
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of azido terpyridine Pd(II) and Pt(II) complexes towards 4,4,4-trifluoro-2-butynoic acid: structural insight into the triazolato coordination mode†
Ahmed M. Mansour *a,
Krzysztof Radackib,
Gamal A. E. Mostafac,
Essam A. Alic and
Ola R. Shehab*d
*a,
Krzysztof Radackib,
Gamal A. E. Mostafac,
Essam A. Alic and
Ola R. Shehab*d
aDepartment of Chemistry, United Arab Emirates University, Al-Ain, United Arab Emirates. E-mail: Mansour_am@uaeu.ac.ae; inorganic_am@yahoo.com
bInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, D-97074 Würzburg, Germany
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia
dDepartment of Chemistry, Faculty of Science, Cairo University, Gamma Street, Giza, 12613, Egypt. E-mail: Olashehab@sci.cu.edu.eg; Olahehab_chem@yahoo.com
First published on 29th November 2023
Abstract
The mono- and binuclear azido terpyridine square-planar complexes of ionic formulas, [Pd2(N3)2L]2+ and [Pt(N3)L]+ (L = 1,4-bis(2,2′:6′,2′′-terpyridin-4′yl)benzene), underwent the catalyst-free [3 + 2] cycloaddition coupling with 4,4,4-trifluoro-2-butynoic acid at ambient temperature affording the corresponding triazolate complexes. A mixture of triazolate isomers was generated by these inorganic click reactions. An increase in the solubility of the compounds was achieved by replacing the azido ligand with a triazolato ligand. By calculating the vibrational modes and comparing the total electronic and zero-point energy values, the local minimum structures of the complexes and the nature of the predominant triazolate isomer were verified. The theoretical work was complemented with natural bond analysis to get an insight into the natural charge and electronic arrangement of the metal ion, the hybridization of M–L bonds and strength of M–N bonds.
1. Introduction
One of the key challenges in the field of bioinorganic chemistry is the chemo-selective functionalization of biomolecules with biologically active coordination and organometallic compounds. Actually, some biomolecules can be modified via 1,3-dipolar cycloaddition in a targeted manner. Azide complexes have thus far been subjected to click coupling with electrophiles such as alkynes and isothiocyanate to produce five-membered rings.1,2 Initially, [Pd(N3)2(PPh3)2] reacted with trifluoro-acetonitrile, under catalyst-free conditions, affording a tetrazolate complex.3 In their study of the correlation between the reaction kinetics and the nature of the substituent R on the nitrile RCN, Beck and Fehlhammer showed that the rate of the reaction is improved as the withdrawing nature of R group increased.4Later, Paul's research team showed the benefits of using a square -planar complex of the general formula [MN3L]+, (L is a tridentate ligand) in the cycloaddition reactions.5 Later on, several investigations on the reactivity of metal azides of Mo,6 W,6 Mn,7 Ru,8 Pd,9,10 Pt,9,11 Re,12–14 and Rh15,16, ions towards some electrophiles were reported. Despite the terminal attachment of the azido ligand to the metal center, the discovered triazolate crystal structures proved unequivocally that the kinetically formed N1 triazolate isomer favored isomerization to the N2 counterpart. The bulkiness of the electrophile, the type of the ancillary ligand, the geometry (square-planar vs. octahedral) of the metal complexes, the synthesis and storage conditions, as well as other factors, all play a role in the N2 triazolate isomerization.1
Kim and coworkers discovered that the click reactions of azido square-planar complexes with isothiocyanates proceeded site-specifically yielding tetrazole-thiolato bound complexes.17,18 Pd(II) thiolato–tetrazolate complexes, with N,N,N-tridentate ligands, have recently shown higher antifungal activity (MIC = 0.31 μM) against Cryptococcus neoformans and Candida albicans than their comparable chlorido complexes (MIC = 0.95–1.90 μM) and the reference medication Fluconazole (MIC = 0.41–26.1 μM).10
Recently, it was examined how four different alkynes reacted to two square-planar Pd(II) and Pt(II) complexes functionalized with the N,N,S-tridentate ligand.19 With the exception of two reactions involving Pt(II) azide and di-alkyl acetylene-dicarboxylate, which resulted in a non-equivalent mixture of N1 and N2-isomers, the kinetically generated N1-triazolate isomers tended to isomerize to N2-analogues. Early in the experiment, according to the 19F NMR kinetics profiles, three triazolate isomers were formed. By the time the experiment was over, all but the dominant N2-triazolate bound isomer remained.19 Next, the reactions of azido Pd(II) and Pt(II) complexes of 2,6-bis(1-ethylbenzimidazol-2′-yl)pyridine with dimethyl acetylene-dicarboxylate and 4,4,4-trifluoro-2-butynoic acid ethyl ester produced a mixture of the two (or three) anticipated triazolate isomers.11 NMR spectroscopy was used to examine the stability of the triazolate complexes, the mechanism of the [3 + 2] cycloaddition coupling and the nature of the intermediate. The isomer ratio of the triazolate-bound Pt(II) complexes appeared to be controlled by the nature of the solvent. The first example of the catalyst-free 1,3-dipolar cycloaddition reactions of (trans-, trans-), diazido dihydroxo dipyridine Pt(IV) complex with some electrophiles was reported by Farrer and Sadler.20
In this contribution, we report the reactivity of azido Pd(II) and Pt(II) complexes of 1,4-bis(2,2′:6′,2′′-terpyridin-4′yl)benzene (Scheme 1) towards 4,4,4-trifluoro-2-butynoic acid at the ambient temperature under the catalyst-free conditions. Using density functional theory calculations, together with spectral tools, it was possible to learn more about the molecular structures, vibrational properties, and the electronic properties of the anticipated triazolate isomers. In order to learn more about the nature, and strength of the hybridization of the bonds in the coordination sphere surrounding the d8 metal ion (Pd(II) or Pt(II)), Weinhold and coworkers' natural bond orbital (NBO) analysis21 was carried out for the triazolate isomers.
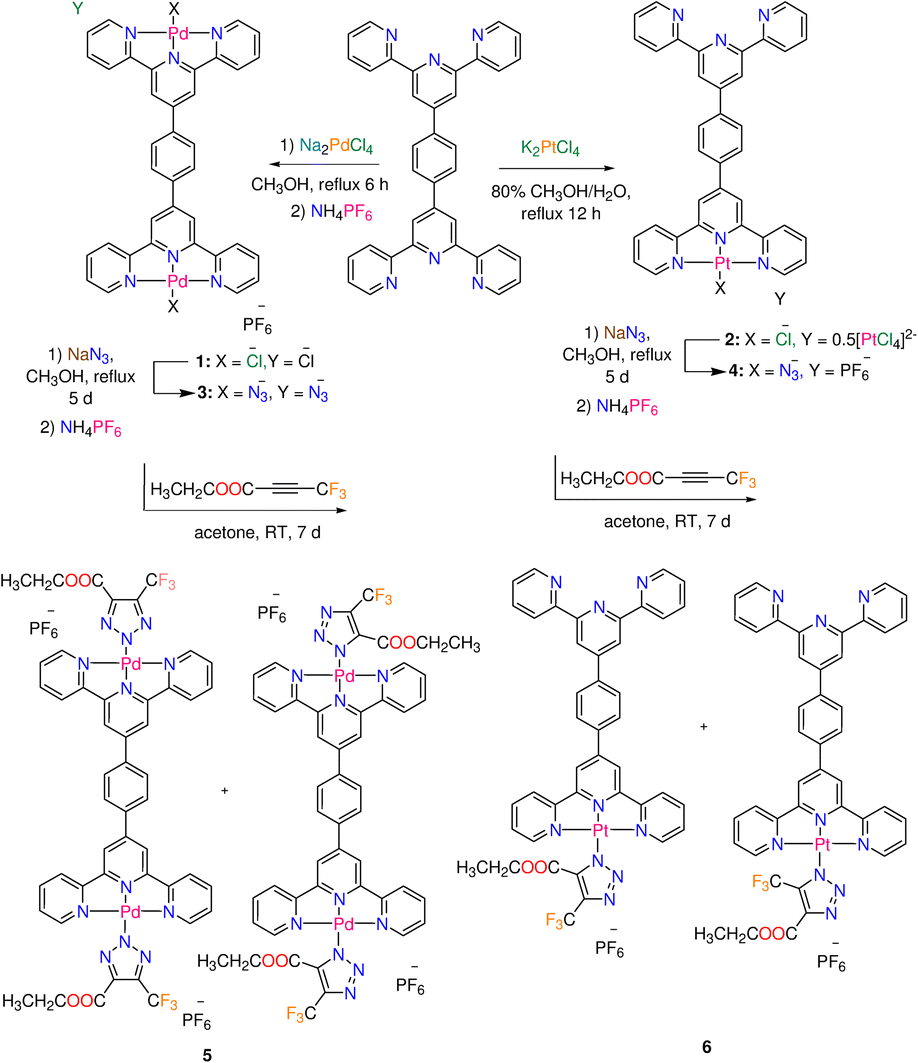 | ||
| Scheme 1 Synthesis of Pd(II) and Pt(II) complexes 1–6 functionalized with terpyridine derivative L (main species are shown in the cases of 5 and 6). | ||
2. Results and discussion
2.1. Synthesis and characterization of complexes
Terephthaldehyde and eight equivalents of 2-acetylpyridine were reacted in a mixture of ammonia and potassium tert-butoxide to produce 1,4-bis (2,2′:6′,2′′-terpyridin-4′yl) benzene (L) in good yield (77%) and purity.22 Square-planar complexes 1 and 2 (Scheme 1), with ionic formulas of [Pd2Cl2L]+ and [PtClL]+, respectively, were obtained by reactions of terpyridine ligand L and either Na2PdCl4 (in methanol) or K2PtCl4 (in methanol/water). Under comparable experimental conditions, it appears that the reaction between the Pd(II) precursor and L afforded a binuclear complex 1, whereas the same reaction with Pt(II) precursor gave a mononuclear complex 2. The IR spectra of the free ligand and complexes (1 and 2) are given in the ESI (Fig. S1–S3†). Complexes 1 and 2 are poorly soluble in DMF, however they breakdown in DMSO, which could be likely attributed to the displacement of chlorido ligands by solvent molecules. These metal-based compounds were therefore subjected to solid state NMR {13C, 15N, 19F, and 31P} analysis (Fig. S4–S8†). In 19F NMR spectrum of 1 (Fig. S6†), the PF6− signal of the counter ion is detected at δ = −69.4 ppm. The 15N NMR spectrum of 1 is characterized by two strong signals at δ = (−154.5, −159.1) ppm as well as three weak signals at δ = (−76.0, −82.7, −85.9) ppm.23 The presence of two metal centers in 1 may complicate the 15N NMR spectrum. The 15N NMR spectrum of 2 (Fig. S8†) displays four signals at δ = −67.5, −71.1, −80.6, and −174.3 ppm. The Δ15Ncoord parameter, the difference between the chemical shifts of free nitrogen atom and M–N bond, gives an idea about the coordination of the heteroatom.23 Compared to 1, the Δ15Ncoord of the signal at δ = −174.3 ppm of 2 is relatively high, which is typical for both Pt central atom and the nitrogen trans- to Cl.23,24 The atmospheric solids analysis probe mass spectrometry (ASAP-MS) spectrum of 1 (in the positive mode, Fig. S9†) shows two distinct fragments at m/z 968.9147 (calcd 968.9150) and m/z 683.0772 (calcd 683.0768) equivalent to {[Pd2Cl2L]PF6}+ and {[PdClL]}+, respectively. The ASAP-MS spectrum of 2 (Fig. S10†) displays a unique fragment at m/z 770.1393 (calcd 770.1393) corresponding to {[PtClL]}+.As shown in Scheme 1, azido Pd(II) and Pt(II) complexes (3 and 4) were obtained by ligand exchange and gathered in pure form. The distinctive vibrational spectral feature in spectra of 3 and 4 is the prominent v(N3) band at 2037 and 2022 cm−1, respectively (Fig. S11 and 12†). The solubility of 3 and 4 in DMF is poor, while both complexes decompose in DMSO. Thus, these metal-azide complexes were analysed with solid-state NMR {13C, 15N, 19F, and 31P} (Fig. S13–S19†). In 19F and 31P NMR spectra of 3 (Fig. S15 and S16†), the PF6− signals are detected at δ = −69.3 and −142.9 ppm, respectively. The 15N NMR spectrum of 3 (Fig. S14†) shows a set of six signals at δ = (−75.9, −84.6, −86.6), and (−158.6, −160.4, −164.8) ppm corresponding to the terminal and central pyridine rings as well as three signals at δ = −122.0, −207.8 and −309.6 ppm equivalent to the coordinated azido ligand.25,26 The most clear 15N signals in the NMR spectrum of 4 (Fig. S18†) are observed at δ = −68.2, −71.6, and −306.4 ppm. The 19F signal of PF6− is seen at δ = −69.4 ppm in the spectrum of 4. Two distinct fragments at m/z 982.9961 (calcd 982.9957) and 688.1186 (calcd 688.1184) matching to {[Pd2(N3)2L]·PF6}+, and {[Pd(N3)L]}+ are visible in the ASAP-MS spectrum of 3 (Fig. S20†). There is a distinct peak (Fig. S21†) at m/z 778.1813 (calcd 778.1799) in the mass spectrum of 4, which corresponds to {[Pt(N3)L]}+. The reactivity of azido complexes (3 and 4) towards CF3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–COOCH2CH3 was examined at the ambient temperature. The inorganic click reactions (iClick) were carried out by suspending the azido complexes in an acetone solution of the additional alkyne, as illustrated in Scheme 1. To precipitate the complexes in their purest form, a saturated solution of ammonium hexafluorophosphate was added to the triazolate reaction mixtures. The formation of triazolate complexes 5 and 6 is clearly determined by the disappearance of the strong v(N3) band, and the growth of the distinctive (C
C–COOCH2CH3 was examined at the ambient temperature. The inorganic click reactions (iClick) were carried out by suspending the azido complexes in an acetone solution of the additional alkyne, as illustrated in Scheme 1. To precipitate the complexes in their purest form, a saturated solution of ammonium hexafluorophosphate was added to the triazolate reaction mixtures. The formation of triazolate complexes 5 and 6 is clearly determined by the disappearance of the strong v(N3) band, and the growth of the distinctive (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band of the triazolate moiety at around 1727 cm−1 (Fig. S22 and S23†). Unlike complexes 1–4, the triazolate derivatives 5 and 6 have good solubility in DMSO. The positively charged mass fragments (Fig. S24†) at m/z 854.1431 (calcd 854.1426) and m/z 1315.0461 (calcd 1315.0440) in the spectrum of 5 are assigned to {[Pd2(triazolato)2L]·PF6}+, and {[Pd(triazolato)L]}+ in that order. The prominent peak at m/z 1110.2287 (calcd 1110.1639) designated to {Na[Pt(triazolato)L]·PF6}+ is what distinguishes the ESI MS spectrum of 6 (Fig. S25†).
O) band of the triazolate moiety at around 1727 cm−1 (Fig. S22 and S23†). Unlike complexes 1–4, the triazolate derivatives 5 and 6 have good solubility in DMSO. The positively charged mass fragments (Fig. S24†) at m/z 854.1431 (calcd 854.1426) and m/z 1315.0461 (calcd 1315.0440) in the spectrum of 5 are assigned to {[Pd2(triazolato)2L]·PF6}+, and {[Pd(triazolato)L]}+ in that order. The prominent peak at m/z 1110.2287 (calcd 1110.1639) designated to {Na[Pt(triazolato)L]·PF6}+ is what distinguishes the ESI MS spectrum of 6 (Fig. S25†).
The literature claims that the N1 or N2 atoms of the triazolato ligand, synthesized from symmetrical alkyne, can bind Mn+ ion. Despite the azido ligand being terminally attached to Pd(II) or Pt(II) ions, X-ray crystallographic examination of numerous triazolate complexes with Mn+ ions other than d8 ions revealed conclusive evidence for the isomerization of the kinetically generated N1 (or N3) isomer into the N2 analogue.12,27,28 A combination of two (or three) isomers can occur when the conversion of N1 (or N3) to N2 is incomplete.29,30 The geometry of the reacting azide complex (square-planar, piano-stool or octahedral), the bulkiness of the electron-poor alkynes, the experimental conditions (coordinating solvents, reactants ratio, temperature, etc.), as well as the nature of the ancillary ligands all have an impact on the final triazolato coordination mode.1 In our situation, the reaction of 3 (or 4) with the asymmetric alkyne is anticipated to result in the formation of three triazolate bound modes (N1, N2 and N3) (Fig. 1). The triazolate isomers bearing –COOCH2CH3 and –CF3 groups close to the ligand framework, respectively, are referred to as N1 and N3, while the name N2 denotes the coordination of the central triazolate N to the metal centre (Fig. 1). The NMR spectra of 5 are shown in Fig. S26–S30.† The 1H NMR spectrum of 5 (Fig. S26†) is broad with the primary feature being two ethyl ester signals at 4.39 and 1.41 ppm as well as few aromatic signals in the range of 7.81–8.74 ppm. Two minor 1H NMR signals for the ethyl ester group are seen clearly at around 4.12 and 1.08 ppm supporting the presence of another isomer. The 13C NMR spectrum of 5 (Fig. S27†) shows the characteristic signals for CF3–C at δ = 138.7 ppm (q, 2JC,F = 39.4 Hz), and 121.7 ppm (q, 1JC,F = 267.0 Hz) as well as three unique signals for CO, –CH2 and –CH3 functional groups at 159.2, 60.9 and 13.7 ppm, respectively. The 19F and 31P signals of PF6− are seen at δ = −70.2 ppm (d, 1JPF = 711 Hz) and δ = −144.2 (sept, 1JPF = 711 Hz) in the spectra of 5, in that order. As indicated by the few tiny signals observed in the 19F NMR spectrum of 5 (Fig. S28†), the presence of two metal centres in 5 undoubtedly increases the number of possible triazolate isomers. According to 19F NMR spectroscopy (Fig. S28†), the cycloaddition reaction of 5 with 4,4,4-trifluoro-2-butynoic acid ethyl ester gave mainly a mixture of N1-, and N2-isomers, with the –CF3 group displaying two singlet signals of different intensities at δ = −59.03, and −58.95 ppm, respectively. If the triazolate N3-isomer is present in the reaction mixture of 5, its 19F NMR signal should be at least 4 ppm apart from those of N1 and N2.11 The up-field change in the –CF3 19F-signal from δ = −52 ppm in the starting coupling agent to higher chemical-shift values in 5 could suggest triazolate formation.12 Complex 5 has low solubility in most organic solvents except DMSO, therefore we couldn't dissolve it in non-coordinating solvents. We discovered in prior contributions to the iClick reaction that performing the NMR measurements in DMSO increases the conversion of N1- into N2-isomer11 and consequently we allocated the highest intensity 19F NMR signal at δ = −58.95 ppm to the N2-isomer. According to 1H and 19F NMR studies, the reaction of 4 with the –CF3 alkyne produced the three expected triazolate isomers (Fig. 1). The presence of isomers and the prospect of rapid isomerization during the NMR experiments make determining its 1H NMR spectrum highly challenging. The crucial spectrum characteristics for the progress of the inorganic click reaction and the formation of the triazolate molecule, on the other hand, were discovered. This includes the appearance of ethyl ester signals in the 1H NMR spectrum, as well as the presence of 19F NMR signals with greater chemical-shift values than the original CF3 alkyne signal. We detected the presence of PF6− counterion by monitoring the 19F and 31P NMR signals, as we had done before with the chlorido and azido complexes.
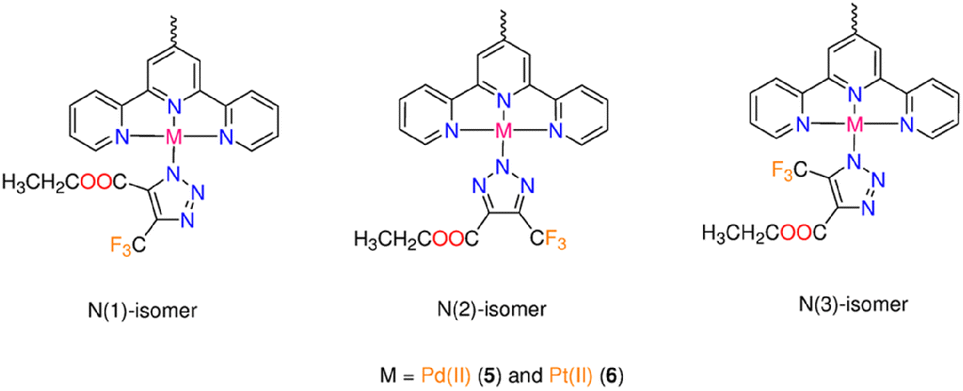 | ||
| Fig. 1 The three triazolate bound isomers formed from reaction of 3 (or 4) with 4,4,4-trifluoro-2-butynoic acid ethyl ester. | ||
2.2. Density functional theory calculations
The analytical and spectroscopic evidence in the preceding section demonstrated the mono- and binuclear nature of the isolated triazolate Pt(II) and Pd(II) complexes. In 5 and 6, tridentate terpyridine derivative and monodentate triazolato ligand are what make up the square-planar arrangement that surrounds the metal core. Utilizing the Becke 3-parameter (exchange) Lee–Yang–Parr functional31,32 and the LANL2DZ basis set,33,34 ground-state geometry optimization and the vibrational analysis of all the isomers were performed in order to get insight into the triazolato binding mode of the formed isomers and their stabilities. The harmonic frequency analysis was used to verify that the obtained geometries were local minima. Except for the N2 isomer of the Pt(II) complex, we could not discover any imaginary modes in any of the optimized geometries. Next, the total electronic and zero-point energy values were compared (Table S1†). Comparatively, it is important to note that the N2-isomer of the Pd(II) complex is more stable than N1 and N3 isomers by 3.5 and 19.7 kJ mol−1, respectively, and that the N1 is therefore about 16.2 kJ mol−1 more stable than the N3 isomer. In terms of the presence of the N1 and N2 isomers, these conclusions agree with the experimental NMR data. The calculated bond lengths and bond angles around the d8 metal ions are presented in Tables S2 and Table S3.† The variation in the bonds and angles of the three isomers were compared to each other. The triazolate rings in the N1 and N3 isomers of 5 are trans to one another and rotated off the plane by 57.0° and 55.0°, respectively, according to the dihedral angle of N73–N69–Pd–N35. The triazolate moiety in N2 isomer is nearly in the plane of the terpyridine ligand system at equal distances from terpyridine framework. The two Pd(II) moieties of the binuclear system in the three isomers are nearly perpendicular to one another and have the same bond length, as illustrated in Fig. 2. As opposed to the N1 and N3 isomers, the two N–N bonds of the triazolate ring are nearly equivalent in the N2 isomer. The distance to the triazolato ligand is 2.027, 2.028 and 2.032 Å in the three isomers of 5, respectively.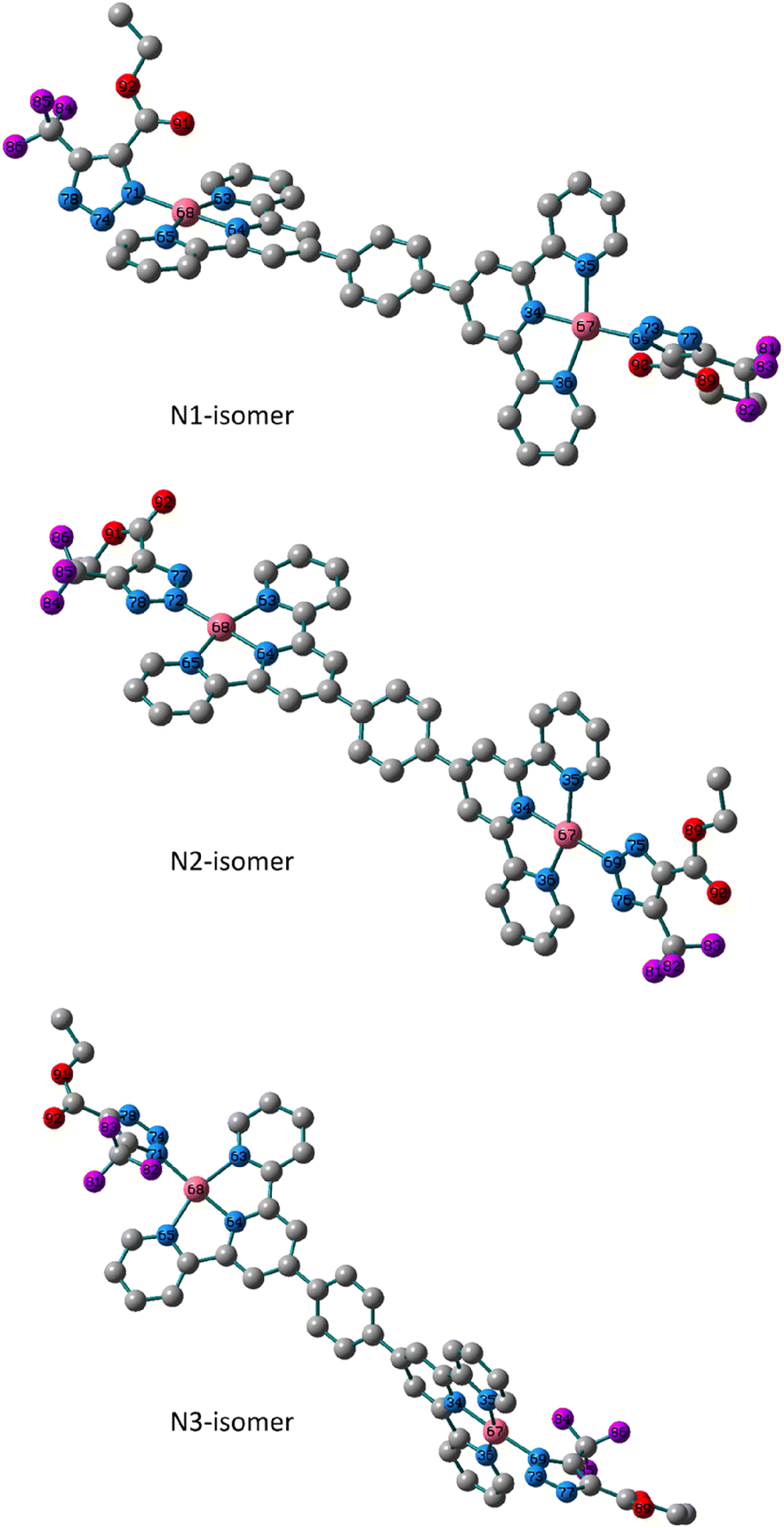 | ||
| Fig. 2 Local minimum structures of the optimized triazolate binuclear Pd(II) isomers of 5 (N1, N2 and N3) calculated at B3LYP/LANL2DZ level of theory. (Hydrogen-atoms were omitted for the clarity). | ||
Likewise, the N1 isomer of the Pt(II) complex is more stable than the N3 analogue by about 4.35 kJ mol−1. Like the N2-isomer of 5, the triazolate moiety of the N2 isomer in 6 is almost in the plane of the terpyridine ligand system with similar distances from the terpyridine framework. The distance to the triazolato ligand is 2.038, 2.033 and 2.042 Å in the three isomers of 6, respectively, that are longer than the equivalent ones in the Pd(II) isomers. The triazolate ring in the N1 and N3 isomers of 6 is rotated from the plane by 55.0° and 52.0° as defined by the dihedral angles of N69–N67–Pt–N35. When compared to those found in the N1 and N3 isomers, the distances between Pt(II) ion and nitrogen atoms of the terpyridine framework in the N2 isomer are shorter (Fig. 3).
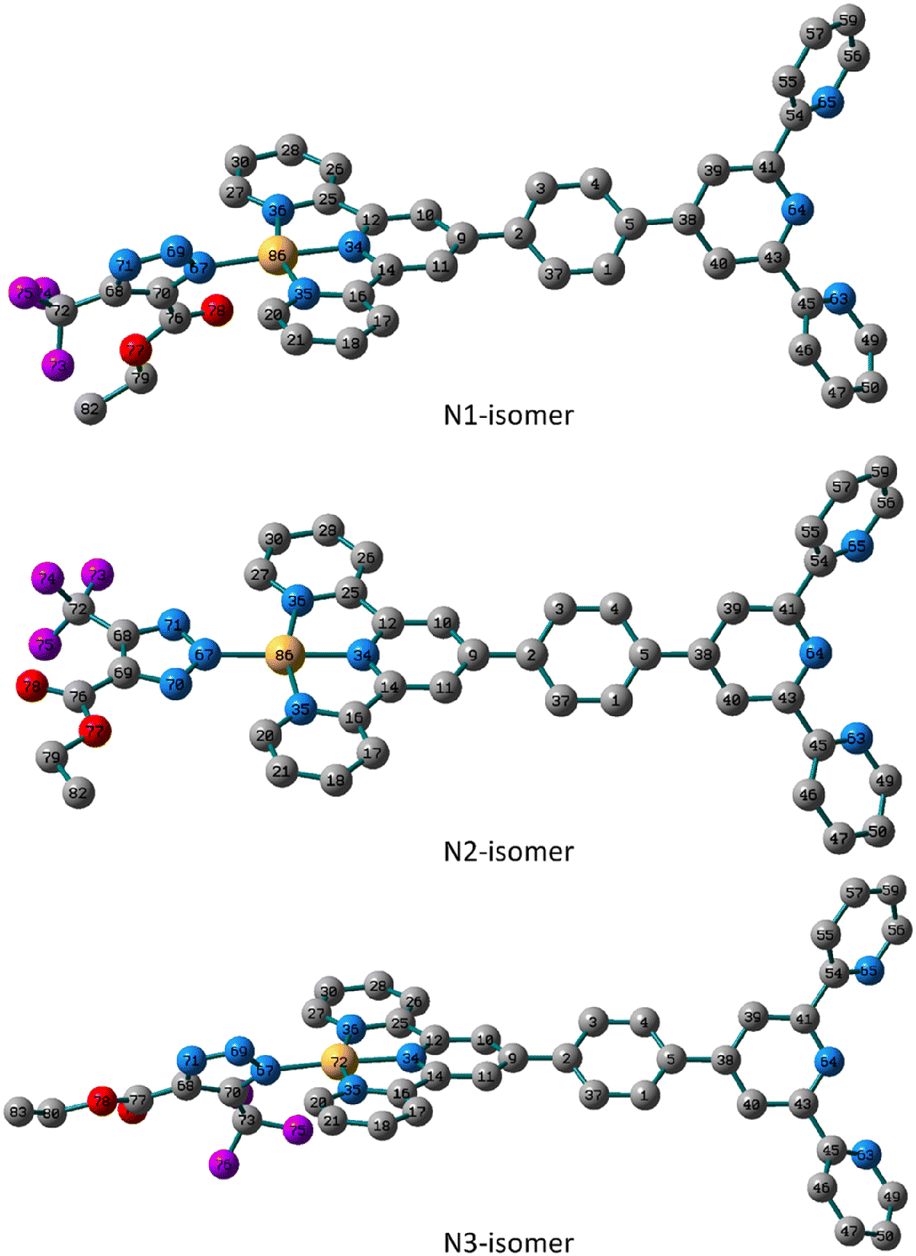 | ||
| Fig. 3 Local minimum structures of the optimized triazolate binuclear Pt(II) isomers of 6 (N1, N2 and N3) calculated at B3LYP/LANL2DZ level of theory. (Hydrogen-atoms were omitted for the clarity). | ||
2.3. Natural bond orbital analyses
It was feasible to determine the natural charge, type of bonding, type of hybridization, and strength of the M−N bonds in all the three triazolate isomers of compounds 5 and 6 using the natural bond orbital (NBO) analysis of Weinhold and co-workers21 and the second order perturbation theory analysis. In addition, the NBO technique has been widely employed in the evaluation of intra- and intermolecular contacts, and it provides a suitable basis for studying charge transfer or conjugative interactions in molecular systems. NBO analysis was carried out to determine the contribution of atomic orbitals (AOs) to NBO σ and π hybrid orbitals for bonded atom pairs. There are three NBO hybrid orbitals defined: bonding orbital (BD), lone pair (LP), and core (CR). The ideal Lewis structure is built from Lewis σ-type (donor) NBOs and non-Lewis σ-type (acceptor) NBOs. Covalency in molecules is defined by filled NBOs, whereas anti-bonds stand for empty valence-shell capacity and span regions of the atomic valence space that are suitably unsaturated by covalent electrons. Weak valence anti-bond occupancies indicate intricate deviation from an idealised localised Lewis picture, i.e., real “delocalization effects.” The electronic configuration of the metal centre (Pd or Pt) of the triazolate isomers (N1, N2 and N3), its 4d (or 5d) electronic distribution and its natural atomic charge are presented in Table S4.† According to the NBO analysis, there appears to be no substantial difference between the electronic structures of the triazolate isomers of 5. For example, the electronic configurations of the Pd ions are essentially identical, and the natural charges of the metal ions have not changed significantly, with all values falling within the range of 0.68069–0.69165. The difference between the two natural charges in the N3 isomer is insignificant (10−4 e), but it is 0.0023e in the N2 isomer. The computed natural charge on Pd atom is much lower than the formal charge (+2) due to electron density donation from the terpyridine and triazolato ligands. The population of 4d(Pd) of 9.33 does not match to the oxidation state Pd(II), which is consistent with ligand-to-d(Pd) electron transfer. On the other hand, the electronic structures of the two metal ions in the same molecule do not differ much. For example, the electronic arrangements of Pd ions in N2-isomer of 5 are nearly the same, [Kr]5s0.324d8.805p0.205d0.01. The calculated natural charges of Pd ions are 0.68069e and 0.68299e. The occupancies of 4d orbitals are as follows: dxy1.97206 dxz1.94229 dyz1.85229 dx2−y21.1085 dz21.92903 and dxy1.95252 dxz1.96486 dyz1.774671 dx2−y21.47997 dz21.65949. In comparison, the difference in the natural charges between the two metal centres in the N2 isomer appears to be attributable mainly to a change in the electron occupancies between dx2−y2 and dz2 orbitals.Hyperconjugation is described as a stabilising effect caused by the overlap of one occupied and another electron deficient orbital. The hyper-conjugative interaction energy between the donor–acceptor orbitals was calculated using the second-order perturbation technique of the Fock Matrix in NBO basis. The stronger the electronic interactions between the donors such as ligand coordination sites and acceptors e.g., metal ions, the higher the value of second order interaction energy (E2). The higher the inclination of electron donors to donate to electron acceptors, and the larger the level of conjugation of the entire system. These interactions can be identified by observing a rise in the electron density of the anti-bonding orbital, which weakens the respective bonds. According to NBO analysis, the four bonds between Pd and the N-atoms can be described as donation of electron density from a lone pair (LP) orbital on each nitrogen atom to palladium molecular orbitals. The E2 values of LP(1)N34→, LP(1)N35→, LP(1)N36→, and LP(1)N69 → RY*(1)Pd are 0.34, 0.88, 0.88, and 0.83 kcal mol−1, respectively. The corresponding E2 values of the other metal centre in the same molecule, LP(1)N63→, LP(1)N64→, LP(1)N65→, and LP(1)N72 → RY*(1)Pd are 0.91, 0.38, 0.91 and 0.90 kcal mol−1, respectively. It can see that the interactions between the terminal nitrogen atoms of the terpyridine ligand and Pd ion are equal, and they are stronger than those of the central terpyridine nitrogen and triazolate nitrogen atom. There is a minor discrepancy in the E2 values of the M–N bonds between the two metal centres.
Like the Pd(II) triazolate complexes, the electronic configuration of the Pt(II) ion in the three isomers ([Xe]6s0.475d8.596p0.206d0.01) are the same (Table S4†).Thus, 67.99 core electrons, 9.26 valence electrons (on 6s, 5d, and 6p atomic orbitals), and 0.01 Rydberg electrons (on 6d orbital) give the 77.25 electrons. The natural charges of the Pt ion have not changed significantly among the three isomers, 0.71638e (N1), 0.73683e (N2) and 0.70854e (N3). The calculated natural charge on Pt atom is substantially lower than the formal charge (+2) due to electron density donation from the surrounding ligands. The population of 5d(Pt) of 9.27 does not match to the oxidation state Pt(II), which is consistent with ligand-to-d(Pt) electron transfer. In the N2 isomer, the σ(Pt–N34) bond is formed from a sp2.85 hybrid on terpyridine N34 atom (which is the mixture of 26.00% s, and 74.00% p atomic orbitals) and sp0.40d5.87 hybrid on platinum atom (which is the mixture of 13.77% s, 5.46% p and 80.77% d atomic orbitals). NBO analysis indicated that the σ(Pt–N34) bond is strongly polarized towards the nitrogen atom with about 75.86% of electron density concentrated on the nitrogen atom. In the most stable triazolate isomer, the N1 isomer, the four bonds formed by Pt and N-atoms are defined as electron density donations from a lone pair (LP) orbital on each nitrogen atom to Pt molecular orbitals. The E2 values of LP(1)N34→, LP(1)N35→, LP(1)N36→, and LP(1)N67 → RY*(4)Pt are 0.15, 0.48, 0.39, and 0.73 kcal mol−1, respectively. It can see that the interaction between the triazolate N atom and Pt ion is stronger than those of the terpyridine nitrogen atoms.
2.4. Time-dependent density functional theory calculations
To get an insight into the nature of electronic transitions anticipated to be seen in the electronic spectra of the three triazolate isomers of complexes 5 and 6, time-dependent density functional theory (TDDFT) calculations were performed in the singlet state using a Becke 3-parameter (exchange) Lee–Yang–Parr functional31,32 with LANL2DZ basis set33,34 and PCM solvation model (DMSO). GAUSSSUM was used to simulate the electronic spectra. A Gaussian convolution with a full-width at half-maximum (FWHM) of 3000 was used to incorporate each excited state. The three triazolate isomers of 5 have virtually identical electronic absorption spectra (Fig. S31†), having two main absorption bands at around 307 and 370 nm. The band at 370 nm shifts slightly when we move from N1, to N3, and finally to N2. The lowest energy band at 307 in all the three isomers is due to HOMO → LUMO. As seen in Fig. 4, HOMO is primarily formed of π(triazolate), whereas LUMO is primarily composed of the terpyridine π-framework with a tiny contribution from d(Pd). Thus, the transition at around 370 nm is assigned to LLCT initiating from the triazolate moiety and terminating at terpyridine ligand. For N1 isomer, the highest energy band at 306 nm is equivalent to HOMO−4 → LUMO+1. This band has a ground state composed of d(Pd)/π(triazolate) and excited state of π(terpyridine) forming LLCT/MLCT. For N2 isomer, the band at 309 nm (HOMO−8 → LUMO/HOMO−1 → LUMO+1) (Table S5†) is LLCT based on the descriptions of the frontier molecular orbitals and the relocation of the electron density of N2 isomer (Fig. 4). In the case of N3-isomer, the band at 305 nm (HOMO−9 → LUMO/HOMO−3 → LUMO+1) is unique as it comprised of π–π* within the triazolate moiety as well as LLCT as that observed in the case of the other isomers. Therefore, all the three isomers of 5 demonstrated two kinds of transitions, LLCT and MLCT.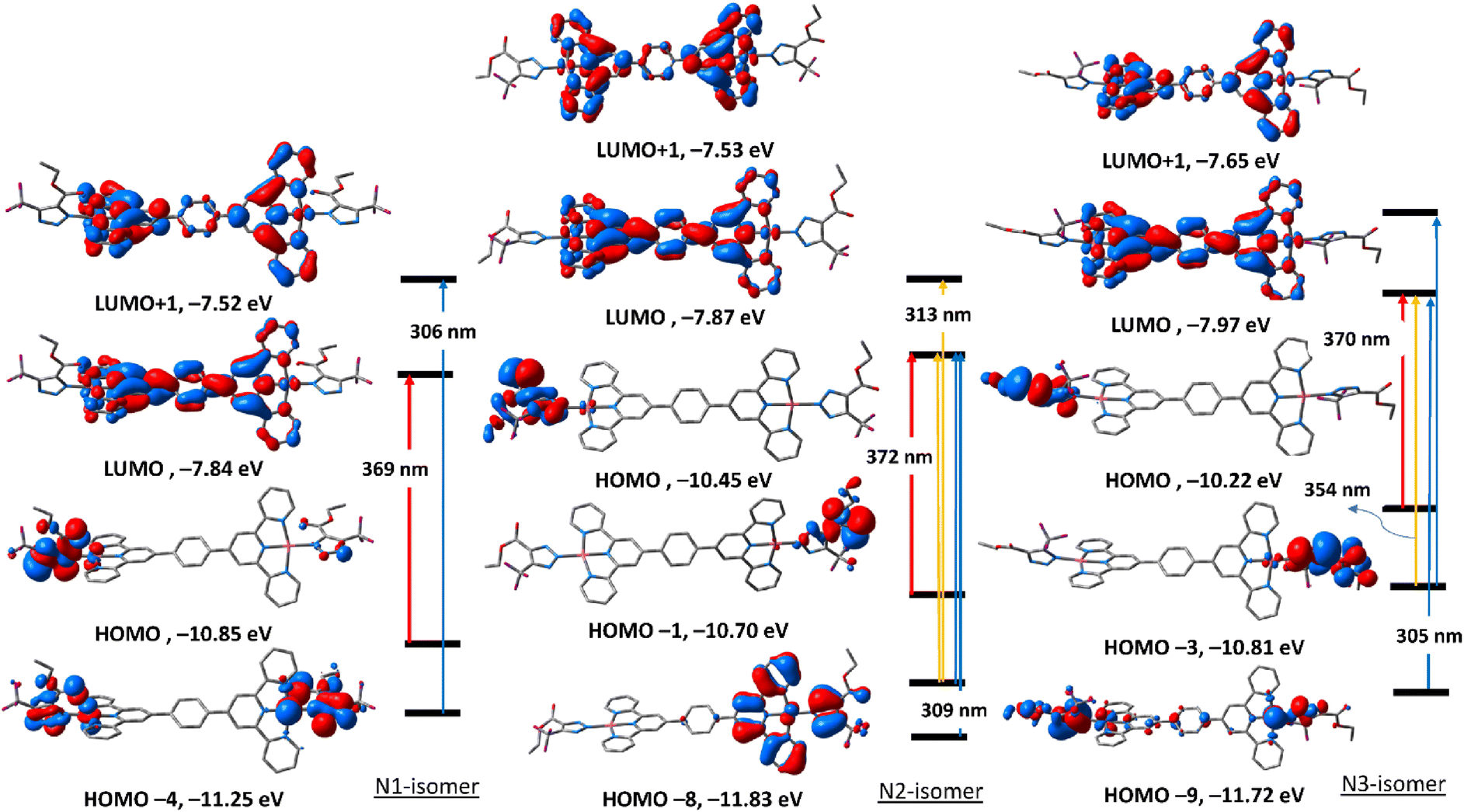 | ||
| Fig. 4 Selected frontier molecular orbitals of the three triazolate isomers (N1, N2 and N3) of 5 obtained by the B3LYP/LANL2DZ method. The electronic transitions were obtained at the same level of theory. | ||
The electronic absorption spectra (Fig. S32†) of the triazolate isomers of 6 are characterised by two major bands at roughly 296 and 400 nm, with the higher energy band having a greater oscillator strength value than the lower energy band. The band at around 400 nm is due to HOMO → LUMO. As shown in Fig. 5, HOMO orbital of N1 isomer is contained upon π-system of the phenyl ring of the terpyridine ligand, and LUMO is of d(Pt). Thus, the band at 400 nm is assigned to LMCT. For N2 and N3-isomers, the transitions at 408 and 403 nm may be assigned to LLCT/MLCT initiating from π(uncoordinated terminal pyridyl rings) and terminating at the electron-deficient π-system of the coordinated pyridyl rings/d(Pt). Changing the coordination mode of the triazolate ring from N1 to N2 or N3 appears to have a significant impact on the overall electronic structure of this class of Pt(II) complexes. The band at 293 nm in N1 isomer may be assigned to LLCT from π(phenyl) → π* (coordinated terminal pyridyl ring). The band at around 295 nm in both N2 and N3-isomers may be attributed to LLCT from the electron donor free-terminal pyridyl rings to π-system of the terpyridine ring. Overall, it seems that the electronic structure of the N2 isomer is more similar to that of N3.
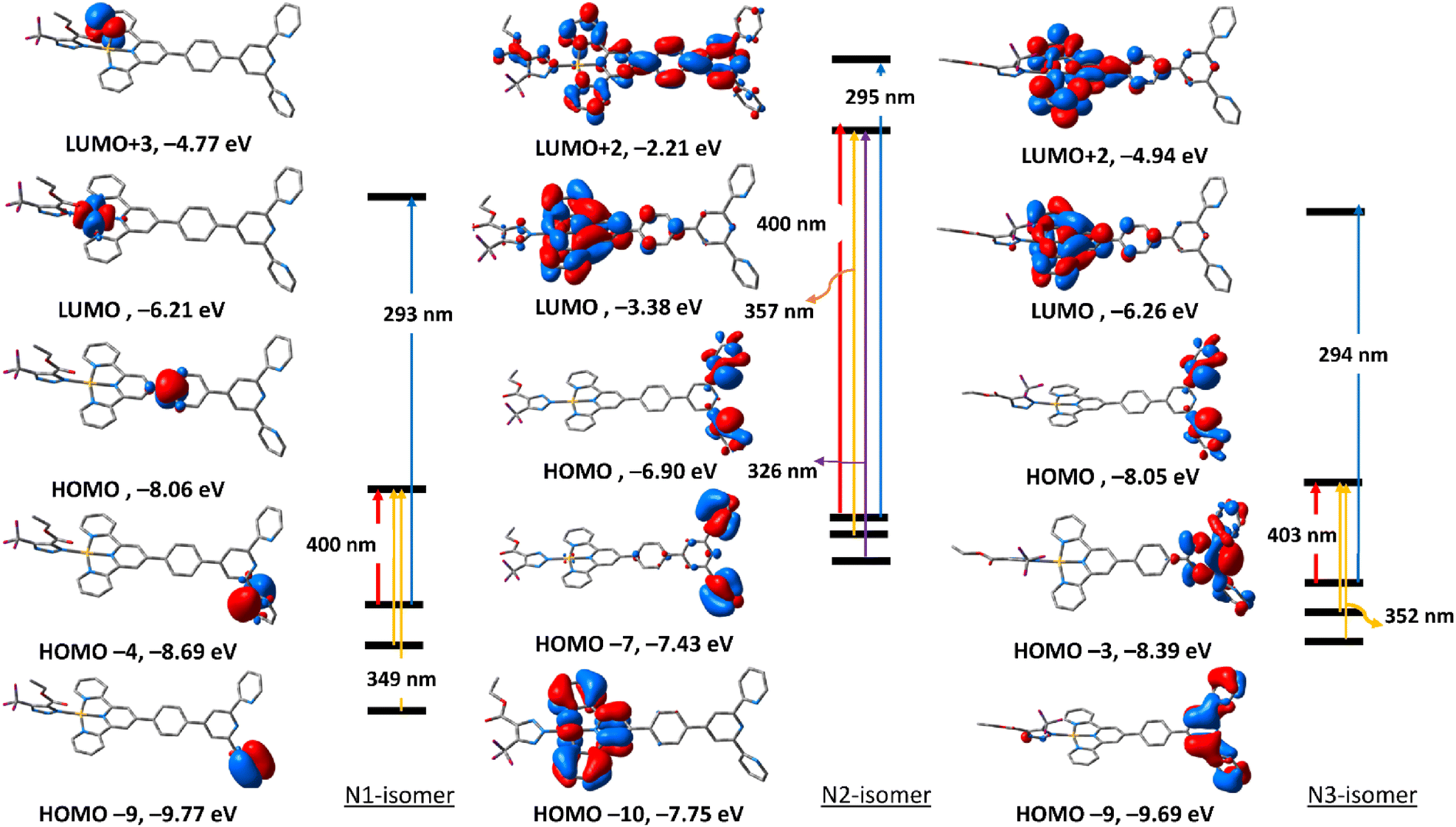 | ||
| Fig. 5 Selected frontier molecular orbitals of the three triazolate isomers (N1, N2 and N3) of 6 obtained by the B3LYP/LANL2DZ method. The electronic transitions were obtained at the same level of theory. | ||
3. Conclusion
To evaluate the opportunity of functionalizing biomolecules with [Pd2(Cl)2L]2+ and [Pt(Cl)L]+ (L = 1,4-bis(2,2′:6′,2′′-terpyridin-4′yl)benzene) through the inorganic click reaction, the chlorido ligand(s) was exchanged with N3− group, which was then coupled with electron poor alkyne (4,4,4-trifluoro-2-butynoic acid) under the catalyst-free conditions, yielding triazolate analogues. Under similar experimental conditions, it appears that the reaction between the Pd(II) precursor and terpyridine ligand afforded a binuclear complex, whereas the same reaction with Pt(II) precursor gave a mononuclear complex. The chloro and azido complexes are insoluble or break down in most organic solvents, however, the corresponding triazolate complexes are soluble in the same solvents. Therefore, the solubility of this class of compounds could be improved by exchanging the chlorido ligand for a triazolate moiety. Solid-state NMR and ASAP-MS were applied to elucidate the structures of insoluble complexes.The reactivity of the azido complexes towards 4,4,4-trifluoro-2-butynoic acid as well as the nature of the isolated triazolate isomers were performed by NMR spectroscopy particularly 19F NMR. In our situation, the reactions of the azido complexes with the asymmetric alkyne is anticipated to result in the formation of three triazolate bound modes (N1, N2 and N3). According to the 19F NMR spectroscopy, the cycloaddition reaction of the azido Pd(II) complex gave mainly a mixture of N1-, and N2-isomers. Parallelly, the total electronic and zero-point energy values of the three isomers of the Pd(II) binuclear complex were compared. The calculations showed that N2-isomer is more stable than N1 and N3 analogues as well as the N1 isomer is more stable than N3 that agrees with the experimental data. In the example of the Pt(II) complex, the comparison of total electronic and zero-point energy values suggested that the most prevalent species of this complex should be either N1 or N3. Time-dependent density functional calculations showed that there is no significant difference in the electronic structures of the three isomers and the observed transitions might be assigned to LLCT or MLCT. According to the natural bond orbital analysis, there appears to be no substantial difference, in terms of natural charge and the populations of the d-subshell, between electronic structures of the three triazolate isomers. It can see that the interactions between the terminal nitrogen atoms of the terpyridine ligand and Pd ion are equal, according to the values of second order interaction energy (E2), and they are stronger than those of the central terpyridine nitrogen and triazolate nitrogen atom. On the contrary, it can see that the interaction between the triazolate N atom and Pt ion is stronger than those of the terpyridine nitrogen atoms. Overall, this contribution sheds light on how to bioconjugate square-planar complexes using the inorganic click reaction, how to improve the solubility of extremely insoluble intriguing complexes, and the nature of the generated species in relation to the nature of the metal ions.
4. Experimental section
4.1. Materials and instruments
On oven-dried glassware, the experiments were conducted in anhydrous solvents. Ammonium hexafluorophosphate, sodium azide, sodium tetrachloropalladate, and potassium tetrachloro- platinate(II), were all purchased from commercial sources and utilised exactly as they were given. 1,4-Bis(2,2′:6′,2′′-terpyridin-4′yl)benzene,22 and 4,4,4-trifluoro-2-butynoic acid ethyl ester,35 were synthesized by following the published schemes. Metal azides can degrade quickly and unexpectedly. Scratching metal azides can result in explosive reactions. Only small-scale synthesis should be carried out, and heating of solid compounds must be avoided at all costs. As a result, handling and purification must be done with the utmost care. The CHN elemental compositions were recorded using either the HEKtech EA 3000 elemental analyzer or the Elementar Vario MICRO cube CHN analyzer. On a Thermo Fisher Exactive Plus Orbitrap mass, electrospray ionisation mass spectra were collected at a resolution of R = 70.000 and a solvent flow rate of 50 L min. To collect vibrational spectra in the solid state, a Nicolet 380 FT-IR spectrometer with a smart iFTR adapter was employed. 1H, 13C, 19F and 31P NMR spectra were recorded with Brucker-Avance 500 (1H, 500.13 MHz; 13C{1H}, 125.77 MHz; 19F, 470.59 MHz; 31P, 202.46 MHz) or Bruker-Avance 400 (1H, 400.40 MHz; 13C{1H}, 100.68 MHz; 31P, 162.08 MHz) spectrometers. Using two-dimensional NMR techniques, {1H, 1H} COS90 and {1H, 13C} HSQC, NMR signal assignments were carried out.4.2. Synthesis
2: A 5 mL aqueous solution of K2PtCl4 (1.0 mmol; 415 mg) was poured to a round-bottomed flask that was already charged with terpyridine derivative (0.5 mmol; 270 mg) and 50 mL of methanol. For 12 h, the reaction mixture was heated to reflux, at which point brown precipitate formed. Filtration was done. The product was cleaned with water and diethyl ether, and then dried under vacuum. Yield: 66% (312 mg, 0.42 mmol). IR (ATR): ν = 3515 (br, OH2), 3060 (w, CH), 1584 (m, CC/CN), 1471, 1394, 1256, 1120, 896, 787, 739 cm−1. Complex 2 decomposes also in DMSO-d6 and did not dissolve in the remaining organic solvents. 13C-NMR (solid-state, 100.68 MHz): δ = 155.3, 154.7, 152.7, 149.5, 145.1, 144.4, 143.9, 142.7, 140.6, 135.4, 134.3, 131.0, 129.1, 127.8, 126.3, 125.7, 124.9, 123.7, 122.6, 120.8, 119.3 ppm. 15N NMR (solid-state, 40.56 MHz): δ = −177.0, −174.3, −80.8, −70.9, −67.3 ppm. ASAP (ESI+): 770.1393 {[M-0.5PtCl4]}+. C36H24ClN6Pt·0.5PtCl4·4H2O: C, 44.32; H, 2.89; N, 8.61, found C, 44.20; H, 2.95; N, 8.60.
4: A round-bottomed flask was filled with 200 mg of compound 2 (0.21 mmol) and 100 mL of a 60% (v/v) acetone–water solution before being charged with sodium azide (0.015 mol; 1 g). For 5 days, the reaction mixture was heated at 50 °C. Then, ammonium hexafluorophosphate (6.13 mmol; 1 g) was added to the reaction mixture. Stirring was continuing for 6 h. A brown precipitate was filtrated off, rinsed with water and diethyl ether, and then dried for a few days under vacuum. Complex 4 is insoluble in most organic solvents and breaks down in DMSO-d6. Yield: 70% (138 mg, 0.15 mmol). IR (ATR): ν = 3062 (w, CH), 2022 (s, N3), 1581 (m, CC/CN), 1468, 1390, 1259, 837, 786, 739 cm−1. 13C-NMR (solid-state, 100.68 MHz): δ = 155.2, 154.6, 152.8, 149.6, 145.2, 144.5, 142.6, 140.7, 136.0, 135.2, 134.1, 130.9, 129.0, 127.9, 126.4, 125.5, 123.7, 122.6, 121.3, 120.7, 119.3 ppm. 15N NMR (solid-state, 40.56 MHz): δ = −68.2, −71.6, −306.4 ppm. 19F-NMR (solid-state, 396.5 MHz): δ = −69.4 (br, PF6−) ppm. ASAP (ESI+): 777.1800 {[PtN3L]}+, 541.2138 {L + H}+. [PtN3L]·Y·H2O (YN3 or PF6−) ((C36H24N9Pt)2·N3·PF6·H2O): C, 49.12; H, 2.86; N, 16.71; found C 49.17, H 3.04, N 15.98.
O), 1606 (m, CC/CN), 1440, 1312, 1248, 1135, 1028, 830, 784, 748, 724 cm−1. 1H NMR (d6-DMSO, 500.13 MHz) (broad spectrum): δ = 8.74, 8.63, 8.47, 8.34, 8.17, 7.81, (aromatic signals), 4.39 (CH2), 1.41 (CH3) ppm. 13C-NMR (d6-DMSO, 125.77 MHz): δ = 159.2 (CO), 156.4, 154.5, 152.4, 142.8, 138.7 (q, 2JC,F = 39.4 Hz, C−CF3), 137.6, 136.5, 129.1, 128.6, 127.8, 121.7 (q, 1JC,F = 267.0 Hz, –CF3), 121.4, 60.9 (–CH2), 13.7 (–CH3) ppm. 19F NMR (d6-DMSO, 490.59 MHz): δ = −70.2 (d, 1JPF = 711 Hz, PF6), −58.95 (s, CF3, major species, I) and −59.04 (s, CF3, minor species, II) ppm. 31P NMR (d6-DMSO, 162.08 MHz): δ = −144.2 (sept, 1JPF = 711 Hz, PF6) ppm. MS (ESI+, DMSO/acetone): m/z = 1315.0461 {[Pd2(triazolato)2L]·PF6}+, 854.1431 {[Pd(triazolato)L]}+. C48H34F18N12O4P2Pd2·4H2O: C 37.64, H 2.76, N 10.97, found C 37.71, H 2.37, N 9.85.6: To a small round flask charged with complex 4 (0.1 mmol; 100 mg) and acetone (10 mL), 4,4,4-trifluoro-2-butynoic acid ethyl ester (0.1 mmol; 17 mg) was added. The reaction mixture was stirred until the solution became clear, which took 7 d. Filtration was done and then one mL of a saturated solution of NH4PF6 was added. Stirring was continuing for 6 h. A brown precipitate was filtrated off, rinsed with water and diethyl ether, and then dried for a few days under vacuum. Yield: 53 (63 mg, 0.05 mmol). IR (ATR): ν = 3325, 2992 (w, CH), 1728 (s, CO), 1606 (s, CC), 1436, 1310, 1139, 1061, 829, 786 cm−1. 31P NMR (d6-DMSO, 162.08 MHz): δ = −144.2 (sept, 1JPF = 712 Hz, PF6) ppm. 19F NMR (d6-DMSO, 490.59 MHz): δ = −70.1 (sept, 1JPF = 699 Hz, PF6), −59.89, −60.13 and −60.26 ppm. MS (ESI+, DMSO/acetone): m/z 1110.2287 {Na[Pt(triazolato)L]·PF6}+. C42H29F3N9O2Pt·3H2O: C 44.14, H 3.09, N 11.03, found C 43.85, H 3.14, N 10.79.
4.3. Density functional theory calculations
The B3LYP31,32/LANL2DZ33,34 approach was used to fully optimize the geometries of the triazolate isomers of 5 and 6 in the ground state. To get an insight into the local minimum structures of the triazolate isomers were verified by computing the vibrational modes at the same level of theory. Natural bond orbital analyses and TDDFT were performed using the previous mentioned DFT approach. All the calculations were carried out using Gaussian03.38 Visualization of the optimized structures and frontier molecular orbitals were done with Gaussview03.39Data availability
Data is in ESI† file.Author contributions
Ahmed Mansour: conceptualization, supervision, investigation, validation, resources, formal analysis, software, writing – original draft, writing – review & editing, resources. Krzysztof Radacki: methodology, investigation, resources. Gamal A. E. Mostafa, writing – review & editing, resources. Essam A. Ali, writing-review & editing, resources. Ola Shehab: investigation, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project Number (RSPD2023R1000), King Saud University Riyadh Saudi Arabia for financial support. The authors wish to thank United Arab Emirates University, Al Ain, UAE for financial support. Funding: This research work was supported by researchers supporting project number (RSPD2023R1000) King Saud University, Riyadh, Saudi Arabia.References
- H.-W. Frühauf, Organotransition metal [3+2] cycloaddition reactions, Coord. Chem. Rev., 2002, 230, 79–96 CrossRef.
- W. P. Fehlhammer and W. Beck, Azide Chemistry–An Inorganic Perspective, Part II [‡][3+2]-Cycloaddition Reactions of Metal Azides and Related Systems, Z. Anorg. Allg. Chem., 2015, 641, 1599–1678 CrossRef CAS.
- W. Beck and W. Fehlhammer, Reaktionsweisen von Azido-Metall-Komplexen, Angew. Chem., 1967, 79, 146–147 CrossRef.
- W. Beck, W. P. Fehlhammer, H. Bock and M. Bauder, Zur Reaktoin von Azido-Metallverbindungen: Tetrazolato-Metallkomplexe, Chem. Ber., 1969, 102, 3637–3646 CrossRef CAS.
- P. Paul and K. Nag, Sulfur-nitrogen-bonded metal chelates. 18. 1,3-Dipolar cycloadditions to coordinated azide in nickel(II) complexes of the types [Ni(SNN)(N3)] and [(SNN)Ni(N3) Ni(NNS)](ClO4), Inorg. Chem., 1987, 26, 2969–2974 CrossRef CAS.
- F.-C. Liu, Y.-L. Lin, P.-S. Yang, G.-H. Lee and S.-M. Peng, [3+2] Cycloadditions of Molybdenum(II) Azide Complexes with Nitriles and an Alkyne, Organometallics, 2010, 29, 4282–4290 CrossRef CAS.
- A. M. Mansour and K. Radacki, Spectroscopic and antimicrobial activity of photoactivatable tricarbonyl Mn(I) terpyridine compounds, Inorg. Chim. Acta, 2020, 511, 119806 CrossRef CAS.
- A. M. Mansour, O. R. Shehab and K. Radacki, Role of Sulfonate Appendage in the Protein Binding Affinity of Half-Sandwich Ruthenium(II)(η6-p-Cym) Complexes, Eur. J. Inorg. Chem., 2020, 2020, 299–307 CrossRef CAS.
- Y.-J. Kim, X. Chang, J.-T. Han, M. S. Lim and S. W. Lee, Cyclometallated Pd(ii) azido complexes containing 6-phenyl-2,2′-bipyridyl or 2-phenylpyridyl derivatives: synthesis and reactivity toward organic isocyanides and isothiocyanates, Dalton Trans., 2004, 3699–3708, 10.1039/B411432A.
- A. M. Mansour, K. Radacki and O. R. Shehab, Role of the ancillary ligand in determining the antimicrobial activity of Pd (II) complexes with N,N,N-tridentate coligand, Polyhedron, 2022, 221, 115857 CrossRef CAS.
- A. M. Mansour and O. R. Shehab, Triazolato Pd (II) and Pt (II) complexes of 2,6-bis (1-ethylbenzimidazol-2′-yl) pyridine formed via catalyst-free [3+2] click reactions, Inorg. Chim. Acta, 2023, 548, 121379 CrossRef CAS.
- A. M. Mansour, K. Radacki and O. R. Shehab, Role of the ancillary ligand in controlling the lysozyme affinity and electronic properties of terpyridine fac-Re (CO) 3 complexes, Dalton Trans., 2021, 50, 1197–1201 RSC.
- A. M. Mansour and K. Radacki, Terpyridine based ReX (CO) 3 compounds (X = Br–, N3– and triazolate): Spectroscopic and DFT studies, Polyhedron, 2021, 194, 114954 CrossRef CAS.
- P. V. Simpson, B. W. Skelton, P. Raiteri and M. Massi, Photophysical and photochemical studies of tricarbonyl rhenium(i) N-heterocyclic carbene complexes containing azide and triazolate ligands, New J. Chem., 2016, 40, 5797–5807 RSC.
- S. M. de Salinas, J. Díez, M. P. Gamasa and E. Lastra, 1, 3-Dipolar Cycloadditions of Rhodium (III) Azido Complexes with Alkynes and Nitriles, ChemistrySelect, 2017, 2, 3172–3177 CrossRef.
- A. M. Mansour, K. Radacki and O. R. Shehab, Half-sandwich triazolato Rh (III) compound of pyridylbenzimidazole ligand with cell selective toxicity towards Cryptococcus neoformans, J. Organomet. Chem., 2021, 949, 121928 CrossRef CAS.
- H.-K. Kim, J.-H. Lee, Y.-J. Kim, Z. Nu Zheng and S. W. Lee, (π-Allyl)Pd Complexes Containing N-Heterocyclic Carbene and Pseudohalogen Ligands – Synthesis, Reactivity toward Organic Isothiocyanates and Isocyanides, and Their Catalytic Activity in Suzuki–Miyaura Cross-Couplings, Eur. J. Inorg. Chem., 2013, 2013, 4958–4969 CrossRef CAS.
- X. Chang, M.-Y. Kim, Y.-J. Kim, H. S. Huh and S. W. Lee, Dinuclear palladium–azido complexes containing thiophene derivatives: reactivity toward organic isocyanides and isothiocyanates, Dalton Trans., 2007, 792–801, 10.1039/B616901E.
- K. Peng, V. Mawamba, E. Schulz, M. Löhr, C. Hagemann and U. Schatzschneider, iClick reactions of square-planar palladium (II) and platinum (II) azido complexes with electron-poor alkynes: metal-dependent preference for N1 vs. N2 triazolate coordination and kinetic studies with 1H and 19F NMR spectroscopy, Inorg. Chem., 2019, 58, 11508–11521 CrossRef CAS PubMed.
- N. J. Farrer, G. Sharma, R. Sayers, E. Shaili and P. J. Sadler, Platinum(IV) azido complexes undergo copper-free click reactions with alkynes, Dalton Trans., 2018, 47, 10553–10560 RSC.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- A. M. Mansour and K. Radacki, Structural Studies, Antimicrobial Activity and Protein Interaction of Photostable Terpyridine Silver(I) Complexes, Eur. J. Inorg. Chem., 2019, 2019, 4020–4030 CrossRef CAS.
- L. Pazderski, in Annual reports on NMR Spectroscopy, Elsevier, 2013, vol. 80, pp. 33–179 Search PubMed.
- W. P. Ozimiński, P. Garnuszek, E. Bednarek and J. C. Dobrowolski, The platinum complexes with histamine: Pt (II)(Hist) Cl2, Pt (II)(Iodo-Hist) Cl2 and Pt (IV)(Hist) 2Cl2, Inorg. Chim. Acta, 2007, 360, 1902–1914 CrossRef.
- J. Mason, L. F. Larkworthy and E. A. Moore, Nitrogen NMR spectroscopy of metal nitrosyls and related compounds, Chem. Rev., 2002, 102, 913–934 CrossRef CAS PubMed.
- W. Beck, T. M. Klapötke, J. Knizek, H. Nöth and T. Schütt, Characterization of Palladium (II) Azide and Crystal Structure of Bis (azido) bis (pyridine) palladium (II) and Tetramminepalladium Tetraazidopalladate (II), Eur. J. Inorg. Chem., 1999, 1999, 523–526 CrossRef.
- F.-C. Liu, Y.-L. Lin, P.-S. Yang, G.-H. Lee and S.-M. Peng, [3+2] Cycloadditions of molybdenum (II) azide complexes with nitriles and an alkyne, Organometallics, 2010, 29, 4282–4290 CrossRef CAS.
- R. Kumar, R. Kishan, J. M. Thomas, S. Chinnappan and N. Thirupathi, Probing the factors that influence the conformation of a guanidinato ligand in [(η 5-C 5 Me 5) M (NN) X](NN= chelating N, N′, N′′-tri (o-substituted aryl) guanidinate (1−); X = chloro, azido and triazolato), New J. Chem., 2018, 42, 1853–1866 RSC.
- K. Peng, V. Mawamba, E. Schulz, M. Löhr, C. Hagemann and U. Schatzschneider, iClick reactions of square-planar palladium (II) and platinum (II) azido complexes with electron-poor alkynes: metal-dependent preference for N1 vs. N2 triazolate coordination and kinetic studies with 1H and 19F NMR spectroscopy, Inorg. Chem., 2019, 58, 11508–11521 CrossRef CAS PubMed.
- P. V. Simpson, B. W. Skelton, P. Raiteri and M. Massi, Photophysical and photochemical studies of tricarbonyl rhenium (i) N-heterocyclic carbene complexes containing azide and triazolate ligands, New J. Chem., 2016, 40, 5797–5807 RSC.
- A. Becke, Density-functional thermochemistry. III. The role of exact exchange, Chem. Phys., 1993, 98, 5648 CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- B. Hamper, T. Harrison and L. Overman, α-Acetylenic esters from α-acylmethylenephosphoranes: ethyl 4, 4, 4-trifluorotetrolate, Org. Synth., 1992, 70, 246–250 CrossRef CAS.
- F. P. Gabbaï, P. J. Chirik, D. E. Fogg, K. Meyer, D. J. Mindiol, L. L. Schafer and S.-L. You, An Editorial About Elemental Analysis, Organometallics, 2016, 35, 3255–3256 CrossRef.
- A. M. Mansour, Terpyridine Zn (II) azide compounds: Spectroscopic and DFT calculations, J. Mol. Struct., 2021, 1242, 130737 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, J. C. B. R. E. Stratmann, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, A. L. G. Liu, I. K. P. Piskorz, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 03 (Revision A.9), Gaussian, Inc., Pittsburgh, 2003 Search PubMed.
- A. Frisch, A. B. Nielson and A. J. Holder, Gaussview User Manual, Gaussian, Inc., Pittsburgh, PA, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06656h |
| This journal is © The Royal Society of Chemistry 2023 |