
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of reaction variables on the surface chemistry of cellulose nanofibers derived from palm oil empty fruit bunches†
Andrea P.
Martínez-Ramírez
 a,
Sergio A.
Rincón-Ortiz
b,
Víctor G.
Baldovino-Medrano
b,
Cristian
Blanco-Tirado
a and
Marianny Y.
Combariza
*a
a,
Sergio A.
Rincón-Ortiz
b,
Víctor G.
Baldovino-Medrano
b,
Cristian
Blanco-Tirado
a and
Marianny Y.
Combariza
*a
aEscuela de Química, Universidad Industrial de Santander, Bucaramanga, 680002, Colombia. E-mail: marianny@uis.edu.co
bLaboratorio de Ciencia de Superficies (#SurfLab-UIS), Parque Tecnológico de Guatiguará, Universidad Industrial de Santander, Piedecuesta, 681011, Colombia
First published on 11th December 2023
Abstract
Nanocellulose, a versatile nanomaterial with a wide range of applications, is gaining significant attention for its sustainable and eco-friendly properties. In this study, we investigate the influence of reaction variables on the surface chemistry of TEMPO-oxidized cellulose nanofibers (TOCN) from palm oil empty fruit bunch (EFB) fibers, a high cellulose content biomass. Reaction time, primary oxidizing agent, and a pretreatment process affect, to various extents, the surface chemistry of EFB-TOCN. Conductometric titrations (CT), X-ray photoelectron spectroscopy (XPS), and statistical analysis indicate a positive and significant influence of reaction time and primary oxidizing agent on EBF-TOCN degree of oxidation and surface charge density. Partial EFB delignification increased EFB-TOCN oxidation and reaction yield compared to EFB without pretreatment. Interestingly, only reaction time has a significant effect on the EFB-TOCN hydrodynamic radii, with a reaction time of over 120 minutes required to obtain nanocellulose less than 100 nm in size. Utilizing palm oil residual biomass for nanocellulose extraction not only valorizes agricultural waste but also enhances the palm oil industry's economic prospects by reducing waste disposal costs and improving material circularity. This research contributes to the growing body of knowledge on nanocellulose production from renewable sources and highlights the potential of palm oil EFB fibers as a valuable raw material for sustainable nanomaterial development.
1. Introduction
The need for renewable, functional, and cost-effective materials has increased due to the depletion of fossil fuel reserves, the rise of synthetic plastic pollution, and the increase of greenhouse gas emissions. One solution is to use agricultural byproducts to produce lignocellulosic materials that can be transformed into biopolymers and platform chemicals via chemical transformations.1 Palm oil (PO) is a high-yield agro-industrial crop in the tropics.2 Palm oil extraction results in aqueous and solid byproducts, with the empty fruit bunch (EFB) being the most abundant solid residue, accounting for about 21 wt% of the fresh fruit bunch (FFB). EFB is typically utilized as a composting material in plantations or as a low-grade fuel in boilers.3 With a yearly production of over sixty-eight million tons, empty fruit bunches (EFB) from PO extraction have the potential to be a valuable source of biopolymers due to their high cellulose (50.9%), lignin (17.84%), and hemicellulose (29.6%) contents.4,5Cellulose, the most abundant biopolymer on earth, is the primary structural component of all plant cell walls.6 Cellulose is an unbranched homopolysaccharide constructed of D-anhydroglucose units linked by β-(1–4)-glycosidic bonds. Each monomer in cellulose has three free hydroxyl groups, one primary (C6) and two secondaries (C2, C3), able to establish intra- and intermolecular hydrogen bonds. These interactions are responsible for cellulose's highly ordered and mechanically robust macrostructure.7 Cellulose biosynthesis occurs at the cell wall level by arrangements of spinnerets incorporating an extraordinary molecular machine called cellulose synthase. During cellulose synthesis, individual strands interact to form fibrils through hydrogen bonds. However, fibril assembly does not always produce perfectly aligned individual cellulose strands, resulting in cellulose fibrils containing amorphous and crystalline regions.8,9
Cellulose extraction from biomass requires chemical, mechanical, or biological deconstruction of the natural biocomposite formed by cellulose, hemicellulose, and lignin. Nanocellulose (NC) is usually derived from pure or partially delignified bulk cellulose, although recent research focuses on nanocelluloses containing lignin.10 NC extracted from agricultural biomass is biodegradable and biocompatible, which are crucial properties for applications in the biomedical field for drug delivery, tissue engineering, and wound dressing. Biomass-based NC exhibits excellent mechanical and barrier properties that make it suitable for composites, lightweight structures, and packaging for food preservation. Also, as a modifier and additive, NC has many potential applications, such as a reinforcing agent, rheological modifier, and oil recovery processes due to its exceptional adsorption properties. NC chemical isolation processes are normally faster and more cost-effective and are usually combined with mechanical pre- or post-treatment. Typically, cellulose nanofibers (CNF) are obtained from the oxidation and complete defibrillation of the cellulose bulk macrostructure, while nanocrystals (CNC) arise from defibrillation and the elimination of the fibril's amorphous regions via hydrolysis reactions.11 The physicochemical properties of CNF and CNC largely depend on the lignocellulosic source and extraction methods.10,12
The TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) oxidation reaction is a pivotal chemical method in CNF production from biomass sources, delivering controlled disintegration, colloidal stability, surface chemistry flexibility, and sustainability.13,14 Requiring sodium hypochlorite (NaClO) as the primary oxidant and the TEMPO radical as a catalyst, the reaction is remarkably selective, targeting only the primary hydroxyl groups of cellulose and sparing the cellulose backbone from significant degradation (Scheme 1).15,16 TEMPO oxidation imparts a substantial surface charge to CNFs, due to the presence of carboxylate units in the form of sodium salts, leading to a significant electrostatic repulsion between individual nanofibers that prevents agglomeration and facilitates the formulation of stable aqueous suspensions.11 The number of carboxylate groups reported as cellulose's degree of oxidation (DO) or charge density (σ) depends on the isolation conditions and biomass source.17 DO and σ are typically calculated from conductimetric titration (CT) measurements. CNFs from TEMPO have a versatile surface chemistry that can be precisely tailored through functionalization, enabling integration into various nanocomposites and hybrid materials. This adaptability expands their utility in nanocomposite development, drug delivery, and biomaterials. The TEMPO reaction also aligns with green chemistry principles, featuring mild reaction conditions and eco-friendly reagents, minimizing waste and environmental impact.14,18 In this work, CNF from EFB was isolated using ultrasound-assisted TEMPO oxidation, followed by mechanical disintegration, using a process previously reported for residual biomass.19,20
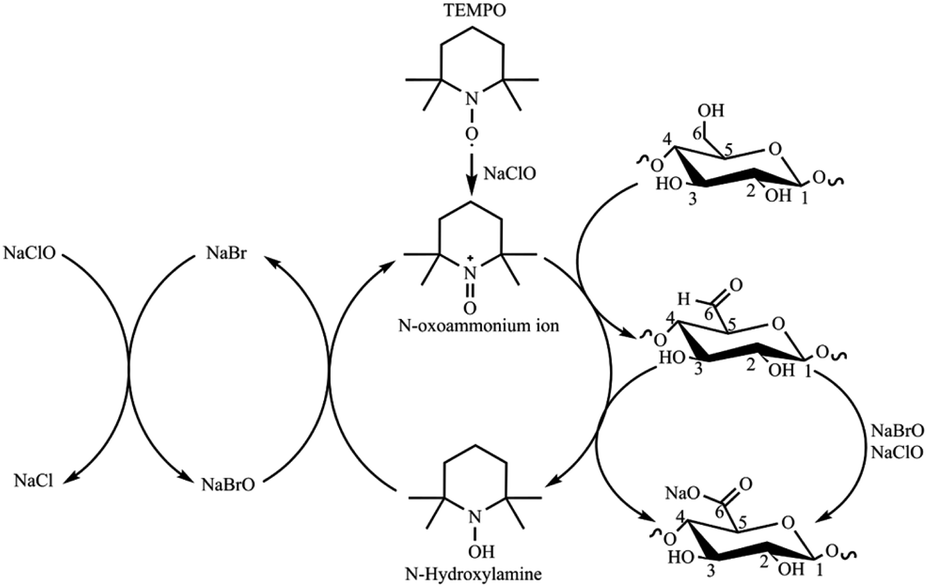 | ||
| Scheme 1 TEMPO-mediated oxidation of cellulose (adapted from ref. 15). | ||
The conditions of the TEMPO reaction and the biomass source significantly influence the physicochemical properties of TOCN. For example, under the same TEMPO oxidation experimental conditions, Norway spruce (a hardwood) and Eucalyptus (a softwood) pulps resulted in TOCN with 1.4 mmol COO− per g and 1.2 mmol COO− per g, respectively. This observation highlights the effect of the biomass source on TOCN properties.21 Huang et al.22 reported that increasing the length of the TEMPO oxidation reaction impacts DO and σ in TOCN extracted from wood pulp. Changing reaction time from 1 to 24 h resulted in increased DO (σ) from 16.4% (1.00 mmol COO− per g) to 25.0% (1.58 mmol COO− per g), respectively. The reaction length also impacted the crystal size, which decreased from 4.3 nm after 1 hour of reaction to 3.8 nm after 24 hours. Additionally, they observed that the kinetics of the TEMPO oxidation reaction is faster in the first hour of the reaction and decreases with time.22 The biomass pretreatment process also affects the yield and physicochemical properties of TOCN. For instance, Kaffashsaie et al.23 tested the influence of a sodium hydroxide/sodium sulfite pretreatment on TOCN extracted from paulownia wood. They observed that carboxylate contents increased from 1.45 mmol COO− per g for untreated biomass to 1.68 mmol COO− per g for the treated material. Performing TEMPO oxidation on untreated matrices results in decreased oxidation yields, likely due to the occurrence of two simultaneous and competing processes, bleaching and oxidation.23 Simultaneously, DO and σ grow as the oxidizing agent (NaClO) increases, as reported by Isogai et al.24 Milanovic et al.25 studied the combined effects of reaction time and amount of NaClO on the TEMPO oxidation of hemp fibers. The authors observed increases in DO and σ as reaction time increases from 1 to 4 hours only at higher oxidant concentrations (9.67 mmol NaClO per g of fibers). For lower oxidant concentrations, there was no significant combined effect.15 Finally, Hastuti et al.20 reported the TEMPO oxidation of EFB at different concentrations of NaClO. Hastuti observed a linear behavior between the increase of NaClO and the degree of oxidation (1.08, 1.34 and 1.50 mmol COO− per g using 10, 20 and 30 mmol NaClO per g cellulose). However, in this work, the reaction time was not varied, and the sample was pre-treated with acid.20
X-ray photoelectron spectroscopy (XPS) is a highly valuable analytical technique that provides crucial insights into the surface chemistry of nanocellulose. While conductimetric and potentiometric data are commonly used in studies related to cellulose surface oxidation, XPS can also offer valuable information on cellulose surface composition, including carbon and oxygen contents and their oxidation states.26 In fact, numerous reports have illustrated the effective use of XPS for quantitative and qualitative analysis of nanocelluloses.27–31 The XPS C 1s band, which contains contributions from bonded carbons exposed to various local environments, can be resolved into four signals attributed to C–H/C–C, C–O, O–C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO.28 Specifically, the intensity of the O–CO signal can be correlated with the acids on the cellulose surface and compared with data from potentiometric and conductimetric measurements.32–34
O, and O–CO.28 Specifically, the intensity of the O–CO signal can be correlated with the acids on the cellulose surface and compared with data from potentiometric and conductimetric measurements.32–34
This study reports the effects of key variables such as reaction time, temperature, and pH on the surface chemistry of cellulose nanofibers isolated by TEMPO oxidation from Palm Oil Empty Fruit Bunches (EFB). Conductimetric and XPS analysis allowed monitoring the oxidation process and precise characterization of reaction kinetics and surface chemistry. In addition, we performed a rigorous comparative evaluation of data from conductimetric and XPS data. This research holds profound significance, given Colombia's status as the world's fourth-largest Palm Oil producer. Yet, the abundant and versatile lignocellulose matrix, EFB, remains underutilized Palm Oil producer. Yet, the abundant and versatile lignocellulose matrix, EFB, remains underutilized.
2. Experimental
2.1 Materials and reagents
Hydrogen peroxide (H2O2), sodium hydroxide (NaOH), hydrochloric acid (HCl 37%), and ethanol (analytical grade) were purchased from Merck (Darmstadt, Germany). TEMPO (2,2,6,6 tetramethylpiperidine-1-oxyl) radical was purchased from Sigma-Aldrich (Saint Louis, MO, USA). A commercial sodium hypochlorite solution (NaClO 15%) was purchased from Máster Químicos (Piedecuesta, Colombia) and standardized according to ASTM D2022. All solutions and suspensions were prepared with distilled water. The mixtures of raw EFBs belonging Elaeis guineensis and the interspecific hybrid OxG were kindly supplied by Extractora Central Palm Oil Mill, Puerto Wilches, Santander, Colombia.2.2 EFB fiber pretreatment
The EFB fibers, as received from the palm oil mill were washed with plenty of distilled water, dried at 60 °C in an oven, and stored for further use. This sample was labeled clean EFB. Clean EFB fibers were delignified using a previously described hydrogen peroxide alkaline (AHP) method.35 Briefly, 2 g of clean EFB fibers were suspended in a 10% (w/v) H2O2 solution brought to pH 11.5 by adding NaOH (4 N); the EFB fibers load was 5% w/v of alkaline solution. The mixture was allowed to react for 2 hours at 70 °C. The treated EFB (AHP-EFB) fibers were washed and dried at room temperature. AHP-EFB fibers were cut into 0.5 cm pieces before TEMPO oxidation. The cellulose, lignin, hemicellulose, moisture, ashes, ethanol–toluene soluble matter contents in EFB and AHP-EFB fibers were determined by the Kurscher and Hoffer, TAPPI 222, Jayme-Wise methods, NREL-TP-510 42621, NREL-TP-510 42622, and ASTM-D1107 standards.2.3 TEMPO-mediated oxidation of EFB and mechanical treatment
The TEMPO-mediated oxidation of EFB fibers followed the process described by Ovalle et al.19Scheme 1 shows the reaction mechanism. In summary, 0.016 g of the TEMPO radical and 0.1 g of NaBr (dissolved in 40 mL of water) were added to a 100 mL aqueous suspension of 1.0 g of AHP-EFB fibers cut into small pieces (2 mm). Gradually, NaClO was added to the suspension; the amount of NaClO added was dependent on the cellulose![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaClO molar ratios 1:1, 1:5, 1:8 and 1:10 (equivalent to 6.17, 30.84, 49.34 and 61.68 mmol NaClO per g dry cellulose respectively). The mixture was stirred, placed, and kept for variable time intervals in an ultrasonic bath (40 kHz, 130 W), where the pH was maintained at 10.5 by adding HCl or NaOH 6 N solutions. Once the pH stabilized (10.5), the reaction mixture was taken from the ultrasonic bath and quenched with ethanol (half the volume of NaClO). The mixture was centrifuged (5000 rpm, 10 minutes), the supernatant was discarded together with the unreacted cellulose, leaving behind the oxidized cellulose pellet. Distilled water was added to the pellet, and the mixture was shaken and centrifuged multiple times until neutral pH. The pellet was then dispersed in 100 mL of water and subjected to sonication with an Ultrasonic Processor (Sonics vibra-cell-VC750, 20 kHz, 750 W, A 40%, 1:1 s cycle for 10 minutes). A direct TEMPO oxidation control experiment was also carried out using raw EFB fibers (without AHP treatment) reacted with TEMPO for 120 minutes using a cellulose:NaClO ratio of 1:8 (49.34 mmol NaClO per g dry cellulose). The suspension concentration was determined using eqn (1), based on the mass of both the wet and dry TOCN suspensions.
:NaClO molar ratios 1:1, 1:5, 1:8 and 1:10 (equivalent to 6.17, 30.84, 49.34 and 61.68 mmol NaClO per g dry cellulose respectively). The mixture was stirred, placed, and kept for variable time intervals in an ultrasonic bath (40 kHz, 130 W), where the pH was maintained at 10.5 by adding HCl or NaOH 6 N solutions. Once the pH stabilized (10.5), the reaction mixture was taken from the ultrasonic bath and quenched with ethanol (half the volume of NaClO). The mixture was centrifuged (5000 rpm, 10 minutes), the supernatant was discarded together with the unreacted cellulose, leaving behind the oxidized cellulose pellet. Distilled water was added to the pellet, and the mixture was shaken and centrifuged multiple times until neutral pH. The pellet was then dispersed in 100 mL of water and subjected to sonication with an Ultrasonic Processor (Sonics vibra-cell-VC750, 20 kHz, 750 W, A 40%, 1:1 s cycle for 10 minutes). A direct TEMPO oxidation control experiment was also carried out using raw EFB fibers (without AHP treatment) reacted with TEMPO for 120 minutes using a cellulose:NaClO ratio of 1:8 (49.34 mmol NaClO per g dry cellulose). The suspension concentration was determined using eqn (1), based on the mass of both the wet and dry TOCN suspensions.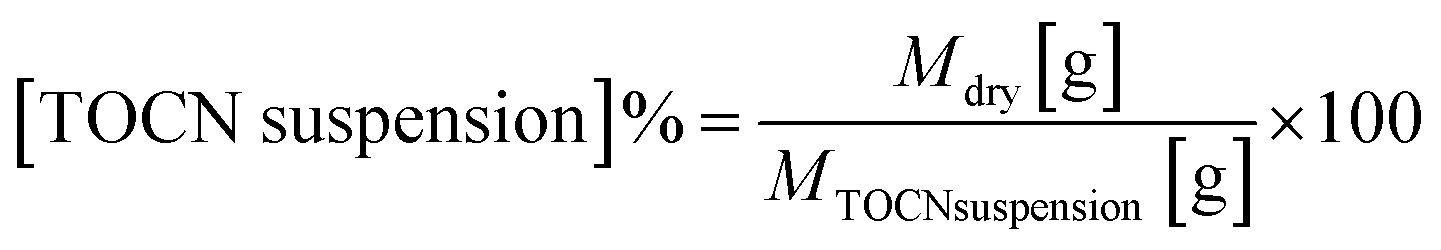 | (1) |
The reaction yield was determined using eqn (2), which considers the suspension concentration, suspension volume, and the fiber mass used for the TEMPO oxidation.
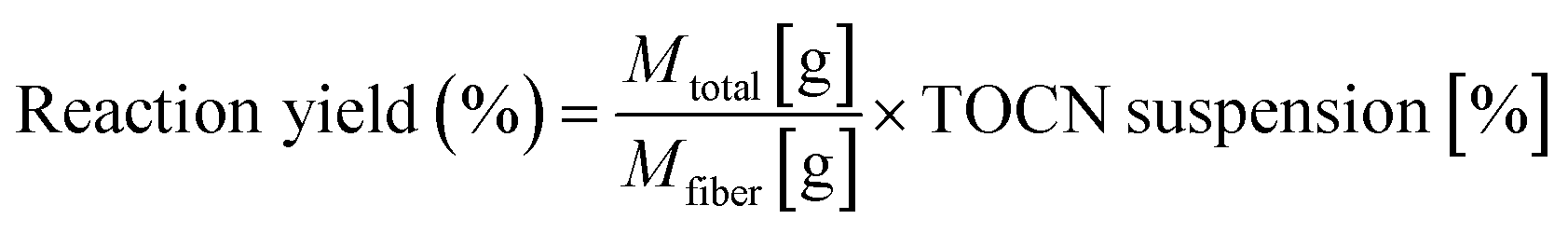 | (2) |
2.4 Statistical analysis
A multilevel factorial design (24) was conducted to test the influence of variables on the TEMPO-oxidation process and reaction yield of AHP-EFB fibers. The design included two independent variables, namely reaction time and cellulose:NaClO molar ratio, each with four levels. The reaction time varied from 30, 60, 90 to 120 minutes, while the cellulose:NaClO molar ratios from 1:1, 1:5, 1:8 to 1:10. The statistical analysis of the design was performed using STATGRAPHICS 18 software using charge density, reaction yield, size distribution, and ζ potential as response variables and the Pareto diagram, main effects plot, estimated response surface and interaction plots as statistical outputs. The labeled samples indicated their reaction time and the cellulose:NaClO ratio (refer to Tables 1 and S1† for sample labels). For example, the sample with a reaction time of 120 minutes and cellulose:NaClO molar ratio of 1:8 was labeled as TOCN 120-1:8 or 120-1:8. Direct TEMPO-oxidized samples were labeled as EFB-TOCN.
| Parameters | Levels | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Reaction time (min) | 30 | 60 | 90 | 120 |
| Cellulose:NaClO molar ratio |
1:1 |
1:5 |
1:8 |
1:10 |
2.5 Materials characterization
Conductimetry was used to determine the TOCN degree of oxidation (DO). First, 0.05 g of each TOCN sample was suspended in 0.01 M HCl solution until pH 2 (excess acid) and stirred for 10 minutes. The suspension was titrated with a previously standardized 0.01 M NaOH solution. The TOCN conductometric titration proceeds through three specific steps. Initially, the mixture's conductivity decreases rapidly due to the excess HCl and NaOH reaction. Secondly, the weak organic acids in the oxidated anhydroglucose units of cellulose react with NaOH, driving a slight and prolonged decrease in conductivity. Finally, conductivity increases sharply due to the excess NaOH. The nanocellulose degree of oxidation (DO) was determined by eqn (3) and the charge density (σ) by eqn (4), as proposed by Habibi.36 Conductimetric titration was performed in triplicate for each TOCN suspension, and error bars indicate standard deviation.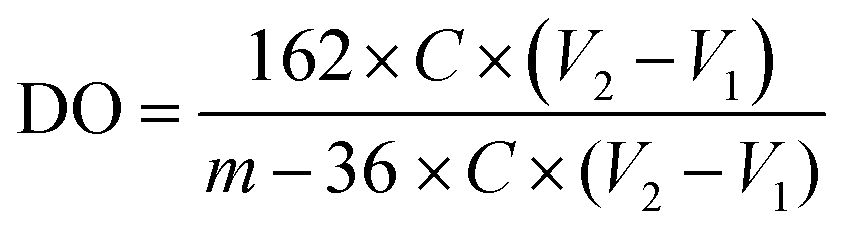 | (3) |
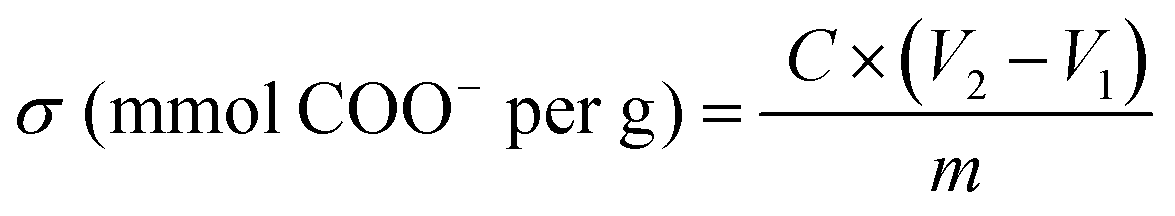 | (4) |
Scanning electron micrographs of the EFB and EFB-AHP fibers were taken at 30 kV on a QUANTA FEG 650 scanning electron microscope equipped with a secondary electron detector (SED), an Everhart Thornley (ETD) detector and a backscattered electron detector (BSE). The samples were affixed with conductive carbon tape and coated with a layer of gold. TOCN micrographs were taken in a Thermo Fisher Scientific Scios 2 DualBeam – FIB SEM equipped with EDX and EDS detectors. 50 mL of the TOCN suspension (0.7%) were placed on conductive carbon tape, dried under vacuum, and coated with gold. Size distribution and ζ potential (colloidal stability) of EFB-TOCN and TOCN suspensions (0.01% w/v) were estimated by Dynamic Light Scattering Analysis (DLS) using a MALVERN Zetasizer ZS90 instrument (Worcestershire, UK)37 DTS-0012 polystyrene and DTS-1070 cells were used for size distribution and ζ potential measurements, respectively. Each measurement of ζ potential was performed in triplicate and error bars indicate standard deviation.
TOCN and EFB-TOCN films were produced by drying 0.8% w/v TOCN suspensions at 60 °C for 24 hours for FTIR-ATR and XPS analysis. FTIR-ATR enables functional group identification and observation of changes caused by the experimental variables tested. FTIR-ATR analysis was performed in a Nicolet iS50 FTIR spectrometer equipped with an integrated diamond ATR module. FTIR-ATR spectra were acquired at a 4 cm−1 resolution and wavenumber range of 4000 to 400 cm−1 using 64 scans.
XPS experiments were recorded using the A-Centeno-XPS/ISS/UPS surface characterization platform produced by SPECS (Germany). The platform is equipped with a PHOIBOS 150 2D-DLD energy analyzer. During XPS analysis, the pressure in the analysis chamber was approximately 1 × 10−9 Pa. The spot size was 2 mm × 2 mm unfocused; the analyzer lens mode was used in medium area, the aperture was 4C, and the calibration resolution was Ag3/2 FWHM 0.05. A monochromatized Al Kα X-ray source (FOCUS 500) operated at 100 W was used for the measurements. The pass energy of the hemispherical analyzer was set at 100 eV for general spectra and 15 eV for high-resolution spectra. Surface charge compensation was controlled using a Flood Gun (FG 15/40-PS FG 500). After each analysis, the C 1s region was re-recorded to verify the sample's surface charge change during the analysis. The CasaXPS software (Casa Software Ltd) was used to process the data using a Shirley-type baseline.38 All spectra were calibrated with the C 1s at 284.8 eV. The signals correspond to C1 284.8 eV (C–H), C2 286.7 eV (O–C–C–C), C3 288.0 eV (O–C–O/CO) and C4 289 eV (OC–O–).39–41 Atomic oxygen (%) was determined after eliminating the contributions of silicon and calcium oxides.
3. Results and discussion
3.1 EFB fiber composition
Fig. 1 displays the EFB and AHP-EFB fibers composition in terms of weight percentage. The EFB fibers were washed only with water and were not degreased prior to TEMPO oxidation. However, less than 4.1% of waxes, fats, resins, and oils that are soluble in ethanol–toluene were present in the raw samples (Fig. 1). After AHP treatment, there was a 76% reduction in the ethanol–toluene soluble content, indicating the removal of surface fats and waxes. The AHP treatment increased the moisture content in the fiber because it makes the fiber surface more hydrophilic and water-accessible by removing lignin and hemicellulose.42 Ash decreased by 46% in AHP-EFB, which suggests the removal of surface minerals. Cellulose, on the other hand, increased by 16%, while lignin decreased by 48% in AHP-EFB. The AHP process aims to remove lignin, leaving cellulose available,35,43 allowing a more efficient TEMPO oxidation than non-delignified EFB. Hemicellulose showed no notable change (less than 5% variation).34,41 ATR-FTIR spectroscopy confirmed chemical changes during EFB fiber delignification and TEMPO oxidation processes. The ATR spectra of EFB and EFB-AHP in Fig. S1† show stretching bands for CH2 (1317 cm−1), C–O–C (1164 cm−1), C–O–C (1109 cm−1), C–O (1030 cm−1), and the C–H anomeric vibration (900 cm−1), characteristic of cellulose functional groups that are common to all samples (Table S2†).23 EFB fibers showed characteristics signals for hemicellulose (CO stretch) and lignin (C–O– deformation) at 1733 and 1230 cm−1, respectively. These bands were not observed in the delignified fibers (AHP-EFB) or the TOCN samples, indicating their removal by the AHP pre-treatment.19 Finally, the TOCN film spectra showed a band at 1602 cm−1, corresponding to sodium carboxylate from TEMPO oxidation.44 The absence of bands at 1733 and 1230 cm−1, associated with the presence of lignin and hemicellulose, has already been reported after cellulose delignification with NaOH.45 On the other hand, TOCN isolated from fique biomass, hyacinths, and cotton stalks also show the band at 1602 cm−1 which indicates the oxidation of the primary alcohol in the anhydroglucose units of cellulose to carboxylate.19,46,47
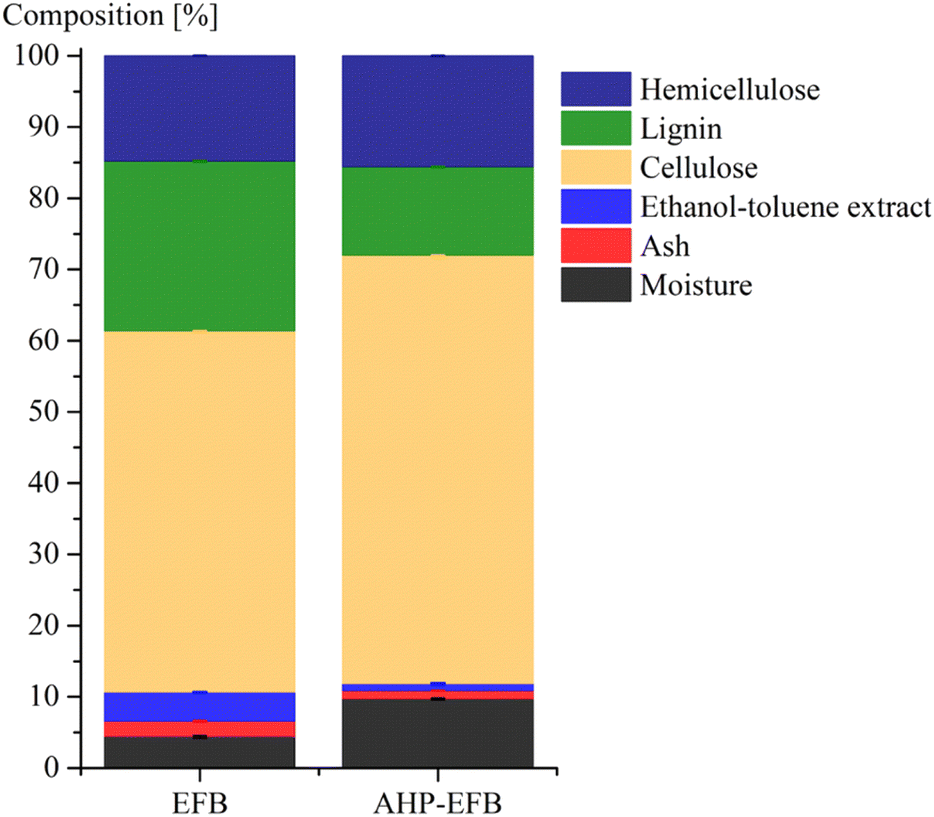 | ||
| Fig. 1 EFB and AHP-EFB fiber composition. | ||
3.2 EFB-TOCN degree of oxidation (DO), charge density (σ), and oxidation yield
Surface charges govern cellulose nanofibers fibrillation and dispersion in water due to electrostatic repulsions. The TEMPO oxidation reaction oxidizes the primary hydroxyl to carboxylate at C6 in the AGU, and the extent of the reaction is determined by both the degree of oxidation (DO) and the surface charge density (σ). These two parameters, determined from conductimetric titrations, exhibit similar trends according to our observations and other authors.48Fig. 2 shows the trends in the degree of oxidation (DO) and charge density (σ, mmoles of COO− per g cellulose) and its standard deviation for TOCN derived from AHP-EFB and EFB fibers. Fig. S2 of the ESI† contains all the conductimetric titration plots. Reaction time and cellulose:NaClO ratios have a directly effect on TOCN surface chemistry. The DO increased by 35% when the reaction time changed from 30 to 120 minutes using a 1:1 cellulose:NaClO ratio. According to the stoichiometry of the TEMPO oxidation reaction (Scheme 1), 2 moles of NaClO oxidize one mole of primary hydroxyl groups.49 Thus, increasing the cellulose:NaClO ratio from 1:1 to 1:10 raised the DO by 52% for 120 minutes of reaction. The maximum DO was 0.27 for the 1:5 cellulose:NaClO at 90 minutes. Interestingly, further increase of the cellulose:NaClO ratio (to 1:8 and 1:10) only slightly raise the DO for 90 and 120 minutes of reaction. Thus, cellulose:NaClO ratios above 1:5 work better for TOCN isolation from AHP-EFB with reaction times above 90 minutes. The relationship between NaClO and the amount of carboxylate groups was reported for TOCN isolated from rice straw at the same reaction time.50
 | ||
| Fig. 2 Effect of experimental conditions (reaction time and cellulose:NaClO ratio) on AHP-EFB (a) DO and σ, (b) cellulose oxidation yield. (c) Optical images of the TOCN isolation process (after the ultrasound bath). (d) Electron micrographs of raw fibers (EFB), delignified fibers (AHP-EFB) and TOCN 120-1:8 film (0.8% w/w). | ||
The DO of TOCN from untreated EFB was 0.24, using a 1:8 cellulose:NaClO ratio and 120 minutes of reaction. Under the same experimental conditions, the DO for the TOCN from AHP-treated EFB was 0.29, representing an increase in oxidation of 21% upon lignin removal from the fibers, as mentioned above. Lignin removal affects the TEMPO oxidation reaction, making the cellulose surface more susceptible to oxidation, as previously reported in the literature.19 Considering the complex structure of lignocellulosic biomass, it is important to note that the efficiency of cellulose oxidation via TEMPO is heavily influenced by the exposure of fibrils to the reaction media. Several pretreatment processes, including alkaline treatment, have been investigated to enhance this exposure. This process is effective in promoting fibril swelling and disrupting intermolecular interactions in lignocellulosic materials, as well as removing waxes, polysaccharides, and lignin.51–53 However, for raw materials with low lignin content (<5%), such as hemp, it may be more advantageous to utilize direct TEMPO oxidation without pretreatment, as it is faster and reduces overall costs.54 Performing TEMPO oxidation on raw EFB fibers (without delignification) affects the reaction yield and degree oxidation because two simultaneous and competing processes consume the primary oxidant: lignin bleaching and carboxylate formation at the cellulose surface. This effect has been previously observed and reported.23 Interestingly, the electron micrographs in Fig. 2d indicate that EFB fibers are abundant in silica and have a porous surface, as reported earlier.55 By partially removing the lignin, the AHP treatment uncovered the cellulosic surface without eliminating the silica microparticles. Even after TEMPO oxidation, the silica particles are still visible within the TOCN, as evidenced by the micrograph. The DO and σ have a positive correlation with reaction time. However, there are two kinetic regimes during the reaction. There is an initial fast oxidation kinetics from zero to thirty minutes. As seen in Fig. 2a, during the first 30 minutes of the reaction, increasing cellulose:NaClO ratios from 1:1, to 1:5, 1:8 and 1:10 raises the AHP-EFB oxidation rate from 0.026 to 0.040, 0.047, and 0.051 mmol COO− per g cell per min, respectively. After 30 minutes of reaction, the process slows down, and the reaction enters a second regime where the oxidation rate is two orders of magnitude lower than during the first 30 minutes. For instance, Fig. 2a shows, for the 1:1 molar ratio, an oxidation rate of 0.004 mmol COO− per g cell per min from 30 to 120 minutes, in contrast with 0.026 COO− per g cell per min for the first 30 minutes.
Interestingly, at higher molar ratios, reaction time influences the oxidation process less, after the initial 30 minutes, as signaled by decreased oxidation rates of 0.003, 0.002, and 0.002 mmol COO− per g cell per min for 1:5, 1:8, and 1:10, respectively. From 30 to 120 minutes, the oxidation reaction can be considered of pseudo zero order for high cellulose:NaClO ratios (1:5, 1:8, 1:10) because the rate constants do not change significantly. Mao et al.51 and Xu et al.52 reported a zero-order reaction order for NaClO concentration during the TEMPO oxidation reaction of cellulose from softwood and eucalyptus wood.56,57 These differences in oxidation rates are explained by assuming that during the first 30 minutes of the reaction, the chemical attack-by the TEMPO catalytic system-is concentrated at the fiber's surface. Increased ratio cellulose:NaClO (1:5, 1:8, 1:10) promotes high oxidation rates, quickly transforming primary hydroxyl groups into carboxylates. Increased carboxylate contents for the fibers reacted at high cellulose:NaClO molar ratios mean more electrostatic repulsion and increased fiber reactivity. Between 30 and 120 minutes, these systems experience a reduced oxidation rate (21 and 15% for the 1:8 and 1:10 molar ratios) due to a decrease in available C-6 hydroxyl groups.57 In contrast, AHP-EFB-TOCN isolated at 1:1 cellulose:NaClO molar ratio presents less surface oxidation during the initial 30 minutes; thus, the second kinetic regime (from 30 to 120 minutes) shows faster oxidation rates (50% for the 1:1).
There is a positive correlation between DO and σ and the amount of NaClO for the low cellulose:NaClO molar ratios. However, as NaClO increases, the carboxylate content levels off (1:8 and 1:10). The TEMPO catalytic cycle involves oxidizing the TEMPO radical to N-oxoammonium and NaBr to NaBrO with NaClO. In turn, the oxidized form of the TEMPO radical (N-oxoammonium) oxidizes the primary hydroxyl groups to carboxyl groups via C6 aldehydes, while reducing TEMPO to N-hydroxylamine. The TEMPO radical is regenerated from N-hydroxilamine by NaBrO (Scheme 1).58 Therefore, NaClO acts as oxidizing agent for both TEMPO and NaBr. As the amount of NaClO increases, the regeneration by oxidation–reduction of the TEMPO radical increases. However, the excess of NaClO does not increase the oxidation, i.e., the number of carboxylate groups on the cellulose, because the disordered regions of the fibers are easily attacked in the first few minutes of the reaction, and most primary alcohols are oxidized fast. The remaining NaClO is then consumed in secondary reactions such as chain scissions.59 Thus, an excessive amount of NaClO leads to cellulose loss, and it also increases the overall cost of the TEMPO reaction.59,60
Fig. 2b also illustrates the percentage of bulk cellulose transformed into nanocellulose (reaction yield %) as a function of reaction time for various cellulose:NaClO molar ratios. Increasing reaction time, from 30 to 120 minutes, resulted in a 28% yield increase for the 1:1 ratio and a 35% increase for the 1:5, 1:8, and 1:10 cellulose:NaClO molar ratios. Once the TEMPO reaction stopped (Fig. 2c, image 120-1:8 bottom right), the oxidized material underwent mechanical treatment using an ultrasonic probe (20 kHz, 175 W) to promote defibrillation and uniform dispersion.
The maximum reaction yield obtained was 61% under the conditions studied using pretreated AHP-EFB. On the other hand, the low yield (32%) of EFB-TOCN, isolated from raw/untreated EFB, deserves attention. Previous studies indicate that performing direct TEMPO oxidation on raw biomass results in higher reaction yields than pretreated biomass but only for low-lignin content materials.23 The differences in reaction yield are caused by the pretreatment process (AHP)-as discussed above- and the mechanical treatment applied to the oxidized material after the TEMPO reaction, as previously reported.23,61,62 The ultrasonic probes work within a confined region of the suspension-around the probe tip-where mechanical stress (cavitation) overcomes the cohesive forces between cellulose fibrils to promote dispersion.63 As a result, there is some size heterogeneity in the isolated NFCs-represented in the presence of large fibers-which were removed during the washing processes. This fact adversely affects the EFB reaction yields.
The statistical analysis indicates that both reaction time and NaClO amount influence charge density and DO (Fig. S3†). The Pareto diagram shows that reaction time and cellulose:NaClO ratios are statistically significant as independent variables and have no significance when combined for AHP-EFB-TOCN charge density and DO. The significance of the two aspects is individually high, and their increment raises the response (DO, σ) according to the main effects plot.64 The response surface produced straight lines, indicating no significant interaction between variables. Approximations were observed between 30–60 and 90–120 minutes in the interaction plots, suggesting that increasing the reaction time from 60 to 90 minutes affects the TOCN charge density. For the reaction yield, the experimental variables time, NaClO, and their combination are individually significant according to the Pareto diagram (Fig. S4†). Reaction yield was more influenced by reaction time than NaClO, as shown by the steeper slope of the main effect plot for time.64 Summarizing, to achieve a charge density greater than 1.5 mmol COO− per g of cell and reaction yields higher than 52%, a cellulose:NaClO molar ratio of 1:8 and a minimum reaction time of 90 minutes are required.
3.3 EFB-TOCN size distribution and ζ potential
Fig. 3 shows the effect of different experimental conditions on AHP-EFB-TOCN dispersion size, expressed as the maxima of the intensity percent distribution, and Fig. S5† shows the particle size distribution from DLS data. The reaction time and cellulose:NaClO ratio affect TOCN size. However, the effect of time is more dramatic. The size distribution range decreases as the time increases. As the cellulose:NaClO ratio increases, the size distribution decreases. When analyzing the maximum intensity values as a function of the size distribution, the AHP-EFB-TOCN size decreased between 76 and 82% when the reaction time increased from 30 to 120 minutes for all cellulose:NaClO molar ratios. In contrast, for any given reaction time, the particle size only decreased by around 40% for increasing cellulose:NaClO ratios. For instance, at 30 minutes, a ten-fold increase in NaClO-from 1:1 to 1:10-reduced the particle size from 842.9 to 576.3 nm. The Pareto diagram (Fig. S6†) indicates that reaction time is the only statistically significant independent variable affecting AHP-EFB-TOCN size. The longer the reaction time, the smaller the AHP-EFB-TOCN size, an effect also measured by other authors using atomic force microscopy and scanning electron microscopy.22,65 The main effect plot shows a steeper slope for reaction time, suggesting an effect of this variable on size distribution. In addition, straight lines on the response surface confirm that reaction time significantly affects size. Finally, the interaction plot intersects at 60, 90, and 120 minutes of reaction, indicating that the amount of NaClO is a variable affecting the size of the suspension.64 Although statistically, there is no significant relationship between TOCN size and NaClO amount, several authors have reported this dependence. Since NaClO initiates the TEMPO catalytic cycle, an increase in NaClO contributes to some extent to more COO− units at the cellulose surface, which implies more electrostatic repulsions, allowing their individualization and thus facilitating size reduction.23,66 However, the size of the TOCN was more influenced by the reaction time and the mechanical disintegration process, as described above.23,68 Finally, EFB–TOCN (isolated from EFB without AHP treatment) exhibited a size distribution maximum of 120.4 nm. Under the same conditions, the delignified fibers afforded TOCN with radii of 100 nm, equivalent to a decrease of 29% in particle size. Thus, it is easier to defibrillate the delignified fibers than the raw fibers, as observed by other researchers.17
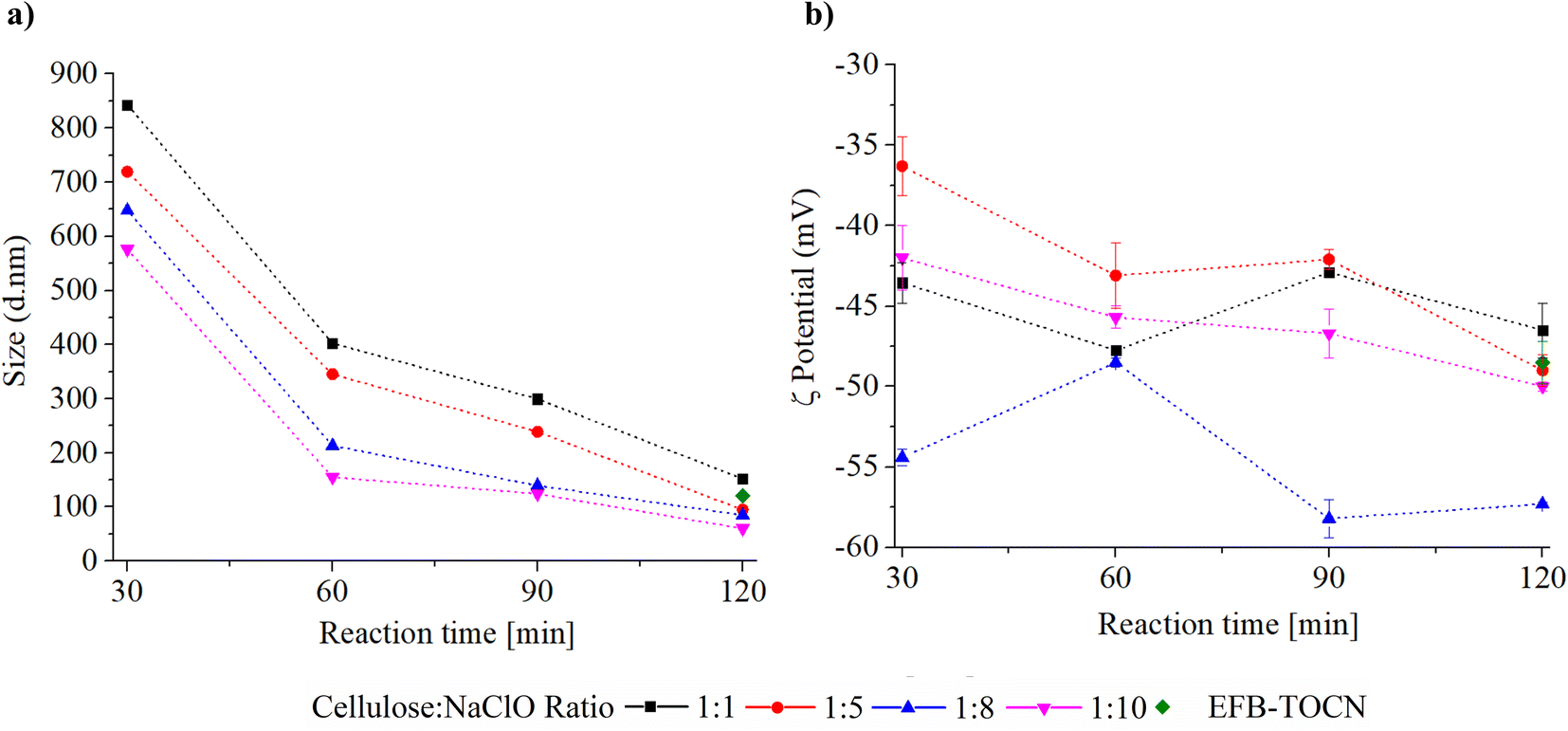 | ||
| Fig. 3 Effect of experimental conditions on AHP-EFB TOCN (a) maximum size distribution and (b) ζ potential. | ||
AHP-EFB-TOCNs intertwined networks composed of elongated cellulose fibers with diameters around 100 nm and micrometric lengths are visible in Fig. 2d. However, this is not likely to be the state of these materials in solution since the use of conductive coatings (carbon, gold) and high vacuum in SEM experiments hinder observation of the native TOCNs structure by increasing the nanomaterial size and promoting aggregation processes by solvent removal.67 Likewise, literature reports show that the size of TOCNs depends on various factors, such as biomass origin, pre- and post-treatments, and reaction conditions. For instance, TOCNs obtained from eucalyptus sawdust pretreated with a homogenizer had an average diameter of 41.0 nm.68 Similarly, TOCNs extracted from bleached and unbleached pineapple stubbles had diameters of 15.5 and 47.9 nm, respectively, as determined by atomic force microscopy; a microfluidizer was used after TEMPO.69 Additionally, mild shearing was used as a pretreatment for bleached and unbleached paulownia wood to obtain TOCNs with diameters of 5.0 and 6.0 nm, respectively, as observed by FESEM.23 It is evident that TOCNs extracted from different biomass and subjected to various pre- and post-treatments exhibit distinct sizes. The bleaching process impacted the size of TOCNs isolated from pineapple stubble, but it did not affect TOCNs obtained from paulownia wood. However, all discussed TOCNs had sizes below 50 nm. In the present work, cellulose:NaClO molar ratios above 1:5 and 120 minutes of reaction with an ultrasonic horn as mechanical treatment afforded TOCNs with sizes below 100 nm. Nonetheless, it is worth noting that the actual size of EFB-TOCNs could be smaller than the values measured by DLS due to nanocellulose aggregation, which could affect the size in this technique.37
Based on the data presented in Fig. 3b, all TOCN suspensions have ζ potential values above ±30 mV, indicating a high level of colloidal stability, likely due to coulombic repulsions between carboxylate groups.70 Interestingly, there does not appear to be any correlation between the experimental parameters (reaction time and amount of NaClO) and the ζ potential values. Isogai et al.24 reported similar results in 2011, as there was no relationship between the carboxylate content and ζ potential of TOCNs isolated from various cellulose sources. However, they did observe ζ potential values around −75 mV for TOCNs with COO− contents ranging from 0.5 to 1.6 mmol per g cellulose.24 Recent theoretical studies have shown that COO− groups in TOCN dispersions can cause a triple screw conformational change, increasing the repulsion between the cellulose particles and their hydrophilicity.71 In our case, TOCN could potentially undergo conformational changes affecting the surface COO− density and the ζ potential value. Another factor to consider is the presence of silica (SiO2) in the TOCN suspensions from the EFB matrix (Fig. 2d). SiO2 has a negative ζ potential, and its presence increases the magnitude of the ζ potential, as previously reported.72 Considering that the concentration of SiO2 in the suspensions is variable, a proportional relationship between the number of carboxylate groups and the ζ potential was therefore not observed. Finally, the Pareto diagrams in Fig. S7† show no influence of the experimental variables on the TOCNs ζ potential value.
3.4 X-ray photoelectron spectroscopy (XPS)
Table 2 presents the elemental surface composition obtained by XPS for AHP-EFB-TOCN, Fig. S8, and Table S3 of the ESI† shows each sample's general spectra and the positions and values of FWHM. All samples contain carbon (284.8 eV), oxygen (532 eV), and calcium (343 eV), characteristic of lignocellulosic compounds.73 Calcium can be in the form of CaCO,74 CaO, or Ca(OH)2.75 The EFB fibers contain silicon as carbon–bonded oxide C–(Si–O) associated with biogenic silica. In plants, silicon is deposited as silica within and between cell walls and fibers in the form of amorphous phytoliths or silica bodies.76 However, silicon only appears in some samples because the AHP-EFB-TOCNs surfaces are heterogeneous. Sodium (1070 eV) and chlorine (198 eV) are in the form of NaCl salts,75 and are attributed to residual compounds of the TEMPO oxidation reaction. For this reason, chlorine and sodium only appear in the oxidized samples (AHP-EFB-TOCN) and not in the raw EFB or the AHP-pretreated fibers. The major component of EFB fibers is carbon, followed by oxygen, silicon, and calcium. After the AHP treatment, the surface silicon was removed, and the amount of oxygen increased.| ID | %C | %O | %Ca | %N | %Si | %Na | %Cl |
|---|---|---|---|---|---|---|---|
| EFB fibers | 84.6 | 11.9 | 1.3 | — | 2.1 | — | — |
| AHP-EFB | 82.3 | 15.6 | 2.1 | — | — | — | — |
| EFB-TOCN | 63.7 | 28.3 | 4.3 | 1.2 | — | 1.2 | 1.3 |
| TOCN30-1:8 | 74.8 | 22.3 | 1.5 | 0.2 | 1.0 | 0.2 | — |
| TOCN 60-1:8 | 71.7 | 24.5 | 2.5 | — | 1.1 | 0.2 | — |
| TOCN 90-1:8 | 70.8 | 25.4 | 1.9 | 0.3 | — | 0.7 | 0.9 |
| TOCN 120-1:8 | 71.1 | 21.7 | 5.6 | 0.4 | 0.4 | 0.8 | — |
| TOCN 120-1:1 | 74.2 | 23.2 | 0.5 | 0.7 | — | 0.6 | 0.8 |
| TOCN 120-1:5 | 72.3 | 24.5 | 1.1 | 0.1 | — | 0.9 | 1.1 |
| TOCN 120-1:10 | 74.8 | 22.9 | 0.9 | 0.2 | — | 0.6 | 0.6 |
During the AHP-EFB-TOCN isolation using different experimental conditions, an increase in the amount of oxygen was expected with increasing reaction time and amount of NaClO, which occurred up to TOCN 90-1:8. However, at TOCN 120-1:8, the percentage of oxygen decreased, and that of calcium increased compared to TOCN 90-1:8. This change can be attributed to a possible diffusion of calcium to the surface of the sample, as a consequence of structural changes (rearrangements) during the chemical treatments (delignification and TEMPO oxidation). Similarly, when the amount of NaClO increased, the percentage of oxygen did not increase after TOCN 120-1:5.
Due to chemical inhomogeneity and roughness on the film surfaces. Depending on the sector in which the interaction with the radiation occurs, the composition will have fluctuations in the compositional percentages.77 Also, the depth of the technique is less than 10 nm,78 limiting the results if diffusion or surface rearrangement processes occur. In a study conducted by Lai et al.79 bacterial cellulose (BC) was oxidized using TEMPO and varying NaClO concentrations and reaction times. XPS analysis revealed an increase in oxygen content with prolonged reaction time, whereas the opposite was observed with a fourfold increase in NaClO concentration. However, these findings conflicted with those obtained through 13C NMR analysis, suggesting that XPS only detects surface changes.79 In our research, XPS analysis demonstrated that the oxidized samples (TOCN) had a greater oxygen content than the raw and delignified fibers (EFB and AHP-EFB), indicating that the oxidation reaction had occurred, as previously reported by other researchers.33,79
Fig. 4 and 5 display the decomposition spectra of the C 1s signals obtained by XPS for the different samples. The EFB, AHP-EFB, and TOCN spectra show 2 (C–H and O–C–O/CO), 3 (C–H, O–C–C, and O–C–O/CO), and 4 (C–H, O–C–C, O–C–O/CO, and C–COO−) signals respectively. The TOCN C–COO− signal in XPS increased with increasing reaction time and the amount of NaClO, following the DO trend observed by conductimetric titration. The C–H, O–C–C–C, and O–C–O/CO signals correspond to alkane carbons, lignin, and hemicellulose,41,80 and the C–COO− signal corresponds to the primary hydroxyl oxidation in the anhydroglucose units (AGU) of cellulose by TEMPO. Therefore, the C–COO− signal only occurs in oxidized samples (TOCN), indicating cellulose oxidation. The decomposed C 1s spectra allowed us to examine the cellulose behavior during the delignification and TEMPO oxidation process. The AGUs in cellulose have a molecular formula C6O5H10, with an O/C ratio of 0.83;81 out of six carbons, five are linked to carbon, hydrogen, and oxygen (C–C–O), while the sixth carbon is bonded to two oxygens. Thus, in XPS 5/6 of the carbon contributes to the C–C–O signal, and 1/6 contributes to the O–C–O/CO signal.82 Benkaddour and Le Gars reported that the C 1s spectrum of TEMPO oxidized cellulose contains 4 components corresponding to C1 (C–C/C–H), C2 (C–O), C3 (O–C–O/CO) and C4 (O–CO), with the C4 signal being associated with oxidation.32,39 The C4 signal is absent in the non-oxidized matrix. In addition, the C2 signal is more intense, followed by C3 and C4, according to the amount of each type of carbon present in the oxidized cellulose.82 Lai et al.79 reported an increase in signal intensity (C–COO−) with increase in reaction time and NaClO concentration in TEMPO oxidation of BC.79
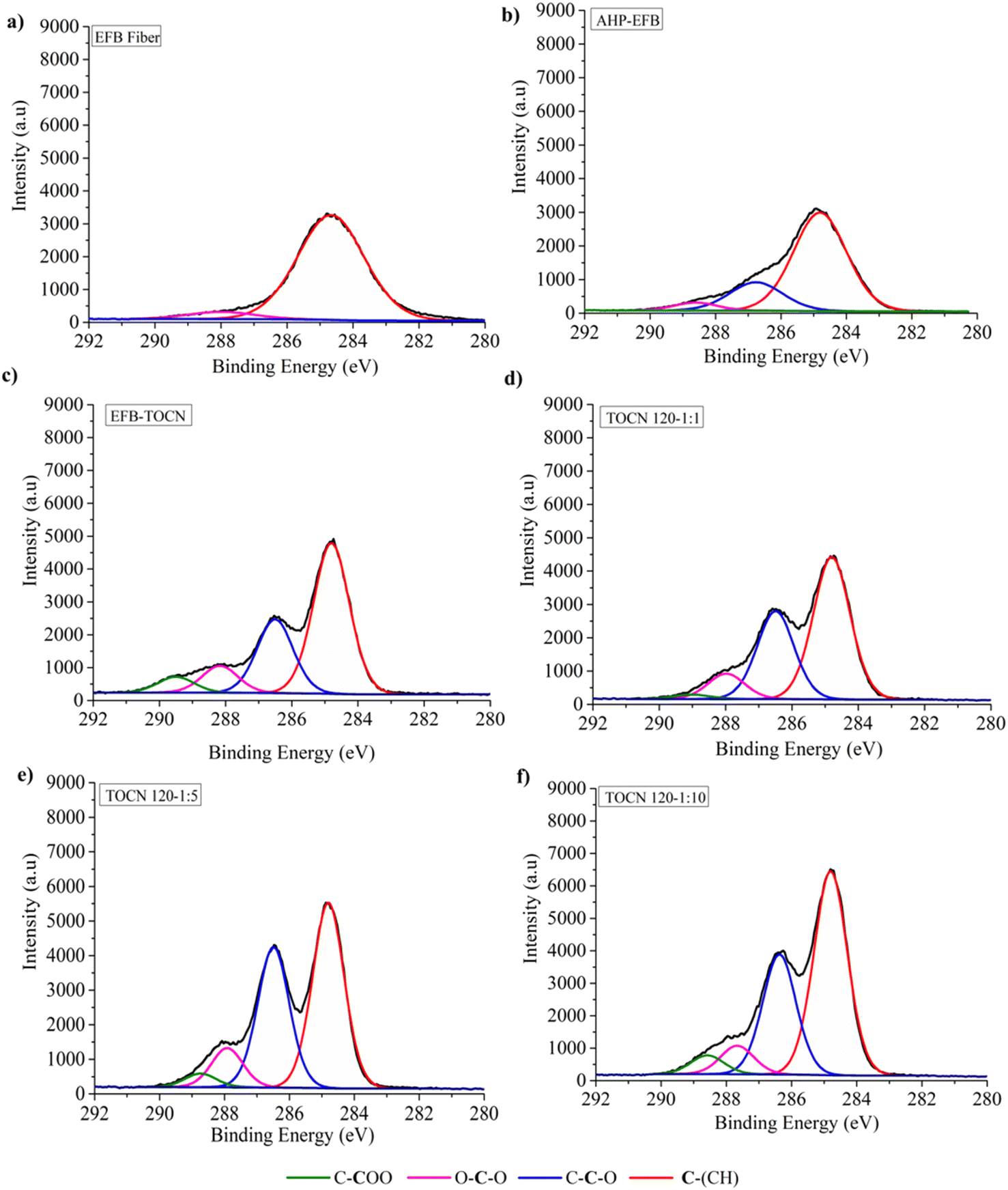 | ||
| Fig. 4 C 1s XPS spectra of (a) EFB, (b) AHP-EFB fibers, (c) direct TEMPO oxidation (EFB-TOCN) and AHP-EFB TOCN isolated using (d) 1:1, (e) 1:5 and (f) 1:10 cellulose:NaClO ratios at 120 minutes of reaction. | ||
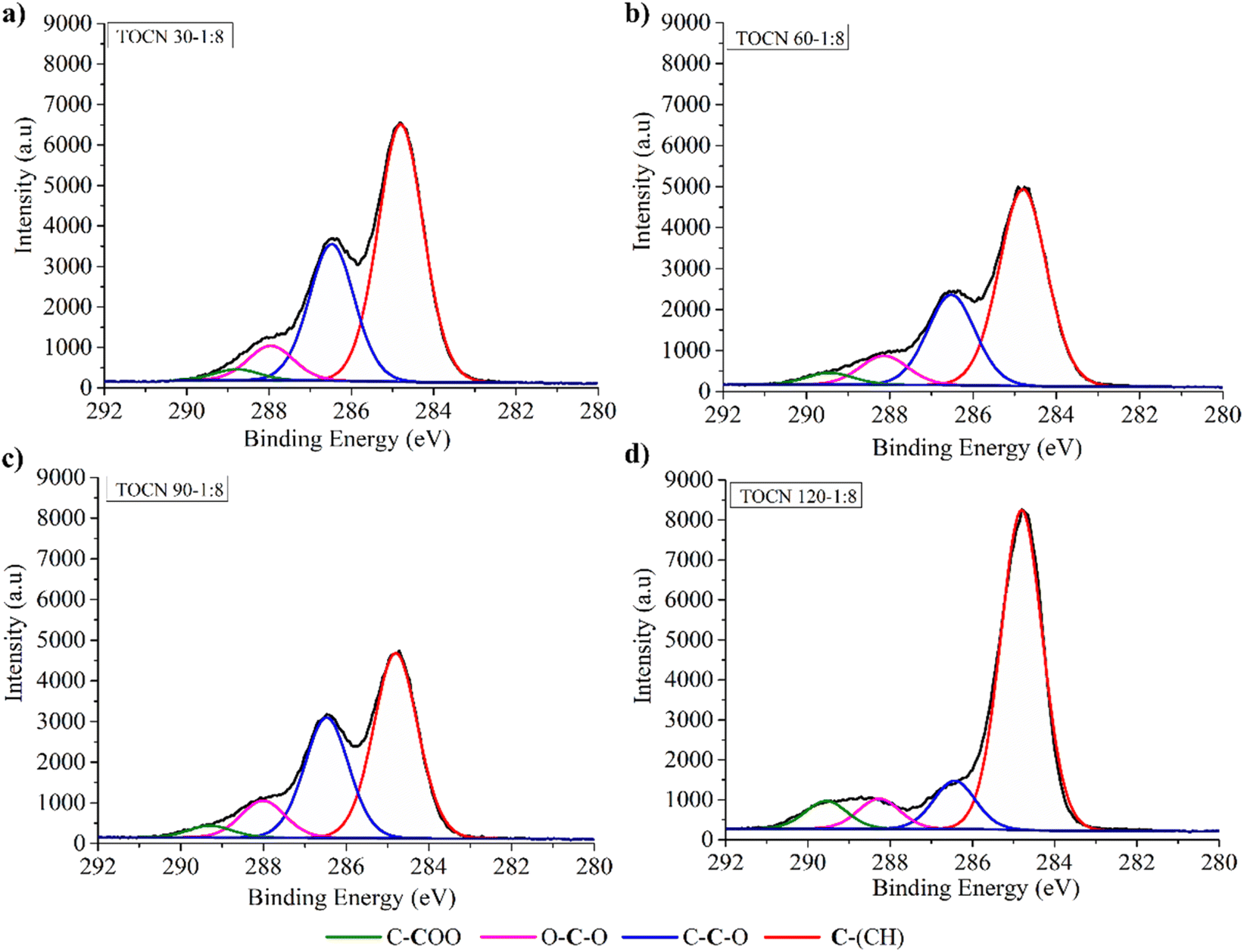 | ||
| Fig. 5 C 1s XPS spectra of AHP-EFB TOCN isolated using 1:8 cellulose:NaClO ratios at (a) 30, (b) 60, (c) 90, and (d) 120 minutes of reaction. | ||
When an AGU in cellulose is oxidized, its chemical formula changes to C6O6H8 (Scheme 1) with an O/C molar ratio of 1. The number of carbons in the structure does not change, but their chemical environment does. Thus, for an oxidized AGU, 4/6 of the carbons contribute to the C–C–O signal, 1/6 of the carbons to the O–C–O/CO signal, and 1/6 of the carbons to the C–COO− signal. Considering the relationships mentioned above, Fig. 6 presents the parity diagrams of the experimental and theoretical values of each type of carbon signal from C 1s by XPS. The theoretical signals were obtained from the sum of the intensities of the carbon signals with oxygen (C–C–O, O–C–O/CO, and C–COO−), using the ratios 5/6 and 1/6 for AHP-EFB, and 4/6, 1/6 and 1/6 for TOCN. An overall coincidence between the experimental and the theoretical values for the C–C–O signal was observed in most samples (Fig. 6a). However, for the O–C–O/CO signal, the experimental value tends to be higher than the theoretical one. This result is because aldehydes were not oxidized to carboxylate during the TEMPO oxidation reaction (Fig. 6b).15 Finally, the experimental C–COO− values were lower than expected, probably due to a heterogeneous carboxylate group distribution on the TOCNs surface (Fig. 6c).34 The data collection of the parity diagram is described in the ESI (Table S4†).
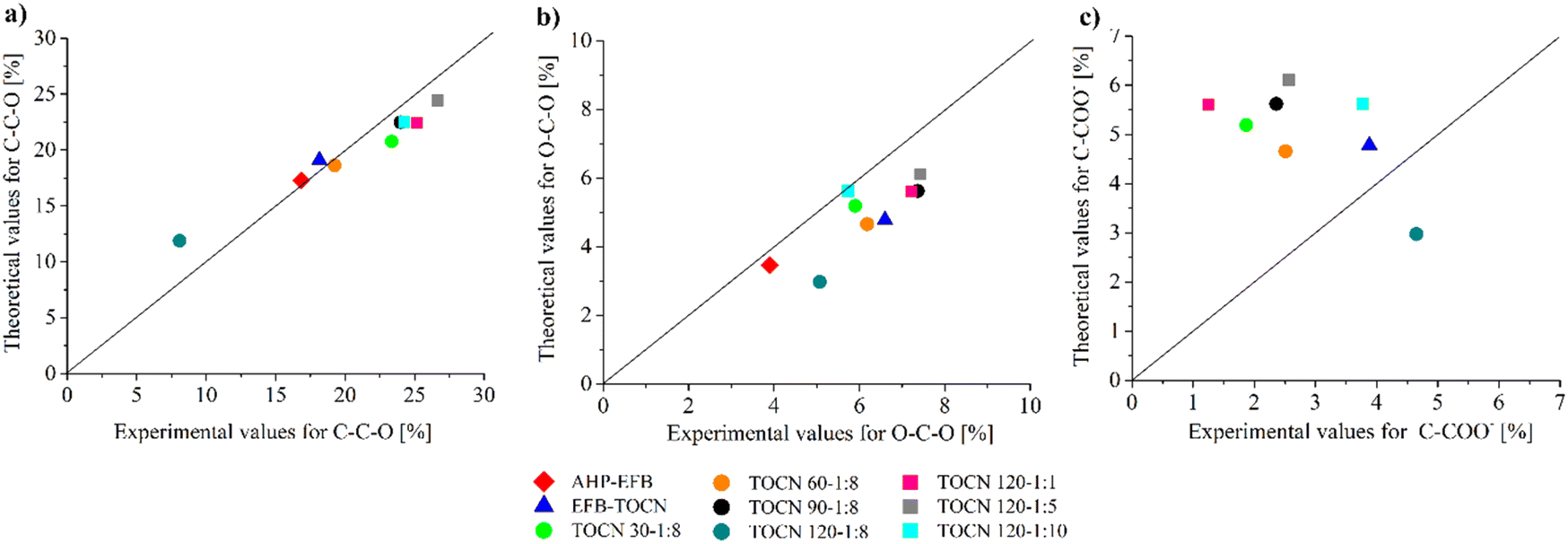 | ||
| Fig. 6 Experimental and theoretical values for each bond type from the C 1s XPS spectra. (a) C–C–O, (b) O–C–O, (c) C–COO−. | ||
The O/C ratios in Table 3 were calculated using data from the C 1s and O 1s spectra. To improve the analysis accuracy, only the C signals with oxygen were considered (C–C–O, O–C–O, and C–COO–) and the contributions of the metals to the oxygen were substracted. The EFB fibers have an O/C ratio of 0.88, 0.05 units higher than the theoretical cellulose value. For the AHP-EFB-TOCN 120:1:8 sample, the O/C ratio was 0.92, the closest to the theoretical value. There was a consistent increase in the O/C ratio as the reaction time increased. However, the trend does not hold for increased cellulose:NaClO ratios. This behavior was associated with a heterogeneous surface morphology and composition in TOCNs.79,83
| Sample | O/C |
|---|---|
| AHP-EFB | 0.88 |
| EFB-TOCN | 0.83 |
| TOCN 30-1:8 | 0.64 |
| TOCN 60-1:8 | 0.76 |
| TOCN 90-1:8 | 0.74 |
| TOCN 120-1:8 | 0.92 |
| TOCN 120-1:1 | 0.72 |
| TOCN 120-1:5 | 0.69 |
| TOCN 120-1:10 | 0.68 |
Fig. 7 shows the correlation between the TOCN charge density (g of COO−/100 g cellulose) data derived from conductimetric titration and the percentage of C 1s carboxylate obtained by XPS. In both techniques, the number of carboxylates increased as the reaction time and the amount of NaClO increased. Fig. 7 shows that the XPS technique supports the results of conductimetric measurements.34 However, the number of carboxylates obtained by conductimetric titration was higher than that of XPS. The difference could be that XPS quantifies the surface concentration of carboxylates and not the overall concentration as conductimetry does. Therefore, the carboxylates obtained by XPS will normally be lower than those obtained by conductimetric titration. As XPS is a surface technique, it gives the amount of COO− available on the surface. In addition, XPS and conductimetric titration show that the amount of carboxylate groups increased with both time and cellulose:NaClO molar ratio.
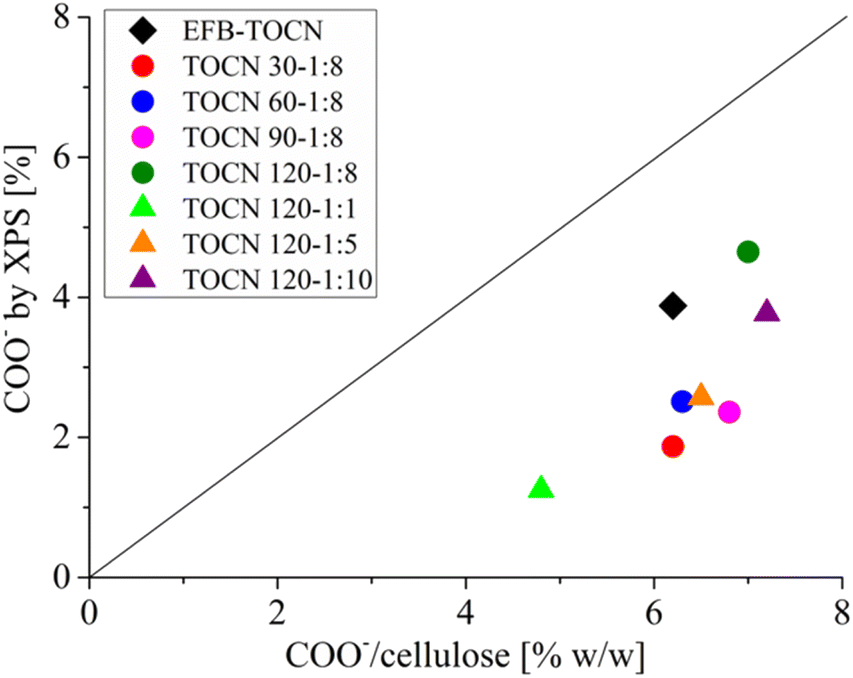 | ||
| Fig. 7 Correlation between charge density (% w/w) from conductimetric titration and C–COO− bonds from C 1s XPS spectra. | ||
4. Conclusions
The alkaline hydrogen peroxide (AHP) pre-treatment of EFB effectively removes surface fats, waxes, and lignin, facilitating the TEMPO oxidation reaction of cellulose. A maximum oxidation yield of 60% was achieved with pretreated AHP-EFB, whereas a low yield (32%) was observed for TOCN isolated from raw/untreated EFB due to the high lignin content of the material (24%). Conductimetric titrations reveal consistent trends in DO and σ for TOCN derived from AHP-EFB fibers. Reaction time and cellulose:NaClO ratios significantly impact TOCN surface chemistry. The TOCN size is most impacted by reaction time, with longer times affording smaller nanofibers. High charge density is related to an increase in NaClO. However, a plateau is reached at a 1:8 cellulose:NaClO ratio. The reaction time and the amount of NaClO do not affect the colloidal stability of the TOCN suspensions. All TOCNs formed stable aqueous suspensions with a ζ potential between −36 and −58 mV due to the electrostatic repulsion of the carboxylate groups. FTIR-ATR, conductimetry, and XPS analysis verified the presence of carboxylate groups on the cellulose nanofibers. Conductimetric and XPS data exhibit similar trends regarding carboxylate contents in AHP-EFB-TOCN. XPS, as a surface analysis technique, provides detailed chemical information, although it underestimates the nanofiber's carboxylate contents. EFB is a potential source for TOCN isolation via TEMPO after pre-treatment involving extractives and lignin removal. While abundant, biomass origin and composition are diverse and require tailored pre-treatments and reaction conditions for nanocellulose isolation using the TEMPO catalytic system.
Data available
Data will be made available on request.Author contributions
APM-R: investigation, methodology, validation, visualization, writing – original draft. SAR-O: investigation, methodology, validation, visualization, writing – original draft. VGB-M: conceptualization, formal analysis, supervision, data curation, writing – original draft. CBT: conceptualization, resources, funding acquisition. MYC: conceptualization, formal analysis, supervision, writing – review and editing, funding acquisition. We confirm that the manuscript has been read and approved by all stipulated authors. We further confirm that all the authors have approved the order of authors listed in the manuscript.Conflicts of interest
Authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank the Guatiguará Technology Park, the Surface Science Laboratory, and the Central Research Laboratory at Universidad Industrial de Santander for infrastructural support. APMR acknowledges Universidad Industrial de Santander for a Graduate Fellowship (ID 2208004).References
- L. H. Saez Zobiole, W. Dantas dos Santos, E. Bonini, O. Ferrase-Filho, R. Kremer, R. Silvério de Oliverira Jr and J. Constantin, in Lignin : Properties and Applications in Biotechnology and Bioenergy, ed. R. J. Paterson, Nova Sicence Publisherts, Inc, United Kingdom, UK, 2012, pp. 419–434 Search PubMed.
- Fedepalma, La palma de aceite en Colombia|Fedepalma, https://web.fedepalma.org/la-palma-de-aceite-en-colombia-departamentos, accessed 8 June 2022.
- P. Boonsawang and W. Youravong, in Sustainability Challenges in the Agrofood Sector, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 596–615 Search PubMed.
- F. N. Mohammad Padzil, S. H. Lee, Z. M. A. ari Ainun, C. H. Lee and L. C. Abdullah, Materials, 2020, 13, 1–26 Search PubMed.
- R. Lou, R. Ma, K. T. Lin, A. Ahamed and X. Zhang, ACS Sustain. Chem. Eng., 2019, 7, 10248–10256 CrossRef CAS.
- A. Farooq, M. K. Patoary, M. Zhang, H. Mussana, M. Li, M. A. Naeem, M. Mushtaq, A. Farooq and L. Liu, Int. J. Biol. Macromol., 2020, 154, 1050–1073 CrossRef CAS PubMed.
- M. Jawaid, S. Boufi and A. Khalil, Cellulose-Reinforced Nanofibre Composites, Elservier, United Kingdom, 2017 Search PubMed.
- H. E. McFarlane, A. Döring and S. Persson, Annu. Rev. Plant Biol., 2014, 65, 69–94 CrossRef CAS PubMed.
- S. Turner and M. Kumar, Philos. Trans. R. Soc., A, 2018, 376, 1–11 Search PubMed.
- W. Huang, in Nanopapers: from Nanochemistry and Nanomanufacturing to Advanced Applications, Elsevier, 2018, pp. 121–173 Search PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- O. J. Rojas, Cellulose Chemistry and Properties: Fibers, Nanocelluloses and Advanced Materials, 2016, vol. 271 Search PubMed.
- G. Pierre, C. Punta, C. Delattre, L. Melone, P. Dubessay, A. Fiorati, N. Pastori, Y. M. Galante and P. Michaud, Carbohydr. Polym., 2017, 165, 71–85 CrossRef CAS PubMed.
- S. Liu, Z. X. Low, Z. Xie and H. Wang, Adv. Mater. Technol., 2021, 6, 1–23 Search PubMed.
- S. P. Mishra, A. S. Manent, B. Chabot and C. Daneault, J. Wood Chem. Technol., 2012, 32, 137–148 CrossRef CAS.
- D. Jaušovec, R. Vogrinčič and V. Kokol, Carbohydr. Polym., 2015, 116, 74–85 CrossRef PubMed.
- K. Benhamou, A. Dufresne, A. Magnin, G. Mortha and H. Kaddami, Carbohydr. Polym., 2014, 99, 74–83 CrossRef CAS PubMed.
- A. Isogai and L. Bergström, Curr. Opin. Green Sustainable Chem., 2018, 12, 15–21 CrossRef.
- S. A. Ovalle-Serrano, F. N. Gómez, C. Blanco-Tirado and M. Y. Combariza, Carbohydr. Polym., 2018, 189, 169–177 CrossRef CAS PubMed.
- N. Hastuti, K. Kanomata and T. Kitaoka, in IOP Conference Series: Earth and Environmental Science, Institute of Physics Publishing, 2019, vol. 359 Search PubMed.
- G. Rodionova, T. Saito, M. Lenes, Ø. Eriksen, Ø. Gregersen, R. Kuramae and A. Isogai, J. Polym. Environ., 2013, 21, 207–214 CrossRef CAS.
- C. F. Huang, C. W. Tu, R. H. Lee, C. H. Yang, W. C. Hung and K. Y. Andrew Lin, Polym. Degrad. Stab., 2019, 161, 206–212 CrossRef CAS.
- E. Kaffashsaie, H. Yousefi, T. Nishino, T. Matsumoto, M. Mashkour, M. Madhoushi and H. Kawaguchi, Carbohydr. Polym., 2021, 262, 117938 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- J. Milanovic, M. Kostic, P. Milanovic and P. Skundric, Ind. Eng. Chem. Res., 2012, 51, 9750–9759 CrossRef CAS.
- H. Huang, Y. Qiao, Y. Yuan and J. Zhang, Reference Module in Materials Science and Materials Engineering, 2023, vol. 2, pp. 407–419 Search PubMed.
- L. Jasmani and S. Adnan, Carbohydr. Polym., 2017, 161, 166–171 CrossRef CAS PubMed.
- R. S. Dassanayake, N. Dissanayake, J. S. Fierro, N. Abidi, E. L. Quitevis, K. Boggavarappu and V. D. Thalangamaarachchige, Appl. Spectrosc. Rev., 2023, 58, 180–205 CrossRef CAS.
- G. Liu, C. Ji, J. Li and X. Pan, Arabian J. Chem., 2022, 15(103964), 1–10 CAS.
- J. H. Jordan, M. W. Easson, B. Dien, S. Thompson and B. D. Condon, Cellulose, 2019, 26, 5959–5979 CrossRef CAS.
- Y. Zhao, C. Jiang, Y. Xiong, Y. Ma, T. Zhang, X. Zhang, J. Qin, X. Shi and G. Zhang, J. Mater. Sci.: Mater. Electron., 2023, 34, 330, DOI:10.1007/s10854-022-09761-x.
- A. Benkaddour, C. Journoux-Lapp, K. Jradi, S. Robert and C. Daneault, J. Mater. Sci., 2014, 49, 2832–2843 CrossRef CAS.
- F. Cheng, C. Liu, X. Wei, T. Yan, H. Li, J. He and Y. Huang, ACS Sustain. Chem. Eng., 2017, 5, 3819–3828 CrossRef CAS.
- L. Fras, L. S. Johansson, P. Stenius, J. Laine, K. Stana-Kleinschek and V. Ribitsch, Colloids Surf., A, 2005, 260, 101–108 CrossRef CAS.
- S. Ovalle-Serrano, C. Blanco-Tirado and Y. Combariza, Cellulose, 2018, 25, 151–165 CrossRef CAS.
- Y. Habibi, H. Chanzy and M. R. Vignon, Cellulose, 2006, 13, 679–687 CrossRef CAS.
- L. Ravindran, M. S. Sreekala and S. Thomas, Int. J. Biol. Macromol., 2019, 131, 858–870 CrossRef CAS PubMed.
- M. S. Hernández-Maya, C. B. Espinosa-Lobo, R. Cabanzo-Hernández, E. Mejía-Ospino and V. G. Baldovino-Medrano, Mol. Catal., 2022, 520, 112158 CrossRef.
- M. Le Gars, A. Delvart, P. Roger, M. N. Belgacem and J. Bras, Colloid Polym. Sci., 2020, 298, 603–617 CrossRef CAS.
- P. G. Rouxhet and M. J. Genet, Surf. Interface Anal., 2011, 43, 1453–1470 CrossRef CAS.
- S. Yu, X. Yang, Q. Li, Y. Zhang and H. Zhou, Green Energy Environ., 2023, 8, 1216–1227 CrossRef CAS.
- M. Asim, M. T. Paridah, M. Chandrasekar, R. M. Shahroze, M. Jawaid, M. Nasir and R. Siakeng, Iran. Polym. J., 2020, 29, 625–648 CrossRef CAS.
- M. C. Ho, V. Z. Ong and T. Y. Wu, Renewable Sustainable Energy Rev., 2019, 112, 75–86 CrossRef CAS.
- S. Fujisawa, Y. Okita, H. Fukuzumi, T. Saito and A. Isogai, Carbohydr. Polym., 2011, 84, 579–583 CrossRef CAS.
- S. N. H. Mustapha, C. W. N. F. C. W. Norizan, R. Roslan and R. Mustapha, J. Nat. Fibers, 2020, 1–14 Search PubMed.
- S. Tanpichai and E. Wimolmala, J. Nat. Fibers, 2022, 19, 10094–10110 CrossRef CAS.
- B. Soni, E. B. Hassan and B. Mahmoud, Carbohydr. Polym., 2015, 134, 581–589 CrossRef CAS PubMed.
- L. Geng, N. Mittal, C. Zhan, F. Ansari, P. R. Sharma, X. Peng, B. S. Hsiao and L. D. Söderberg, Macromolecules, 2018, 51, 1498–1506 CrossRef CAS.
- A. Rattaz, S. P. Mishra, B. Chabot and C. Daneault, Cellulose, 2011, 18, 585–593 CrossRef CAS.
- F. Jiang, S. Han and Y. Lo Hsieh, RSC Adv., 2013, 3, 12366–12375 RSC.
- C. Alvarez-Vasco and X. Zhang, Biomass Bioenergy, 2017, 96, 96–102 CrossRef CAS.
- A. Balea, N. Merayo, E. De La Fuente, C. Negro and Á. Blanco, Ind. Crops Prod., 2017, 97, 374–387 CrossRef CAS.
- R. Pönni, T. Pääkkönen, M. Nuopponen, J. Pere and T. Vuorinen, Cellulose, 2014, 21, 2859–2869 CrossRef.
- B. Puangsin, H. Soeta, T. Saito and A. Isogai, Cellulose, 2017, 24, 3767–3775 CrossRef CAS.
- P. Rama Rao and G. Ramakrishna, Cleaner Mater., 2022, 6, 100144 CrossRef.
- L. Mao, P. Ma, K. Law, C. Daneault and F. Brouillette, Ind. Eng. Chem. Res., 2010, 49, 113–116 CrossRef CAS.
- H. Xu, J. L. Sanchez-Salvador, A. Balea, A. Blanco and C. Negro, Cellulose, 2022, 29, 6611–6627 CrossRef CAS.
- A. Isogai, T. Hänninen, S. Fujisawa and T. Saito, Prog. Polym. Sci., 2018, 86, 122–148 CrossRef CAS.
- H. Xu, J. L. Sanchez-Salvador, A. Balea, A. Blanco and C. Negro, Cellulose, 2022, 29, 6611–6627 CrossRef CAS.
- S. S. Lal and S. T. Mhaske, Cellulose, 2019, 26, 6099–6118 CrossRef CAS.
- S. A. Chew, V. A. Hinojosa and M. A. Arriaga, in Bioresorbable Polymers for Biomedical Applications: from Fundamentals to Translational Medicine, Elsevier, 2017, pp. 229–264 Search PubMed.
- J. Levanič, K. Svedström, V. Liljeström, M. Šernek, I. G. Osojnik Črnivec, N. Poklar Ulrih and A. Haapala, Cellulose, 2022, 29, 9121–9142 CrossRef.
- J. D. Redlinger-Pohn, M. Petkovšek, K. Gordeyeva, M. Zupanc, A. Gordeeva, Q. Zhang, M. Dular and L. D. Söderberg, Biomacromolecules, 2022, 23, 847–862 CrossRef CAS PubMed.
- A. Jiju, in Design of Experiments for Engineers and Scientists, 2nd edn, 2014, pp. 33–50 Search PubMed.
- L. Van Hai, L. Zhai, H. C. Kim, J. W. Kim, E. S. Choi and J. Kim, Carbohydr. Polym., 2018, 191, 65–70 CrossRef CAS PubMed.
- A. Isogai, in High-Performance and Specialty Fibers Concepts, Technology and Modern Applications of Man-Made Fibers for the Future, 2016, pp. 297–311 Search PubMed.
- C. Liu, B. Li, H. Du, D. Lv, Y. Zhang, G. Yu, X. Mu and H. Peng, Carbohydr. Polym., 2016, 151, 716–724 CrossRef CAS PubMed.
- M. E. Vallejos, F. E. Felissia, M. C. Area, N. V. Ehman, Q. Tarrés and P. Mutjé, Carbohydr. Polym., 2016, 139, 99–105 CrossRef CAS PubMed.
- K. Araya-Chavarría, R. Rojas, K. Ramírez-Amador, B. Sulbarán-Rangel, O. Rojas and M. Esquivel-Alfaro, Waste Biomass Valorization, 2022, 13, 1749–1758 CrossRef.
- H. D. Silva, M. Â. Cerqueira and A. A. Vicente, Food Bioprocess Technol., 2012, 5, 854–867 CrossRef CAS.
- M. Asgarpour Khansary, P. Pouresmaeel-Selakjani, M. Ali Aroon, A. Hallahisani, J. Cookman and S. Shirazian, Heliyon, 2020, 6, e05776 CrossRef CAS PubMed.
- J. J. Carlson and S. K. Kawatra, Miner. Process. Extr. Metall. Rev., 2013, 34, 269–303 CrossRef CAS.
- S. H. Gong, H. S. Im, M. Um, H. W. Lee and J. W. Lee, Fuel, 2019, 239, 693–700 CrossRef CAS.
- M. Genet, C. Dupont-Gillain and P. Rouxhet, in Medical Applications of Colloids, Springer, New York, 2008, pp. 177–291 Search PubMed.
- P. Ghods, O. B. Isgor, J. R. Brown, F. Bensebaa and D. Kingston, Appl. Surf. Sci., 2011, 257, 4669–4677 CrossRef CAS.
- K.-N. Law, W. Rosli, W. Daud and A. Ghazali, BioResources, 2007, 2(3), 351–362 CAS.
- D. G. Gray, M. Weller, N. Ulkem and A. Lejeune, Cellulose, 2010, 17, 117–124 CrossRef CAS.
- G. Pintori and E. Cattaruzza, Opt. Mater.: X, 2022, 13, 100108 CAS.
- C. Lai, L. Sheng, S. Liao, T. Xi and Z. Zhang, Surf. Interface Anal., 2013, 45, 1673–1679 CrossRef CAS.
- E. Espino-Pérez, S. Domenek, N. Belgacem, C. Sillard and J. Bras, Biomacromolecules, 2014, 15, 4551–4560 CrossRef PubMed.
- K. Kolářová, V. Vosmanská, S. Rimpelová and V. Švorčík, Cellulose, 2013, 20, 953–961 CrossRef.
- A. Sbiai, A. Maazouz, E. Fleury, H. Sautereau and H. Kaddami, in Composites and Their Applications, InTech, 2012. DOI:10.5772/47763.
- S. Barazzouk and C. Daneault, Cellulose, 2012, 19, 481–493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06933h |
| This journal is © The Royal Society of Chemistry 2023 |