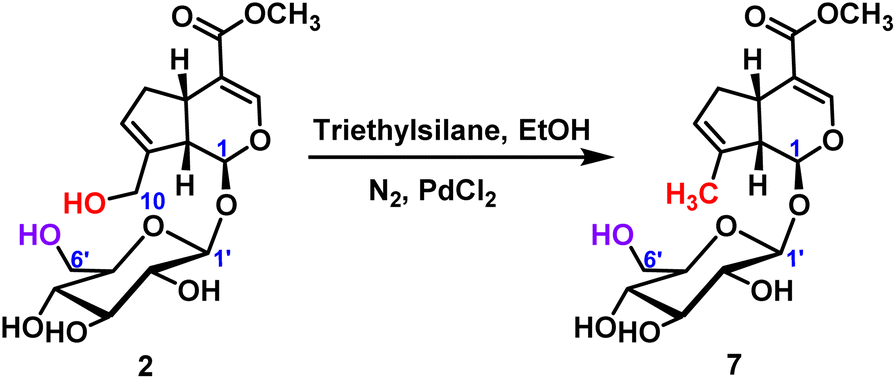 | ||
| Scheme 4 Reduction reaction of C10-hydroxyl of geniposide. | ||
Through the iodination experiments, oxidation experiments and reduction experiments in chemical reactions, it was verified that the hydroxyl chemical reactivity at the C10-position in different iridoid glycosides was higher than that at the C6′-position.
3 Conclusions
The chemical reactivity of the two primary hydroxyl groups in iridoid glycosides can be obtained by computer simulation results that the chemical reactivity of the C10-position hydroxyl group is higher than that of the C6′-position hydroxyl group, and it is further proved by the experiments of iodination, oxidation and reduction of the iridoid glycosides. This important discovery lays the foundation on polyhydroxy iridoid glycosides compounds in the future.4 Experimental
4.1 Reagents and instruments
All reagents and solvents were obtained from commercial sources and used without further purification unless otherwise indicated. All reactions were performed in oven-dried glass ware and were monitored for completeness by thin-layer chromatography (TLC) using silica gel (visualized by UV light or developed by treatment with anisaldehyde stain or iodine stain). 1H NMR and 13C-NMR spectra were recorded on a Bruker AV-400 spectrometer at 500 and 125 MHz, in DMSO-d6, using DMSO-d6 as the reference standard (2.50 ppm). Chemical shifts are reported in ppm (δ) relative to tetramethylsilane (TMS) as an internal standard. Multiplicities were given as s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet), p (pentet) and m (multiplet). Coupling constants (J) are reported in Hz. All the experiments were recorded and data were processed using standard Bruker software. MS data were obtained on a Mainer System Saimofei LCQ fleet mass spectrometer. Thin-layer chromatography was performed on silica gel 60 F254 (Qingdao Marine Chemical Ltd, P. R. China). Column chromatography purification was conducted on silica gel (200–300 mesh, Qingdao Marine Chemical Ltd, P. R. China).4.2 Experimental methods
Author contributions
Y. F. K. supervised chemistry experiments, interpreted the data, participated to the writing of the manuscript. J. D. X. and B. Y. conducted chemical experiments. P. B. Z. and S. P. W. carried out computer simulation. J. Y. Z. and C. H. D. performed the project administration. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Henan Provincial Science and Technology Research Project (222102310231) and the Zhongjing Scholars Research Funding of Henan University of Chinese Medicine (00104311-2023-9).Notes and references
- D. Hai, Z. Pei and O. Ramstroem, Cheminform, 2008, 39, 1359 Search PubMed.
- D. Deredas and A. Frankowski, Carbohydr. Res., 1994, 252, 275 CAS.
- P. Wuts, Greene's protective groups in organic synthesis, 2007 Search PubMed.
- D. Lee and M. S. Taylor, Org. Biomol. Chem., 2013, 11, 5409 RSC.
- A. Santra, G. Guchhait and A. K. Misra, Green Chem., 2011, 13, 1345 RSC.
- N. Kasuya, K. Iiyama, G. Meshitsuka and A. Ishizu, Carbohydr. Res., 1994, 260, 251 CrossRef CAS.
- A. A. Sherman, O. N. Yudina, Y. V. Mironov, E. V. Sukhova, A. S. Shashkov, V. M. Menshov and N. E. Nifantiev, Carbohydr. Res., 2001, 336, 13 CrossRef CAS PubMed.
- B. Dinda, D. R. Chowdhury and B. C. Mohanta, Chem. Pharm. Bull., 2009, 57, 765 CrossRef CAS PubMed.
- M. Shan, S. Yu, H. Yan, S. Guo, W. Xiao, Z. Wang, L. Zhang, A. Ding, Q. Wu and S. F. Y. Li, Molecules, 2017, 22, 1689 CrossRef PubMed.
- X. Zeng, F. Guo and D. Ouyang, Fitoterapia, 2020, 140, 104443 CrossRef CAS PubMed.
- C. R. Pungitore, L. G. León, C. García, V. S. Martín, C. E. Tonn and J. M. Padrón, Bioorg. Med. Chem. Lett., 2007, 17, 1332 CrossRef CAS PubMed.
- C. García, L. G. León, C. R. Pungitore, C. Ríos-Luci, A. H. Daranas, J. C. Montero, A. Pandiella, C. E. Tonn, V. S. Martín and J. M. Padrón, Bioorg. Med. Chem., 2010, 18, 2515 CrossRef PubMed.
- H. A. Garro, C. García, V. S. Martín, C. E. Tonn and C. R. Pungitore, Bioorg. Med. Chem. Lett., 2015, 25, 914 CrossRef CAS PubMed.
- C. R. Pungitore, M. J. Ayub, M. García, E. J. Borkowski, M. E. Sosa, G. Ciuffo, O. S. Giordano and C. E. Tonn, J. Nat. Prod., 2004, 67, 357 CrossRef CAS PubMed.
- O. A. Martin, H. A. Garro, M. B. Kurina Sanz, C. R. Pungitore and C. E. Tonn, J. Mol. Model., 2011, 17, 2717 CrossRef CAS PubMed.
- L. Q. Zhang, H. B. Xue, G. H. Ma, Y. M. Li, K. X. Chen and Q. Jia, US Pat., CN109734759A, 2019 Search PubMed.
- S. Lei, D. Zhang, Y. Qi, S. R. Chowdhury, R. Sun, J. Wang, Y. Du, L. Fu and F. Jiang, Eur. J. Med. Chem., 2020, 205, 112508 CrossRef CAS PubMed.
- M. Wang, J. Chen, R. Zhang, X. Guo, D. Chen, X. Guo, Y. Chen, Y. Wu, J. Sun, Y. Liu and C. Liu, Bioorg. Chem., 2021, 116, 105321 CrossRef CAS PubMed.
- V. C. Rakotondramasy, R. Laschiazza, M. Lecsö-Bornet, M. Koch, F. Tillequin and B. Deguin, J. Nat. Prod., 2007, 70, 19 CrossRef CAS PubMed.
- S. Liu, X. Cheng, X. Li, Y. Kong, S. Jiang, C. Dong and G. Wang, Sci. Rep., 2020, 10, 20415 CrossRef CAS PubMed.
- S. Liu, Y. Kong, J. Cai and C. Dong, ChemistrySelect, 2021, 6, 13520 CrossRef CAS.
- C. Dong, S. Liu, X. Cheng, Q. Wang, S. Jiang and G. Wang, BMC Chem., 2019, 13, 109 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- Y. Kong, S. Liu, S. Wang, B. Yang, W. He, H. Li, S. Yang, G. Wang and C. Dong, Sci. Rep., 2023, 13, 7756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of all new compounds are given in the Supporting Information. See DOI: https://doi.org/10.1039/d3ra05674k |
| This journal is © The Royal Society of Chemistry 2023 |