
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time in situ monitoring using visible spectrophotometry as a tool for probing electrochemical advanced oxidation processes for dye decolorisation†
Chelsea M. Schroeder ,
Taylor M. Koehler,
Kristiane K. Ohlhorst and
Nicholas E. Leadbeater*
,
Taylor M. Koehler,
Kristiane K. Ohlhorst and
Nicholas E. Leadbeater*
Department of Chemistry, University of Connecticut, 55 North Eagleville Road, Storrs, Connecticut 06269, USA. E-mail: nicholas.leadbeater@uconn.edu
First published on 16th November 2023
Abstract
An apparatus for real-time in situ monitoring of electrochemical processes using UV-visible spectrophotometry has been used to optimise the electrochemically-activated persulfate decolorisation of Acid Orange 7. The impacts of varying electrode composition, current density, persulfate loading, and stirring speed on the rate of decolorisation have been probed. Decolorisation through this activated persulfate approach was compared to that using anodic oxidation for nine dyes; three from each of the azo, triarylmethane, and xanthene families. The core structure and presence of functional groups have a significant impact on the rate of decolorisation. Azo and xanthene dyes decolorise faster than triarylmethane dyes, while electron-withdrawing groups and halogens are especially detrimental to the rate of decolorisation. Electrochemically-activated persulfate resulted in faster decolorization than anodic oxidation for almost every dye, an effect that was enhanced with the electron-deficient substrates. This type of systematic structural comparison study is essential for designing electrochemical degradation procedures for the remediation of real wastewater.
Introduction
Despite increased environmental awareness of the toxicity of organic pollutants in wastewater, the rapid growth of industrial processes with minimal regulation often allows these pollutants to be released in significant quantities. A major source of organic pollutants is synthetic dyes. Over 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of synthetic dyes with wide-ranging structures are produced every year.1 These dyes are used in a range of manufactured goods, particularly those from the textile and food industries. Release of these dyes into the water stream severely endangers the health of both aquatic life and humans. As a result, water remediation is an essential area of study.1–4
000 tons of synthetic dyes with wide-ranging structures are produced every year.1 These dyes are used in a range of manufactured goods, particularly those from the textile and food industries. Release of these dyes into the water stream severely endangers the health of both aquatic life and humans. As a result, water remediation is an essential area of study.1–4
Azo compounds are the most common type of synthetic dye. They contain a (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) functionality in the chromophore of the molecule (Fig. 1). Azo dyes are ideal for nearly every industry because they are inexpensive, easy to use, and, with the broad range that are available, can be any color. However, they are widely considered to be mutagenic.1,2,4,5 Two other common dye types are triarylmethanes and xanthenes (Fig. 1). Triarylmethane dyes have three aromatic groups bonded to one central carbon. Despite their toxic, corrosive, and mutagenic properties, they are popular for their bright blue, green, and red colors, and are also used as antimicrobial agents. These dyes are most dangerous when protonated because they can pass through lipid membranes and cause cell death.2,4,5 Xanthene dyes have a dibenzopyran core that is usually densely functionalised. They have wide utility as textile dyes, cosmetics, printer inks, paints, medical diagnosis tools, and photocatalysts. However, like many dyes, their hazards are not well understood.2,5,6
N–) functionality in the chromophore of the molecule (Fig. 1). Azo dyes are ideal for nearly every industry because they are inexpensive, easy to use, and, with the broad range that are available, can be any color. However, they are widely considered to be mutagenic.1,2,4,5 Two other common dye types are triarylmethanes and xanthenes (Fig. 1). Triarylmethane dyes have three aromatic groups bonded to one central carbon. Despite their toxic, corrosive, and mutagenic properties, they are popular for their bright blue, green, and red colors, and are also used as antimicrobial agents. These dyes are most dangerous when protonated because they can pass through lipid membranes and cause cell death.2,4,5 Xanthene dyes have a dibenzopyran core that is usually densely functionalised. They have wide utility as textile dyes, cosmetics, printer inks, paints, medical diagnosis tools, and photocatalysts. However, like many dyes, their hazards are not well understood.2,5,6
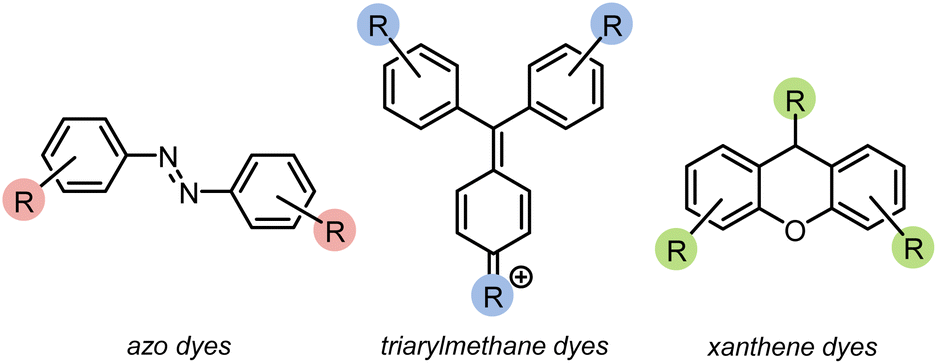 | ||
| Fig. 1 Structural motifs for three classes of common dyes. | ||
There is much debate regarding the best approach for dye wastewater treatment, but electrochemically-mediated strategies have shown particular promise. Two examples are anodic oxidation (AO) and electrochemically activated persulfate oxidation (EAP). They are distinguished by their efficient decolorisation, minimal use of chemical additives, broad applicability to all classes of pollutants, and ease of use.1,3,7–12 In AO, organic pollutants can be removed from water by two different processes: direct and indirect oxidation. In the case of direct oxidation, contaminants are absorbed and oxidised through an electron-transfer mechanism directly on the anode surface. Meanwhile, highly oxidising (1.9–2.7 V vs. standard hydrogen electrode) but short-lived (1 μs) hydroxyl radicals are generated at the anode and also contribute to the mineralisation of contaminants (Fig. 2).8,9,13,14 In EAP, the oxidising power of hydroxyl radicals is supplemented by longer-lived (30–40 μs) sulfate radicals. Sodium persulfate, a stable, green, inexpensive, and readily available peroxide salt, is often used to generate these radicals.3,7,9 Persulfate can be split into a sulfate radical and sulfate anion at the cathode. The sulfate anion can either be converted to a radical or combined with another sulfate anion at the anode to reform a persulfate molecule.3,4,7,9,12 Persulfate is also adsorbed onto the anode, activating the peroxide bond. This transition state structure effectively oxidises pollutants in a process termed nonradical oxidation (Fig. 3). However, the increased efficiency of EAP over AO comes with the requirement for more complicated and expensive anode materials like boron-doped diamond (BDD), lead oxide, or Ti/Pt.1,7,9,15
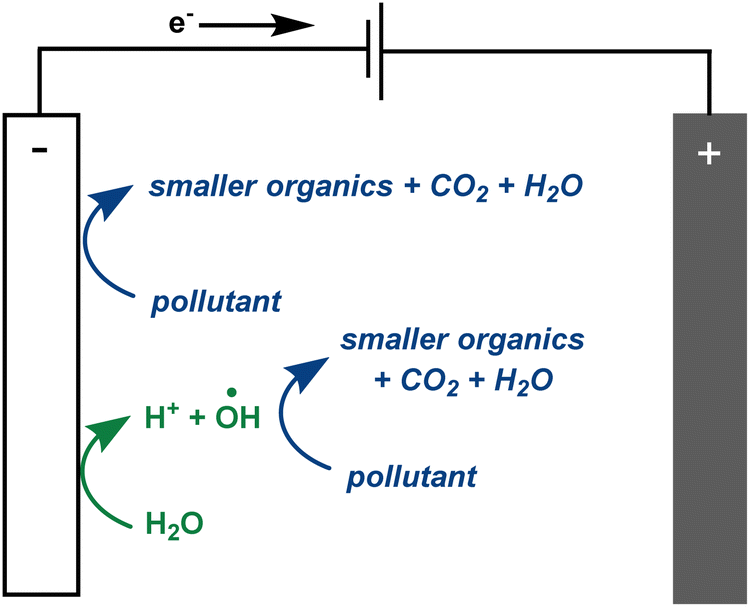 | ||
| Fig. 2 Anodic oxidation of pollutants. | ||
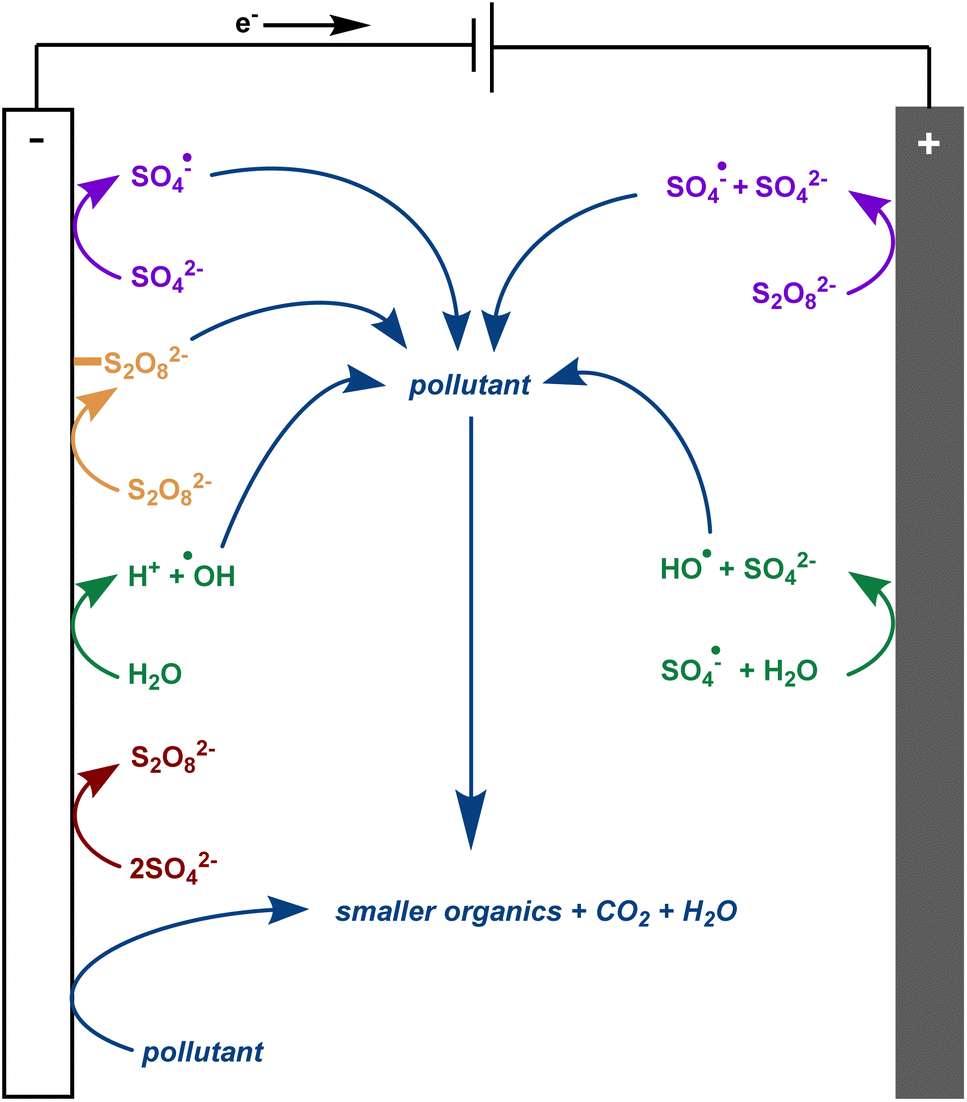 | ||
| Fig. 3 Electrochemically activated persulfate oxidation of pollutants. | ||
The most common detection method for dye decolorisation is UV-visible (UV-vis) spectrophotometry. Monitoring the rate of decolorisation in this manner is time-consuming, so thorough investigations of important parameters like stirring speed, electrolyte, electrodes, and constant solution volume are often neglected in favor of fast data collection. To overcome this challenge, our group has developed an in situ real-time monitoring apparatus which we have used to optimise the AO of Acid Orange 7, a common azo hair dye. Our approach, shown in Fig. 4, features a 3D-printed cuvette sleeve that blocks out extraneous light while aligning an LED with a collimating lens on opposite sides of the reaction vial. An optical fiber connects the apparatus to a UV-vis spectrophotometer, which can record data on the millisecond timescale.14 The closest technology to ours is a system comprising of a flow-through cuvette where measurements are taken every 2–5 min.16
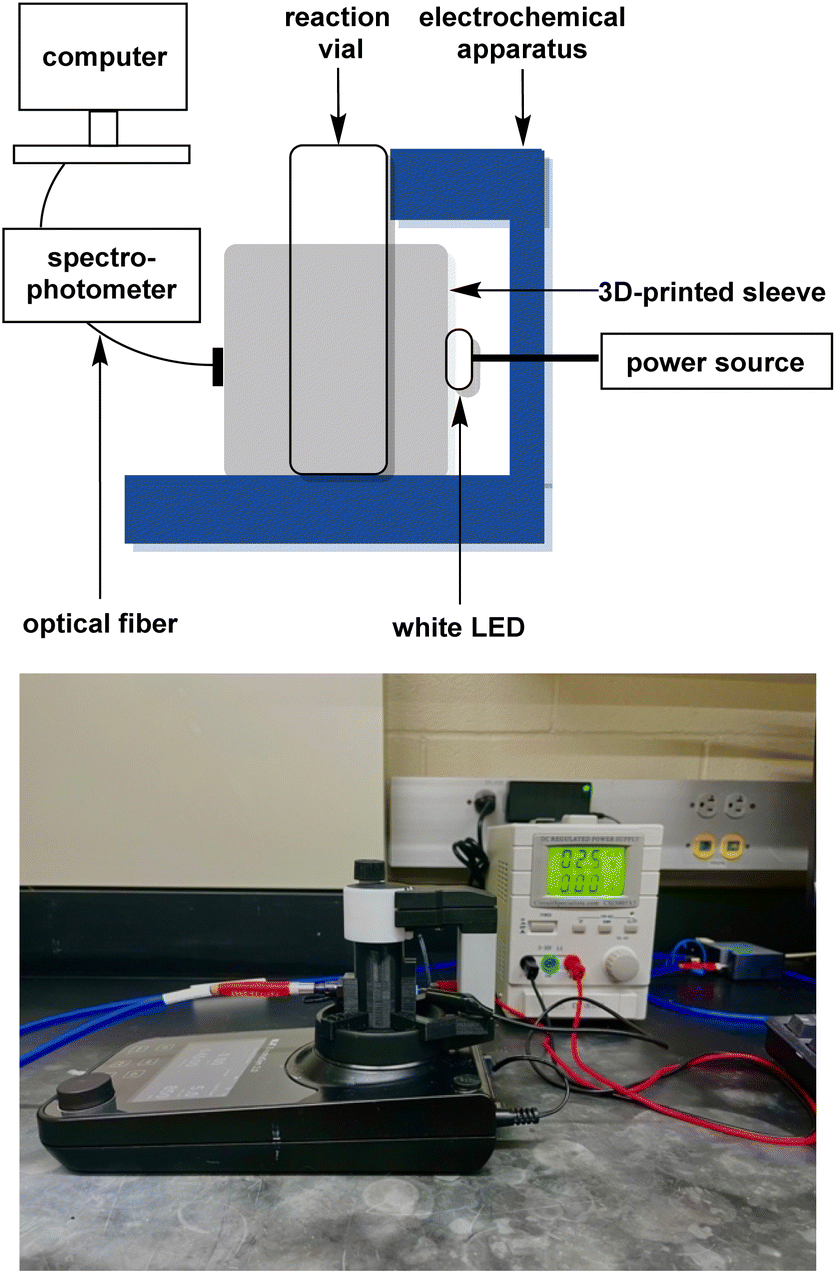 | ||
| Fig. 4 Top-schematic of the apparatus designed for in situ real-time monitoring of electrochemical processes. Bottom-apparatus for real-time in situ monitoring of electrochemical reactions using a UV-visible spectrophotometer. | ||
In the field of wastewater remediation, it would be of great value to be able to compare results for dye persistence based on the structural characteristics of the dye.3,7,9,11,17,18 However, this is not a trivial exercise. The disparate nature of the scientific literature on dye degradation means that it is a challenge to make meaningful comparisons between different reports. It is rare for researchers to assess the degradation of more than one dye, and when multiple dyes are studied, their structures are typically too dissimilar to be able to draw any meaningful comparisons.7,16,17,19–26 To address this, we have employed our continuous-monitoring apparatus to study the AO and EAP decolorisation of nine electronically-diverse dyes from the common azo, triarylmethane, and xanthene structural families, and we present our results here. We have probed the impact of dye structure and oxidant identity on the rate of decolorisation. To the best of our knowledge, this is the first systematic study to compare both a hydroxyl radical-based (AO) and a persulfate-based (EAP) approach across a range of structurally-diverse dyes. This type of study is essential for designing electrochemical degradation procedures for the remediation of real wastewater.
Results and discussion
Optimisation of electrochemically activated persulfate decolorisation
We previously used our in situ real-time monitoring interface for probing the AO driven decolorisation of Acid Orange 7 (AO7), an azo dye.14 As our first step in this current study, we opted to examine the EAP mediated decolorisation of the same dye. To optimise the EAP approach, we decided to use our AO conditions of 50 ppm Acid Orange 7 in water (8 mL), graphite electrodes, sodium persulfate (14 mM), applied current (5 mA), and 1000 rpm stirring as a starting point. Our objective was to probe the impact of varying electrode composition, the quantity of sodium persulfate used, applied current density, and stirring speed on the rate of decolorisation.Boron-doped diamond (BDD) is widely considered one of the most efficient electrode materials for advanced oxidation processes (AOPs) because of its high oxygen evolution potential and its ability to adsorb pollutants. We decided to probe this in detail and opted to screen three combinations of BDD and graphite as the electrodes for the EAP mediated decolorisation of AO7. Reactions were monitored as a function of time and graphed as the percentage of dye remaining. As seen in Fig. 5, decolorisation using a BDD anode and graphite cathode was the fastest. Changing the configuration to use a graphite anode and BDD cathode led to the poorest result. Graphite is an active electrode with a low oxygen evolution potential, making it incapable of activating persulfate when used as an anode. Using BDD as both the anode and cathode was also less effective, indicating that BDD, while an excellent anode, is not an optimal cathode choice.7,8
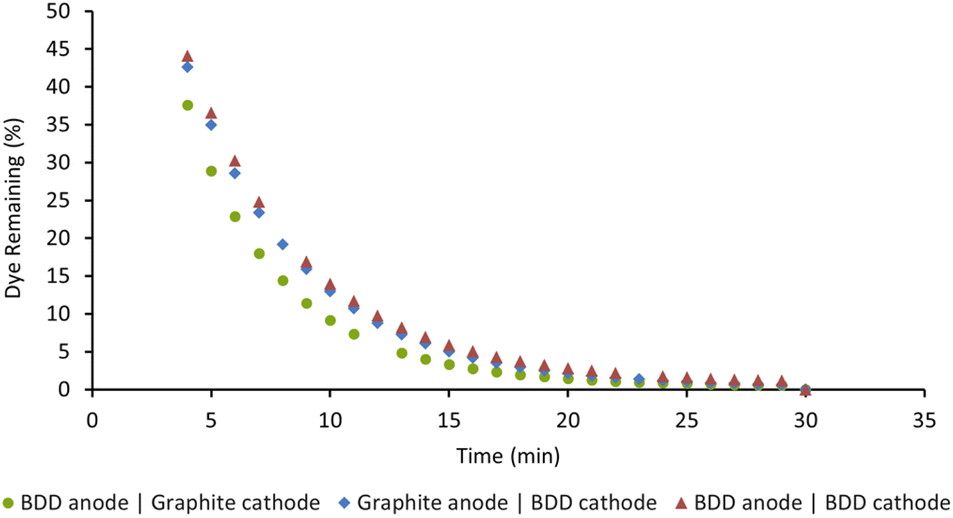 | ||
| Fig. 5 Effect of electrode composition on the decolorisation of AO7. Conditions: [AO7] = 100 μM, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm, for 30 min. | ||
In AOPs, sulfates can act both as an oxidant and an electrolyte, making their identity and concentration important parameters for optimisation. We wanted to probe this and chose sodium persulfate for our experiments because of its high oxidation potential, reactive peroxide bond, ability to split into more reactive species, and ease of storage on the industrial scale.3,7 Furthermore, it can be used as an additive in textile production and may be present at low concentrations in some effluent. We probed the relationship between sodium persulfate concentration and the rate of decolorisation of AO7. As shown in Fig. 6, the first-order rate constants for decolorisation decrease with increasing sodium persulfate concentration from 3.99 × 10−3 s−1 at 14 mM to 2.37 × 10−3 s−1 at 112 mM. This is likely due to an increase in radical termination events at higher oxidant concentrations.3,12 A lower sodium persulfate concentration was not tested to accuracy limitations of available balances.
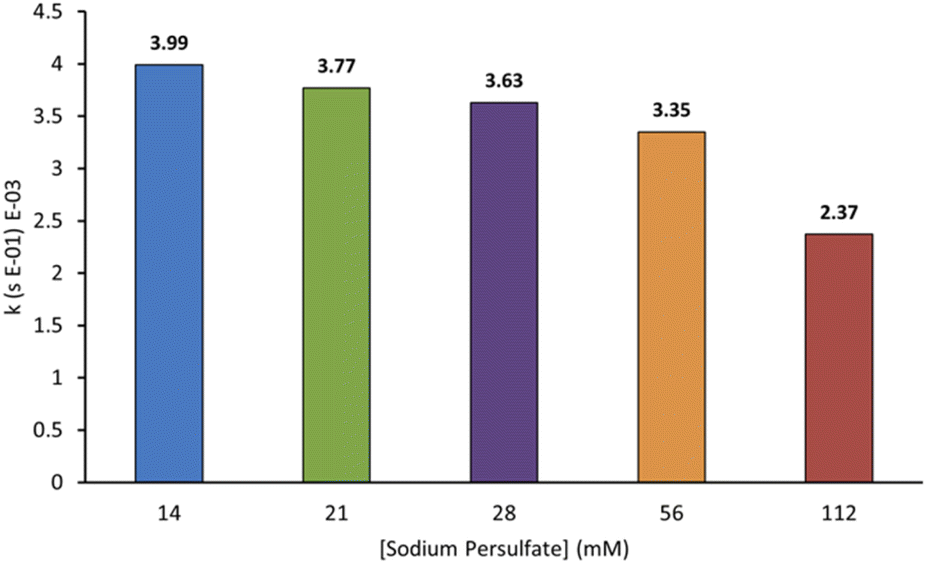 | ||
| Fig. 6 Effect of varying the concentration of sodium persulfate on the first-order rate constants for the decolorisation of AO7 at 20 min. Conditions: [AO7] = 100 μM, boron-doped diamond anode, graphite cathode, I = 5 mA, stirring = 1000 rpm. | ||
We next probed the impact of applied current on the rate of decolorisation of AO7 (Fig. 7). As expected, essentially no decolorisation is observed in the absence of applied current. The first-order rate constant of decolorisation for 1 mA was slower than that of 5 mA and 10 mA which were themselves nearly identical at 3.99 × 10−3 s−1 and 4.26 × 10−3 s−1 respectively. An applied current of 25 mA was also probed, but significant anode fouling occurred, so experiments were not continued. Operating at a higher current also increases the number of off-target reactions, mainly hydrogen and oxygen evolution, which decreases the efficiency of the system. As a result, an applied current of 5 mA appeared optimal.3,12
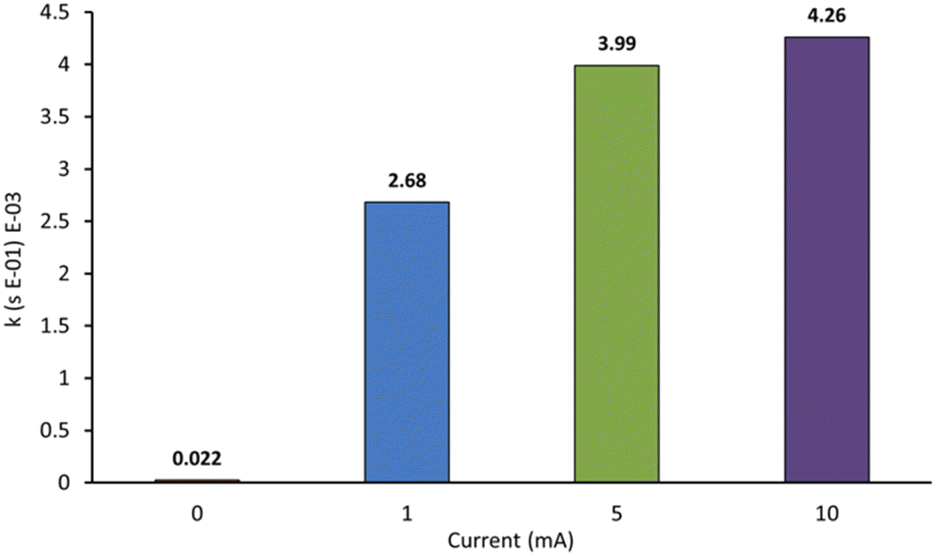 | ||
| Fig. 7 Effect of varying applied current on the first-order rate constants for the decolorisation of AO7 at 20 min. Conditions: [AO7] = 100 μM, boron-doped diamond anode, graphite cathode, [Na2S2O8] = 14 mM, stirring = 1000 rpm. | ||
One variable that is often neglected in the optimisation of electrochemical AOPs is stirring speed. High stirring rates are essential for efficient mass transport as electrochemistry is typically diffusion-limited.14 To confirm this, we probed the rate of decolorisation of AO7 as a function of stirring speed. As expected, there is a linear relationship between the first-order rate constant for decolorisation and stirring speed (Fig. 8).
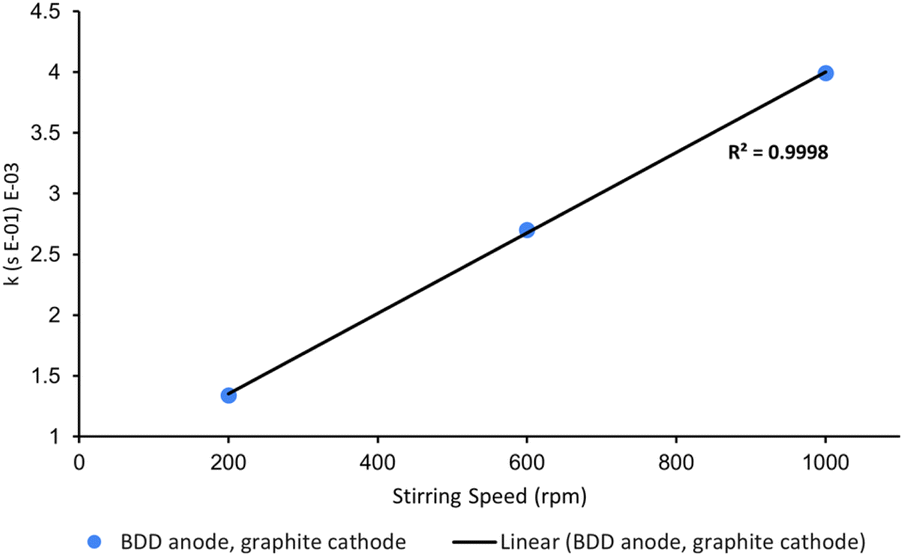 | ||
| Fig. 8 Effect of varying stirring speed on the first-order rate constants for the decolorisation of AO7 at 10 min. Conditions: [AO7] = 100 μM, boron-doped diamond anode, graphite cathode, [Na2S2O8] = 14 mM, I = 5 mA. | ||
Effects of varying the dye class and substructure on the rate of decolorisation
With optimised EAP conditions in hand, we set out to determine how varying the class of dye and the functional group structure within the dye impacts the rate of decolorisation. Three dyes from each the azo, triarylmethane, and xanthene families were selected, for a total of nine dyes (Fig. 9). From the azo family, we selected: Acid Orange 7 (AO7), Acid Orange 10 (AO10), and Acid Red 18 (AR18). From the triarylmethane family, we selected: Acid Blue 1 (AB1), Basic Violet 3 (BV3), and Acid Violet 19 (AV19). From the xanthene family, we selected: Acid Red 51 (AR51), Acid Red 87 (AR87), and Acid Red 91 (AR91). | ||
| Fig. 9 Structures of azo, triarylmethane, and xanthene dyes compared in the study. | ||
Since sulfate and hydroxyl radicals differ in reactivity towards distinct functional groups, each dye was tested in both our currently optimised EAP and previously developed AO protocols.14 First-order rate constants are presented in Fig. 10 and further graphical visualisation is provided in the ESI.† Overall, AO7, with a first-order rate constant of 3.99 × 10−3 s−1, was the easiest to decolorise, and AV19, with a first-order rate constant of 1.15 × 10−3 s−1, was the most difficult. It follows that the category that became decolorised most quickly is the azo family, followed by the xanthenes, and finally the triarylmethane dyes. This is because azo dye degradation usually occurs by cleavage of the azo bond to form two aromatic amines.1,20,22,24,27 The azo dyes examined in this work have a resonance structure bearing a nitrogen–nitrogen single bond, making it easier to cleave and hence resulting in faster decolorisation than the other dye classes.
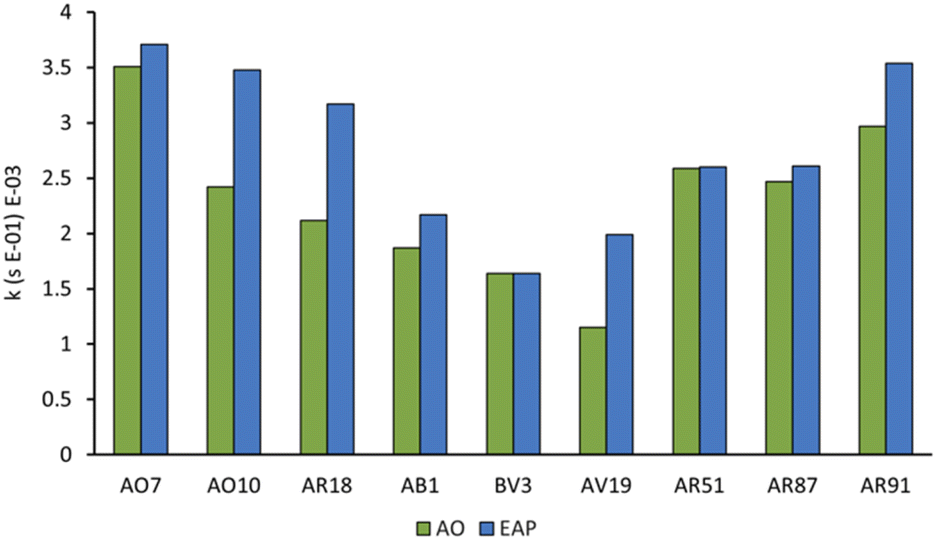 | ||
| Fig. 10 First-order rate constants at 20 min for the decolorisation of nine dyes under both AO and EAP protocols. AO conditions: [dye] = 100 μM graphite anode and cathode, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm. EAP conditions: [dye] = 100 μM, boron-doped diamond anode, graphite cathode, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm. Azo dyes: AO7 – Acid Orange 7, AO10 – Acid Orange 10, AR 18 – Acid Red 18. Triarylmethane dyes: AB1 – Acid Blue 1, BV3 – Basic Violet 3, AV19 – Acid Violet 19. Xanthene dyes: AR 51 – Acid Red 51, AR 87 – Acid Red 87, AR 91 – Acid Red 91. | ||
Within each family, electron-rich dyes were decolorised faster than their electron-poor counterparts. EAP usually resulted in a faster rate of decolorisation than AO, indicating that activated persulfate has a positive impact on rate of decolorisation. However, the magnitude of the rate increase varies significantly based on the structure of the dye in question (Fig. 10). This can be explained by the ability of the sulfate radical and persulfate anion, and non-radical oxidation to attack electron-deficient species faster than the indiscriminate hydroxyl radical.9 With no activated persulfate available in AO, the rate of decolorisation suffers even more with electron-deficient dyes. For example, azo dyes AO7, AO10, and AR18 vary by having one, two, and three sulfate groups, respectively. Of all the dyes tested, AO10 shows the most significant difference between the first-order rate constants of AO (2.12 × 10−3 s−1) and EAP (3.71 × 10−3 s−1). This is followed closely by AR18. However, the first-order rate constants for decolorisation of AO7 by AO and EAP are similar. This indicates that changing the number of electron-withdrawing groups from one to two has a significant impact on the rate of decolorisation, but increasing from two to three has much less of an impact.
Further evidence for the advantage of generating activated persulfate species when decolorising electron-deficient dyes is seen in the results of the triarylmethane family. In the case of BV3, which does not have any electron-withdrawing groups, the rate of decolorisation is essentially equivalent for AO and EAP. However, AB1 with two sulfate groups shows a difference in the rate of decolorisation between the two methods (AO: 1.87 × 10−3 s−1; EAP: 2.17 × 10−3 s−1) as does AV19 (AO: 1.15 × 10−3 s−1; EAP: 1.99 × 10−3 s−1). Triarylmethane dyes are typically decolorised by protonation at their central carbon,2 so the number and location of substituents as well as the pH of the solution may play an important role, but further study is required.
The xanthene dyes AR51, AR87, and AR91 vary by the number and identity of halogen atoms in the molecule. The role of free halogen (X−) in AOPs is complex. Small amounts can slow the rate of degradation by forming XOH˙− species, larger quantities can change the identity of the major oxidant and, depending on the dye, accelerate the decolorisation process.9 Analysis of the first-order rate constant at 20 min (Fig. 10) suggests that having a different halogen (bromine in AR87 or iodine in AR51) does not make a difference in the rate of decolorisation. However, replacing two halogens with electron-withdrawing nitro groups speeds it up. Plots of percent dye remaining vs. time for AR51 and AR87 in Fig. 11 and 12 provide more information about this phenomenon. In both tetra-halogenated species, decolorisation occurs rapidly before plateauing for a time, and then decreasing again. When the dye becomes dehalogenated, X− can react with a hydroxyl radical to form XOH˙− which can only react with very electron-rich substrates and prevents the original oxidising species from acting. This, in turn, retards the rate of decolorisation. Since the XOH˙− species is unstable, it decomposes back into X− and OH˙. The rate of decomposition is much faster for bromine than iodine,9 causing the plateau of AR51 to be more pronounced than that of AR87. AR91 with half as many halogens as the other two dyes studied does not exhibit this phenomenon.
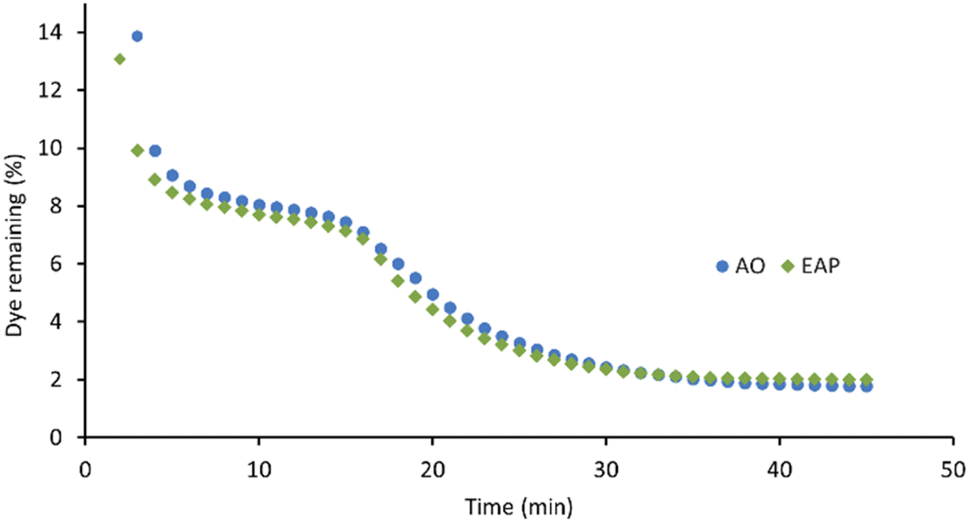 | ||
| Fig. 11 AO- and EAP-mediated decolorisation of Acid Red 87 as a function of time. AO conditions: [AR87] = 100 μM, graphite anode and cathode, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm. EAP conditions: [AR87] = 100 μM, boron-doped diamond anode, graphite cathode, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm. | ||
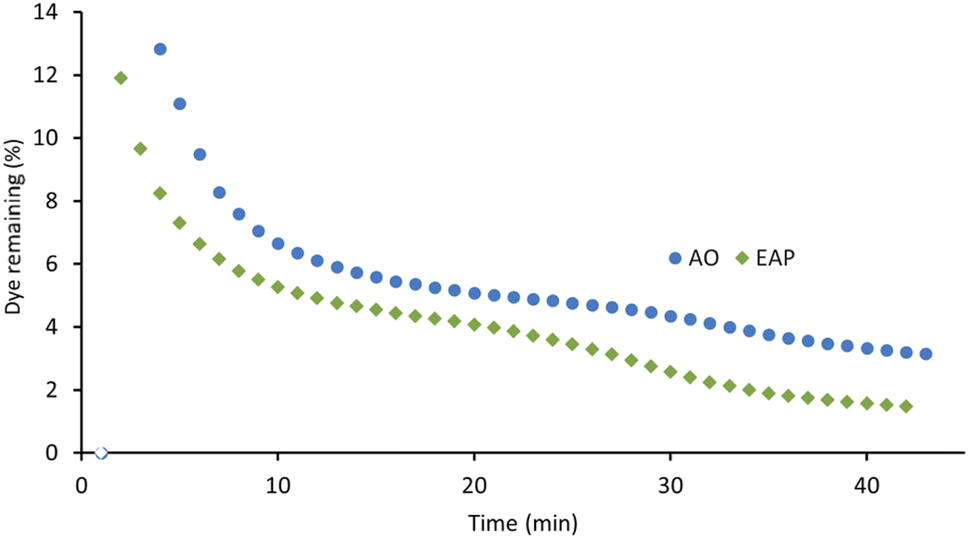 | ||
| Fig. 12 AO- and EAP-mediated decolorisation of Acid Red 51 as a function of time. AO conditions: [AR51] = 100 μM, graphite anode and cathode, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm. EAP conditions: [AR51] = 100 μM, boron-doped diamond anode, graphite cathode, [Na2S2O8] = 14 mM, I = 5 mA, stirring = 1000 rpm. | ||
Experimental section
General considerations
An IKA ElectraSyn 2.0 with a standard 10 mL vial was interfaced with an Ocean Insight SR-2UVV400-25 spectrophotometer with a range of 183–909 nm, optical resolution of 1.33 nm, and signal to noise ratio of 380:1. The spectrophotometer was attached to an Ocean Insight P400-1-SR 400 μm diameter optical fiber (200 nm to 1.1 μm) with a 74-UV collimating lens (185 nm to 2.5 μm). A diffuse white LED with a range of 420–700 nm was employed as the light source. Data was collected using OceanView 2.0 from Ocean Insight. Graphite and boron-doped diamond electrodes with dimensions of 52 mm L × 8 mm W × 2 mm H were obtained from IKA. The 3D-printed interface (STL file available in the ESI†) was fabricated from black polylactic acid using an Entina Tina2S 3D-printer.
Chemicals
Acid Orange 7, Acid Blue 1, and Avid Violet 19 were purchased from Sigma-Aldrich. Acid Orange 10, Basic Violet 3, and Acid Red 91 were purchased from Thermo Fisher Scientific. Acid Red 18, Acid Red 51, and Acid Red 87 were purchased from TCI America. Sodium persulfate was purchased from Oakwood Chemicals.General procedure
The 3D-printed sleeve was placed on the stage of the IKA ElectraSyn 2.0 single-vial holder. An optical fiber was attached to the front cavity in the 3D-printed sleeve by means of a collimating lens. The other end of the optical fiber was attached to the spectrophotometer. A standard 5 mm diffuse white LED was placed in the slot at the back of the 3D-printed sleeve and connected to a bench power supply using alligator clips. The power supply was set to 2.7 V and allowed to warm up for 10 min until the absorbance spectrum of the LED remained at a constant intensity. To calibrate the spectrophotometer, a standard 10 mL capacity ElectraSyn 2.0 vial containing deionised water (8 mL) was placed inside of the assembled electrochemical/spectrophotometer interface, and the resulting absorbance spectrum was set to zero.To prepare a reaction, a 10 mL capacity vial was charged with 8 mL of a 100 μM dye solution in deionised water (pH = 7), the desired amount of sodium persulfate (14, 28, 56, or 112 mM) as the electrolyte, and a magnetic stir bar. An ElectraSyn 2.0 vial cap was equipped with the desired anode and cathode then screwed onto the vial so the submerged surface area of each electrode was 6.72 cm2. The reaction vessel was placed inside the assembled electrochemical/spectrophotometer interface, and the contents electrolysed at a constant current of 0, 1, 5, or 10 mA while stirring the reaction mixture at 1000 rpm. Absorbance spectra were collected every minute with an automated clicker until the oxidation was deemed complete by in situ UV-visible spectrophotometer detection. Cartesian points of absorbance vs. time at the λmax were transferred to a microsoft excel workbook. All reactions were performed in triplicate, absorbances were averaged and then converted to μM values by employing a calibration curve. Finally, μM values were converted to percentage dye remaining and plotted as a function of time.
To clean the boron-doped diamond electrodes between trials, they were rinsed with water, then acetone, and then dried with lab air; the graphite electrodes were rinsed with water, polished on 2000 grit sandpaper, and then rinsed with water and acetone.
Conclusions
The optimisation of an electrochemically activated persulfate protocol for the decolorisation of Acid Orange 7 has been performed rapidly and efficiently by means of a real-time in situ monitoring apparatus. The effect of electrode composition, current density, persulfate loading, and stirring speed were all probed. The optimal conditions were then employed for a study of three dyes in each of three classes (azo, triarylmethane, and xanthene), totaling nine dyes. To the best of our knowledge, this is the first time such a thorough comparison has been reported. Azo dyes decolorised fastest, followed by xanthene, and then triarylmethane dyes. Electron-rich substrates were easier to decolorise than electron-deficient ones. For nearly every substrate, the EAP protocol was more expeditious than the AO analogue. The enhanced rate of EAP over AO became more pronounced with more electron-deficient dyes. The rate of decolorisation of halogenated dyes shows a more complex dependence on the number and identity of the halogens. Overall, structural category, functional groups, and oxidant identity play crucial roles in electrochemical dye decolorisation. More systematic studies comparing molecules that vary by only one functional group are currently underway to understand further the utility of EAP and AO protocols for the degradation of genuine wastewater samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Connecticut Office of Undergraduate Research and the University of Connecticut Department of Chemistry Charles Waring Fund are thanked for funding.Notes and references
- R. Al-Tohamy, S. S. Ali, F. Li, K. M. Okasha, Y. A.-G. Mahmoud, T. Elsamahy, H. Jiao, Y. Fu and J. Sun, Ecotoxicol. Environ. Saf., 2022, 231, 113160 CrossRef CAS PubMed
.
- L. D. Ardila-Leal, R. A. Poutou-Piñales, A. M. Pedroza-Rodríguez and B. E. Quevedo-Hidalgo, Molecules, 2021, 26, 3813 CrossRef CAS PubMed
- A. Fernandes, M. J. Nunes, A. S. Rodrigues, M. J. Pacheco, L. Ciríaco and A. Lopes, Molecules, 2021, 26, 4821 CrossRef CAS PubMed
- A. B. Isaev, N. S. Shabanov, A. G. Magomedova, P. V. Nidheesh and M. A. Oturan, Environ. Chem. Lett., 2023, 21, 2863–2911 CrossRef CAS
- A. Iqbal, A. Yusaf, M. Usman, T. Hussain Bokhari and A. Mansha, Int. J. Environ. Anal. Chem., 2023, 1–35 CrossRef
- P. Vithalani and N. Bhatt, Gis Sci. J., 2023, 10, 1428–1445 Search PubMed
- K. C. Araújo, E. V. Dos Santos, P. V. Nidheesh and C. A. Martínez-Huitle, Curr. Opin. Chem. Eng., 2022, 38, 100870 CrossRef
- Y. Jiang, H. Zhao, J. Liang, L. Yue, T. Li, Y. Luo, Q. Liu, S. Lu, A. M. Asiri, Z. Gong and X. Sun, Electrochem. Commun., 2021, 123, 106912 CrossRef CAS
- J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed
- J. Li, Y. Li, Z. Xiong, G. Yao and B. Lai, Chin. Chem. Lett., 2019, 30, 2139–2146 CrossRef CAS
- L. W. Matzek and K. E. Carter, Chemosphere, 2016, 151, 178–188 CrossRef CAS PubMed
- D. Zhi, Y. Lin, L. Jiang, Y. Zhou, A. Huang, J. Yang and L. Luo, J. Environ. Manage., 2020, 260, 110125 CrossRef CAS PubMed
- M. H. Abdel-Aziz, M. Bassyouni, M. S. Zoromba and A. A. Alshehri, Ind. Eng. Chem. Res., 2019, 58, 1004–1018 CrossRef CAS
- C. M. Schroeder, A. León Sandoval, K. K. Ohlhorst and N. E. Leadbeater, Chem.: Methods, 2023, e202300014 Search PubMed
- J. Yao, Y. Zhang and Z. Dong, Chem. Eng. J., 2021, 415, 128938 CrossRef CAS
- G. Kuchtová, P. Mikulášek and L. Dušek, Monatsh. Chem., 2022, 153, 237–243 CrossRef
- J. Alagesan, M. Jaisankar, S. Muthuramalingam, E. Mousset and P. V. Chellam, Chemosphere, 2021, 262, 128381 CrossRef CAS PubMed
- C. Gutiérrez-Bouzán and M. Pepió, Ind. Eng. Chem. Res., 2014, 53, 18993–19000 CrossRef
- N. E. H. Abdessamad, H. Akrout and L. Bousselmi, Desalin. Water Treat., 2014, 52, 1735–1744 CrossRef CAS
- H. Chao, D. Xue, W. Jiang, D. Li, Z. Hu, J. Kang and D. Liu, Water Environ. Res., 2020, 92, 779–788 CrossRef CAS PubMed
- F. Mahmoudian, F. Nabizadeh Chianeh and S. M. Sajjadi, J. Electroanal. Chem., 2021, 884, 115066 CrossRef CAS
- M. A. Sandoval, N. Zúñiga-Mallea, L. C. Espinoza, J. Vidal, P. Jara-Ulloa and R. Salazar, ChemistrySelect, 2019, 4, 13856–13866 CrossRef CAS
- M. Cerón-Rivera, M. M. Dávila-Jiménez and M. P. Elizalde-González, Chemosphere, 2004, 55, 1–10 CrossRef PubMed
- E. Kusmierek, E. Chrzescijanska, M. Szadkowska-Nicze and J. Kaluzna-Czaplinska, J. Appl. Electrochem., 2011, 41, 51–62 CrossRef CAS
- I. M. Sasidharan Pillai and A. K. Gupta, J. Electroanal. Chem., 2016, 762, 66–72 CrossRef CAS
- A. Sakalis, K. Mpoulmpasakos, U. Nickel, K. Fytianos and A. Voulgaropoulos, Chem. Eng. J., 2005, 111, 63–70 CrossRef CAS
- M. Nashat, M. Mossad, H. K. El-Etriby and M. Gar Alalm, Chemosphere, 2022, 286, 131579 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06634g |
| This journal is © The Royal Society of Chemistry 2023 |