
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization of Fe3O4@SiO2 nanoparticles with Cu(I)-thiosemicarbazone complex as a robust and efficient heterogeneous nanocatalyst for N-arylation of N-heterocycles with aryl halides†
Narjes Kaviania,
Somayeh Behrouz *b,
Abbas Ali Jafari*a and
Mohammad Navid Soltani Radb
*b,
Abbas Ali Jafari*a and
Mohammad Navid Soltani Radb
aDepartment of Chemistry, Faculty of Science, Yazd University, Yazd, Iran. E-mail: Jafari@yazd.ac.ir
bDepartment of Chemistry, Shiraz University of Technology, Shiraz 71555-313, Iran. E-mail: behrouz@sutech.ac.ir; Fax: +98 71 3735 4520; Tel: +98 71 3735 4500
First published on 16th October 2023
Abstract
In this research, the functionalized silica-coated magnetite nanoparticles with Cu(I)-thiosemicarbazone complex (Fe3O4@SiO2-[CuL]) has been designed and synthesized as a magnetically retrievable nanocatalyst. Different techniques were employed to characterize the structure of Fe3O4@SiO2-[CuL] comprising FT-IR, FE-SEM, TEM, DLS, XRD, EDX, TGA, AAS, and VSM analysis. The catalytic performance of Fe3O4@SiO2-[CuL] was perused in Ullmann-type N-arylation of nucleobases, xanthines, and other N-heterocycles with diverse aryl halides which acquired the desired N-aryl products in good to excellent yields. Fe3O4@SiO2-[CuL] is a thermal and chemical stable, easy to prepare and recyclable, inexpensive, and ecofriendly catalyst that needs no additional additive or ligand as promoters. This catalyst could be separated without difficulty by a simple magnet and reused for at least seven sequential runs without a significant decline in its catalytic performance.
1. Introduction
Indeed, the C–N bond formation is one of the most significant reactions in organic synthesis since it provides numerous attractive and important substrates which have plenty of utilizations in different scientific areas and human life. Amongst, the N-heterocyclic compounds have found immense research interest especially due to their key roles in chemistry, medicine, and material sciences.1,2 Therefore, the synthesis of N-heterocycles and modification of the N-heterocyclic cores are of massive research interest. The N-arylation of the N-heterocycles is an important approach to modify the parent N-heterocyclic unit since it not only increases the lipophilicity nature of the N-heterocyclic unit but also can mask the nucleophilic nitrogen. The N-arylated N-heterocycles have found diverse applications in agriculture, organic, and medicinal chemistry. Many herbicides and approved drugs are involved in the N-aryl N-heterocycle residues in their structures (Fig. 1). The N-aryl nucleobases are used as agrochemicals,3 molecular tools, and probes to study biological systems.4 They exhibit some biological profiles such as anticancer,5 antibacterial,6 and antiviral.7 Some N-aryl xanthine derivatives inhibit proliferation in T47D tumor cells and phosphoinositide 3-kinases (PI3Ks).8 The N-aryl azoles and N-aryl indoles are useful precursors for the synthesis of drugs, natural products, and bioactive agents.2,9–11 Furthermore, they display various biological activities such as anti-HIV1,12 antiallergic,13 melatonin receptor MT1 agonists,14 COX-2 inhibitors,15 and antipsychotic agents.16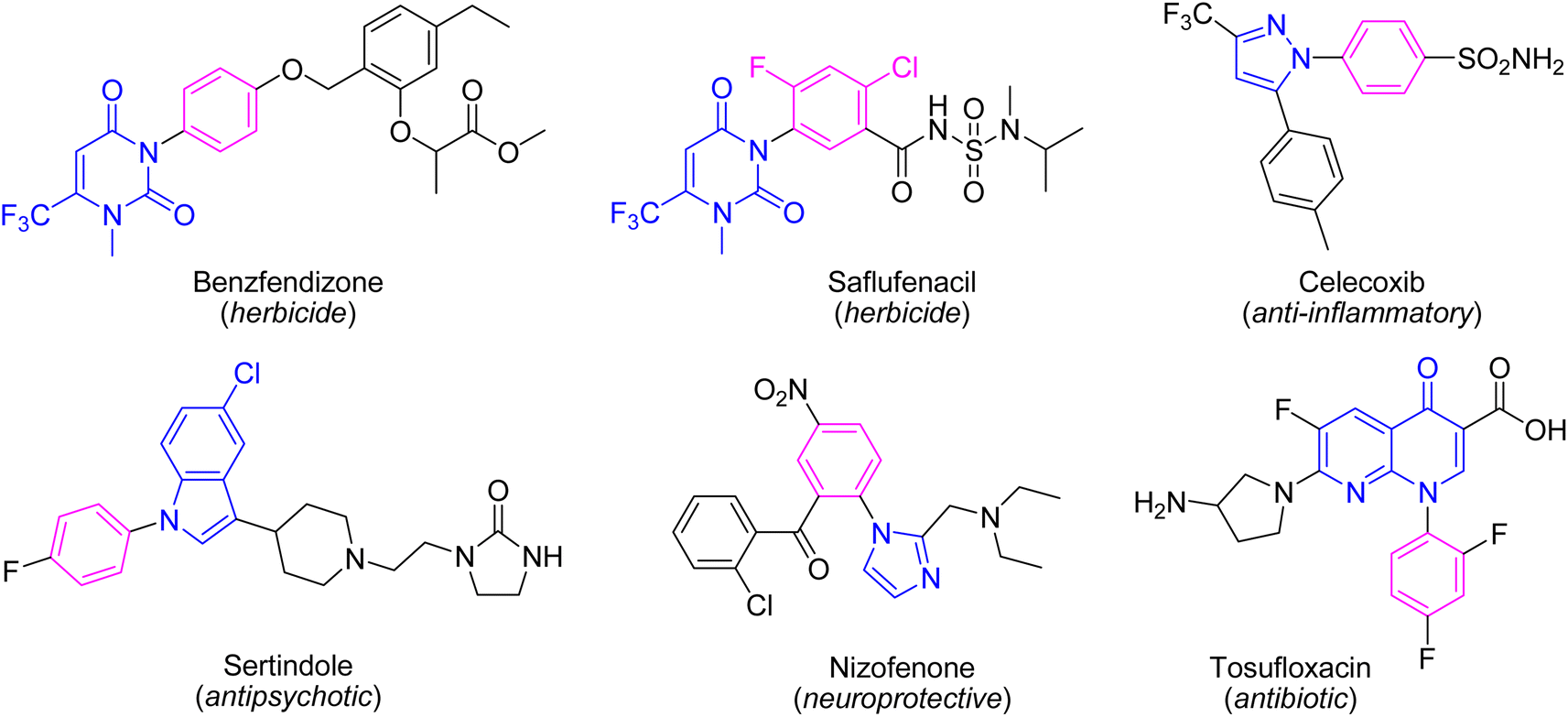 | ||
| Fig. 1 The structure of some approved drugs and herbicides containing the N-aryl N-heterocycle residue. | ||
Owing to the unique properties and applications of N-aryl N-heterocycles, different synthetic methodologies have been established to afford these important compounds.17–19 These general approaches are involved (i) the nucleophilic aromatic substitution (SNAr) reactions of electron-deficient aryl halides with N-heterocycles,20 (ii) the treatment of N-heterocycles with aryliodonium salts,21 (iii) the heterocyclization of appropriate precursors,22 (iv) the N-arylation of N-heterocycles with aryl boronic acids,8,23 (v) Ullmann-type C–N cross coupling reaction between aryl halides and heterocycles.24,25 However, these protocols are often associated with numerous shortcomings such as the use of the stoichiometric amount of Cu catalyst, the relative instability and high cost of boronic acids, the use of toxic and expensive ligands, harsh reaction conditions, unavailability of some precursors, and limited scope. In this connection, the Ullmann-type reaction is traditionally preferred since more starting materials are available compared to other methods. However, the classical Ullmann-type reaction was often carried out at very high temperatures and in the presence of the stoichiometric amount of copper catalyst which is not adequate for the scale-up synthesis.26 Buchwald et al.27 and Hartwig et al.28 have developed the Pd-catalyzed C–N coupling reactions using aryl halides. However, the use of toxic and expensive Pd catalysts and ligands has restricted its applicability in scale up synthesis and/or industrial processes. Thus, the use of less toxic, safe, inexpensive, and environmentally benign metals instead of Pd is extensively preferred. A striking breakthrough was made by Taillefer29 and Buchwald30 in the Cu-catalyzed C–N cross coupling reactions using aryl halides under mild reaction conditions. In this context, numerous Cu-catalyzed Ullmann-type C–N cross coupling reactions have been described over the years.18,31 Nevertheless, several problems were associated with these methods such as less activity because of the low surface area of the catalyst, long reaction time, the use of toxic and sensitive ligands, by-product formation, and the low yields causing a remarkable need for further development of more efficient catalytic systems to achieve the desired N-aryl N-heterocycles.
The heterogeneous catalysts exhibit many environmental and economic benefits such as recoverability, reusability, and stability of the catalyst, waste minimization and ease of handling.32 Practically, in some cases, the main products are insoluble in the reaction media and hence the catalyst cannot be recovered by simple centrifugation or filtration. On the other hand, the purification of the product and the work-up process will be difficult without initial separation of the catalyst. To avoid these shortcomings, using magnetic nanoparticle (MNPs) based catalysts would be an ideal strategy since the catalyst could be removed from the reaction media using an external magnet field.33 To prevent the agglomeration and coagulation of MNPs and conserve their activity, they usually coated with a protecting layer such as carbon, polymer, metal oxide, and silica.34 Among various Fe3O4 core–shell composites, silica-coated Fe3O4 MNPs (Fe3O4@SiO2) are popular, in particular for preparing sustainable and powerful heterogeneous catalysts.33,35 It is well known and fully established that silica has large pore volumes, high surface area, and biocompatibility as well as high chemical and thermal stability. Additionally, the modification of the silica surface could be simply achieved with different functional groups to afford the desired core–shell structures. Moreover, applying an external magnetic field is solely enough to separate the modified Fe3O4@SiO2 nanoparticles from the reaction media. In this regard, some Cu complex-functionalized Fe3O4@SiO2 nanoparticles were developed for the Ullmann-type N-arylation of N-heterocycles such as imidazole, benzimidazole, and indole.36–38 However, to the best of our knowledge, there has been no report yet on the Ullmann-type N-arylation of nucleobase and xanthine derivatives in the presence of Cu complex-functionalized Fe3O4@SiO2 nanoparticles. Hence, there is still ongoing interest in preparing and applying such catalysts having broader applicability for Ullmann-type N-arylation of N-heterocycles. To this end and in pursuit of our research interest on copper-catalyzed C–N bond coupling reactions,39–42 herein we introduce the functionalized silica-coated magnetite nanoparticles with Cu(I)-thiosemicarbazone complex (Fe3O4@SiO2-[CuL]) as a new magnetically recoverable nano catalyst for Ullmann-type N-arylation of nucleobases, xanthines, and other N-heterocycles with aryl halides in DMF at 110 °C (Scheme 1).
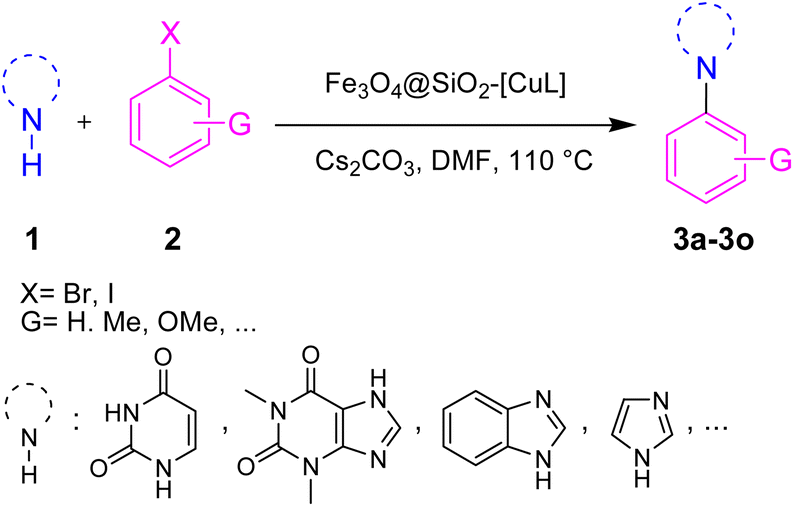 | ||
| Scheme 1 N-Arylation of N-heterocycles with aryl halides using Fe3O4@SiO2-[CuL]. | ||
2. Results and discussion
2.1. Preparation of Fe3O4@SiO2-[CuL]
The preparation of Fe3O4@SiO2-[CuL] as a new magnetically recoverable nano catalyst was achieved through 6 steps as indicated in Scheme 2. The nanoparticles I–III were synthesized according to the described procedures in the literature.43,44 In brief, the co-precipitation of ferrous and ferric ions under the nitrogen gas afforded Fe3O4 nanoparticles (I). The reaction was carried out under the nitrogen gas since nanoparticles I are susceptible to oxidation.45 Hence, magnetically separable Fe3O4 nanoparticles (I) were acquired via the treatment of FeCl3·6H2O and FeCl2·4H2O under N2 gas in NH4OH (25 wt%) and deionized water. The black solid I was attained after washing the solid with water and ethanol and drying it in a vacuum oven (80 °C). Tetraethyl orthosilicate (TEOS) was employed for improving the chemical stability of I with silica shell according to the Stöber process.43,44 The solid I and TEOS were reacted together in the presence of NH4OH (25 wt%) in a deionized H2O/EtOH solution. After 12 h, the solid was washed several times with deionized water and EtOH. Then, it was dried at 70 °C to attain the silica-coated Fe3O4 nanoparticles II (Fe3O4@SiO2). Next, the functionalization of Fe3O4@SiO2 (II) was done in EtOH using (3-chloropropyl)trimethoxysilane (CPTMS). After 24 h, the solid was collected using an external magnet field, washed with EtOH, and dried in an oven which affords chloropropyl-functionalized Fe3O4@SiO2 nanoparticles (III). CPTMS was selected for this step since the electrophilic sites were needed for the nucleophilic substitution reactions (SN2) with 4-hydroxyacetophenone as the chosen nucleophile. In the fourth step, Fe3O4@SiO2-hydroxyacetophenone intermediate (IV) was prepared through the SN2 reaction between 4-hydroxyacetophenone and adduct III in refluxing dry toluene. After 48 h, the solid IV was separated by a magnet field, washed with dry toluene, and dried in an oven (50 °C).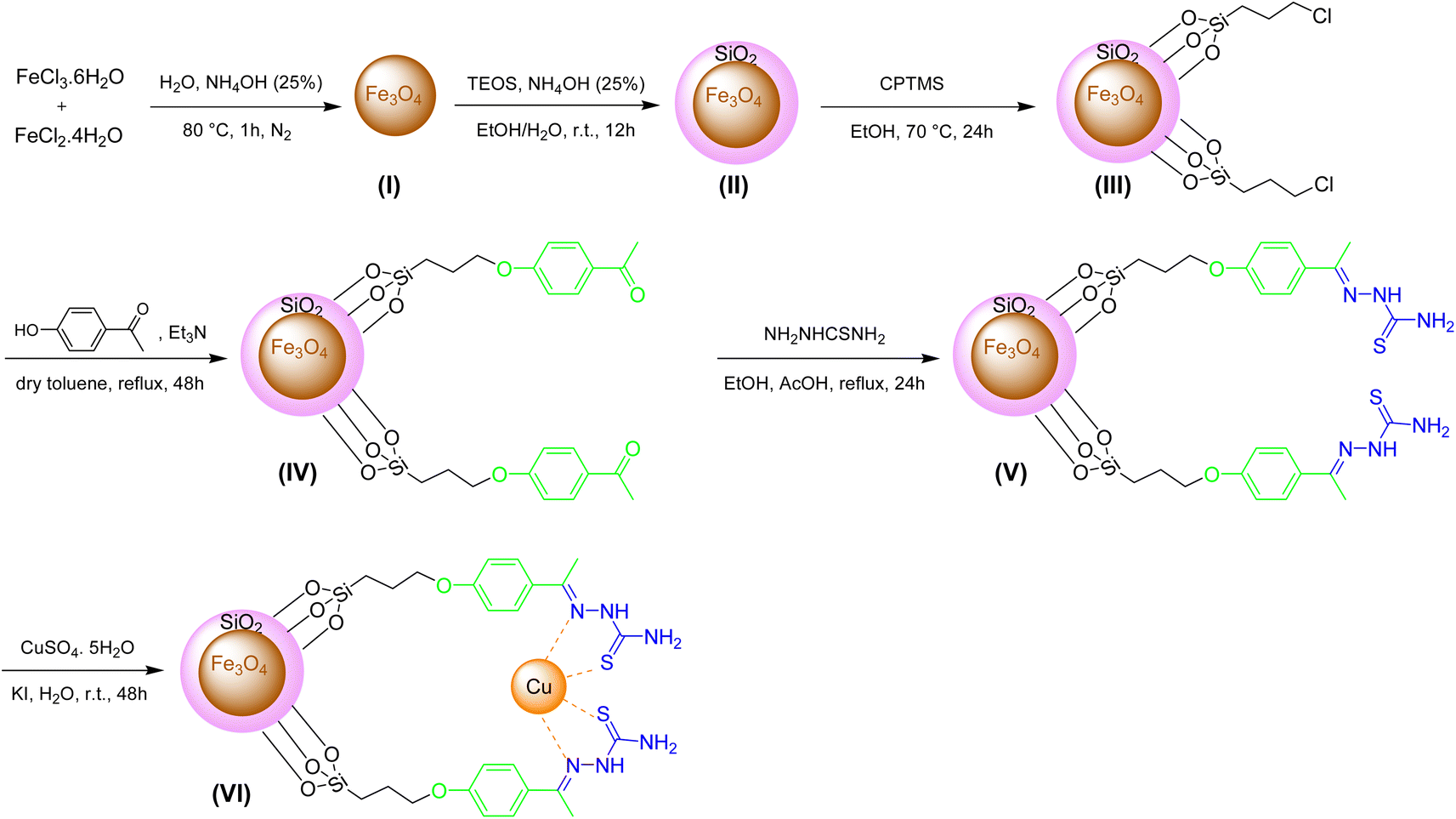 | ||
| Scheme 2 The procedures for synthesis of Fe3O4@SiO2-[CuL]. | ||
In the subsequent step, a mixture containing thiosemicarbazide, adduct IV, and acetic acid (a few drops) was refluxed in EtOH for 24 h. The thiosemicarbazone adduct V (creamy solid) was acquired after separating the solid by an external magnet, washing (deionized water and EtOH), and drying in a vacuum oven (50 °C). In the last step, thiosemicarbazone adduct V was reacted with copper sulfate pentahydrate and potassium iodide in deionized water. After 2 days, the solid was separated using an external magnet, washed with deionized water, and dried in an oven (70 °C) to afford Fe3O4@SiO2-[CuL] (VI) as a new magnetically separable nano catalyst.
2.2. Characterization of Fe3O4@SiO2-[CuL]
The characterization of magnetic Fe3O4@SiO2-[CuL] was achieved using various techniques including Fourier-transform infrared spectroscopy (FT-IR), field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), dynamic laser scattering (DLS), X-ray diffraction spectroscopy (XRD), energy dispersive X-ray spectroscopy (EDX), thermogravimetric analysis (TGA), atomic absorption spectroscopy (AAS), and vibrating sample magnetometer (VSM).Fig. 2 represents the FT-IR spectra of adducts III–VI. Apparently, the formation of Fe3O4@SiO2-[CuL] was confirmed by the FT-IR spectra. In Fig. 2a, the absorption bands at 590 and 618 cm−1 correspond to the vibration of Fe–O (stretching). The peak that appeared at 796 cm−1 is ascribed to C–Cl vibration (stretching). The peaks in the region of 1099, 894, and 464 cm−1 are related to the vibration of Si–O–Si (asymmetric and symmetric stretching), and the vibration of Fe–O–Si (stretching), respectively. The peak in the region of 1633 cm−1 is attributed to the O–H vibration (stretching) absorbed on the surface of Fe nanostructures. The band at 2952 cm−1 is assigned to the vibration of aliphatic C–H groups (stretching) which is followed by a broad band related to the O–H vibration in the surface of nanoparticles (stretching). Accordingly, CPTMS was grafted on the surface of magnetic Fe3O4@SiO2 nanoparticles. Treatment of 4-hydroxyacetophenone with adduct III led to the synthesis of adduct IV which shows the new peaks in the FT-IR spectra (Fig. 2b). Hence, the newly appeared band at 1435 cm−1 is attributed to the vibration of methyl moiety in 4-hydroxyacetophenone (bending). The bands that appeared at 1156 and 1219 cm−1 are related to the presence of C–O stretching. The bands at 1504 and 1597 cm−1 correspond to the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO (stretching), respectively. The aromatic C–H vibration (stretching) is expected to be around 3000–3100 cm−1 which is covered by the vibration of O–H (stretching) on the surface of nanoparticles. Fig. 2c displays the FT-IR spectra of thiosemicarbazone intermediate V. The peaks at 1280, 1359, and 1657 cm−1 are assigned to the vibration of CS, C–NH2, and CN bonds, respectively. The peaks around 3304 cm−1 correspond to the amine groups. A comparison between the FT-IR spectra of intermediate V and Fe3O4@SiO2-[CuL] confirmed the preparation of the desired catalyst (Fig. 2c and d).
C and CO (stretching), respectively. The aromatic C–H vibration (stretching) is expected to be around 3000–3100 cm−1 which is covered by the vibration of O–H (stretching) on the surface of nanoparticles. Fig. 2c displays the FT-IR spectra of thiosemicarbazone intermediate V. The peaks at 1280, 1359, and 1657 cm−1 are assigned to the vibration of CS, C–NH2, and CN bonds, respectively. The peaks around 3304 cm−1 correspond to the amine groups. A comparison between the FT-IR spectra of intermediate V and Fe3O4@SiO2-[CuL] confirmed the preparation of the desired catalyst (Fig. 2c and d).
 | ||
| Fig. 2 FT-IR spectra of adducts III (a), IV (b), V (c), and Fe3O4@SiO2-[CuL] (d). | ||
The size and surface morphology of Fe3O4@SiO2-[CuL] were explored by the FE-SEM images as shown in Fig. 3. Consequently, the fabricated nanoparticles have almost a sphere-shaped morphology with a slight agglomeration. The synthesized particles of Fe3O4@SiO2-[CuL] were also in a nanoscale size. To have better insight into the morphology and size of Fe3O4@SiO2-[CuL], the TEM image was also assessed (Fig. 4, left). Apparently, the obtained nanoparticles have a spherical-like shape with a core–shell structure. This structure involves the light amorphous silica shell and the ligand that coated and wrapped the dark magnetic Fe3O4.
 | ||
| Fig. 3 FE-SEM images of Fe3O4@SiO2-[CuL]. | ||
 | ||
| Fig. 4 TEM image (left) and DLS analysis (right) of Fe3O4@SiO2-[CuL]. | ||
Dynamic light scattering (DLS) technique was employed to study the hydrodynamic diameter of Fe3O4@SiO2-[CuL] and the distribution of its particle size. The results are depicted in Fig. 4 (right). The size distribution with a mean diameter of 151 nm was observed for the particles. It is established that the hydrodynamic diameter by the DLS technique is larger than the obtained diameter by other techniques such as TEM and SEM. The rational reason for this observation could be the inability of DLS to differentiate between constituent and agglomerate particles.46
The XRD pattern of Fe3O4@SiO2-[CuL] is illustrated in Fig. 5. The characteristic diffraction peaks at 2θ = 30.13°, 35.45°, 43.17°, 53.41°, 57.17°, and 62.65° were related to the (220), (311), (400), (422), (511) and (440) indices, respectively. These results correspond to the standard XRD pattern for Fe3O4 (JCPDS card no. 19-0629) having a cubic spinel structure of the magnetite. Therefore, the crystal structure of the synthesized magnetic Fe3O4 nanoparticles remained unaffected during the synthesis of Fe3O4@SiO2-[CuL]. The broad peaks below 30° are ascribed to the amorphous silica wrapping the Fe3O4 nanoparticles. The presence of broad peaks in the XRD pattern and the scattering of Cu ions in the catalyst structure caused some difficulties in the detection of the copper species. In general, an amorphous shape could be suggested for the copper species.
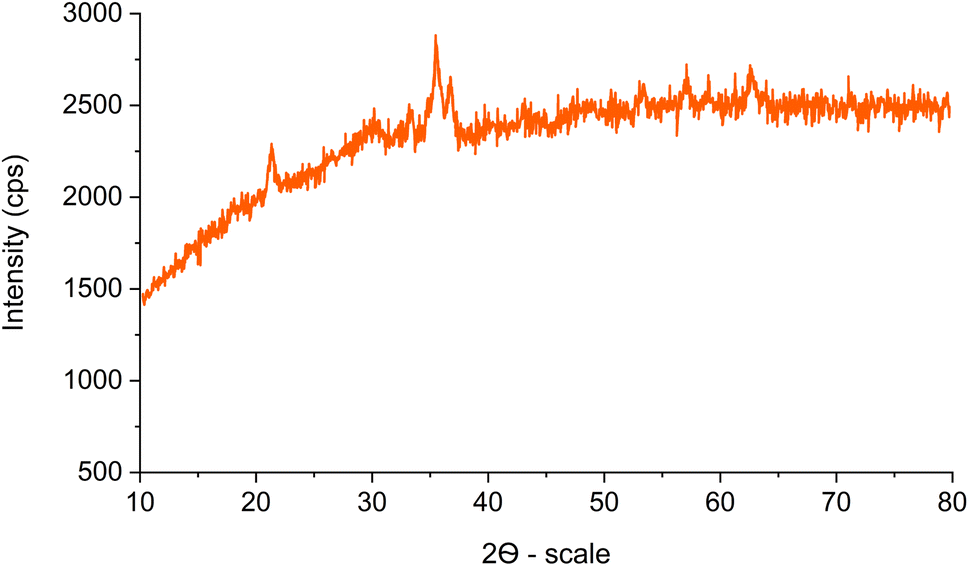 | ||
| Fig. 5 The XRD pattern of Fe3O4@SiO2-[CuL]. | ||
The energy-dispersive X-ray spectroscopy (EDX) was employed to determine the chemical composition of magnetic Fe3O4@SiO2-[CuL] nanoparticles (Fig. 6). The resulting spectrum from EDX analysis endorses the presence of Fe, O, Si, C, N, S, and Cu in the structure of Fe3O4@SiO2-[CuL]. Consequently, the functionalization of Fe3O4@SiO2 with [CuL] was confirmed.
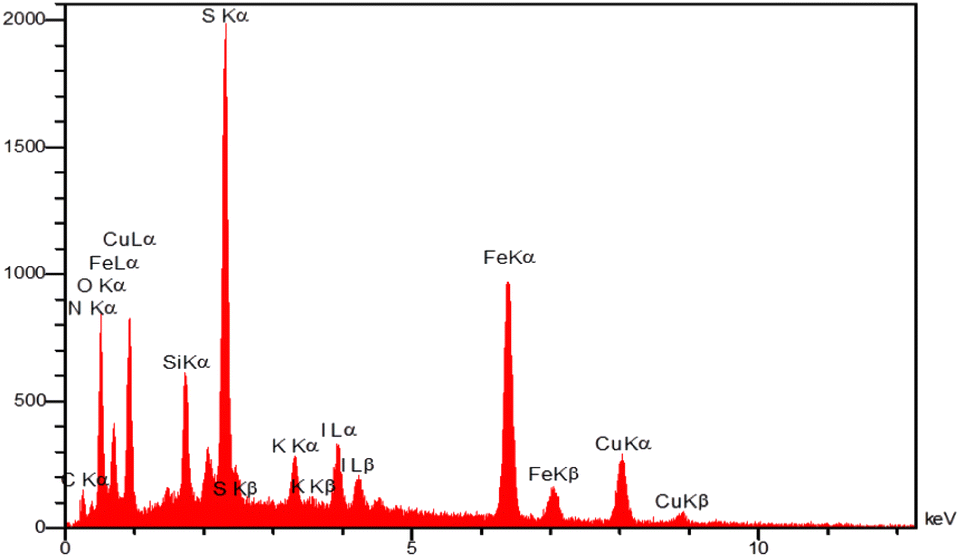 | ||
| Fig. 6 EDX analysis of Fe3O4@SiO2-[CuL]. | ||
The thermal stability of Fe3O4@SiO2-[CuL] and the weight loss of its material upon heating were investigated by TGA analysis (Fig. 7). As shown in Fig. 7, around 0.3% weight loss was detected below 200 °C which is ascribed to the removal of adsorbed water from the catalyst pores. Hence, Fe3O4@SiO2-[CuL] is thermally stable up to 200 °C. A gentle weight loss (∼4.7%) was observed in a range of 200–800 °C which corresponds to the thermal degradation of organic residue tethered to the surface of Fe3O4@SiO2 nanoparticles and decomposition of the copper–thiosemicarbazone complex. Consequently, grafting the organic residue on the Fe3O4@SiO2 surface was confirmed by TGA analysis.
 | ||
| Fig. 7 TGA of Fe3O4@SiO2-[CuL]. | ||
Atomic absorption spectroscopy (AAS) was conducted to investigate the amount of Cu content supported on Fe3O4@SiO2-Ligand (adduct V). According to the AAS analysis, the loading amount of copper in Fe3O4@SiO2-[CuL] was determined to be 1.04 mmol g−1. Thus, the synthesized nanoparticles V have a high capacity to form chelates with the Cu species which could be attributed to the presence of thiosemicarbazone residues.
The magnetic properties of Fe3O4@SiO2 and Fe3O4@SiO2-[CuL] were assessed with magnetic hysteresis measurements. The magnetization curves of both studied nanoparticles are shown in Fig. 8. According to the obtained results, the saturation magnetization (Ms) values of Fe3O4@SiO2-[CuL] and Fe3O4@SiO2 proved to be 20.12 and 30.96 emu g−1, respectively. The hysteresis loops characteristic displays the magnetic property of the studied substrates. As a result, the magnetic Fe3O4@SiO2-[CuL] nanoparticles may simply be collected from the reaction mixture using a magnet field. Clearly, the Ms value of Fe3O4@SiO2-[CuL] is lower than that of Fe3O4@SiO2 since the organic moiety and the silica shell on the Fe3O4 surface are non-magnetic materials.
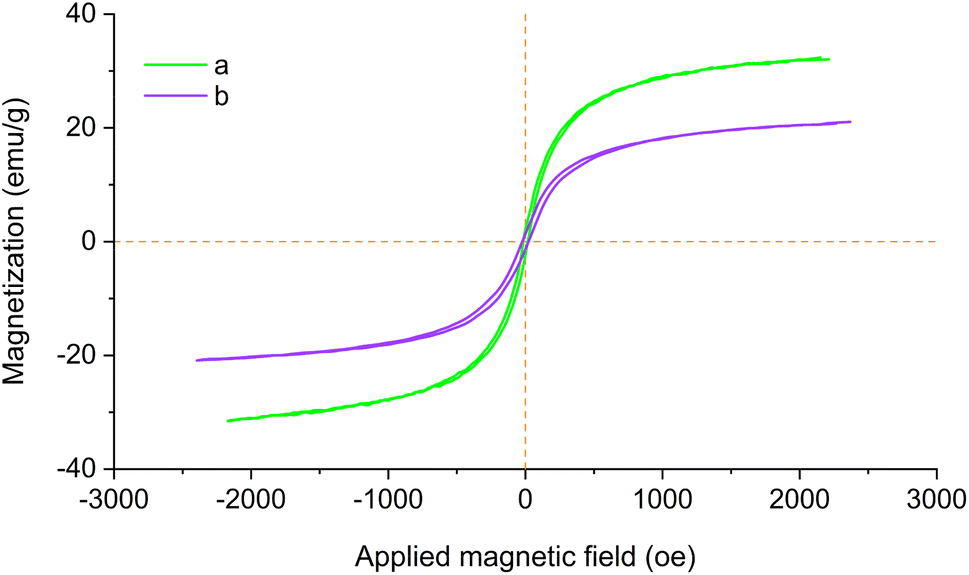 | ||
| Fig. 8 Magnetization curves of Fe3O4@SiO2 (a) and Fe3O4@SiO2-[CuL] (b). | ||
2.3. Ullmann-type N-arylation of N-heterocycles with aryl halides using Fe3O4@SiO2-[CuL]
After the preparation and characterization of Fe3O4@SiO2-[CuL], its efficiency was explored by its application in the Ullmann-type N-arylation of N-heterocycles with diverse aryl halides (3a–3o). Our initial attempt was conducted to find out the optimized reaction conditions. Therefore, the reaction of imidazole with iodobenzene in the presence of 0.01 g (1 mol%) of Fe3O4@SiO2-[CuL] was chosen as the sample reaction to attain 1-phenyl-1H-imidazole (3m). In this regard, the influence of key factors including solvent, base, catalyst amount, and temperature was assessed on the progress of the sample reaction (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Catalyst amount (mol%) | T (°C) | Time (h) | Yieldb (%) |
| a Imidazole (1.0 mmol), iodobenzene (1.2 mmol), base (2.0 mmol), Fe3O4@SiO2-[CuL] (X mol%), and solvent (4 mL).b Isolated yield.c No reaction. | ||||||
| 1 | H2O | Cs2CO3 | 1 | Reflux | 24 | NRc |
| 2 | EtOH | Cs2CO3 | 1 | Reflux | 10 | 30 |
| 3 | MeCN | Cs2CO3 | 1 | Reflux | 10 | 48 |
| 4 | THF | Cs2CO3 | 1 | Reflux | 12 | 26 |
| 5 | Toluene | Cs2CO3 | 1 | Reflux | 10 | 54 |
| 6 | DMF | Cs2CO3 | 1 | 110 | 5 | 93 |
| 7 | DMF | K2CO3 | 1 | 110 | 8 | 79 |
| 8 | DMF | Et3N | 1 | 110 | 12 | 36 |
| 9 | DMF | NaOH | 1 | 110 | 10 | 68 |
| 10 | DMF | NaOAc | 1 | 110 | 12 | 40 |
| 11 | DMF | MgO | 1 | 110 | 18 | Trace |
| 12 | DMF | DMAP | 1 | 110 | 16 | 45 |
| 13 | DMF | Cs2CO3 | 1 | r.t. | 24 | Trace |
| 14 | DMF | Cs2CO3 | 1 | 80 | 8 | 71 |
| 15 | DMF | Cs2CO3 | 1 | 100 | 6 | 83 |
| 16 | DMF | Cs2CO3 | 1 | 120 | 5 | 93 |
| 17 | DMF | Cs2CO3 | — | 110 | 24 | NR |
| 18 | DMF | Cs2CO3 | 0.5 | 110 | 12 | 47 |
| 19 | DMF | Cs2CO3 | 0.7 | 110 | 8 | 76 |
| 20 | DMF | Cs2CO3 | 0.9 | 110 | 5 | 90 |
| 21 | DMF | Cs2CO3 | 1.1 | 110 | 5 | 93 |

Primarily, it was attempted to determine the best solvent for the progress of the reaction. In this connection, several protic and aprotic solvents were employed to perform the sample reaction (Table 1, entries 1–6). When the reaction was carried out in water as a green and protic solvent, 3m was not prepared even after prolonging the reaction time up to 24 h (entry 1). The use of EtOH as another protic solvent afforded 3m in 30% yield after 10 h (entry 2). Low to moderate yields of 3m were obtained when MeCN, THF, and toluene were applied as the reaction media (entries 3–5). Practically, the best result was achieved when DMF was used as the reaction solvent (entry 6). The next endeavor was carried out to find an efficient base to perform the reaction (Table 1, entries 6–12). As indicated in Table 1, Et3N, NaOAc, MgO, and DMAP didn't influence adequately on the reaction time and yield (entries 8 and 10–12). A considerable improvement in yield of 3m was acquired when K2CO3 and NaOH were employed as the reaction base (entries 7 and 9). The Cs2CO3 proved to be an adequate base for enhancing the yield and the completion of the reaction (entry 6), hence it was selected base for further optimization experiments.
Temperature is undeniably important factor in determining the optimized condition. To this end, the synthesis of 3m was carried out at different temperatures (Table 1, entries 6 and 13–16). Practically, enhancing the reaction temperature from room temperature up to 100 °C remarkably increased the reaction efficacy (entries 13–15). The highest yield of 3m in the shortest reaction time was obtained when the sample reaction was conducted at 110 °C (entry 6). However, more elevation in the reaction temperature did not lead to further improvement and progress of the reaction (entry 16). The last attempt was focused on the assessment of loading the catalyst amount on the reaction progress (Table 1, entries 6 and 17–21). Firstly, the sample reaction was achieved in the absence of Fe3O4@SiO2-[CuL]. However, as expected no reaction happened and 3m was not acquired even in trace amount (entry 17). As depicted in Table 1, the reaction efficacy was improved by enhancing the amount of Fe3O4@SiO2-[CuL]. The best result was observed when 1 mol% Fe3O4@SiO2-[CuL] was employed for the preparation of 3m (entry 6). Loading more catalyst amounts didn't improve the reaction progress. Overall, the optimized conditions for this current Ullmann-type N-arylation of N-heterocycles proved to be the use of 1 mol% Fe3O4@SiO2-[CuL] in the presence of Cs2CO3 and DMF at 110 °C. The optimized stoichiometric ratios of N-heterocycle/aryl halide/Cs2CO3 for preparation of 3m were determined to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2:2. It is also worth mentioning that while a higher amount of aryl halides was employed, the formation of byproduct like biaryl compounds was observed.
:1.2:2. It is also worth mentioning that while a higher amount of aryl halides was employed, the formation of byproduct like biaryl compounds was observed.
Having determined the optimized condition, the generality and limitations of this procedure were explored by its application to Ullmann-type N-arylation of some N-heterocycle compounds comprising pyrimidine nucleobases (uracil, thymine), xanthines (theophylline, theobromine), azoles (imidazole, 2-phenylimidazole), and the other analogs (benzimidazole, indole) with structurally diverse aryl halides. In this regard, the different aryl halides including iodobenzene, bromobenzene, chlorobenzene, 4-iodoanisole, 2-iodoanisole, 4-bromotoluene, and 1-bromonaphthalene were employed. The starting materials, yields, and structure of the synthesized compounds 3a–3o are depicted in Table 2. The structures of 3a–3o were confirmed by 1H-, 13C-NMR, and IR spectroscopy methods. The synthesized N-aryl N-heterocycles 3a–3o were obtained in good to excellent yields depending on the nature of examined aryl halides and N-heterocycles. In the case of uracil and thymine, products 3a–3d were obtained in 54–67% yields (Table 2, entries 1–4). For nucleobases, the lower yield of products is attributed to the weaker nucleophilic power of nucleobases compared to other screened N-heterocycles. It is worth mentioning that good regioselectivity was observed at the site of nucleobase N-arylation using the present protocol. As a consequence, the N1-aryl adducts 3a–3d were majorly prepared while the N1,N3-diaryl products were attained in trace amounts. Theophylline and theobromine were also examined under the optimized conditions to afford products 3e–3h in 74–78% yields (Table 2, entries 5–8). Indole, benzimidazole, and imidazole derivatives were successfully performed in the Ullmann-type N-arylation reaction with aryl halides. It seems that the reaction is sensitive to the steric hindrance on both aryl halide and N-heterocycle. Practically, a lower yield of products was attained when 2-iodoanisole, 1-bromonaphthalene, and 2-phenylimidazole were used as the sterically hindered starting materials (Table 2, entries 10, 12, and 17). The influence of the steric factor is well recognized by comparing the reaction efficiency for 4-iodoanisole and 2-iodoanisole as the starting materials (Table 2, entries 9 and 10). In addition, the type of halogen atom in aryl halide influenced the reactivity in the Ullmann-type N-arylation reaction. In this connection, the iodobenzene, bromobenzene, and chlorobenzene were reacted with imidazole under the optimized conditions to acquire product 3m (entries 13–15). The reactivity of bromobenzene is lower than that of iodobenzene. While bromobenzene affords a comparable yield of 3m, however, the reaction took a longer time to be completed. Among pointed out halobenzenes, chlorobenzene has the least reactivity due to the higher bond dissociation energy. Consequently, 3m was attained in trace amount when chlorobenzene was employed as the starting aryl halide.
| Entry | Aryl halide | N-Heterocycle | Product | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: aryl halide (1.2 mmol), N-heterocycle (1 mmol), Cs2CO3 (2 mmol), Fe3O4@SiO2-[CuL] (0.01 g, 1 mol%), DMF (4 mL), 110 °C.b Isolated yield. | |||||
| 1 | C6H5I | Uracil |  |
6 | 67 |
| 2 | 4-MeO-C6H4I | Uracil | 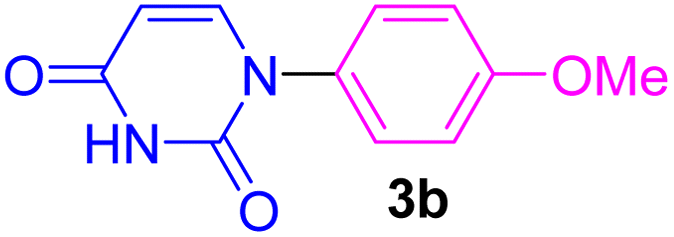 |
7 | 65 |
| 3 | C6H5I | Thymine | 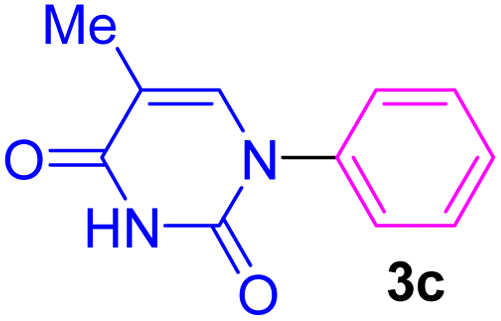 |
7 | 59 |
| 4 | 4-Me-C6H4Br | Thymine | 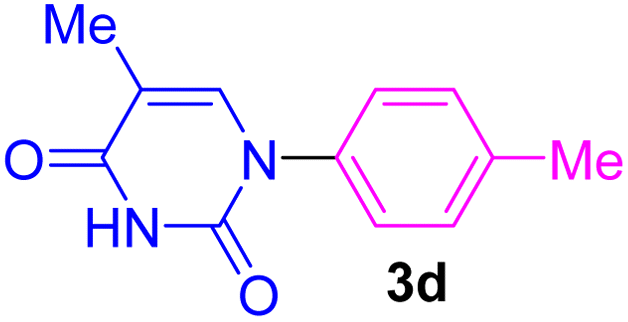 |
9 | 54 |
| 5 | C6H5I | Theophylline |  |
5 | 77 |
| 6 | 4-MeO-C6H4I | Theophylline | 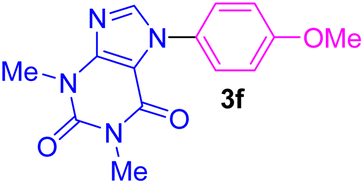 |
5 | 74 |
| 7 | C6H5I | Theobromine | 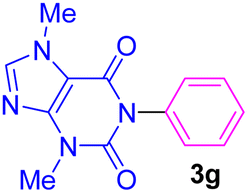 |
5.5 | 78 |
| 8 | 4-MeO-C6H4I | Theobromine | 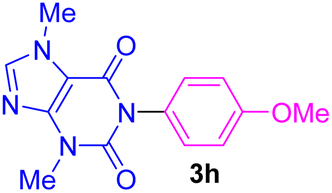 |
5.5 | 76 |
| 9 | 4-MeO-C6H4I | Indole | 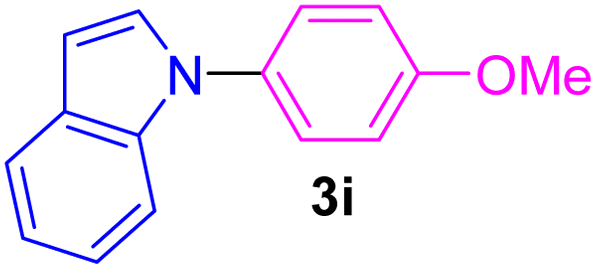 |
6 | 88 |
| 10 | 2-MeO-C6H4I | Indole |  |
9 | 73 |
| 11 | C6H5I | Benzimidazole |  |
4.5 | 92 |
| 12 | 1-Br-C10H7 | Imidazole |  |
7 | 82 |
| 13 | C6H5I | Imidazole | 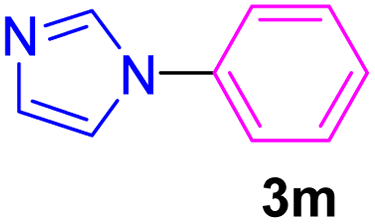 |
5 | 93 |
| 14 | C6H5Br | Imidazole | 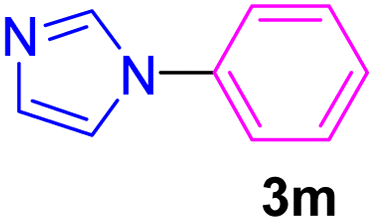 |
7 | 88 |
| 15 | C6H5Cl | Imidazole |  |
24 | Trace |
| 16 | 4-MeO-C6H4I | Imidazole | 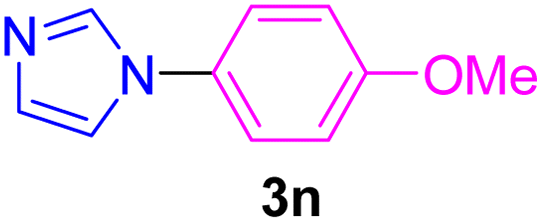 |
5 | 91 |
| 17 | C6H5I | 2-Phenylimidazole |  |
6 | 79 |
To realize the efficacy and potency of Fe3O4@SiO2-[CuL], the present catalytic system was compared with several previously reported methods for Ullmann-type N-arylation of imidazole with iodobenzene to afford 3m (Table 3). As can be seen in Table 3, the use of Fe3O4@SiO2-[CuL] is associated with several benefits such as short reaction time, excellent yield, low amount of catalyst, simple recovery, and separability from the reaction media.
|
||||
|---|---|---|---|---|
| Entry (ref.) | Catalyst | Conditions | Time (h) | Yield (%) |
| 1 (ref. 47) | Cu2O (10 mol%) | DMSO, KOH, 110 °C | 24 | 90 |
| 2 (ref. 48) | Cu–Y zeolite (10.8 mol%) | DMF, K2CO3, 120 °C | 24 | 99 |
| 3 (ref. 49) | CuFAP (12.5 mol%) | DMSO, K2CO3, 110 °C | 6 | 92 |
| 4 (ref. 50) | Nano-CuO (2.5 mol%) | DMSO, KOH, 110 °C | 24 | 91 |
| 5 (ref. 51) | SiO2-Py-Cu(OAc)2 (5 mol%) | Toluene, Cs2CO3, 100 °C | 8 | 92 |
| 6 (ref. 37) | Fe3O4@SiO2-PVA-Cu(II) (0.6 mol%) | DMF, t-BuONa, 100 °C | 5 | 94 |
| 7 (this work) | Fe3O4@SiO2-[CuL] (1 mol%) | DMF, Cs2CO3, 110 °C | 5 | 93 |
The recovery and recycling of a catalyst are essential issues from both economic and green chemistry aspects. In this context, the application of heterogeneous catalysts is highly preferred in comparison with homogeneous catalysts. In this regard, the consecutive synthesis of 1-phenyl-1H-imidazole (3m) via the Ullmann-type N-arylation of imidazole with iodobenzene using 0.01 g (1 mol%) Fe3O4@SiO2-[CuL] was considered. The heterogeneity and recoverability of Fe3O4@SiO2-[CuL] was evaluated for seven sequential reactions in a model reaction. After the achievement of each reaction run, Fe3O4@SiO2-[CuL] was separated and retrieved from the reaction by a magnet. After washing the separated catalyst with deionized water and ethanol, it was dried in an oven at 70 °C. The dried Fe3O4@SiO2-[CuL] was directly used for achieving the subsequent reaction in the absence of any fresh catalyst. All successive reaction runs were quenched after 5 h (Fig. 9). As illustrated in Fig. 9, the performance and efficacy of Fe3O4@SiO2-[CuL] are remarkable for at least up to seven consecutive runs with no significant deterioration in its reactivity. Among the synthesized compounds shown in Table 2, the lowest yield was related to 5-methyl-1-p-tolylpyrimidine-2,4(1H,3H)-dione (3d). The recycling test was also performed for the synthesis of 3d via the same procedure that was used for 3m. Accordingly, product 3d was obtained in 54% to 47% yields after the first and seventh consecutive reaction runs, respectively. These experiments have verified the heterogeneous nature, stability, and recyclability of Fe3O4@SiO2-[CuL].
 | ||
| Fig. 9 Catalytic potency of Fe3O4@SiO2-[CuL] in 7 repeated runs for preparation of 3m. | ||
The structure of recovered Fe3O4@SiO2-[CuL] was studied by the FT-IR spectrum after the seventh reaction run which is shown in Fig. 10. As can be understood from Fig. 10, there is no significant difference between the FT-IR spectrum of the recycled Fe3O4@SiO2-[CuL] with that of the fresh catalyst. This evidence proves that the chemical structure of Fe3O4@SiO2-[CuL] is remaining intact after seven runs. Besides, the hot-filtration test (split test) was performed on the sample reaction to explore the heterogeneous nature and stability of Fe3O4@SiO2-[CuL].52 In this context, the sample reaction was achieved for synthesis of 3m and after 2.5 h the catalyst was retrieved from the reaction media using a magnet. The remaining residue was then allowed to continue the reaction for a further 2.5 h. As expected, further conversion and improvement of the sample reaction was paused which is good evidence for negligible leaching of copper species from the surface of Fe3O4@SiO2-[CuL]. Overall, the heterogeneity, strong binding of Cu specious on the catalyst surface, and remarkable stability of Fe3O4@SiO2-[CuL] were endorsed by these experiments.
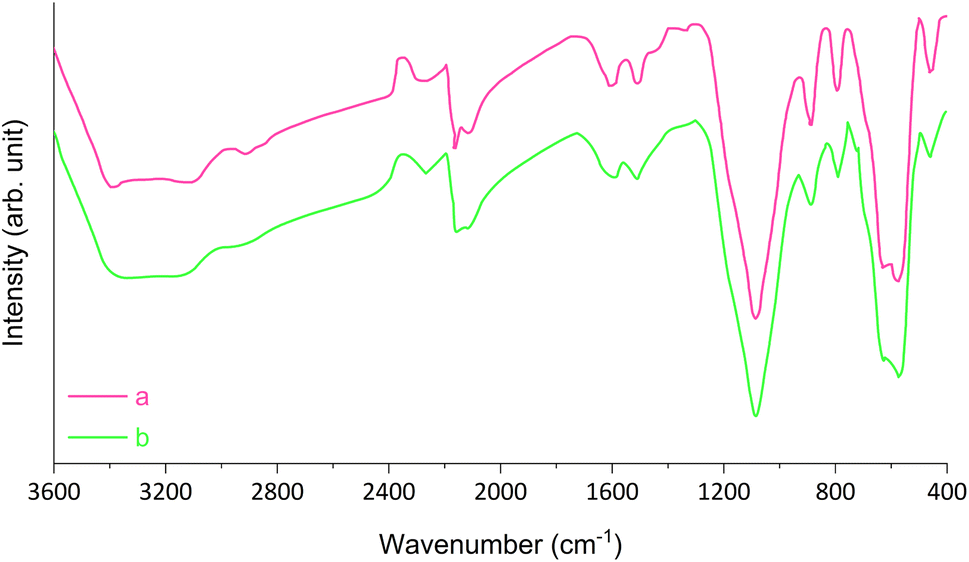 | ||
| Fig. 10 FT-IR spectra of fresh (a) and recovered (b) Fe3O4@SiO2-[CuL]. | ||
Based on the previously reported mechanism for Ullmann-type N-arylation of N-heterocycles with aryl halides,53,54 we proposed a plausible mechanism for the synthesis of 3m in the presence of Fe3O4@SiO2-[CuL] (Scheme 3). Initially, the oxidative addition of iodobenzene to Fe3O4@SiO2-[CuL] occurs to produce a transient Cu(III) species (B). Then, the treatment of adduct B with the in situ base-activated imidazole generates adduct C. Eventually, the C–N coupling reaction and synthesis of 3m are conducted via the reductive elimination from adduct C. Simultaneously, the active Fe3O4@SiO2-[CuL] is regenerated to proceed to the next catalytic cycle.
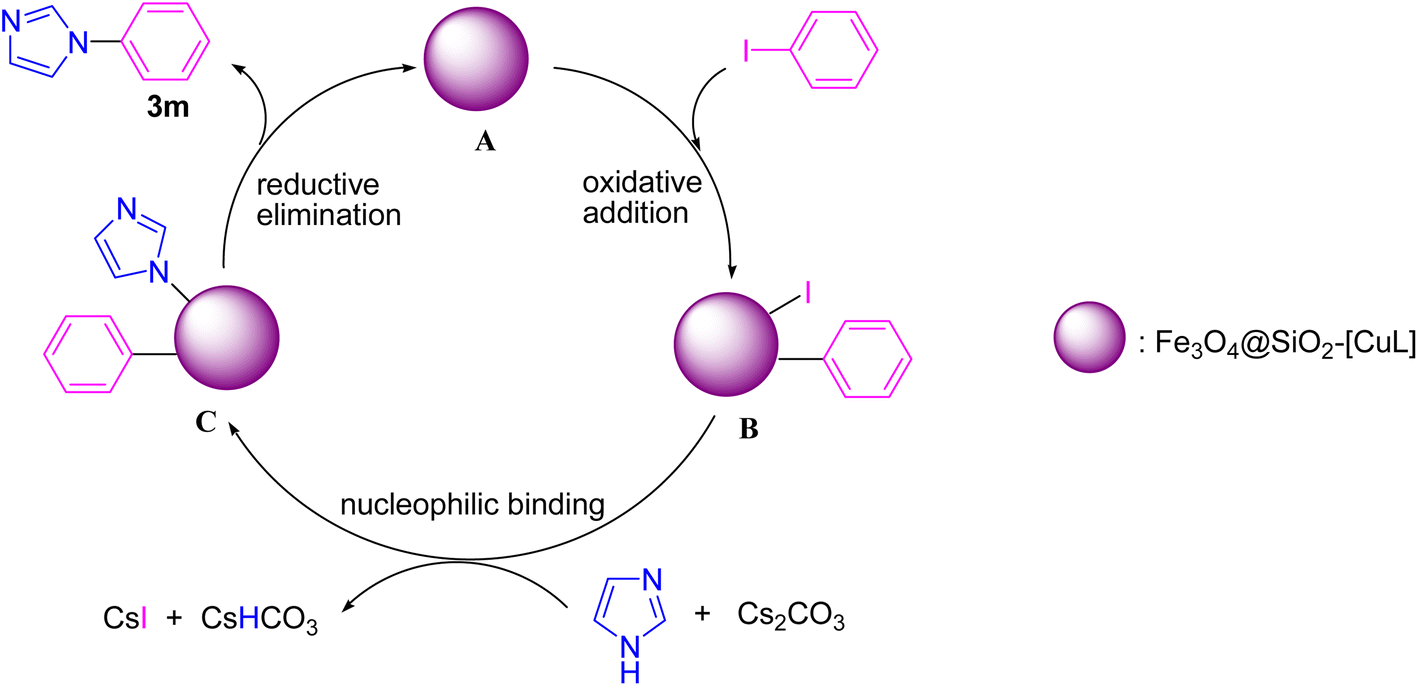 | ||
| Scheme 3 A plausible mechanism for synthesis of 3m using Fe3O4@SiO2-[CuL]. | ||
3. Conclusion
In this study, we have described the synthesis of the functionalized silica-coated magnetite nanoparticles with Cu(I)-thiosemicarbazone complex (Fe3O4@SiO2-[CuL]) as a new magnetically retrievable nano catalyst. The Fe3O4@SiO2-[CuL] was efficiently employed for the synthesis of N-aryl N-heterocycles via Ullmann-type N-arylation of nucleobases, xanthines, and other N-heterocycles with aryl halides. The structure of Fe3O4@SiO2-[CuL] was characterized by several techniques including FT-IR, FE-SEM, TEM, DLS, XRD, EDX, TGA, AAS, and VSM analysis. The significant benefits associated with this catalytic system are: (i) the stability and the facile synthesis of Fe3O4@SiO2-[CuL], (ii) the cheapness and the heterogeneous nature of Fe3O4@SiO2-[CuL], (iii) the ease of catalyst separation and collection by an external magnet field, (iv) the reusability of Fe3O4@SiO2-[CuL] for at least 7 sequential reaction runs, (v) the ease of handling, operation and simple work-up process, (vi) good to excellent yields of the N-aryl adducts, and (vii) no need to use external promoters such as ligands or additives. Altogether, Fe3O4@SiO2-[CuL] could be considered for the synthesis of industrially and medicinally important compounds as well as other copper-catalyzed Ullmann-type coupling reactions.4. Experimental
4.1. General
The solvents and substrates were purchased from Merck and other chemical vendors. The chemicals were directly applied without further purification. The following instruments were employed for characterizing the structure and morphology of Fe3O4@SiO2-[CuL] as well as products. XRD: Brüker D8 Advance X-ray diffractometer; EDX: SAMX; FE-SEM: MIRA3 TESCAN-XMU; TEM: 912 AB (120 kV) microscope; DLS: Horiba SZ-100; TGA: Shimadzu (TG-50), heating rate = 14 °C min−1; VSM: Lake Shore VSM-7400; Atomic absorption spectrometer (AAS): GBC-932 AA; FT-IR: Thermo Nicolet Avatar 370; Melting points: Electrothermal IA 9000, uncorrected data; 1 H- and 13C-NMR: Brüker Avance (DPX-300 MHz and DPX-250 MHz), internal standard: tetramethylsilane, abbreviations: coupling constant (J in Hz), singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), multiplet (m), broad (br).4.2. General procedure for synthesis of Fe3O4@SiO2-[CuL]
Fe3O4@SiO2-[CuL] was synthesized via 6 steps as shown in Scheme 2. The nanoparticles I–III were synthesized through the procedures reported in the literature.43,44 Initially, the co-precipitation method was employed to prepare the Fe3O4 nanoparticles. Thus, a mixture of FeCl2·4H2O (20 mmol, 3.976 g) and FeCl3·6H2O (40 mmol, 10.812 g) was added to the deionized water (140 mL) which was preheated to 80 °C in an oil bath. A solution of NH4OH (0.64 mol, 25.4 mL, 25 wt%) was added dropwise to vigorously stirred the reaction mixture under the N2 (g) atmosphere. The Fe3O4 nanoparticles were prepared after 1 h which was magnetically separated and collected from the reaction container. The black solid was then washed with deionized water (4 × 15 mL) and EtOH (4 × 15 mL) and dried in a vacuum oven at 80 °C to attain Fe3O4 (I) as a black powder. Afterward, the Fe3O4 nanoparticles (I, 2.0 g) were added to a solution of deionized water and EtOH (1:3, 80 mL). After the sonication of the reaction mixture for half an hour, NH4OH (6.0 mL, 25 wt%) was added. The mixture was stirred at room temperature while tetraethyl orthosilicate (TEOS, 10 mL) was added portionwise. After 12 h, the obtained solid was separated by an external magnet field, washed with deionized water (4 × 15 mL) and EtOH (4 × 15 mL), and dried in a vacuum oven at 60 °C to afford the silica-coated Fe3O4 nanoparticles (Fe3O4@SiO2, II). In the third step, (3-chloropropyl)trimethoxysilane (CPTMS) was used to functionalize the silica surface of II. Therefore, CPTMS (3.64 mL, 20 mmol) and II (2.0 g) were reacted together in EtOH (70 mL) at 70 °C. The magnetic solid III was salvaged from the reaction media after 24 h using a magnet field, washed with EtOH (4 × 15 mL), and kept in a vacuum oven at 60 °C to be dried. In step 4, a mixture of Et3N (4.04 g, 40 mmol), 4-hydroxyacetophenone (4.08 g, 30 mmol), and freshly prepared III (2.0 g) was added in a double-necked round bottom flask containing dry toluene (70 mL). The reaction was conducted in refluxing toluene using an oil bath. After 48 h, the solid was magnetically collected and separated from the reaction flask and washed with dry toluene (4 × 15 mL). Then, it was placed and dried in a vacuum oven at 50 °C to give the functionalized Fe3O4@SiO2-hydroxyacetophenone intermediate (IV). To prepare the thiosemicarbazone adduct V in step 5, thiosemicarbazide (2.42 g, 26 mmol), adduct IV (2.0 g), and AcOH (few drops) was added in a double-necked round bottom flask containing EtOH (40 mL) and the flask was heated to reflux in an oil bath for 24 h. The thiosemicarbazone adduct V was obtained as a creamy solid after magnetic separation of the solid, washing with deionized water (4 × 15 mL) and EtOH (4 × 15 mL), and drying in a vacuum oven at 50 °C. Finally, a mixture of KI (1.5 g, 9 mmol), CuSO4·5H2O (2.5 g, 10 mmol), and thiosemicarbazone adduct V (2.0 g) was dispersed in a flask containing deionized water (30 mL). After stirring the mixture at room temperature for 48 h, the dark-brown solid was separated from the flask by a magnet, washed with deionized water (4 × 15 mL), and kept in a vacuum oven at 70 °C to be dried. The obtained solid which is Fe3O4@SiO2-[CuL] (VI) was corked and stored in a desiccator at room temperature.
4.3. General procedure for the Ullmann-type N-arylation of N-heterocycles
In a round bottom flask containing DMF (4 mL), it was charged with the desired N-heterocycle (1 mmol), aryl halide (1.2 mmol), Cs2CO3 (2 mmol), and Fe3O4@SiO2-[CuL] (0.01 g, 1 mol%). The mixture was heated to 110 °C and the reaction progress was checked by TLC. When the reaction was completed (the indicated times in Table 2), the crude mixture was cooled to room temperature. Then, Fe3O4@SiO2-[CuL] was separated by a magnet from the reaction mixture and DMF was evaporated under reduced pressure. The remaining residue was dissolved in EtOAC (10 mL), washed twice with water (20 mL), and dried using sodium sulfate. Afterward, the extracted EtOAC was evaporated using a rotary evaporator under a vacuum to afford the crude product. The desired N-aryl N-heterocycle was obtained after purification via column chromatography on silica gel eluted with the proper solvent.Note: The characterization data of 3a–3o can be found in the ESI.†
4.4. Recycling of Fe3O4@SiO2-[CuL]
The separated solid by a magnet was washed with deionized water (2 × 30 mL) and ethanol (2 × 30 mL). Afterward, the catalyst was kept in a vacuum oven to be dried at 70 °C and reused for subsequent reactions.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors wish to thank Shiraz University of Technology and Yazd University research councils for their partial support of this work.References
- A. R. Katritzky and C. W. Rees, Comprehensive Heterocyclic Chemistry II, Elsevier, Oxford, 1996 Search PubMed.
- A. Kleeman, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances, Thieme, Stuttgart, 5th edn, 2009 Search PubMed.
- D.-W. Wang, L. Liang, Z.-Y. Xue, S.-Y. Yu, R.-B. Zhang, X. Wang, H. Xu, X. Wen and Z. Xi, Discovery of N-phenylaminomethylthioacetylpyrimidine-2,4-diones as protoporphyrinogen IX oxidase inhibitors through a reaction intermediate derivation approach, J. Agric. Food Chem., 2021, 69, 4081–4092, DOI:10.1021/acs.jafc.1c00796.
- M. Legraverend and D. S. Grierson, The purines: potent and versatile small molecule inhibitors and modulators of key biological targets, Bioorg. Med. Chem., 2006, 14, 3987–4006, DOI:10.1016/j.bmc.2005.12.060.
- N. Kode, L. Chen, D. Murthy, D. Adewumi and S. Phadtare, New bis-N9-(methylphenylmethyl)purine derivatives: synthesis and antitumor activity, Eur. J. Med. Chem., 2007, 42, 327–333, DOI:10.1016/j.ejmech.2006.10.017.
- A. K. Bakkestuen, L.-L. Gundersen and B. T. Utenova, Synthesis, biological activity, and SAR of antimycobacterial 9-aryl-, 9-arylsulfonyl-, and 9-benzyl-6-(2-furyl)purines, J. Med. Chem., 2005, 48, 2710–2723, DOI:10.1021/jm0408924.
- L. Aguado, H. J. Thibaut, E.-M. Priego, M.-L. Jimeno, M.-J. Camarasa, J. Neyts and M.-J. Pérez-Pérez, 9-Arylpurines as a novel class of enterovirus inhibitors, J. Med. Chem., 2009, 53, 316–324, DOI:10.1021/jm901240p.
- D. Kim, H. Lee, H. Jun, S.-S. Hong and S. Hong, Fluorescent phosphoinositide 3-kinase inhibitors suitable for monitoring of intracellular distribution, Bioorg. Med. Chem., 2011, 19, 2508–2516, DOI:10.1016/j.bmc.2011.03.025.
- J. B. Buckingham, in Dictionary of Natural Products, CRC Press, Boca Raton, 1994, vol. 1 Search PubMed.
- R. A. Helmick, A. E. Fletcher, A. M. Gardner, C. R. Gessner, A. N. Hvitved, M. C. Gustin and P. R. Gardner, Imidazole antibiotics inhibit the nitric oxide dioxygenase function of microbial flavohemoglobin, Antimicrob. Agents Chemother., 2005, 49, 1837–1843, DOI:10.1128/AAC.49.5.1837-1843.2005.
- D. G. Hulcoop and M. Lautens, Palladium-catalyzed annulation of aryl heterocycles with strained alkenes, Org. Lett., 2007, 9, 1761–1764, DOI:10.1021/ol070475w.
- H. Xu, W.-Q. Liu, L.-L. Fan, Y. Chen, L.-M. Yang, L. Lv and Y.-T. Zheng, Synthesis and HIV-1 integrase inhibition activity of some N-arylindoles, Chem. Pharm. Bull., 2008, 56, 720–722, DOI:10.1248/cpb.56.720.
- P. C. Unangst, D. T. Connor, S. R. Stabler, R. J. Weikert, M. E. Carethers, J. A. Kennedy, D. O. Thueson, J. C. Chestnut, R. L. Adolphson and M. C. Conroy, Novel indolecarboxamidotetrazoles as potential antiallergy agents, J. Med. Chem., 1989, 32, 1360–1366, DOI:10.1021/jm00126a036.
- G. Spadoni, C. Balsamini, A. Bedini, G. Diamantini, B. D. Giacomo, A. Tontini, G. Tarzia, M. Mor, P. V. Plazzi, S. Rivara, R. Nonno, M. Pannacci, V. Lucini, F. Fraschini and B. M. Stankov, 2-[N-Acylamino(C1−C3)alkyl]indoles as MT1 melatonin receptor partial agonists, antagonists, and putative inverse agonists, J. Med. Chem., 1998, 41, 3624–3634, DOI:10.1021/jm970721h.
- H. Sano, T. Noguchi, A. Tanatani, Y. Hashimoto and H. Miyachi, Design and synthesis of subtype-selective cyclooxygenase (COX) inhibitors derived from thalidomide, Bioorg. Med. Chem., 2005, 13, 3079–3091, DOI:10.1016/j.bmc.2005.03.002.
- K. Andersen, T. Liljefors, J. Hyttel and J. Perregaard, Serotonin 5-HT2 receptor, dopamine D2 receptor, and α1 adrenoceptor antagonists. Conformationally flexible analogues of the atypical antipsychotic sertindole, J. Med. Chem., 1996, 39, 3723–3738, DOI:10.1021/jm960159f.
- P. Ruiz-Castillo and S. L. Buchwald, Applications of palladium-catalyzed C−N cross-coupling reactions, Chem. Rev., 2016, 116, 12564–12649, DOI:10.1021/acs.chemrev.6b00512.
- T. Weidlich, M. Špryncová and A. Cegan, Copper-catalyzed reactions of aryl halides with N-nucleophiles and their possible application for degradation of halogenated aromatic contaminants, Catalysts, 2022, 12, 911, DOI:10.3390/catal12080911.
- C. Fischer and B. Koenig, Palladium- and copper-mediated N-aryl bond formation reactions for the synthesis of biological active compounds, Beilstein J. Org. Chem., 2011, 7, 59–74, DOI:10.3762/bjoc.7.10.
- A. Gondela and K. Walczak, N-Functionalization of uracil derivatives: synthesis of chiral 2-(3-methyl-5-nitro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)alkanoic acids and their methyl esters, Tetrahedron: Asymmetry, 2005, 16, 2107–2112, DOI:10.1016/j.tetasy.2005.05.009.
- T. Zhou, T.-C. Li and Z.-C. Chen, Hypervalent iodine in synthesis. Part 86: Selective copper-catalyzed n-monoarylation and N1,N3-diarylation of uracil and its derivatives with diaryliodonium salts, Helv. Chim. Acta, 2005, 88, 290–296, DOI:10.1002/hlca.200590010.
- M. Hocek, Syntheses of purines bearing carbon substituents in positions 2, 6 or 8 by metal- or organometal-mediated C–C bond-forming reactions, Eur. J. Org Chem., 2003, 2003, 245–254, DOI:10.1002/ejoc.200390025.
- N. Devarajan and P. Suresh, Framework-copper-catalyzed C-N cross-coupling of arylboronic acids with imidazole: Convenient and ligand-free synthesis of N-arylimidazoles, ChemCatChem, 2016, 8, 2953–2960, DOI:10.1002/cctc.201600480.
- M. N. Soltani Rad, S. Behrouz, M. M. Doroodmand and N. Moghtaderi, Copper nanoparticle-doped silica cuprous sulfate as a highly efficient and reusable heterogeneous catalysis for N-arylation of nucleobases and N-heterocyclic compounds, Synthesis, 2011, 23, 3915–3924, DOI:10.1055/s-0030-1260236.
- Q. Zhou, F. Du, Y. Chen, Y. Fu, W. Sun, Y. Wu and G. Chen, L-(–)-Quebrachitol as a ligand for selective copper(0)-catalyzed N-arylation of nitrogen-containing heterocycles, J. Org. Chem., 2019, 84, 8160–8167, DOI:10.1021/acs.joc.9b00997.
- I. P. Beletskaya and A. V. Cheprakov, Copper in cross-coupling reactions: The post-Ullmann chemistry, Coord. Chem. Rev., 2004, 248, 2337–2364, DOI:10.1016/j.ccr.2004.09.014.
- J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Rational development of practical catalysts for aromatic carbon−nitrogen bond formation, Acc. Chem. Res., 1998, 31, 805–818, DOI:10.1021/ar9600650.
- J. F. Hartwig, Transition metal catalyzed synthesis of arylamines and aryl ethers from aryl halides and triflates: Scope and mechanism, Angew. Chem., Int. Ed., 1998, 37, 2046–2067, DOI:10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L.
- H.-J. Cristau, P. P. Cellier, J.-F. Spindler and M. Taillefer, Mild conditions for copper-catalysed N-arylation of pyrazoles, Eur. J. Org Chem., 2004, 2004, 695–709, DOI:10.1002/ejoc.200300709.
- A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, A general and efficient copper catalyst for the amidation of aryl halides and the N-arylation of nitrogen heterocycles, J. Am. Chem. Soc., 2001, 123, 7727–7729, DOI:10.1021/ja016226z.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Selected copper-based reactions for C–N, C–O, C–S, and C–C bond formation, Angew. Chem., Int. Ed., 2017, 56, 16136–16179, DOI:10.1002/anie.201701690.
- U. Díaz, M. Boronat and A. Corma, Hybrid organic-inorganic structured materials as single-site heterogeneous catalysts, Proc. R. Soc. A, 2012, 468, 1927–1954, DOI:10.1098/rspa.2012.0066.
- R. B. NasirBaig and R. S. Varma, Magnetically retrievable catalysts for organic synthesis, Chem. Commun., 2013, 49, 752–770, 10.1039/C2CC35663E.
- B. Liu and Z. Zhang, Catalytic conversion of biomass into chemicals and fuels over magnetic catalysts, ACS Catal., 2016, 6, 326–338, DOI:10.1021/acscatal.5b02094.
- P. K. Sharma, S. Dutta, S. Sharma, R. Zboril, R. S. Varma and M. B. Gawande, Fe3O4 (iron oxide)-supported nanocatalysts: synthesis, characterization and applications in coupling reactions, Green Chem., 2016, 18, 3184–3209, 10.1039/C6GC00864J.
- S. Zahmatkesh, M. Esmaeilpour and A. Mollaiy Poli, Ligand complex of copper (II) supported on superparamagnetic Fe3O4@SiO2 nanoparticles: An efficient and magnetically separable catalyst for N-arylation of nitrogen-containing heterocycles with aryl halides, Inorg. Nano-Met. Chem., 2019, 49, 323–334, DOI:10.1080/24701556.2019.1618326.
- A. R. Sardarian, H. Eslahi and M. Esmaeilpour, Copper(II) complex supported on Fe3O4@SiO2 coated by polyvinyl alcohol as reusable nanocatalyst in N-arylation of amines and N(H)- heterocycles and green synthesis of 1H-tetrazoles, ChemistrySelect, 2018, 3, 1499–1511, DOI:10.1002/slct.201702452.
- A. R. Sardarian, M. Kazemnejadi and M. Esmaeilpour, Functionalization of superparamagnetic Fe3O4@SiO2 nanoparticles with a Cu(II) binuclear Schiff base complex as an efficient and reusable nanomagnetic catalyst for N-arylation of α-amino acids and nitrogen-containing heterocycles with aryl halides, Appl. Organomet. Chem., 2020, 35, e6051, DOI:10.1002/aoc.6051.
- S. Behrouz, M. N. Soltani Rad, Z. Ganji, M. Behrouz, E. Zarenezhad and M. Agholi, Design, synthesis, antigiardial and in silico assessments of novel propargylamines containing nitroimidazole core, Tetrahedron, 2022, 124, 133007, DOI:10.1016/j.tet.2022.133007.
- M. N. Soltani Rad, S. Behrouz, M. Mohammad-Javadi and E. Zarenezhad, Synthesis of fish scale derived hydroxyapatite silica propyl bis aminoethoxy ethane cuprous complex (HASPBAEECC) as a novel hybrid nano-catalyst for highly efficient synthesis of new benzimidazole-1,2,3-triazole hybrid analogues as antifungal agents, Mol. Diversity, 2022, 26, 2503–2521, DOI:10.1007/s11030-021-10346-9.
- S. Behrouz, Copper-doped silica cuprous sulfate: A highly efficient heterogeneous nano-catalyst for one-pot three-component synthesis of 1-H-2-substituted benzimidazoles from 2-bromoanilines, aldehydes, and [bmim]N3, J. Saudi Chem. Soc., 2018, 22, 261–268, DOI:10.1016/j.jscs.2016.07.003.
- S. Behrouz, Highly efficient one pot-three component synthesis of 2H-indazoles by consecutive condensation, C–N and N–N bond formations using Cu/aminoclay/reduced graphene oxide nanohybrid, J. Heterocycl. Chem., 2017, 54, 1863–1871, DOI:10.1002/jhet.
- H. Rajabi-Moghaddam, M. R. Naimi-Jamal and M. Tajbakhsh, Fabrication of copper(II)-coated magnetic core-shell nanoparticles Fe3O4@SiO2-2-aminobenzohydrazide and investigation of its catalytic application in the synthesis of 1,2,3-triazole compounds, Sci. Rep., 2021, 11, 2073, DOI:10.1038/s41598-021-81632-7.
- W. Stöber, A. Fink and E. Bohn, Controlled growth of monodisperse silica spheres in the micron size range, J. Colloid Interface Sci., 1968, 26, 62–69, DOI:10.1016/0021-9797(68)90272-5.
- F. Godarzbod, Z. Mirjafary, H. Saeidian and M. Rouhani, Highly efficient synthesis of silica-coated magnetic nanoparticles modified with iminodiacetic acid applied to synthesis of 1,2,3-triazoles, Appl. Organomet. Chem., 2021, 35, e6132, DOI:10.1002/aoc.6132.
- G. Zanchettin, G. da Silva Falk, S. Y. G. González and D. Hotza, High performance magnetically recoverable Fe3O4 nanocatalysts: fast microwave synthesis and photo-fenton catalysis under visible-light, Chem. Eng. Process., 2021, 166, 108438, DOI:10.1016/j.cep.2021.108438.
- Y. Z. Huang, H. Miao, Q. H. Zhang, C. Chen and J. Xu, Cu2O: A simple and efficient reusable catalyst for N-arylation of nitrogen-containing heterocycles with aryl halides, Catal. Lett., 2008, 122, 344–348, DOI:10.1007/s10562-007-9386-0.
- M. L. Kantam, B. P. C. Rao, B. M. Choudary and R. S. Reddy, A mild and efficient method for N-arylation of nitrogen heterocycles with aryl halides catalyzed by Cu(II)-NaY zeolite, Synlett, 2006, 14, 2195–2198, DOI:10.1055/s-2006-949615.
- M. Lakshmi Kantam, G. T. Venkanna, C. Sridhar and K. B. Shiva Kumar, Copper fluorapatite catalyzed N-arylation of heterocycles with bromo and iodoarenes, Tetrahedron Lett., 2006, 47, 3897–3899, DOI:10.1016/j.tetlet.2006.03.173.
- L. Rout, S. Jammi and T. Punniyamurthy, Novel CuO nanoparticle catalyzed C-N cross coupling of amines with iodobenzene, Org. Lett., 2007, 9, 3397–3399, DOI:10.1021/ol0713887.
- P. R. Likhar, S. Roy, M. Roy, M. Lakshmi Kantam and R. Lal De, Silica immobilized copper complexes: efficient and reusable catalysts for N-arylation of N(H)-heterocycles and benzyl amines with aryl halides and arylboronic acids, J. Mol. Catal. A: Chem., 2007, 271, 57–62, DOI:10.1016/j.molcata.2007.02.036.
- A. Bourouina, V. Meille and C. de Bellefon, About solid phase vs. liquid phase in Suzuki-Miyaura reaction, Catalysts, 2019, 9, 60, DOI:10.3390/catal9010060.
- D. Wang, F. Zhang, D. Kuang, J. Yu and J. Li, A highly efficient Cu-catalyst system for N-arylation of azoles in water, Green Chem., 2012, 14, 1268–1271, 10.1039/C2GC35077G.
- M. Gopiraman, S. G. Babu, Z. Khatri, W. Kai, Y. A. Kim, M. Endo, R. Karvembu and I. S. Kim, An efficient, reusable copper-oxide/carbonnanotube catalyst for N-arylation of imidazole, Carbon, 2013, 62, 135–148, DOI:10.1016/j.carbon.2013.06.005.
Footnote |
| † Electronic supplementary information (ESI) available: Data and copies of NMR spectra (1H and 13C NMR) for synthesized compounds. See DOI: https://doi.org/10.1039/d3ra06327e |
| This journal is © The Royal Society of Chemistry 2023 |