
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel benzopyrimido[4,5-d]azoninone analogs catalyzed by biosynthesized Ag-TiO2 core/shell magnetic nanocatalyst and assessment of their antioxidant activity†
Mohamed M. Hammouda *ab,
Kamal Shalabiab,
Abdulaziz A. Alanazia,
Khaled M. Elattarc,
Maged A. Azzamad and
Marwa M. Rashede
*ab,
Kamal Shalabiab,
Abdulaziz A. Alanazia,
Khaled M. Elattarc,
Maged A. Azzamad and
Marwa M. Rashede
aDepartment of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia. E-mail: m.elmettwaly@psau.edu.sa; Tel: +966 570260397
bChemistry Department, Faculty of Science, Mansoura University, El-Gomhoria Street, Mansoura, 35516, Egypt. E-mail: elmhammouda@mans.edu.eg
cUnit of Genetic Engineering and Biotechnology, Faculty of Science, Mansoura University, El-Gomhoria Street, Mansoura, 35516, Egypt. E-mail: khaledelattar2@yahoo.com
dDepartment of Chemistry, Faculty of Science, Menoufia University, Shibin El Kom, Egypt
eToxicology Department, Mansoura Hospital, Faculty of Medicine, Mansoura University, El-Gomhoria Street, Mansoura, 35516, Egypt. E-mail: marwarashed@mans.edu.eg
First published on 6th November 2023
Abstract
The present work reported the synthesis of novel benzopyrimido[4,5-d]azoninone analogs using a biosynthesized Ag-TiO2 core/shell magnetic nanocatalyst. Accordingly, three-component one-pot reactions of benzoazonine-dione with thiourea and aryl aldehyde derivatives under nanocatalytic and optimized conditions afforded reasonable to brilliant yields of the target products (57–91%). The nanocatalyst was synthesized by a facile method using turmeric ethanol extract as a reducing and chelating agent. The synthesized nanocatalyst was verified by FT-IR, XRD, zeta potential, EDX, SEM, and TEM. The nanocatalyst presented remarkable catalytic activity for the synthesis of the target products. The procedure provided biosynthesis of the nanocatalyst, accessible reagents, high yields and rates of the reactions, nanocatalyst recyclability, and ease of product isolation and purification. The novel products were characterized by FT-IR, 1H-NMR, 13C-NMR, mass spectra, and 2D NMR analysis (COSY, NOESY, HMQC & HSQC) spectral analyses. The antioxidant activity was assessed by DPPH and phosphomolybdate assays, in which the compounds exhibited excellent potency. Overall, this approach could be used to develop new and sustainable methods for the synthesis of antioxidants and other biologically active molecules.
1. Introduction
Biosynthesis of nanomaterials is a process that uses eco-friendly and sustainable methods to produce nanomaterials.1,2 It is a rapidly growing field of research, as there is a growing demand for nanomaterials that are safe for human health and the environment.3,4 There are a variety of different biosynthesis methods that can be used to produce different types of nanomaterials.5,6 Some of the most common methods include plant-mediated synthesis,7 which involves the use of plant extracts to reduce and stabilize metal ions into nanoparticles based on the capacity of biomolecules, and reactive species of the plant extract; microbial synthesis,8 which involves the use of microorganisms, such as bacteria, yeasts, and fungi, to synthesize nanomaterials; and biomimetic synthesis,9 which involves the use of natural processes to inspire the synthesis of nanomaterials. For example, researchers have developed methods to synthesize nanomaterials that mimic the way that seashells and bones are formed. The biosynthesis protocols of the nanocatalysts have several advantages over traditional methods of synthesis, including safety, more sustainable materials, less waste, and being more cost-effective.10 The biosynthetic scope was also extended to include biosynthesis, solvent-free synthesis, microwave-assisted synthesis, and ultrasound-assisted synthesis.11,12 The field of biosynthesis is rapidly growing, with many innovative new methods being developed.13 With its many advantages over traditional methods, biosynthesis is likely to become the standard for synthesizing nanomaterials in the future.14On the other hand, nine-membered nitrogen heterocycles such as azonines are difficult to synthesize with specific structures and configurations. Azoninones are a category of organic molecules containing a nine-membered ring containing a nitrogen atom and a carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonded at the ring system. However, azonines are magnificent scaffolds of natural and pharmaceutical products.15 One of the most notable features of azonines is their reactivity, in which these heterocycles can undergo a variety of reactions, including cycloadditions, cyclizations, and rearrangements.16 These reactions can be used to synthesize a wide range of complex molecules. Another important feature of azonines is their ability to form hydrogen bonds.17 This ability allows azonines to bind to a variety of biomolecules, including proteins and DNA. This binding can be exploited to develop new drugs and diagnostic tools. Despite their importance, azonines are relatively difficult to synthesize. This is because the nine-membered ring is prone to ring-opening reactions. However, in recent years, chemists have developed a number of new methods for synthesizing azonines.18,19 These methods have made it possible to access a wider range of azonine derivatives.
O) bonded at the ring system. However, azonines are magnificent scaffolds of natural and pharmaceutical products.15 One of the most notable features of azonines is their reactivity, in which these heterocycles can undergo a variety of reactions, including cycloadditions, cyclizations, and rearrangements.16 These reactions can be used to synthesize a wide range of complex molecules. Another important feature of azonines is their ability to form hydrogen bonds.17 This ability allows azonines to bind to a variety of biomolecules, including proteins and DNA. This binding can be exploited to develop new drugs and diagnostic tools. Despite their importance, azonines are relatively difficult to synthesize. This is because the nine-membered ring is prone to ring-opening reactions. However, in recent years, chemists have developed a number of new methods for synthesizing azonines.18,19 These methods have made it possible to access a wider range of azonine derivatives.
Azoninones typically synthesized by periodate oxidation of 2,3,4,9-tetrahydro-1H-carbazole or via Friedel–Crafts ring closures, and Claisen rearrangements as previously reported.20–24 Azoninones are also known to undergo a variety of other reactions, including cycloaddition and transannular reactions,25 and transannular ring contraction.26 The reactivity of azoninones is still under investigation, and new reactions are being discovered all the time. The stereochemistry of azoninones was reported through the use of planar diastereomer azoninones in the syntheses of the azonanones via cycloaddition reactions.27 The potential applications of azoninones are wide-ranging, in which these heterocycles have been used in the synthesis of a diversity of alkaloids such as indolizidinones.28–30 Azoninones have also been investigated for their biological activity, for example, selective glycosidase inhibitors,30 and apoptotic inducers.31 As research into azoninones continues, they will likely find even more applications in a variety of fields.
The Biginelli reaction is a multicomponent reaction that involves the condensation of an aldehyde, a β-ketoester (active methylene group), and urea or thiourea to form a dihydropyrimidinone. It is a versatile and efficient reaction for the synthesis of a wide range of heterocyclic compounds with potential pharmaceutical applications. Recoverable magnetic nanoparticles can be used as catalysts for the Biginelli reaction, offering several advantages over traditional catalysts, such as ease of recyclability, improved catalytic activity, and broad substrate scope. Recoverable magnetic nanoparticles have been successfully used to catalyze the Biginelli reaction under a variety of conditions, including solvent-free, microwave-assisted, and ultrasonic conditions.32–34 Alvim et al.35 showed that nanocatalyst could be used to catalyze the Biginelli reaction under solvent-free conditions at room temperature. The reaction was completed in as little as 10 minutes, and the dihydropyrimidinone products were obtained in excellent yields. Yadav et al.36 showed that nanocatalyst could be used to catalyze the Biginelli reaction under microwave-assisted conditions. Safari, and Zarnegar37 showed that nanocatalyst could be used to catalyze the Biginelli reaction under ultrasonic conditions.
The current work aims to synthesize a novel series of benzopyrimido[4,5-d]azoninone derivatives using a biosynthesized Ag-TiO2 core/shell magnetic nanocatalyst through a developed protocol. The nanocatalyst is more efficient than traditional metal catalysts because it has a larger surface area. This means that more of the catalyst is exposed to the reactants, which leads to a faster reaction rate. The nanocatalyst provided a high reaction rate, improved yields with high purity, reduced reaction rate, and recyclability. The work also attempted to investigate the antioxidant capacity of the newly synthesized compounds by DPPH and phosphomolybdate assays.
2. Results and discussion
2.1. Characterization of the nanocatalyst
O group disappeared in the analysis of Ag-TiO2 bmNPs. This specified the contribution of this group in the bioreduction process of metal ions in the solution. The characteristic absorption bands at ν = 1653, and 1652 cm−1 are ascribed to the stretching vibrations of carbonyl groups in both samples, in which the shift in the values identified the interaction with metal ions. The absorption bands at ν = 1540, and 1507 cm−1 are identified as the stretching vibrations of phenyl groups. In addition, the bending vibrations of C–H bending groups appeared within ν = 1456–1411 cm−1 with a slight shift in these values in both samples. It is worth stating that a new absorption band appeared in the analysis of Ag-TiO2 bmNPs at ν = 382.68 cm−1 is ascribed to the stretching vibration of M-O bond as an indication for the formation of metal oxides in the solution.38
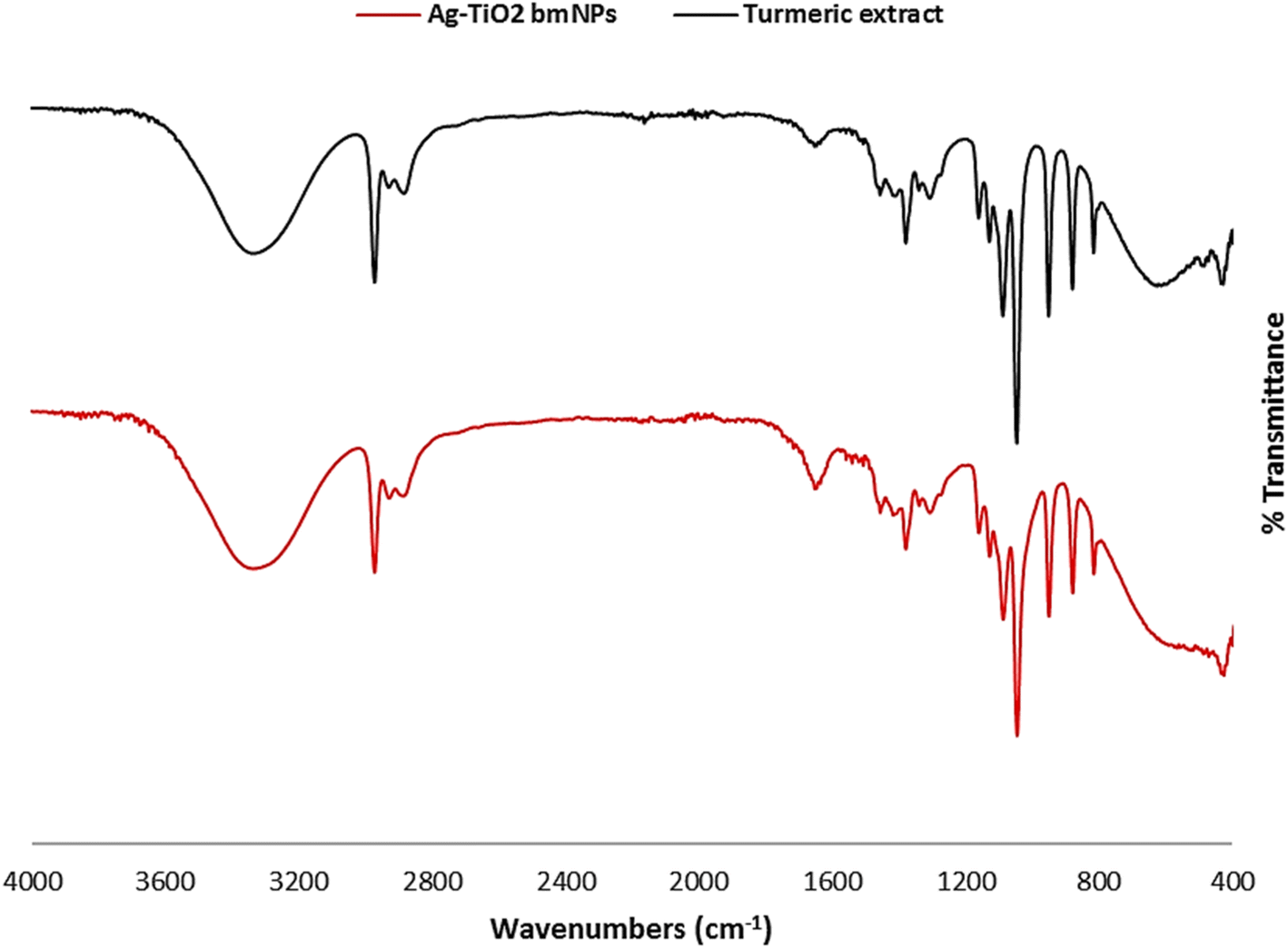 | ||
| Fig. 1 The FT-IR spectral analyses of turmeric extract, and Ag-TiO2 bmNPs. | ||
XRD analysis was run on a Pan Analytical Philips instrument to explore material type, phase, crystallographic, and physical characteristics of Ag-TiO2 bmNPs nanocatalyst (Fig. 2). The analysis depended upon constructive meddling concerning the crystallinity of the nanocatalyst and the monochromatic X-rays. It was noticed that various diffraction peaks of the nanocomposite appeared at 2θ = 25.4282, 27.9393, 32.3090, 37.8764, 46.2968, 48.1503, 54.0300, 55.1796, 57.5827, 62.7931, 68.8952, 70.3194, 75.1116, and 82.9333 in the core–shell. The diffraction peaks are related to (294.02), (51.89), (81.99), (72.28), (45.27), (75.29), (46.50), (50.89), (12.51), (28.94), (10.12), (10.57), (25.78), and (6.21) of lattice planes, respectively.
 | ||
| Fig. 2 XRD analysis of the Ag-TiO2 bmNPs nanocatalyst. | ||
Hong et al.,39 recently reported that the diffraction peaks around 37.8764, 46.2968, 62.7931, and 75.1116 conceivably will be attributed to the silver nanoparticles, which resembled (72.28), (45.27), (28.94), and (25.78) lattice planes of the face-centered-cubic of silver crystal structure, respectively. The results had consistency with the JCPDS database # 01-073-1764, and 00-006-0480. The results indicated the high stability of silver nanoparticles in Ag-TiO2.
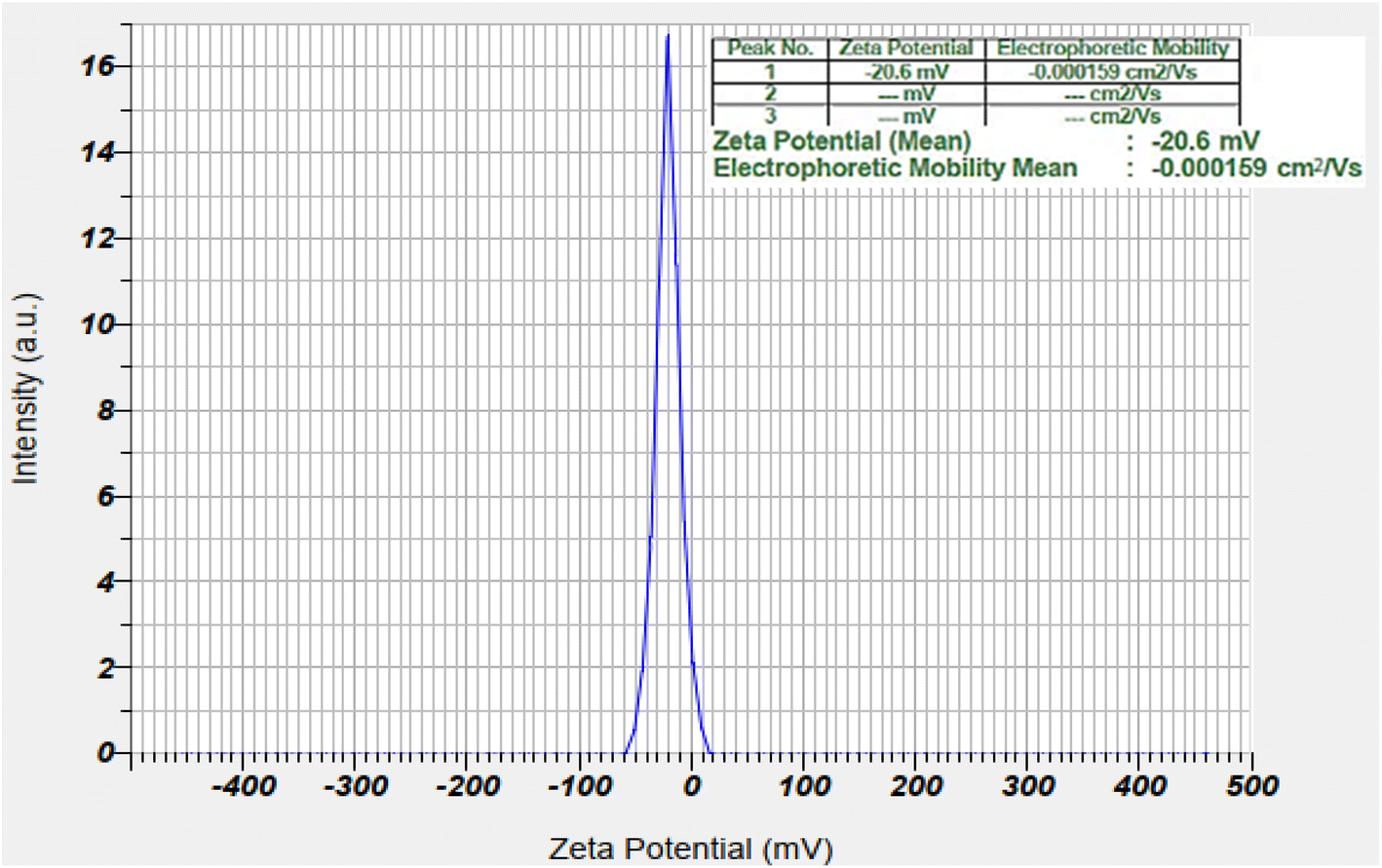 | ||
| Fig. 3 Zeta potential analyses of the bimetallic nanocatalyst. | ||
EDX analysis was applied to characterize the elemental content of magnetic Ag-TiO2 bmNPs as demonstrated in Fig. 4. The results certified the presence of carbon (53.13%), oxygen (18.65%), silver (12.35%), and titanium (15.87%) as the total weight of the nanocatalyst composition. The presence of a high ratio of oxygen element is a sign of the formation of metal oxide nanoparticles, as well as the oxygen has other sources from the turmeric compounds in the plant extract. The analysis verified the high purity, and stability of the sample.41
 | ||
| Fig. 4 EDX, and zeta potential analyses of the bimetallic nanocatalyst. | ||
 | ||
| Fig. 5 SEM micrographs with different magnifications of the bimetallic nanocatalyst. | ||
The TEM analysis was investigated to study the morphology of the bimetallic nanocatalyst in the solution. As publicized in Fig. 6, the average size of the nanoparticles of bimetallic Ag-TiO2 bmNPs commonly raged from 8 to 33 nm in the nanoscale range.
 | ||
| Fig. 6 TEM micrograph and selected area diffraction (SED) of the bimetallic nanocatalyst. | ||
The micrograph of TEM (Fig. 6) indicated the spherical shapes of the nanoparticles, which delivered the large surface area of the nanocatalyst. The large surface area indicated the high efficiency of the nanocatalyst in the organic reactions during the synthesis of the target compounds. As it appeared from the TEM micrograph, two types of nanoparticles with different size ranges are referred to bimetallic nature of the nanocatalyst. The nanoparticles tended to be more aggregated, this performance is efficient for the catalyst activation of the reacted substrates. The reality of the active phases of the bimetallic nanoparticles is beneficial to offer the nanocatalyst plentiful additional reactive sites.43
2.2. Synthesis of benzo[b]pyrimido[4,5-d]azoninones
As outlined in Scheme 1, the synthesis of the target products, benzo[b]pyrimido[4,5-d]azoninone derivatives 3a–k was achieved through a simple procedure. Fischer indole synthesis was utilized for the synthesis of carbazole 1 via reactions of phenyl hydrazine with cyclohexanone.20–22 Subsequently, compound 1 was applied as a reactive synthon for the synthesis of benzoazonine-dione 2 through periodate oxidation.44 Therefore, one-pot three-component reactions (Biginelli type reactions) of 2 with thiourea and diverse aryl aldehydes under nanocatalytic and optimized conditions gave moderate to brilliant yields of the respective target products (57–91%). | ||
| Scheme 1 Synthesis of benzopyrimido[4,5-d]azoninone series. | ||
After fruitful biosynthesis and characterization of Ag-TiO2 bmNPs using turmeric ethanol extract, we have patterned its nanocatalytic activity for the synthesis of 8H-benzo[b]pyrimido[4,5-d]azoninones 3a–k. Firstly, we selected product 3a as an example to study the various optimized conditions on the product yield. We have applied the use of different solvents, and nanocatalysis amounts to investigate the most reliable conditions that achieved the developed yield. The results of the optimization conditions, product yield, and reaction time are publicized in Table 1. Initially, we carried out the three-component one-pot reaction of compound 2 with thiourea and benzaldehyde as a reaction model using 2.5 mg of the nanocatalyst in ethanol as a solvent under heating conditions. It was noticed that product 3a was formed with a yield of 65% after a long reaction time. After that, we carried out the same reaction using an increased amount of the nanocatalyst (5 mg) in a route to increase the product yield and reduce the reaction time. The product 3a was acquired with a 68% yield after a slightly improved reaction time (9 h). Subsequently, we increased the amount of the nanocatalyst (10, 15, 20, and 25 mg) in the next trials to achieve improved product yield and reaction rate. The yield of product 3a was increased by increasing the nanocatalyst amount from 73% to 86%, in addition, the reaction time was decreased from 7.5 to 6 h. Next, we performed the reaction under various conditions using other solvents such as DMF, and acetonitrile as well as under solvent-free conditions using the same effective amount of the nanocatalyst (25.0 mg). The carrying out of the reaction under solvent-free conditions produced a very low yield of compound 3a (26%), while the use of a solvent improved the product yield as the solvent enabled the increased reactivity of the nanocatalyst. The use of DMF produced product 3a with a 67% yield, while the use of acetonitrile produced product 3a with a lower yield (34%).
| Entry | Ag-TiO2 bmNPs (mg) | Solvent | Reaction time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Equimolar amounts (0.001 mmol) of compound 2, thiourea, and benzaldehyde were applied.b Isolated yield of the product. | ||||
| 1 | 2.5 | EtOH | 10 | 65 |
| 2 | 5.0 | EtOH | 9 | 68 |
| 3 | 10.0 | EtOH | 7.5 | 73 |
| 4 | 15.0 | EtOH | 7 | 76 |
| 5 | 20.0 | EtOH | 6.5 | 81 |
| 6 | 25.0 | EtOH | 6 | 86 |
| 7 | 25.0 | Solvent-free | 9 | 26 |
| 8 | 25.0 | DMF | 7 | 67 |
| 9 | 25.0 | Acetonitrile | 8 | 34 |
| 10 | 30 | Solvent-free | 8 | 28 |
| 11 | Nanocatalyst-free | EtOH | 9 | 25 |
In a trial to improve the product yield under solvent-free conditions, we implemented a model reaction applying an increased amount of the nanocatalyst, but the yield of the product still gained a lower yield (28%), while the reaction rate was not significantly affected. In a trial to explore the reactivity of the nanocatalyst, the reaction was run under free-nanocatalytic conditions in ethanol. The reaction in this case produced product 3a with a poor yield (25%) and a long reaction time. Accordingly, the improved product yield in this reaction is concerned by the solvent type, and nanocatalyst amount (Table 1).
The scope of the reaction was investigated using various aldehydes to synthesize a variety of benzo[b]pyrimido[4,5-d]azonin-8-one analogs 3a–k using Ag-TiO2 bmNPs (25.0 mg) nanocatalyst in ethanol (Table 2). In particular, the products 3a–k were prepared in 57–91% yields through a one-pot three-component synthetic procedure. The reaction rate as well as the product yield was influenced by the reactivity of the applied aryl aldehyde, in which the other conditions and reactants are the same in all reactions. Thus, the varied substituents at the phenyl ring of the aryl aldehydes or the aryl skeleton, in general, controlled the product yield and reaction rate. The most improved yield was acquired in the case of using 1-naphthaldehyde to prepare compound 3k (91%). In addition, benzaldehyde provided also improved yield of the product 3a (ArC6H5; 86%). Correspondingly, compounds 3e, 3h, and 3i were prepared with very good yields higher than 81%. Thus, the nature of substituents in this reaction type is preferred to use aldehydes with phenyl or naphthyl rings as well as the phenyl ring substituted with electron-donating groups such as p-dimethylamino-, and p-methoxy groups. The reaction of 6-bromobenzo[d][1,3]dioxole-5-carbaldehyde with compound 2 and thiourea provided improved yield of compound 3i (81%).
| Entry | Ar | Product | Yieldb (%) | M.p. (°C) |
|---|---|---|---|---|
| a Reaction conditions: 3,4,5,6-tetrahydro-1H-benzo[b]azonine-2,7-dione (1.0 mmol), thiourea (1.0 mmol), and aryl aldehydes (1.0 mmol); AgTiO2 bmNPs (25.0 mg); 10 mL EtOH; room temperature (25 °C).b Isolated yield. | ||||
| 1 | C6H5 | 3a | 86 | 192–4 |
| 2 | 2,4-(Cl)2-C6H3 | 3b | 57 | 175–7 |
| 3 | 4-Cl-C6H4 | 3c | 79 | 187–9 |
| 4 | 2,6-(Cl)2-C6H3 | 3d | 61 | 168–70 |
| 5 | 4-NMe2-C6H4 | 3e | 84 | 194–7 |
| 6 | 2-OH-C6H4 | 3f | 74 | 185–8 |
| 7 | 3,5-(Br)2-C6H3 | 3g | 67 | 172–4 |
| 8 | 4-OMe-C6H4 | 3h | 81 | 178–80 |
| 9 | 5-Br-benzo[d][1,3]dioxol-6-yl | 3i | 81 | 208–10 |
| 10 | 2-Pyridyl | 3j | 75 | 202–5 |
| 11 | 1-Naphthyl | 3k | 91 | 210–3 |
The factor that controlled the improved product yield could be noticed through the introduction of phenyl- or naphthyl- or phenyl ring p-substituted with electron-donating groups. On the other hand, p-chlorobanzaldehyde also produced compound 3c with an honorable yield (79%) (Table 2). The dichloro-substitution of the phenyl ring of the aldehyde drives the formation of the products with reduced yields (3b: 57%, 3d: 61%). The dibromo-substitution at the phenyl ring of the aldehyde provided a slightly improved yield of the product 3g (67%) than the substitution with dichloro-substituents. Thus, it seems to be noted that the unsubstituted phenyl rings of the aldehyde, as well as the phenyl ring substituted with electron-donating groups at the p-position, are preferred for enhanced product yield, while the substitution with electron-withdrawing groups slightly affected the product yield.
The structures of the novel synthesized benzo[b]pyrimido[4,5-d]azonin-8-one analogs 3a–k were established by accurate elemental and spectroscopic analyses (FT-IR, 1H-NMR, 13C-NMR, 2D NMR analysis (COSY NMR, COSY/HMQC, HSQC, NOESY) and mass spectra) (c.f. Experimental section & ESI†).
The 2D-NMR spectral analysis (COSY) of compound 3d (Fig. 7) was used to identify the protons that are coupled to each other and to establish the connectivity between protons in the molecule. Compound 3d includes seven aromatic CH protons, three protons of three NH groups, and protons of three CH2 groups. The aromatic CH protons will be coupled to each other in a variety of ways, depending on their relative positions on the aromatic ring. For example, protons that are ortho to each other will be coupled by a strong coupling constant, while protons that are meta to each other will be coupled by a weaker coupling constant. In addition, the CH2 protons will be coupled to each other in the region of aliphatic.
 | ||
| Fig. 7 2D-NMR spectrum (COSY) of compound 3d. | ||
Once the diagonal peaks have been identified, the cross-peaks can be analyzed to determine which protons are coupled to each other. The following are some examples of the cross-peaks that might be observed in the COSY spectrum of compound 3d (Fig. 7): (1) cross-peaks between adjacent aromatic CH protons. (2) Cross-peaks between ortho aromatic CH protons. (3) Cross-peaks between adjacent CH2 protons. The intensity of the cross-peaks will be proportional to the coupling constant between the two protons. The coupling constant will depend on the distance between the two protons and the dihedral angle between the proton-carbon bonds. By analyzing the COSY spectrum, it is possible to elucidate the structure of the compound up to the relative stereochemistry of the protons. To determine the relative stereochemistry of the protons, it is necessary to use other NMR techniques, such as HSQC.
The HSQC (heteronuclear single quantum correlation) is generated by applying a pulse sequence that excites the protons in a molecule. To analyze the HSQC spectrum of compound 3d (Fig. 8), the first step is to identify the cross-peaks between the aromatic CH protons and the carbon atoms that they are bonded to. These cross-peaks will be located in the region of the spectrum where the aromatic CH protons are resonating (6–8 ppm) and the aromatic carbon atoms are resonating (120–140 ppm). The HSQC spectrum can also be used to identify the cross-peaks between the NH protons and the carbon atoms that they are bonded to. These cross-peaks will be located in the region of the spectrum where the NH protons are resonating (7–9 ppm) and the nitrogen-bonded carbon atoms are resonating (120–140 ppm). The HSQC spectrum of compound 3d can also be used to identify the cross-peaks between (1) the aromatic CH protons and the carbon atoms that are bonded to identify the carbon atoms in the aromatic ring. (2) The NH protons and the carbon atoms that they are bonded to identified the carbon atoms that are bonded to the nitrogen atoms. (3) The cross-peaks between the CH2 protons and the carbon atoms that they are bonded to identify the carbon atoms in the aliphatic chain (Fig. 8).
 | ||
| Fig. 8 2D-NMR spectrum (HSQC) of compound 3d. | ||
The stereochemistry of the final products indicated that these compounds contain one chiral center at the 4-position, which can be either R or S. The R configuration corresponds to the aryl group being oriented towards the front of the molecule, while the S configuration corresponds to the aryl group being oriented towards the back of the molecule. The stereochemistry of the final product will depend on the stereochemistry of the starting materials and the reaction conditions. If the starting materials are both achiral, then the final product will be a racemic mixture of R and S enantiomers. However, if one of the starting materials is chiral, then the final product can be enantioselectively synthesized. For example, if a chiral active methylene compound is used in the synthesis of the target products, then the final product will be a mixture of two diastereomers. The ratio of diastereomers will depend on the stereochemistry of the active methylene compound and the reaction conditions. The stereochemistry of the final product can also be controlled by using chiral catalysts. For example, the use of chiral Brønsted acid catalysts can lead to the enantioselective synthesis of the target products. The enantioselective synthesis of the target products is important for the development of new pharmaceutical drugs. Many pharmaceutical drugs are chiral, and only one enantiomer is typically biologically active. The enantioselective synthesis of the target products allows chemists to produce the desired enantiomer of the drug, which can improve the efficacy and safety of the drug. However, it is important to note that the synthesis of the target products is still an area of active research, and the enantioselective synthesis of these products is challenging.
2.3. The proposed reaction mechanism
The proposed mechanism of the entitled three-component reactions for the synthesis of benzo[b]pyrimido[4,5-d]azonin-8-one analogs 3a–k could be assembled as described in Scheme 2. In the first step, the nanocatalyst activated the carbonyl group of the aldehyde for Knoevenagel condensation with benzoazonine-dione 2 to generate the intermediate A. Subsequently, the interaction of thiourea with the generated intermediate A through a nucleophilic attack of the amino group of thiourea at the carbonyl group adjacent to the arylidene moiety of the intermediate A produced intermediate B. This step is also supported by the nanocatalyst, which enables the nucleophilic attack of the amino group at the carbonyl group by electron donation to the carbonyl group. Next, the proton transfer reactions of intermediate B followed by a condensation step produced intermediate D. Intramolecular cyclization of intermediate D via the attack of the terminal amino group activated by the nanocatalyst at the arylidene carbon produced intermediate E. In this step, the Ag-TiO2 nanocatalyst plays an important role in the activation of the CC bond and the formation of the carbon–nitrogen bond. The silver atoms in the nanocatalyst are thought to act as Lewis acids, which electron-rich CC bonds are attracted to. The titanium dioxide atoms in the nanocatalyst are thought to act as Brønsted acids, which can donate protons to the nitrogen atom in the amino group. The proton transfers of intermediate E generated intermediate F, in which the nanocatalyst was released in the final step with the neutralization of the charged by the proton–transfer reaction to give the final adducts 3a–k. The use of a metal nanoparticle catalyst can significantly improve the rate of the reaction owing to the various benefits of the nanocatalyst including increased reactivity of the reactants, selectivity for the desired product, yields, and reduced reaction time. The use of nanocatalysis is a promising new methodology for the synthesis of benzopyrimidoazoninone analogs and other organic compounds. It is an adaptable performance that can be utilized to catalyze a widespread diversity of reactions.
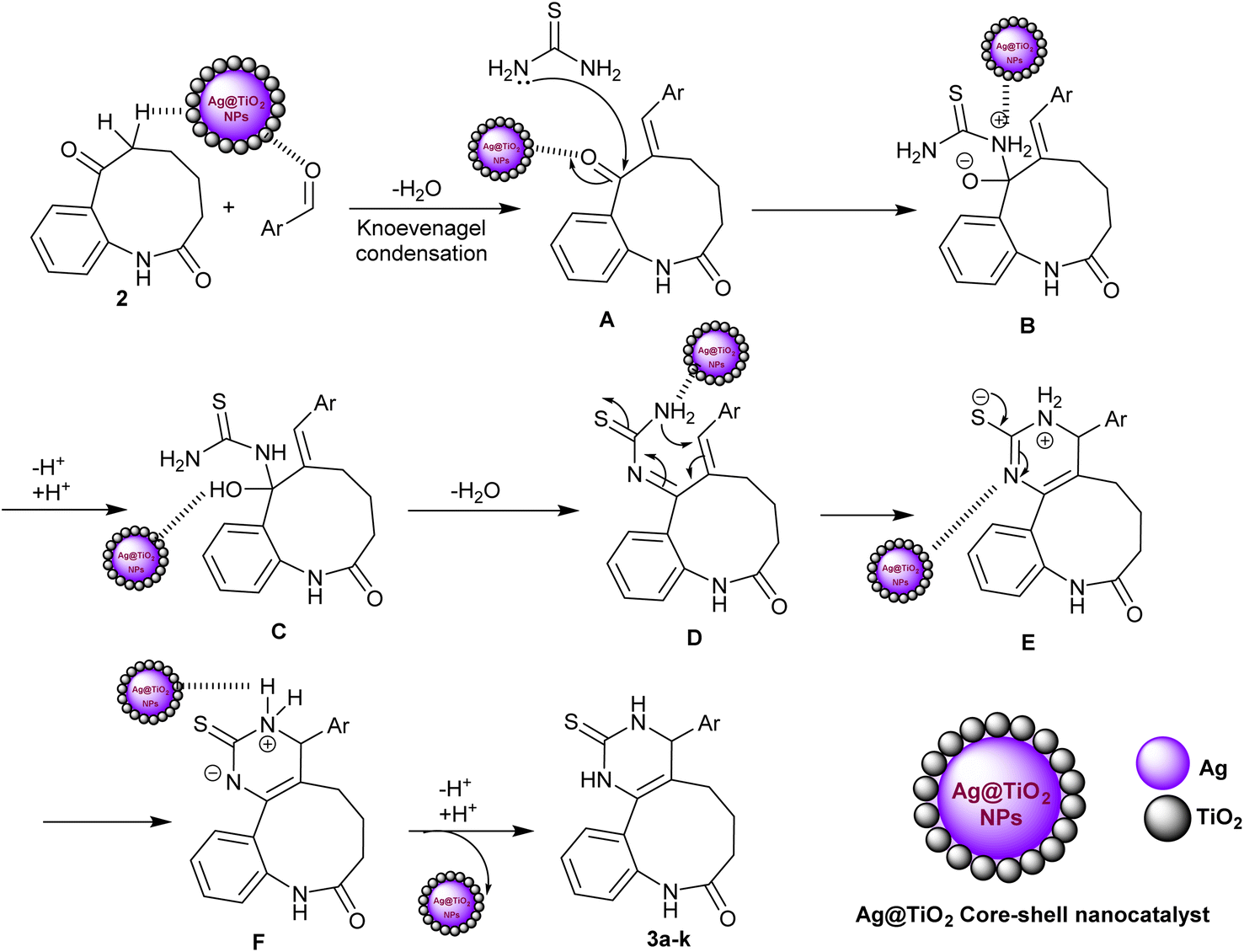 | ||
| Scheme 2 Plausible mechanistic route for the synthesis of benzopyrimido[4,5-d]azoninone derivatives. | ||
2.4. Nanocatalyst recyclability
The rare solubility of the Ag-TiO2 bimetallic nanoparticles in comparison to the alkaline earth oxides enables the recyclability of the nanocatalyst several times (Fig. 9).45 Thus, we inspected the recyclability of the nanocatalyst in the model reaction for the synthesis of compound 3a. The insolubility of the nanocatalyst enables the ease of its isolation from the reaction mixture by filtration. The reaction was run several times (merely 5 runs) using the same filtrated nanocatalyst that was washed with ethanol after each run, dried in the air, and utilized precisely for the subsequent round of reactions. Thus, the catalytic Ag-TiO2 bimetallic NPs delivered a high reaction rate, and reduced time, as well as good yields of the products owing to the excellent catalytic activity of the nanocatalyst without notable loss in its activity.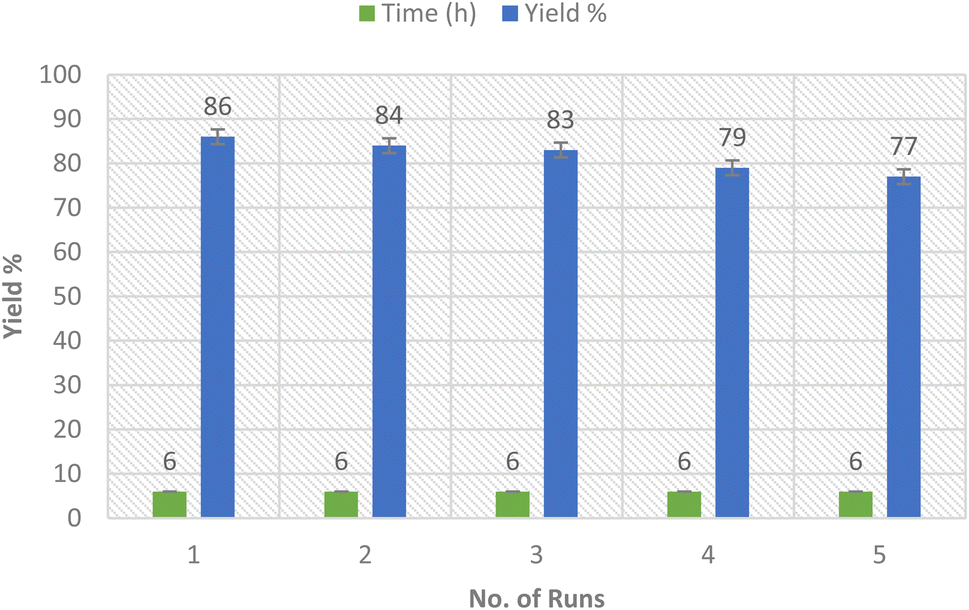 | ||
| Fig. 9 Recyclability of Ag-TiO2 bmNPs for the synthesis of compound 3a. | ||
2.5. Antioxidant activity
The antioxidant activity of the newly synthesized benzopyrimido[4,5-d]azoninone derivatives was assessed by DPPH free radical assay46 using ascorbic acid as a reference standard. The results as shown in Fig. 10 demonstrated the increase in the percentages of the scavenging activity by increasing the sample concentration. The most potent antioxidant result was recorded by compound 3f with 88.49% of the percentage of scavenging at 0.096 mg mL−1. This result is comparable to that of ascorbic acid, which revealed 84.73% inhibition at 0.062 mg mL−1. Also, compounds 3e (81.65% at 0.313 mg mL−1), and 3h (81.03% at 0.214 mg mL−1) revealed remarkable scavenging activities at the higher concentrations. It was noticed that compounds 3c (72.78%), 3d (69.05%), and 3g (69.36%) were at the most used concentration ranges of the samples (0.49–1.104 mg mL−1). The analogs with halogen substituents such as compounds 3b (Ar = 2,4-(Cl)2-C6H3), 3c (Ar = 4-Cl-C6H4), 3d (Ar = 2,6-(Cl)2-C6H3), and 3g (Ar = 3,5-(Br)2-C6H3) revealed the least order of antioxidant potency, e.g. compound 3b revealed antioxidant activity with 60.96% inhibition percentage at 0.424 mg mL−1, as well as compounds 3c (72.78%, 0.524 mg mL−1), 3d (69.05%, 1.104 mg mL−1), and 3g (69.36%, 0.49 mg mL−1).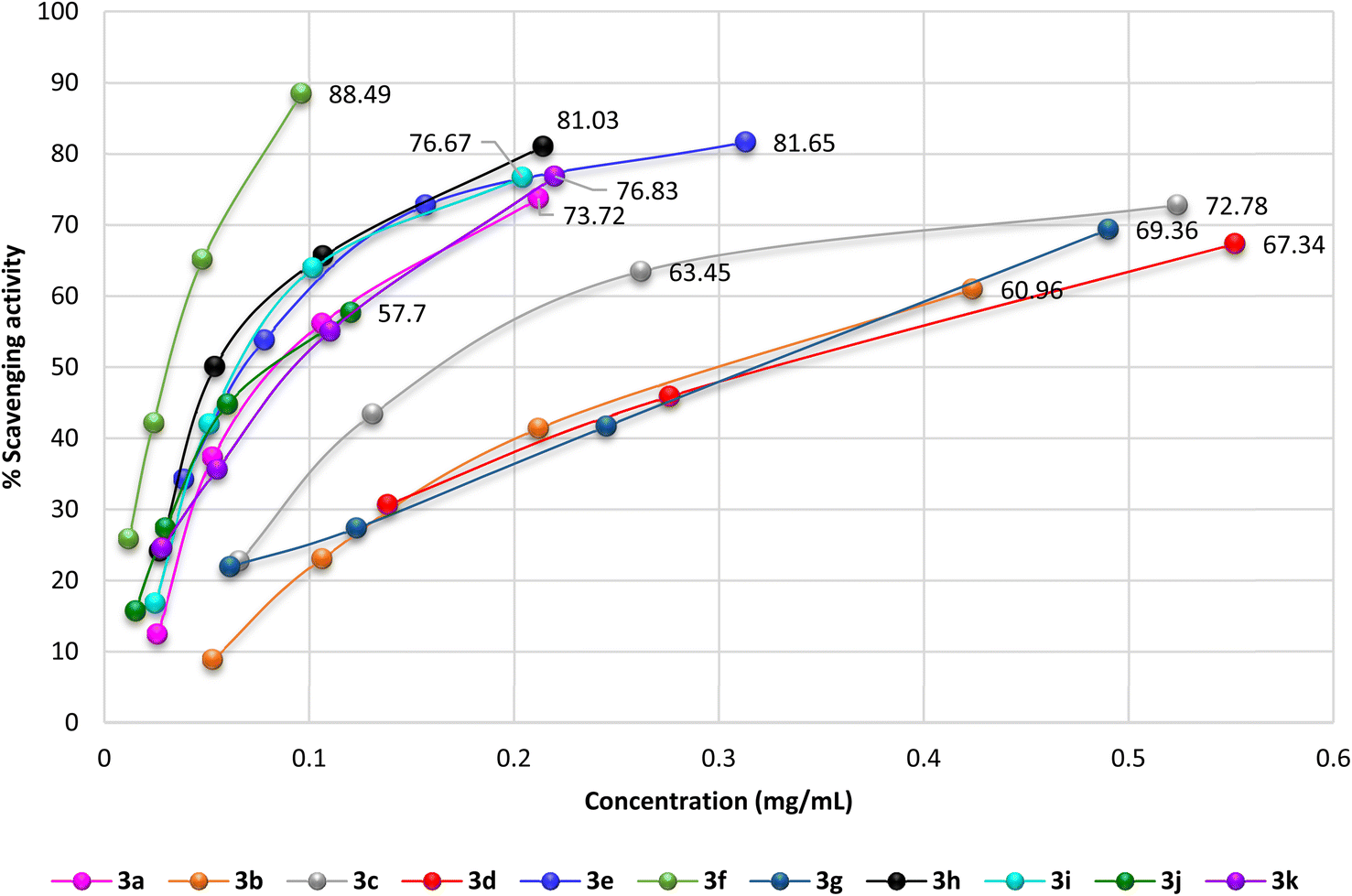 | ||
| Fig. 10 A diagram plotted % scavenging activity versus the various sample concentrations (mg mL−1) by DPPH assay. | ||
The IC50 values specified that compound 3f exhibited the most potent antioxidant activity (IC50 = 0.03 ± 0.45 mg mL−1). In addition, compounds 3h (IC50 = 0.067 ± 2.01 mg mL−1), 3e (IC50 = 0.068 ± 3.36 mg mL−1), 3i (IC50 = 0.08 ± 1.56 mg mL−1), 3j (IC50 = 0.088 ± 1.08 mg mL−1), 3k (IC50 = 0.095 ± 2.16 mg mL−1), and 3a (IC50 = 0.099 ± 1.34 mg mL−1) revealed significant activities for scavenging DPPH radicals in the reaction mixture.
On the other side, compounds 3d (IC50 = 0.362 ± 0.97 mg mL−1), 3b (IC50 = 0.304 ± 3.02 mg mL−1), 3g (IC50 = 0.284 ± 1.57 mg mL−1), and 3c (IC50 = 0.204 ± 1.52 mg mL−1) displayed moderate antioxidant activities in comparison with the results of the other tested samples. It was perceived that the introduction of substituents such as the hydroxyl group is favorable for potent activity. The SARs verified that the induction of electron-donating substituents, such as hydroxyl and dimethylamino groups, into the basic skeleton of benzopyrimido[4,5-d]azoninone are more likely to donate a hydrogen atom to DPPH˙ than compounds with electron-withdrawing substituents, such as halogen atoms. The formation of a stable free radical from the tested sample enables the interaction with radicals of DPPH˙ in the solution and hence terminates the free radical reaction. As a result, compounds with electron-donating substituents will show a greater decrease in absorbance at 517 nm than compounds with electron-withdrawing substituents. This is because the greater the decrease in absorbance, the more DPPH˙ molecules have been converted to DPPH-H, which is colorless. The following is a diagram of the mechanism of the DPPH assay:
| DPPH˙ + AOH → DPPH-H + A˙ | (1) |
where AOH is an antioxidant with an electron-donating substituent and A˙ is the radical formed when AOH donates a hydrogen atom to DPPH˙.
When a phenolic compound reacts with DPPH (DPPH assay), it can donate a hydrogen atom or electron to the DPPH molecule (Fig. 11). This reduces the DPPH molecule to a stable non-radical form, resulting in a decrease in the absorbance of the DPPH solution at 517 nm. The extent of the decrease in absorbance is proportional to the antioxidant activity of the phenolic compound. The reaction mechanism of phenolic compounds with DPPH is thought to involve a combination of hydrogen atom transfer and single electron transfer reactions. The hydrogen atom transfer mechanism involves the donation of a hydrogen atom from the phenolic compound to the DPPH molecule, resulting in the formation of a phenoxyl radical and a reduced DPPH molecule. The single electron transfer mechanism involves the transfer of an electron from the phenolic compound to the DPPH molecule, resulting in the formation of a phenolate anion and a DPPH radical anion. The relative importance of the hydrogen atom transfer and single electron transfer mechanisms in the reaction of phenolic compounds with DPPH depends on several factors such as the structure of the phenolic compound. Generally, phenolic compounds with multiple hydroxyl groups and electron-donating substituents are more likely to react with DPPH via the single electron transfer mechanism (Fig. 11).
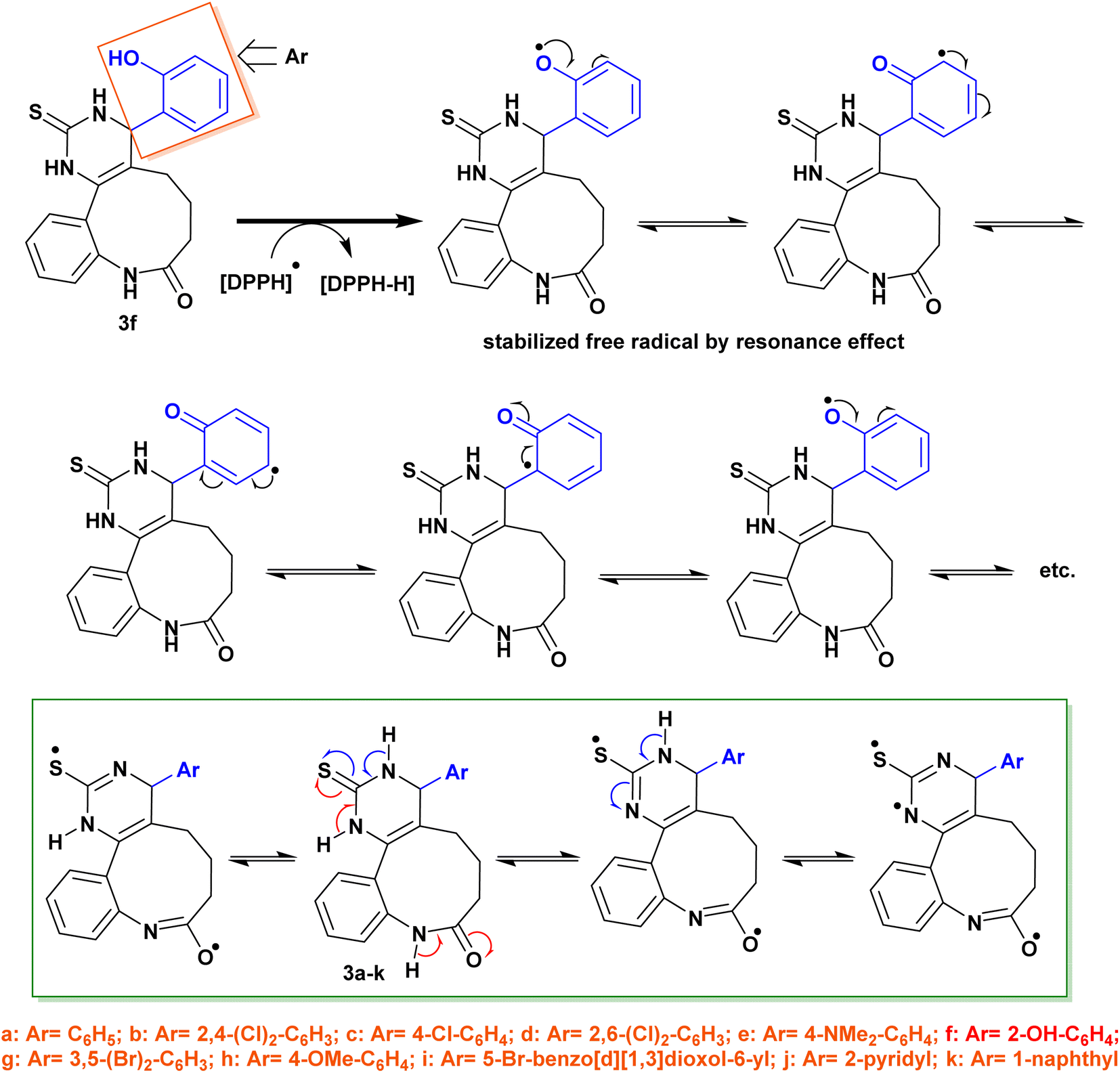 | ||
| Fig. 11 The planned reaction mechanism of phenolic compounds with DPPH. | ||
Alternatively, the antioxidant activity of benzopyrimido[4,5-d]azoninone derivatives was correspondingly evaluated by phosphomolybdate assay (Fig. 12). The results revealed that compound 3f is the most potent antioxidant agent with a total antioxidant capacity of 7894.563 ± 2.92 (mg AAE/100 g DS). Subsequently, most of the other tested compounds revealed good antioxidant activity in comparison to the ascorbic acid result, e.g. compound 3h revealed the antioxidant capacity with 7575.59 ± 2.86 (mg AAE/100 g DS), compound 3e exhibited the third order of potency with 7017.389 ± 3.38 (mg AAE/100 g DS), compound 3i displayed good antioxidant activity with 6379.444 ± 4.18 (mg AAE/100 g DS), and compound 3j shown good activity with 5422.528 ± 3.36 (mg AAE/100 g DS). Moreover, the reduced antioxidant activities were recorded by compounds 3b (1966.205 ± 3.74 mg AAE/100 g DS), 3c (2392.291 ± 1.98 mg AAE/100 g DS), 3d (1412.344 ± 3.76 mg AAE/100 g DS), and 3g (2104.67 ± 3.49 mg AAE/100 g DS) in comparison to the results of the other tested compounds and ascorbic acid (Fig. 12).
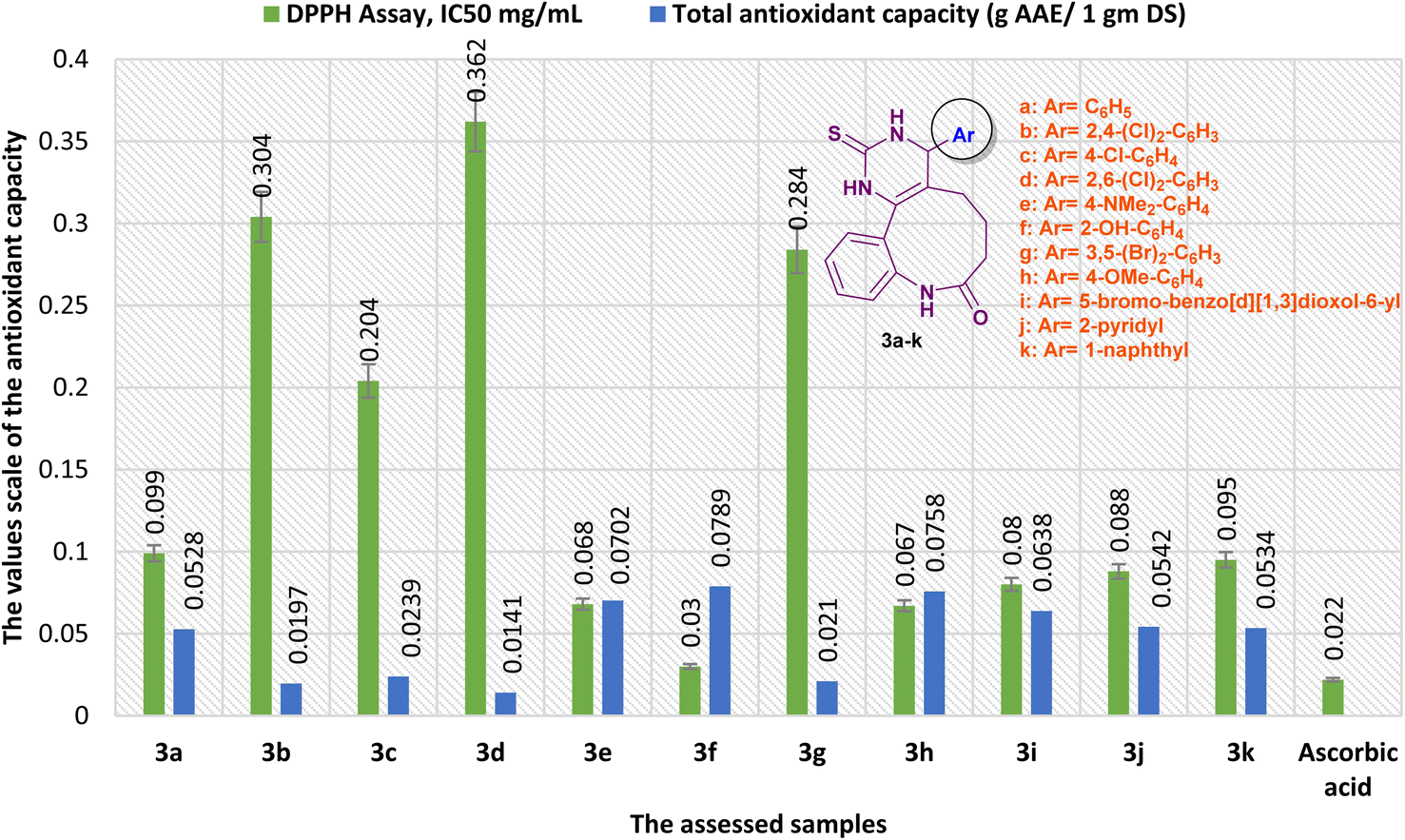 | ||
| Fig. 12 A comparison of the antioxidant results obtained by DPPH, and total antioxidant capacity assays. | ||
Our results of the antioxidant activity obtained by the phosphomolybdate assay agree with the results obtained by the DPPH free radical assay. The mechanism of action during this method depends on the interaction between the sample with phosphomolybdate ions leading to the reduction of these ions in the solution by the influence of the antioxidants, e.g. the tested samples, to form a green phosphate/MoV complex. The change in color of the solution was measured at a specific wavelength (λ = 695 nm), in which the color depth depended on the antioxidant capacity of the tested samples. In particular, the ions of (MoO4)2- were reduced to Mo(V) with the generation of a green phosphate/Mo(V) complex with phosphate ions (PO43−). The rich electron source substituents will provide a high capacity for the antioxidant sample, in which these electron donating groups are responsible for the antioxidant activity of the tested sample.
3. Conclusion
We have advanced an efficient and applied protocol for the synthesis of novel benzopyrimido[4,5-d]azoninones using Ag-TiO2 core/shell magnetic nanocatalyst. The appealing features of this procedure are the simplicity of the synthesis of the nanocatalyst from biosource, accessible materials and chemicals, ease of workup of the reaction, high reaction rate, and yields, low reaction time, respectable stability of the nanocatalyst as well as the nanocatalyst recyclability. The compounds revealed comparable and privileged antioxidant activities by DPPH and phosphomolybdate assays. Specifically, compound 3f (Ar = 2-OH-C6H4) revealed the most potent antioxidant activity by both methods, this is related to the stable free radical of the phenolic substituent that stabilized the free radicals of DPPH in the solution as well as the formed stable free radicals enabling the reduction of (MoO4)2 ions into Mo(V) with the formation of a green phosphate/Mo(V) complex with phosphate ions (PO43−). The green procedure is an attractive strategy in the synthesis of this class of compounds leading to worthy inputs to the heterocyclic chemistry.4. Experimental
4.1. Instruments
All melting points were determined on a Gallenkamp electric melting point apparatus and were uncorrected. Bruker DRX500 was used for NMR analyses, in which the measurements were performed at 400 MHz for 1H-NMR analyses and 100 MHz for 13C-NMR analyses. All samples were dissolved in CDCl3 as a solvent for NMR analyses except for compound 3d (13C-NMR: CD3COCD3), compound 3f (1H-NMR, 13C-NMR: DMSO-d6), 3g (13C-NMR: MeOD-d4), 3h (1H-NMR, 13C-NMR: MeOD-d4), and 3j (1H-NMR, 13C-NMR: DMSO-d6). Chemical shifts were recorded in ppm using the signals of the residual solvents as a reference value (CDCl3: 7.26/77.0 ppm; DMSO-d6: 2.50/39.51 ppm; MeOD-d4: 3.33/49.15 ppm).4.2. Biosynthesis of bimetallic nanocatalyst
20 g of turmeric “Curcuma longa” powder was placed in a conical flask (250 mL) and mixed with 100 mL ethanol absolute. The mixture was soaked overnight at 25 °C, filtered, and stored at 0–5 °C. In a distinctive experimental route, to prepare the bimetallic Ag-TiO2 NPs: 50 mL of the turmeric extract was added dropwise with continuous stirring at 25 °C to a solution of silver nitrate (2 mM, 25 mL) (99% Sigma-Aldrich), and titanium dioxide (2 mM) (97% Sigma-Aldrich). The pH of the solution was adjusted at 7 using a 0.1 M sodium hydroxide solution. The mixture was stirred for an additional 12 hours using a magnetic stirrer. The color change of the plant extract was noticed by continuous stirring from orange to gray-brown. Sonication of the mixture for 30 min was attained and the mixture was transferred into an autoclave, sealed, and heated up to 70 °C for 24 h. The flask was next cooled using tap water instantly and the mixture was filtered with Whatman no. 1 filter paper. The obtained Ag-TiO2 NPs nanocatalyst was washed with distilled water and dried at 100 °C for 3 h. The nanocatalyst was treated in an electrical furnace at 400 °C for 9 h to remove the residual organic components and a gray-brown powder was acquired.47–494.3. General procedure for the synthesis of the target compounds
To a solution of compound 2 (0.001 mmol), thiourea (0.001 mmol), and aryl aldehydes (0.001 mmol), in ethanol (10 mL), the nanocatalyst “Ag-TiO2 bmNPs” (25.0 mg) was added. The reaction mixture was refluxed for 3–7 h and monitored by thin-layer chromatography. The reaction mixture was left to cool to room temperature (approximately 30 °C) after the product formed and no spots were observed on the TLC paper. The mixture was poured into ice water with continuous stirring. The extraction process was continued three times using CH2Cl2, and the extracted component was dried over anhydrous Na2SO4 under a vacuum. The residue of the solvent was evaporated and refined by recrystallization in ethanol.O, amide), 1035 (CS). 1H-NMR (CDCl3, 300 MHz): 1.91–1.99 (m, 2H, CH2), 2.22–2.26 (m, 2H, CH2), 2.56–2.65 (m, 2H, CH2), 5.35 (s, 1H, CH-pyrimidine), 6.65–7.57 (m, 9H, Ar–H), 7.84 (s, 1H, NH), 7.87 (s, 1H, NH), 7.90 (s, 1H, NH) ppm; 13C-NMR (CDCl3, 75 MHz): δ 175.53 (1C, CS), 156.17 (1C, CO), 140.38–120.05 (13C, 12-Ar-C, C6-pyrimidine), 115.33 (1C, C5-pyrimidine), 77.0 (1C, C4-pyrimidine), 32.94 (1C, CH2), 27.7 (1C, CH2), 23.74 (1C, CH2) ppm. MS (m/z, %): 349.58 (M+, 19.53). Calcd for C20H19N3OS (349.45): C, 68.74; H, 5.48; N, 12.02%. Found: C, 68.87; H, 5.54; N, 12.11%.O, amide), 1172 (CS). 1H-NMR (CDCl3, 300 MHz): δ 1.84 (m, 2H, CH2, J = 1.98 Hz), 2.23 (m, 2H, CH2, J = 1.99 Hz), 2.55 (m, 2H, CH2, J = 1.98 Hz), 5.29 (s, 1H, CH-pyrimidine), 7.09–7.70 (m, 7H, Ar–H), 8.48, 8.65 (s, 3H, 3NH) ppm; 13C-NMR (CDCl3, 75 MHz): δ 198.95 (1C, CS), 175.56 (1C, CO), 142.10–126.98 (13C, 12-Ar-C, C6-pyrimidine), 126.58 (1C, C5-pyrimidine), 68.1 (1C, C4-pyrimidine), 31.93 (1C, CH2), 26.66 (1C, CH2), 22.74 (1C, CH2) ppm. MS (m/z, %): 418.69 (M+, 25.61). Calcd for C20H17Cl2N3OS (418.34): C, 57.42; H, 4.10; N, 10.04%. Found: C, 57.53; H, 4.17; N, 10.18%.O, amide), 1191 (CS). 1H-NMR (CDCl3, 300 MHz): δ 1.67 (m, 2H, CH2), 2.08–2.20 (m, 2H, CH2), 2.48 (m, 2H, CH2), 5.30 (s, 1H, CH-pyrimidine), 6.61–7.52 (m, 8H, Ar–H), 7.85 (s, 1H, NH), 7.87 (s, 1H, NH), 7.98 (s, 1H, NH) ppm; 13C-NMR (CDCl3, 75 MHz): δ 198.84 (1C, CS), 175.61 (1C, CO), 141.02–128.35 (13C, 12-Ar-C, C6-pyrimidine), 128.29 (1C, C5-pyrimidine), 32.17 (1C, C4-pyrimidine), 31.81 (1C, CH2), 30.90 (1C, CH2), 25.59 (1C, CH2) ppm. MS (m/z, %): 383.19 (M+, 17.86). Calcd for C20H18ClN3OS (383.89): C, 62.57; H, 4.73; N, 10.95%. Found: C, 62.69; H, 4.68; N, 10.87%.O, amide), 1173 (CS). 1H-NMR (CDCl3, 300 MHz): δ 1.48–1.77 (m, 2H, CH2, J = 1.55 Hz), 1.86–1.93 (m, 2H, CH2, J = 2.18 Hz), 2.14–2.46 (m, 2H, CH2, J = 2.06 Hz), 4.45 (s, 1H, CH-pyrimidine), 6.60–7.75 (m, 7H, Ar–H), 8.05 (s, 1H, NH), 8.35 (s, 1H, NH), 8.77 (s, 1H, NH) ppm; 13C-NMR (CD3COCD3, 75 MHz): δ 198.17 (1C, CS), 175.45 (1C, CO), 156.47–119.54 (13C, 12-Ar-C, C6-pyrimidine), 115.30 (1C, C5-pyrimidine), 73.76 (1C, C4-pyrimidine), 32.76 (1C, CH2), 30.77 (1C, CH2), 20.27 (1C, CH2) ppm. MS (m/z, %): 418.77 (M+, 12.80). Calcd for C20H17Cl2N3OS (418.34): C, 57.42; H, 4.10; N, 10.04%. Found: C, 57.54; H, 4.22; N, 10.13%.O, amide), 1182 (CS). 1H-NMR (CDCl3, 300 MHz): δ 2.05–2.07 (m, 2H, CH2, J = 2.09 Hz), 2.24–2.27 (m, 2H, CH2, J = 2.0 Hz), 2.89 (m, 2H, CH2, J = 2.01 Hz), 3.04 (s, 6H, 2CH3), 3.10 (s, 1H, CH-pyrimidine), 6.47–7.76 (m, 8H, Ar–H), 9.75 (s, 3H, 3NH) ppm; 13C-NMR (CDCl3, 75 MHz): δ 175.53 (1C, CS), 151.14 (1C, CO), 141.24–11.65 (13C, 12-Ar-C, C6-pyrimidine), 110.98 (1C, C5-pyrimidine), 77.32 (1C, C4-pyrimidine), 40.19 (2C, 2CH3), 40.02 (1C, CH2), 28.72 (1C, CH2), 24.72 (1C, CH2) ppm. MS (m/z, %): 392.27 (M+, 16.59). Calcd for C22H24N4OS (392.52): C, 67.32; H, 6.16; N, 14.27%. Found: C, 67.38; H, 6.26; N, 14.35%.O, amide), 1186 (CS). 1H-NMR (DMSO-d6, 300 MHz): δ 1.39–1.44 (m, 2H, CH2, J = 1.0 Hz), 1.63–1.84 (m, 2H, CH2, J = 4.05 Hz), 2.40–2.43 (m, 2H, CH2, J = 0.98 Hz), 5.76 (s, 1H, CH-pyrimidine), 6.56–7.99 (m, 8H, Ar–H), 8.23 (s, 1H, OH), 8.65 (s, 2H, 2NH), 8.78 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6, 75 MHz): δ 173.09 (1C, CS), 150.42 (1C, CO), 141.98–116.07 (13C, 12-Ar-C, C6-pyrimidine), 114.39 (1C, C5-pyrimidine), 39.51 (1C, C4-pyrimidine), 32.69 (1C, CH2), 29.47 (1C, CH2), 26.16 (1C, CH2) ppm. MS (m/z, %): 365.43 (M+, 18.79). Calcd for C20H19N3O2S (365.45): C, 65.73; H, 5.24; N, 11.50%. Found: C, 65.82; H, 5.36; N, 11.57%.O, amide), 1198 (CS). 1H-NMR (CDCl3, 300 MHz): δ 1.83–2.05 (m, 2H, CH2, J = 2.04 Hz), 2.17–2.28 (m, 2H, CH2, J = 2.0 Hz), 2.40–2.69 (m, 2H, CH2, J = 1.97 Hz), 5.30 (s, 1H, CH-pyrimidine), 6.62–7.63 (m, 7H, Ar–H), 7.85 (s, 1H, NH), 7.99 (s, 1H, NH), 8.12 (s, 1H, NH) ppm; 13C-NMR (MeOD-d4, 75 MHz): δ 175.68 (2C, CS, CO), 141.85–123.08 (13C, 12-Ar-C, C6-pyrimidine), 122.91 (1C, C5-pyrimidine), 77.42 (1C, C4-pyrimidine), 32.37 (1C, CH2), 27.22 (1C, CH2), 22.95 (1C, CH2) ppm. MS (m/z, %): 507.18 (M+, 16.84). Calcd for C20H17Br2N3OS (507.24): C, 47.36; H, 3.38; N, 8.28%. Found: C, 47.43; H, 3.43; N, 8.39%.O, amide), 1184 (CS). 1H-NMR (MeOD-d4, 300 MHz): δ 1.62–1.66 (m, 2H, CH2, J = 3.96 Hz), 2.21–2.23 (m, 2H, CH2, J = 2.04 Hz), 2.71–2.74 (m, 2H, CH2, J = 0.98 Hz), 3.33 (s, 3H, OCH3), 4.78 (s, 1H, CH-pyrimidine), 6.62–7.35 (m, 8H, Ar–H), 7.66 (s, 1H, NH), 7.68 (s, 1H, NH), 7.69 (s, 1H, NH) ppm; 13C-NMR (MeOD-d4, 75 MHz): δ 198.37 (1C, CS), 177.35 (1C, CO), 160.94–116.91 (13C, 12-Ar-C, C6-pyrimidine), 115.17 (1C, C5-pyrimidine), 61.49 (1C, C4-pyrimidine), 55.85 (1C, OCH3), 35.20 (1C, CH2), 28.68 (1C, CH2), 23.69 (1C, CH2) ppm. MS (m/z, %): 379.88 (M+, 21.17). Calcd for C21H21N3O2S (379.48): C, 66.47; H, 5.58; N, 11.07%. Found: C, 66.52; H, 5.49; N, 11.16%.O, amide), 1187 (CS). 1H-NMR (CDCl3, 300 MHz): δ 1.74–1.80 (m, 2H, CH2, J = 1.11 Hz), 2.06–2.32 (m, 2H, CH2, J = 3.16 Hz), 2.66–2.86 (m, 2H, CH2, J = 2.07 Hz), 5.30 (s, 1H, CH-pyrimidine), 6.55 (s, 2H, –O–CH2–O–), 6.88–7.53 (m, 6H, Ar–H), 8.22 (s, 1H, NH), 8.24 (s, 2H, 2NH) ppm; 13C-NMR (CDCl3, 75 MHz): δ 200.71 (1C, CS), 169.13 (1C, CO), 151.99–123.37 (13C, 12-Ar-C, C6-pyrimidine), 118.03 (1C, C5-pyrimidine), 112.03 (1C, –O–CH2–O), 70.81 (1C, C4-pyrimidine), 41.50 (1C, CH2), 30.97 (1C, CH2), 27.55 (1C, CH2) ppm. MS (m/z, %): 472.16 (M+, 11.64). Calcd for C21H18BrN3O3S (472.36): C, 53.40; H, 3.84; N, 8.90%. Found: C, 53.57; H, 3.92; N, 8.98%.O, amide), 1182 (CS). 1H-NMR (DMSO-d6, 300 MHz): δ 1.31–1.39 (m, 2H, CH2, J = 2.66 Hz), 2.05–2.09 (m, 2H, CH2, J = 2.40 Hz), 2.23–2.27 (m, 2H, CH2, J = 2.49 Hz), 4.06 (s, 1H, CH-pyrimidine), 6.53–6.83 (m, 8H, Ar–H), 7.43 (s, 2H, 2NH), 7.45 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6, 75 MHz): δ 167.71 (2C, CS, CO), 148.30 (1C, C2′-pyridine), 132.07–112.36 (11C, 10-Ar-C, C6-pyrimidine), 109.64 (1C, C5-pyrimidine), 40.14 (1C, C4-pyrimidine), 23.64 (1C, CH2), 19.10 (1C, CH2), 13.48 (1C, CH2) ppm. MS (m/z, %): 350.71 (M+, 15.43). Calcd for C19H18N4OS (350.44): C, 65.12; H, 5.18; N, 15.99%. Found: C, 65.25; H, 5.26; N, 16.04%.O, amide), 1187 (CS). 1H-NMR (CDCl3, 300 MHz): δ 1.26–1.83 (m, 2H, CH2), 2.02–2.04 (m, 2H, CH2), 2.40–2.44 (m, 2H, CH2), 5.30 (s, 1H, CH-pyrimidine), 6.47–7.68 (m, 11H, Ar–H), 7.70 (s, 1H, NH), 7.78 (s, 1H, NH), 7.80 (s, 1H, NH) ppm; 13C-NMR (CDCl3, 75 MHz): δ 177.15 (1C, CS), 171.21 (1C, CO), 145.45–125.29 (17C, 12-Ar-C, C6-pyrimidine), 122.56 (1C, C5-pyrimidine), 76.68 (1C, C4-pyrimidine), 33.60 (1C, CH2), 25.22 (1C, CH2), 21.04 (1C, CH2) ppm. MS (m/z, %): 399.17 (M+, 23.68). Calcd for C24H21N3OS (399.51): C, 72.15; H, 5.30; N, 10.52%. Found: C, 72.23; H, 5.39; N, 10.64%.4.4. Antioxidant procedures
The methods of DPPH and phosphomolybedate assays were applied as previously reported50–53 and outlined in the ESI.†Conflicts of interest
The authors assert no conflict of interest.Acknowledgements
This project was supported by the Deanship of Scientific Research at Prince Sattan Bin Abdulaziz University under the project (PSAU-2022/01/22235).References
- P. Kurhade, S. Kodape and R. Choudhury, Chem. Pap., 2021, 75(10), 5187–5222 CrossRef CAS.
- N. M. Noah and P. M. Ndangili, Sens. Int., 2022, 3, 100166 CrossRef.
- X. Qu, J. Brame, Q. Li and P. J. Alvarez, Acc. Chem. Res., 2013, 46(3), 834–843 CrossRef CAS PubMed.
- M. I. Sohail, A. A. Waris, M. A. Ayub, M. Usman, M. Z. ur Rehman, M. Sabir and T. Faiz, Environmental application of nanomaterials: A promise to sustainable future, Compr. Anal. Chem., 2019, 87, 1–54 CAS.
- N. B. Singh, P. Jain, A. De and R. Tomar, Curr. Pharm. Biotechnol., 2021, 22(13), 1705–1747 CAS.
- I. Ijaz, E. Gilani, A. Nazir and A. Bukhari, Green Chem. Lett. Rev., 2020, 13(3), 223–245 CrossRef CAS.
- I. M. Chung, I. Park, K. Seung-Hyun, M. Thiruvengadam and G. Rajakumar, Nanoscale Res. Lett., 2016, 11, 1–14 CrossRef CAS PubMed.
- H. Bahrulolum, S. Nooraei, N. Javanshir, H. Tarrahimofrad, V. S. Mirbagheri, A. J. Easton and G. Ahmadian, J. Nanobiotechnol., 2021, 19(1), 1–26 CrossRef PubMed.
- R. R. Nasaruddin, T. Chen, Q. Yao, S. Zang and J. Xie, Coord. Chem. Rev., 2021, 426, 213540 CrossRef CAS.
- M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41(4), 1415–1427 RSC.
- S. Amin, M. Sher, A. Ali, M. F. Rehman, A. Hayat, M. Ikram, A. Abbas and H. M. Amin, Environ. Nanotechnol., 2022, 18, 100735 CAS.
- A. Hussain, F. Attique, S. A. R. Naqvi, A. Ali, M. Ibrahim, H. Hussain, F. Zafar, R. S. Iqbal, M. A. Ayub, M. A. Assiri and M. Imran, ACS Omega, 2022, 8(1), 1088–1096 CrossRef PubMed.
- Cellulose fibers: bio-and nano-polymer composites: green chemistry and technology, ed. S. Kalia, B. S. Kaith and I. Kaur, Springer Science & Business Media, 2011 Search PubMed.
- M. Khalaj, M. Kamali, M. E. V. Costa and I. Capela, J. Cleaner Prod., 2020, 267, 122036 CrossRef.
- P. A. Evans and A. B. Holmes, Tetrahedron, 1991, 47, 9131 CrossRef CAS.
- F. Bohland, I. Erlin, L. Platte, M. Schröder, D. Schollmeyer and U. Nubbemeyer, Eur. J. Org Chem., 2014, 2014(28), 6272–6284 CrossRef CAS.
- N. P. Atmuri and W. D. Lubell, J. Org. Chem., 2019, 85(3), 1340–1351 CrossRef PubMed.
- K. Sun, D. Zhao, Q. Li, S. Ni, G. Zheng and Q. Zhang, Sci. China: Chem., 2023, 1–8 Search PubMed.
- B. Chakraborty and U. Jana, Org. Biomol. Chem., 2021, 19(48), 10549–10553 RSC.
- E. M. Afsah, A. A. Fadda, S. Bondock and M. M. Hammouda, Z. Naturforsch., 2009, 64, 415–422 CrossRef CAS.
- M. M. Hammouda, H. M. Metwally, A. Fekri and J. Van der Eycken, Polycyclic Aromat. Compd., 2021, 41(6), 1223–1240 CrossRef CAS.
- E. M. Afsah, A. A. Fadda, S. Bondock and M. M. Hammouda, Z. Naturforsch., 2015, 70(6), 385–391 CrossRef CAS.
- H. A. Abd El-Aal, A. A. Khalaf and A. M. El-Khawaga, Aust. J. Chem., 2014, 68(3), 404–415 CrossRef.
- H. A. Abd El-Aal and A. A. Khalaf, Aust. J. Chem., 2015, 69(6), 652–661 CrossRef.
- A. Sudau, W. Münch, J. W. Bats and U. Nubbemeyer, Chem.–Eur. J., 2001, 7(3), 611–621 CrossRef CAS PubMed.
- U. Nubbemeyer, Eur. J. Org Chem., 2001, 2001(10), 1801–1816 CrossRef.
- A. Levai, J. Heterocycl. Chem., 2004, 41(3), 299 CrossRef CAS.
- A. Sudau, W. Münch, U. Nubbemeyer and J. W. Bats, J. Org. Chem., 2000, 65, 1710–1720 CrossRef CAS PubMed.
- J. Jung, N. Choi and Y. Suh, Arch. Pharm. Res., 2004, 27(10), 985–989 CrossRef CAS PubMed.
- H. Yun, J. Kim, J. Sim, S. Lee, Y. T. Han, D. J. Chang, D. D. Kim and Y. G. Suh, J. Org. Chem., 2012, 77(12), 5389–5393 CrossRef CAS PubMed.
- M. M. Hammouda, A. A. Elmaaty, M. S. Nafie, M. Abdel-Motaal, N. S. Mohamed, M. A. Tantawy, A. Belal, R. Alnajjar, W. M. Eldehna and A. A. Al-Karmalawy, Bioorg. Chem., 2022, 127, 105995 CrossRef CAS PubMed.
- D. Girija, H. B. Naik, B. V. Kumar, C. N. Sudhamani and K. N. Harish, Arab. J. Chem., 2019, 12(3), 420–428 CrossRef CAS.
- C. G. Lima, S. Silva, R. H. Goncalves, E. R. Leite, R. S. Schwab, A. G. Correa and M. W. Paixao, ChemCatChem, 2014, 6(12), 3455–3463 CrossRef CAS.
- J. Mondal, T. Sen and A. Bhaumik, Dalton Trans., 2012, 41(20), 6173–6181 RSC.
- H. G. Alvim, D. L. Pinheiro, V. H. Carvalho-Silva, M. Fioramonte, F. C. Gozzo, W. A. da Silva, G. W. Amarante and B. A. Neto, J. Org. Chem., 2018, 83(19), 12143–12153 CrossRef CAS PubMed.
- M. Yadav, M. Dutta, P. Tanwar, R. Jain, A. Srivastava and R. K. Sharma, Curr. Microwave Chem., 2021, 8(2), 96–116 CrossRef CAS.
- J. Safari and Z. Zarnegar, RSC Adv., 2013, 3(39), 17962–17967 RSC.
- P. K. Sudeep, K. Takechi and P. V. Kamat, J. Phys. Chem. C, 2007, 111(1), 488–494 CrossRef CAS.
- D. Hong, L. M. Lyu, K. Koga, Y. Shimoyama and Y. Kon, ACS Sustain. Chem. Eng., 2019, 7(23), 18955–18964 CrossRef CAS.
- H. Y. Chuang and D. H. Chen, Nanotechnology, 2009, 20(10), 105704 CrossRef PubMed.
- S. F. Chin, S. C. Pang and F. E. I. Dom, Mater. Lett., 2011, 65(17–18), 2673–2675 CrossRef CAS.
- J. Y. Song and B. S. Kim, Korean J. Chem. Eng., 2008, 25, 808–811 CrossRef CAS.
- Y. Dai, Y. Wang, B. Liu and Y. Yang, Small, 2015, 11(3), 268–289 CrossRef CAS PubMed.
- L. J. Dolby and D. L. Booth, J. Am. Chem. Soc., 1966, 88(5), 1049 CrossRef CAS.
- C. Xu, J. K. Bartley, D. I. Enache, D. W. Knight, M. Lunn, M. Lok and G. J. Hutchings, Tetrahedron Lett., 2008, 49, 2454–2456 CrossRef CAS.
- J. Kumar, N. Kumar, N. Sati and P. K. Hota, New J. Chem., 2020, 44(21), 8960–8970 RSC.
- K. M. Elattar, A. A. Ghoniem, F. O. Al-Otibi, M. S. El-Hersh, Y. A. Helmy and W. E. I. A. Saber, Appl. Sci., 2023, 13(18), 10110 CrossRef CAS.
- M. Maham, M. Nasrollahzadeh, S. M. Sajadi and M. Nekoei, J. Colloid Interface Sci., 2017, 497, 33–42 CrossRef CAS PubMed.
- M. Khashaei, L. Kafi-Ahmadi, S. Khademinia, A. Poursattar Marjani and E. Nozad, Sci. Rep., 2022, 12(1), 8585 CrossRef CAS PubMed.
- D. D. Kitts, A. N. Wijewickreme and C. Hu, Molec. Cellular Biochem., 2000, 203, 1–10 CrossRef CAS PubMed.
- I. Parejo, C. Codina, C. Petrakis and P. Kefalas, J. Pharmacol. Toxicol. Methods, 2000, 44, 507–512 CrossRef CAS PubMed.
- P. Prieto, M. Pineda and M. Aguilar, Anal. Biochem., 1999, 269, 337–341 CrossRef CAS PubMed.
- N. Belyagoubi-Benhammou, L. Belyagoubi, A. Gismondi, G. Di Marco, A. Canini and F. Atik Bekkara, Med. Chem. Res., 2019, 28, 754–767 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reagents & instruments, procedures & results of antioxidant activity, FT-IR charts & XRD data of the nanocatalyst, structures of the products, and charts of the spectroscopic analyses. See DOI: https://doi.org/10.1039/d3ra06404b |
| This journal is © The Royal Society of Chemistry 2023 |