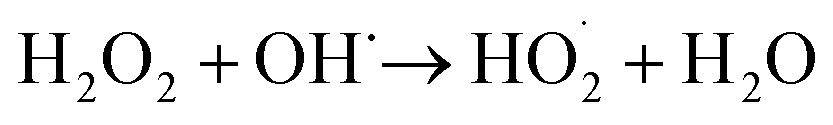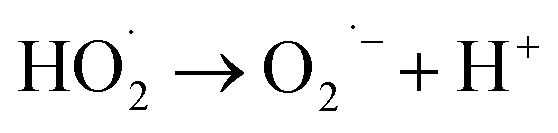DOI:
10.1039/D3RA06986A
(Paper)
RSC Adv., 2023,
13, 32833-32841
Degradation of phenol by perborate in the presence of iron-bearing and carbonaceous materials†
Received
13th October 2023
, Accepted 31st October 2023
First published on 7th November 2023
Abstract
We investigated the oxidation of phenol by perborate—a newly proposed oxidant—in the presence of iron-bearing and carbonaceous materials through batch experiments. We hypothesized that the oxidation of phenol by perborate was enhanced due to the formation of reactive oxygen species (ROS) in the presence of iron-bearing or carbonaceous materials. Zero-valent iron and ferrous iron (Fe2+) promoted the oxidation of phenol by perborate. Biochar, granular activated carbon, an anode carbonaceous material recovered from a spent Li-ion battery, and graphite also accelerated the oxidation of phenol by perborate. Quenching experiments with radical scavengers and electron paramagnetic resonance (EPR) analysis revealed that hydroxyl (˙OH) and superoxide (O2˙−) radicals were generated and enhanced the degradation of phenol in the perborate systems. Singlet oxygen (1O2) was involved in the iron-bearing material–perborate systems. Moreover, we found that Persil®, a commercial perborate detergent, enhances the oxidation of phenol in the presence of iron-bearing and carbonaceous materials. Our results suggest that perborate can be used for advanced oxidation processes to remediate recalcitrant organic contaminants in natural environments and engineered systems.
1. Introduction
In the last decades, in situ or ex situ chemical oxidation of organic contaminants has become one of the most common remediation processes for natural environments. Depending on the properties of the contaminants, such as their molecular size or solubility in water, the types of oxidants, and experimental conditions (e.g., solution pH, initial amount of contaminant, and dosage of oxidant), as well as the kinetics and pathways of oxidative degradation vary and have been intensely examined. Some oxidants are non-selective and their oxidation rate is insufficient to achieve a substantial removal of contaminants within days or weeks. To enhance the kinetics and degree of degradation by oxidants like hydrogen peroxide (H2O2) and ozone (O3), radicals can be inductively generated via activation processes using UV or transition metals (i.e., advanced oxidation processes (AOPs)).1,2 Because of the high oxidation potential of radicals, the chemical oxidation of contaminants could be greatly promoted. However, AOPs may have drawbacks. The high reactivity and low stability of oxidants may result in the rapid consumption of oxidants in natural environments or engineered systems before AOPs are achieved. Therefore, managing oxidants and activating agents in natural environments is essential to remediate contaminated water and soils.
There have been many efforts to develop new oxidants that can be safely and effectively used for chemical oxidation processes in environmental applications. For example, persulfate (S2O82−) was proposed as an alternative oxidant because it is fairly stable and easy to handle.3,4 Persulfate can be activated by heat, UV, and transition metals such as Fe2+.5 Moreover, iron-bearing materials (e.g., zero-valent iron and iron sulfide) and carbonaceous materials (e.g., activated carbon, carbon nanotubes, and graphene) can also activate persulfate to generate sulfate or hydroxyl radicals to enhance the oxidation of contaminants by persulfate.6–11 Because of the graphitic structure and surface functional groups of carbonaceous materials, the oxidation of contaminants is promoted. Various types of carbon nanomaterials such as multi-walled carbon nanotubes, N-doped carbon nanotubes, N-graphene, N-graphene oxide, g-C3N4, and O-doped g-C3N4 have also been intensively examined as catalysts.12 Following free radical (OH˙ and SO4−˙) and non-radical pathways (1O2 or electron transfer), different types of organic pollutants were rapidly and effectively degraded.12 Various types of reactive oxygen species (ROS) have been identified, and oxidation pathways have been suggested for carbonaceous material–oxidant systems.7,11
Other types of oxidants have also been suggested as alternatives (e.g., perborate for chemical oxidation and AOPs). A few attempts have been made to use perborate for oxidation of reagents in chemical engineering. Kabalka et al.13 suggested that perborate was a mild and convenient reagent for the oxidation of organoboranes. The oxidation of iodide by perborate in combination with molybdenum and zirconium was investigated.14,15 The oxidation of iodine is first order with respect to perborate and is independent of the iodide and hydrogen concentrations. Perborate was used for selective nucleophilic oxidation of sulfides with microwave irradiation to obtain sulfone.16 Perborate showed a similar oxidation capability to that of hydrogen peroxide for repulping, deinking, and pre-bleaching of wastepaper.17 Perborate was also used as an alternative to hydrogen peroxide for chemical oscillations.18 Devi et al.19 demonstrated that perborate oxidation of substituted 5-oxoacids was first order with respect to perborate and second order regarding pH. The results revealed that perborate oxidation was faster than hydrogen peroxide oxidation. In addition, an effort was made to use perborate as an alternative to hydrogen peroxide for AOP with UV to degrade natural organic matter.20 The results revealed that perborate was as effective as hydrogen peroxide in the AOP system. Although several attempts have been made to take advantage of the oxidative properties of perborate, studies on the application of perborate as an alternative oxidant for the degradation of organic contaminants are still limited.
In this study, we explored the application of perborate, which is used as an ingredient in a commercial detergent (Persil®), for an AOP to remove recalcitrant organic contaminants. We hypothesized that perborate may be a good oxidant so that radicals can be readily generated in AOPs and that iron-bearing materials and carbonaceous materials may enhance the oxidation by perborate. Phenol was chosen as a model contaminant because of its toxicity and classification as a primary pollutant by the Environmental Protection Agency of the United States (US EPA). Zero-valent iron and iron sulfate were chosen as iron-bearing materials, and biochar, granular activated carbon (GAC), graphite, and carbonaceous materials recovered from the anode of a spent Li-ion battery were selected as carbonaceous materials to activate perborate. The objectives of this study were to determine how perborate and activating agents enhanced phenol degradation and identify the radicals generated, using scavenging experiments and electron paramagnetic resonance (EPR) analysis to elucidate the oxidation mechanisms. In addition, the perborate oxidation of phenol in the presence of iron-bearing and carbonaceous materials was examined using the commercial perborate-containing detergent Persil®.
2. Materials and methods
2.1. Chemicals
Sodium perborate monohydrate (NaBO3·H2O, 96%) and the spin trap reagents for EPR, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO, >99.0%) and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, >98%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phenol (C6H5OH, 99%), iron(II) sulfate heptahydrate (FeSO4·7H2O), furfuryl alcohol (FFA, 98.1%), monosodium phosphate (NaH2PO4, >98%), and disodium hydrogen phosphate dodecahydrate (Na2HPO4·12H2O, >99%) were purchased from Daejung Chemical (Gyeonggi, South Korea). Tert-Butyl alcohol (TBA, 99.0%) and sodium azide (NaN3, 99%) were obtained from Samchun Chemical (Seoul, South Korea) and 1,4-benzoquinone (p-BQ, ≥98%) was purchased from Junsei Chemical (Tokyo, Japan). Methanol (HPLC-grade) was purchased from Honeywell Burdick and Jackson (Muskegon, MI, USA). Iron powder (99%, <212 μm) was purchased from Acros (Geel, Belgium).
Graphite (<20 μm) and GAC (CP500G, 8–20 mesh) were purchased from Sigma-Aldrich and OCI (Seoul, South Korea), respectively. Anode carbonaceous materials (ACM) were provided by SungEel Hitech Company (Chonbuk, South Korea). Biochar was synthesized through pyrolysis of rice straw collected from a rice farm in Ulsan, South Korea. The sampled rice straw was dried in an oven at 105 ± 5 °C for at least 2 h. After storing in a desiccator overnight, the dried rice straw was pulverized to smaller sizes (<5 mm) using an electric mixer. Then, rice straw was pyrolyzed for 4 h at 550 °C, using a tube-type electrical furnace under N2 at 1000 mL min−1. To prevent the production of ash from biomass during pyrolysis, a high nitrogen flow rate was applied. After cooling down to room temperature, the biochar was put in a desiccator for thorough drying. The synthesized biochar was stored in a plastic bag for batch experiments. The properties of biochar and other carbonaceous materials are summarized in Table 1.
Table 1 Properties of carbonaceous materials used in the present study
| |
pHa |
BET S.A.b (m2 g−1) |
Cation exchange capacity (CEC)c (cmol kg−1) |
Point of zero charge (PZC)d |
Elemental contentse (%) |
| C |
H |
O |
N |
| Determined by the method of Rump and Krist21 using deionized water. The Brunauer–Emmett–Teller (BET) surface area was analyzed using a nanoPOROSITY-XQ (Mirae Scientific Instruments, Korea) with N2. The cation exchange capacity (CEC) was determined using the method of Hesse.22 Point of zero charge (PZC) was determined using the method of Faria et al.23 Analyzed using a Vario EL elemental analyzer (Elementar, GmbH, Germany). |
| Biochar |
9.08 |
16.7 |
3.08 |
8.19 |
56.1 |
2.77 |
12.7 |
1.92 |
| ACM |
6.73 |
6.70 |
15.7 |
75.8 |
89.9 |
0.26 |
3.03 |
0.03 |
| GAC |
6.42 |
739 |
11.6 |
6.89 |
79.8 |
0.56 |
2.81 |
0.59 |
| Graphite |
3.73 |
13.6 |
5.40 |
4.94 |
97.5 |
0.06 |
<0.01 |
<0.01 |
2.2. Batch experiments
Experiments on phenol oxidation by perborate in the presence of iron-bearing and carbonaceous materials were conducted using 500 mL Erlenmeyer flasks covered with aluminum foil. In a typical run, 250 mL of sodium perborate solution (250 mg L−1) and 1 g of iron-bearing or carbonaceous materials, except for GAC, were added to the flasks. Because of its high surface area, 0.3 g of GAC was used to decrease the effect of sorption removal. The batch experiment was started by adding predetermined amounts (0.25 mmol) of phenol to obtain an initial concentration of 1 mM. Duplicate flasks were shaken at 180 rpm by a reciprocal shaker (Hanbaek, Gyeonggi, South Korea) at room temperature. At each sampling time (1, 3, 5, 7, and 10 h), 1 mL of the solution was taken using a glass syringe pipette and filtered through a 0.22 μm membrane (Millipore, Burlington, MA, USA). The material left on the membrane was chemically analyzed. For each batch, control experiments were conducted in parallel under identical conditions without one of the elements (250 mg L−1 perborate, iron-bearing, or carbonaceous materials).
To indirectly determine the formation of hydroxyl and superoxide radicals (˙OH and O2˙−) and singlet oxygen (1O2), as well as the transfer of electrons in the reactions, quenching reagents were added to the solution under identical conditions. p-BQ (0.05 M), TBA (0.4 M), FFA (0.2 M), and sodium azide (NaN3, 0.2 M) were added to quench O2˙−, ˙OH, 1O2, and 1O2 as well as electron transfer reactions, respectively.24–27
2.3. Chemical analysis
Phenol was analyzed using an UltiMate™ 3000 HPLC system (Dionex, Sunnyvale, CA, USA) equipped with an Acclaim® 120 guard column (4.3 × 10 mm, Dionex) and an Acclaim® 120 C18 column (4.6 × 250 mm, 5 μm, Dionex). A methanol–water mixture (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, v/v) was used as the mobile phase at a flow rate of 1.0 mL min−1, and the sample injection volume was 100 μL. The wavelength of the UV detector was set at 224 nm. The phenol retention time was 5.3 min. For quantification purposes, a calibration curve was plotted within the range of the experimental concentrations used (coefficient of determination > 0.99). A standard phenol solution was used to confirm the accuracy and precision of the phenol concentration in the HPLC analysis. Total organic carbon (TOC) concentration was determined using a TOC analyzer (TOC-L, Shimadzu, Kyoto, Japan).
:40, v/v) was used as the mobile phase at a flow rate of 1.0 mL min−1, and the sample injection volume was 100 μL. The wavelength of the UV detector was set at 224 nm. The phenol retention time was 5.3 min. For quantification purposes, a calibration curve was plotted within the range of the experimental concentrations used (coefficient of determination > 0.99). A standard phenol solution was used to confirm the accuracy and precision of the phenol concentration in the HPLC analysis. Total organic carbon (TOC) concentration was determined using a TOC analyzer (TOC-L, Shimadzu, Kyoto, Japan).
Free radicals in the perborate systems were determined by EPR.24 As a pre-treatment, DMPO was used to capture the radical species, and 0.04 g of iron-bearing or carbonaceous material, 0.1 mM phosphate buffer solution (pH = 7.4), and perborate (250 mg L−1) were added to a 10 mL phenol solution (approximately 100 mg L−1). Thereafter, 10 mM of DMPO was introduced into the solution, which was allowed to react for 1–1.5 min before filtering through a 0.45 μm membrane. To identify the singlet oxygen (1O2), 5 mM of TEMPO was used as a trapping reagent under identical conditions. The resulting filtrate was analyzed with EPR equipment (E580, Bucker Co., Germany). The microwave frequency, resonance, and modulation were 20.00 mW, 9.4 GHz, and 100 kHz, respectively.
3. Results and discussion
3.1. Oxidation by perborate in the presence of iron-bearing materials
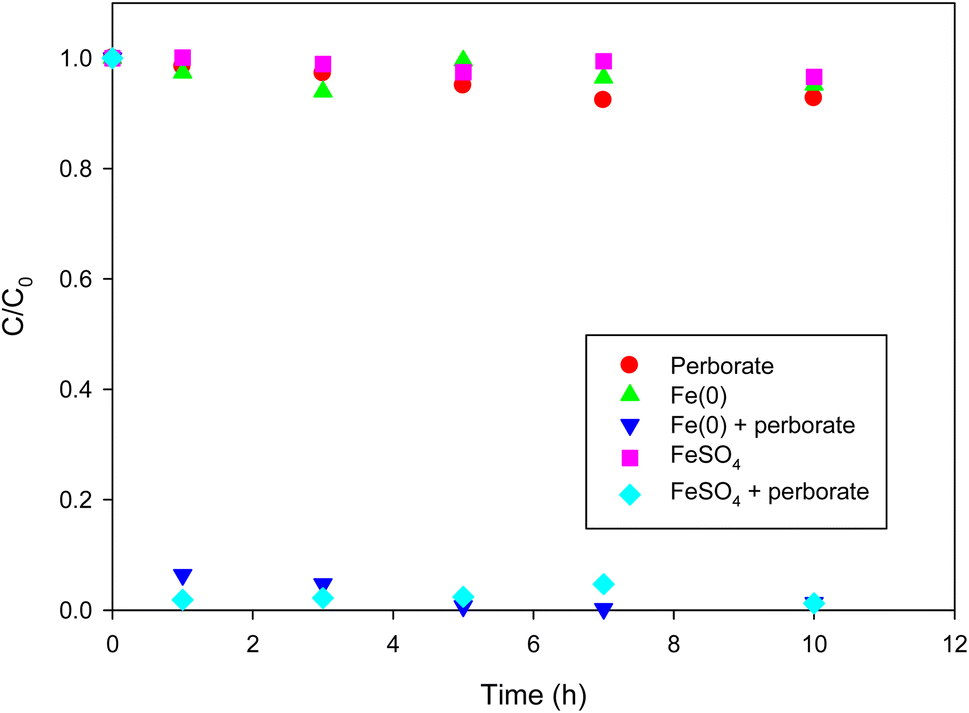
Direct oxidation of phenol by perborate resulted in less than 10% of phenol removal in 10 h (Fig. 1). Under the given conditions, the removal of phenol using only Fe(0) or FeSO4 was not substantial (less than 5% in 10 h). By contrast, the introduction of Fe(0) or FeSO4 into the perborate solution markedly enhanced the removal of phenol (Fig. 1), resulting in 93% and 98% removal in 1 h, respectively. After 7 h, complete removal of phenol was observed in the perborate system. The results indicate that perborate was activated by the iron-bearing materials to generate highly reactive radicals. Oxidation was likely enhanced by the formation of hydrogen peroxide when sodium perborate monohydrate (NaBO3·H2O) was fully dissociated in water.28,29| | |
2(NaBO3·H2O) → 2Na+ + H4B2O82−
| (1) |
| | |
H4B2O82− + 2H2O → 2[B(OH)3·(HO2)]−
| (2) |
| | |
[B(OH)3·(HO2)]− → B(OH)3 + HO2−
| (3) |
| | |
B(OH)3 + HO2− + H2O → B(OH)4− + H2O2
| (4) |
| | |
B(OH)3 + H2O → B(OH)4− + H+
| (5) |
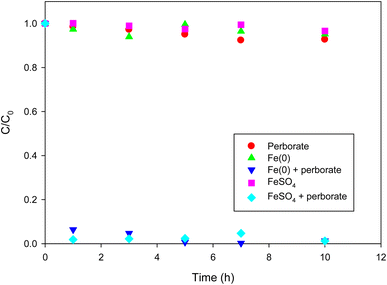 |
| | Fig. 1 Degradation of phenol with perborate in the presence of iron-bearing materials. The concentration of perborate, the initial concentration of phenol, the amounts of iron-bearing materials, the volume of solution, and initial pH are 250 mg L−1, 1 mM, 1 g, 250 mL, and pH 3, respectively. | |
As shown in eqn (1)–(4), hydrogen peroxide was produced from the dissociation of sodium perborate in a stepwise way. According to eqn (5), orthoborate (B(OH)3 = H3BO3) can, as a weak acid, produce hydrogen ions in water when the pH of the solution is higher than 9.14 (the pKa of orthoborate). As long as the pH of the solution is lower than 9.14, orthoborate remains the dominant species, and hydrogen peroxide can be formed according to eqn (4). Under the given conditions, the initial and final pH was in the range of 2.6–3.8, suggesting that the formation of hydrogen peroxide may not be affected by alkaline pH. The formation of hydrogen peroxide was confirmed by UV-vis spectrometry analysis.30 Therefore, the activation of perborate by iron (Fe2+ or F(0)) can be expressed as follows.
| | |
H4B2O82− + 4H2O + Fe2+ (or 0.5Fe(0)) → 2B(OH)4− + 2H2O2 + Fe2+ (or 0.5Fe(0)) → B(OH)4− + 2OH− + 2OH˙ + Fe3+ (or 0.5Fe2+)
| (6) |
According to eqn (6), hydroxyl radicals, which can also be transformed into other radicals such as superoxide radicals (O2˙−), were formed to enhance the oxidation of phenol in the perborate systems.
| |
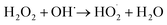 | (7) |
| |
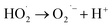 | (8) |
The radicals formed in the presence of iron-bearing materials can promote the oxidation of phenol in the perborate systems. In addition, the radicals were also terminated according to the following chain of reactions in the presence of hydrogen peroxide and Fe2+ (i.e., a Fenton oxidation).31
| | |
Fe2+ + OH˙ → Fe3+ + OH−
| (9) |
| | |
Fe3+ + H2O2 → Fe2+ + H+ + HO2−
| (10) |
| | |
Fe3+ + O2˙− → Fe2+ + O2
| (11) |
| | |
2H+ + O2˙− + O2˙− → H2O2 + O2
| (12) |
| | |
H+ + O2˙− + OH˙ → H2O + O2
| (13) |
3.2. Oxidation by perborate in the presence of carbonaceous materials
To determine whether carbonaceous materials can enhance the oxidation of phenol by perborate, various types of such materials were evaluated under identical conditions. Without perborate, phenol was removed from the solution via sorption to carbonaceous materials. The use of biochar, GAC, ACM, and graphite resulted in 27%, 44%, 18%, and 20% phenol removal, respectively, after 10 h (Fig. 2). Because of the high surface area of GAC, it achieved the greatest removal even though only 0.3 g of it was added to the phenol solution. However, sorptive removal was limited because the sorption capacity of the carbonaceous materials was saturated. The addition of perborate to carbonaceous materials greatly enhanced the oxidation of phenol, regardless of the type of carbonaceous material. After 10 h, the oxidation of phenol by perborate was enhanced by 11%, 16%, 49%, and 31% in the presence of biochar, GAC, ACM, and graphite, respectively (Fig. 2). The results confirmed that carbonaceous materials played the role of catalysts to enhance the oxidation of phenol by perborate.
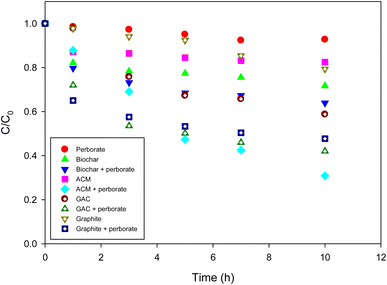 |
| | Fig. 2 Degradation of phenol with perborate in the presence of carbonaceous materials. The concentration of perborate, the initial concentration of phenol, the amounts of carbonaceous material, the volume of solution, and initial pH are 250 mg L−1, 1 mM, 1 g (except for GAC: 0.3 g), 250 mL, and pH 3, respectively. | |
It was previously reported that both the surface functional groups and graphitic structure of carbonaceous materials account for the catalytic characteristic of these materials in an oxidation reaction. The oxygen-containing surface functional groups of carbonaceous materials (e.g., –OH and –COOH) are persistent free radicals and are known to generate radicals with oxidants such as H2O2 and S2O82−. Similarly, oxygen-containing functional groups on the surface of carbonaceous materials can act as electron shuttles to mediate electron transfer reactions between persistent free radicals and perborate32–36 according to the following equations:
| | |
Asurface–OOH + H4B2O82− + 3H2O → Asurface–OO˙ + 2B(OH)4− + OH− + 2OH˙
| (15) |
| | |
Asurface–OH + H4B2O82− + 3H2O → Asurface–O˙ + 2B(OH)4− + OH− + 2OH˙
| (16) |
| |
Asurface![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + H4B2O82− + 3H2O → Asurface–O˙ + 2B(OH)4− + OH− + 2OH˙ O + H4B2O82− + 3H2O → Asurface–O˙ + 2B(OH)4− + OH− + 2OH˙
| (17) |
(A: carbonaceous material; A
surface–OO˙, A
surface–O˙: radicals).
Interestingly, compared with biochar and GAC, the oxidation enhancement by perborate was much greater in the presence of ACM and graphite (Fig. 2), suggesting that the graphitic structure may have a greater effect on promoting oxidation than surface functional groups. According to the properties of carbonaceous materials (Table 1) and a previous study,37 ACM is mostly graphite and its surface is slightly oxidized. Obviously, graphite does not have oxygen-containing functional groups to mediate the electron transfer to generate radicals. Therefore, the graphitic moiety should account for the enhanced oxidation of phenol by perborate. Two possible mechanisms could be responsible for the enhancement produced by the graphitic structure. First, delocalized electrons may be generated by defects in the graphitic structure. Electron-rich or -deficient particles resulting from the delocalized electrons on the surface of carbonaceous materials can activate perborate to generate radicals. Carbonaceous materials contain sp2-hybridized graphitic structures, which have numerous free-flowing π electrons.38 It has been reported that hydrogen peroxide, peroxymonosulfate (HSO5−), and persulfate can be decomposed into radicals on the surface of graphitic carbonaceous materials because of the delocalization of π-electrons from the graphitic layers, thereby enhancing the oxidation of organic compounds in an aqueous solution.37,39,40 The XRD analysis confirmed the clean development of the graphitic structures in GAC and ACM.41 Phenol oxidation by perborate accelerated when using GAC, ACM, and graphite (Fig. 2), suggesting that graphitic structures (C-π) may be responsible for enhancing carbonaceous material–perborate systems. Therefore, heterogeneous phenol oxidation in a graphitic carbonaceous material–perborate system can be represented by the equation below:
| | |
C-π + H4B2O82− + 3H2O → C-π+ + 2B(OH)4− + OH− + 2OH˙
| (18) |
In addition to the delocalized electrons, the facilitative role of the graphitic structure can be explained by carbonated material-mediated electron transfer reactions. In a previous study, we suggested that carbonaceous materials may serve as electron transfer mediators between phenol and persulfate.37 Similarly, when carbonaceous materials contain a graphitic structure, electrons from phenol can be readily transferred to perborate. The enhancement of phenol oxidation by perborate in the presence of graphite, GAC, ACM, and biochar confirms the possible acceleration of electron transfer via graphitic structures in carbonaceous materials (Fig. 2). Electrons can also be transferred between phenol and perborate via oxygen-containing surface functional groups of carbonaceous materials.
Another possible ROS that may be involved in the perborate system is singlet oxygen (1O2). The defects in carbon nanotubes may generate singlet oxygen to enhance the oxidation of phenol by persulfate.26 As a result of the presence of perborate and the development of graphitic structures in graphite, GAC, ACM, and biochar, the formation of singlet oxygen and its involvement in the oxidation of phenol oxidation by perborate cannot be completely ruled out. Therefore, the ROS in the iron-bearing and carbonaceous material–perborate systems should be identified.
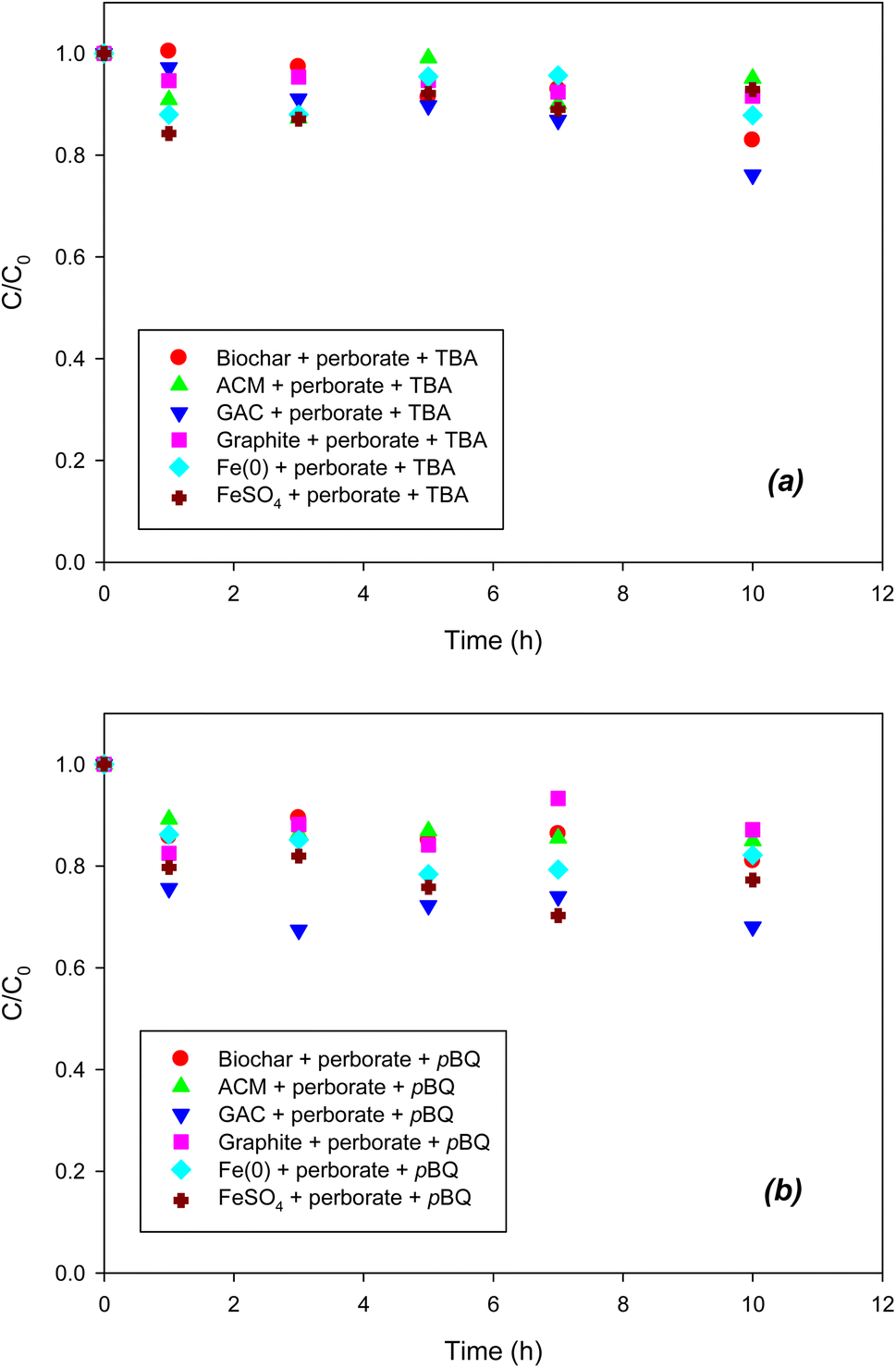
3.3. Quenching of reactive oxygen species with radical scavengers
To indirectly determine the ROS, radical scavengers were used for scavenging each radical species in the perborate systems. As shown in Fig. 3a, compared with the oxidation of phenol by perborate in the presence of iron-bearing or carbonaceous materials (Fig. 1 and 2), the addition of TBA substantially decreased the oxidation of phenol in the iron/carbonaceous material–perborate systems. Compared to the sorption to iron and carbonaceous materials and direct oxidation by perborate in the control experiments (Fig. 1 and 2), the quenching of hydroxyl radicals (OH˙) did not show enhanced oxidative removal of phenol in the iron/carbonaceous material–perborate systems. These results indicate that hydroxyl radicals were mostly responsible for the enhancement of phenol oxidation by perborate. By contrast, the addition of p-BQ substantially decreased the oxidation of phenol by perborate in the presence of iron-bearing or carbonaceous materials (Fig. 3b). Compared to the removal of phenol in the sorption and direct oxidation control (Fig. 1 and 2), phenol oxidation by perborate in the presence of iron-bearing or carbonaceous materials was slightly enhanced in the presence of p-BQ, revealing that superoxide radical (O2˙−) was predominantly involved in phenol oxidation through the chain reactions (eqn (7), (8) and (11)–(13)) once the hydroxyl radicals were generated from perborate by the effects of iron-bearing or carbonaceous materials.
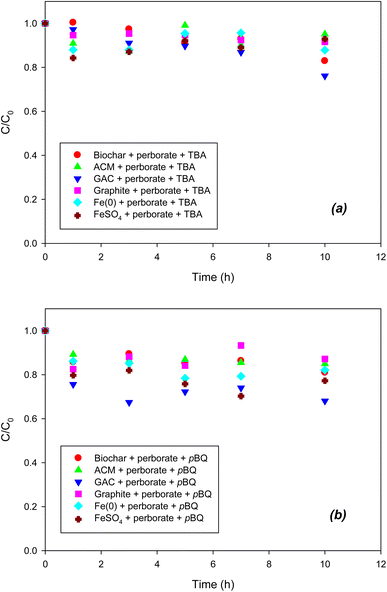 |
| | Fig. 3 Effects of (a) TBA (˙OH scavenger) and (b) p-BQ (O2˙− scavenger) on the degradation of phenol with perborate in the presence of iron-bearing and carbonaceous materials. The concentration of perborate, the initial concentration of phenol, the amount of iron-bearing and carbonaceous materials, the volume of solution, and initial pH are 250 mg L−1, 1 mM, 1 g (except for GAC: 0.3 g), 250 mL, and pH 3, respectively. | |
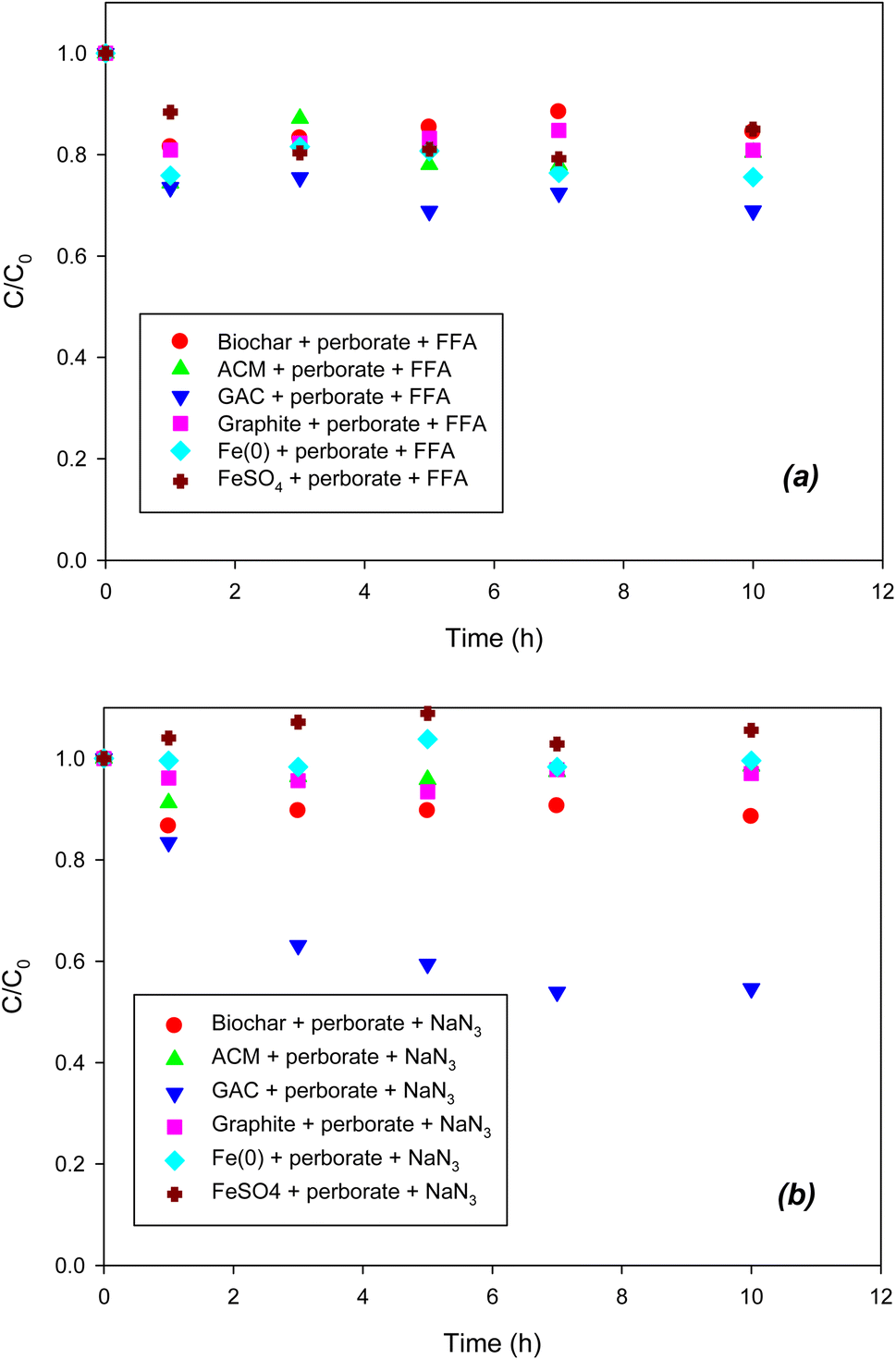
To determine the formation of singlet oxygen (1O2) in the iron/carbonaceous material–perborate systems, FFA was added to the latter. It was reported that the defects of graphitic structures result in the formation of singlet oxygen from the O–O bonding of persulfate.26 Likewise, the graphitic structure of carbonaceous materials may generate singlet oxygen from perborate. However, as shown in Fig. 4a, the addition of FFA did not inhibit the oxidation of phenol by perborate in the presence of iron-bearing or carbonaceous materials, revealing that singlet oxygen may not be involved in this process. By contrast, the addition of NaN3 substantially decreased the oxidation of phenol by perborate in the presence of iron-bearing or carbonaceous materials (Fig. 4b). The difference in inhibition between NaN3 and FFA suggests that electron transfer reactions may occur in homogeneous solutions or heterogeneous systems including iron-bearing or carbonaceous materials.26 In iron material–perborate systems, electron transfer from Fe2+ to perborate in solution and electron transfer from Fe(0) to perborate at the iron surface was quenched to inhibit the formation of radicals. As a result, the oxidation of phenol by perborate was markedly decreased. In the carbonaceous material–perborate systems, both the oxygen-containing surface functional groups26 and graphitic structure account for the electron transfer between phenol and perborate to promote phenol oxidation. Overall, the quenching experiments reveal that radical (˙OH and O2˙−) and non-radical (direct electron transfer) reactions are involved in the iron-bearing and carbonaceous material–perborate systems.
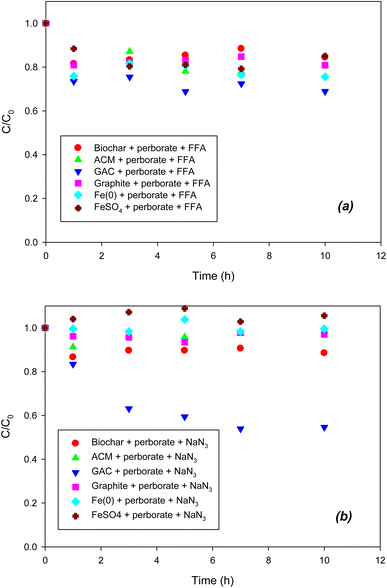 |
| | Fig. 4 Effects of (a) FFA (1O2 scavenger) and (b) NaN3 (1O2 and electron transfer scavenger) on the degradation of phenol with perborate in the presence of iron-bearing and carbonaceous materials. The concentration of perborate, the initial concentration of phenol, the amount of iron-bearing and carbonaceous materials, the volume of solution, and initial pH are 250 mg L−1, 1 mM, 1 g (except for GAC: 0.3 g), 250 mL, and pH 3, respectively. | |
3.4. Determination of reactive oxygen species by EPR analysis
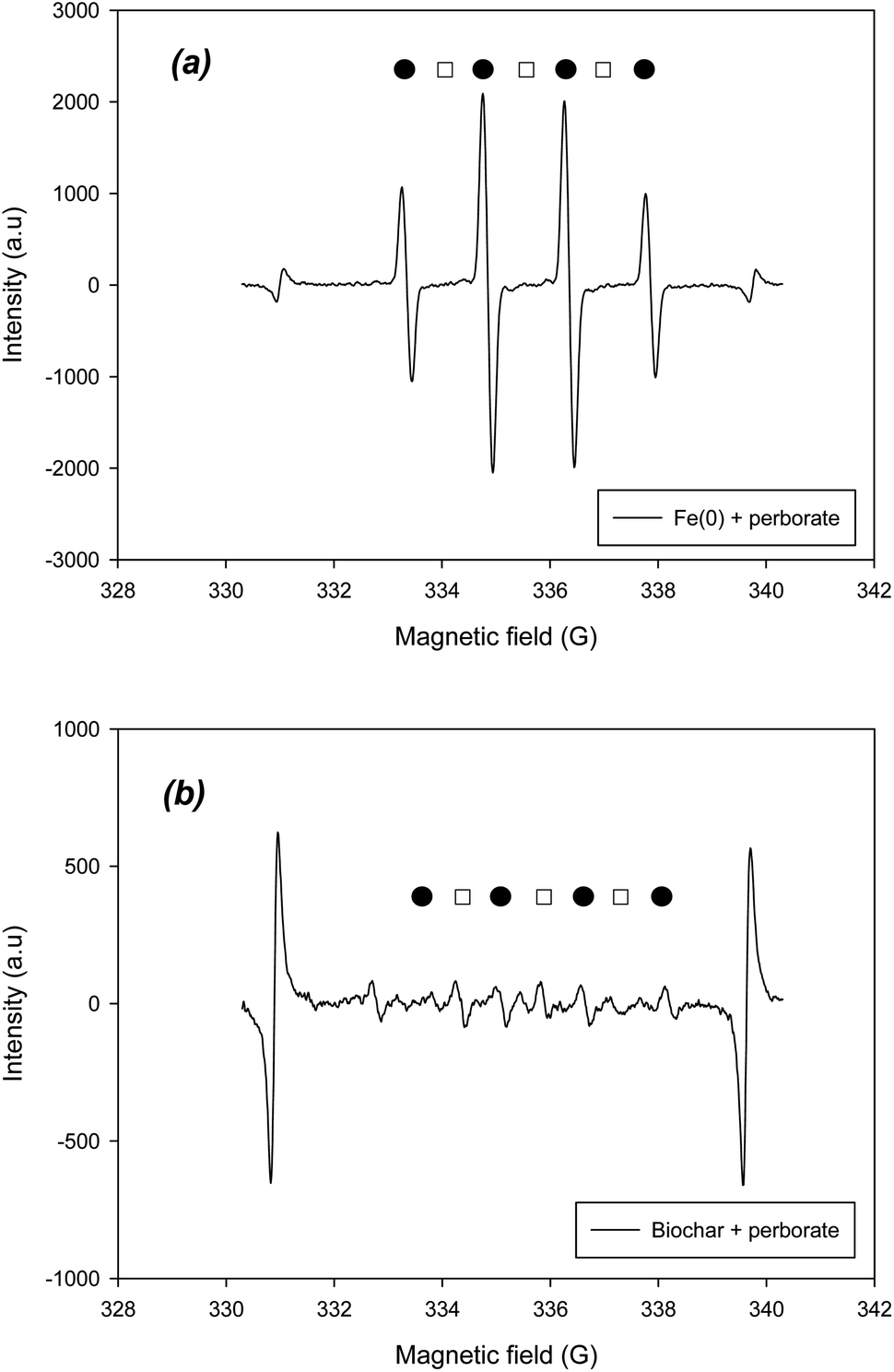
To verify the radical and non-radical reactions, the radical and non-radical ROS should be determined using EPR. To identify the hydroxyl and superoxide radicals, DMPO was added to the systems. Based on the hyperfine splitting constants of DMPO–˙OH and DMPO–O2˙−, the existence of two radicals was confirmed in the EPR spectra of Fe(0)– and biochar–perborate systems (Fig. 5). According to the intensity of the peaks in the spectra, the hydroxyl radical (˙OH) was predominant, whereas the superoxide radical was barely observed in the Fe(0)–perborate system (Fig. 5a). These results indicate that hydroxyl radicals are the predominant species in the oxidation of phenol by perborate in the presence of iron materials.
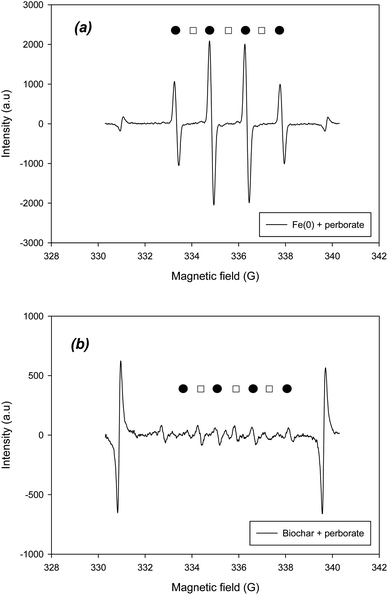 |
| | Fig. 5 EPR spectra of ˙OH and O2˙− (●: DMPO–˙OH; □: DMPO–O2˙−) obtained from (a) Fe(0)-perborate and (b) biochar-perborate systems. The reaction time and reactor volume are 1 min and 20 mL, respectively. The initial concentrations of phenol and DMPO are 1.06 mM and 10 mM, respectively. The dosage of carbonaceous material is 200 mg. The pH is 7.0, adjusted with 0.1 mM phosphate buffer. | |
On the other hand, the addition of biochar to perborate slightly increased the intensity of the DMPO–˙OH and DMPO–O2˙− peaks, confirming that biochar increased the formation of oxygen-containing radicals (Fig. 5b). The addition of GAC, ACM, and graphite also resulted in the appearance of the peaks of the two radicals in the EPR spectra (Fig. S1†), suggesting that hydroxyl and superoxide radicals are responsible for the oxidation of phenol by perborate in the presence of carbonaceous materials. Compared with that of the iron material–perborate system, the intensity of the two radical peaks was much lower and somewhat ambiguous (Fig. 5b), suggesting that, besides the two radicals, non-radical reactions such as electron transfer through the graphitic structure and surface functional groups of carbonaceous materials may be involved in the oxidation of phenol by perborate. The identification of singlet oxygen by EPR was conducted using TEMPO. The EPR spectra did not clearly show the development of TEMPO–1O2 peaks during the oxidation of phenol by perborate in the iron– or biochar–perborate systems (Fig. S2†), which is consistent with the results of the quenching experiments with FFA (Fig. 4).
The results of the EPR analysis confirmed that ˙OH and O2˙− are involved in the oxidation of phenol in the presence of carbonaceous materials. Based on the results of the batch experiments and EPR analysis, in addition to direct oxidation by perborate, oxidation by hydroxyl and superoxide radicals generated by the chain reactions from perborate (eqn (1)–(14)) may be a primary pathway in the oxidation of phenol by perborate in the presence of iron-bearing or carbonaceous materials. The quenching experiments also revealed the direct transfer of electrons between phenol and perborate when carbonaceous materials oxidize phenol in the carbonaceous material–perborate system. The electron-transfer oxidation pathways may be explained by the graphitic structures and oxygen-containing surface functional groups of carbonaceous materials. Oxidation products were determined by GC-MS analysis. Ethyl ether, n-hexane, and dichloromethane were used to extract the organic products after the perborate oxidation. However, no products were detected in the chromatograms, suggesting that phenol may have oxidatively transformed into highly hydrophilic compounds or CO2 in the iron– or carbonaceous material–perborate systems under the given conditions. Benzoquinone, a possible oxidation product was not detected in the perborate systems. After 10 h, TOC removal in the iron material–perborate systems reached 85–90%, suggesting that the transformation of CO2 may predominate under the given conditions. By contrast, less than 30% of phenol in the carbonaceous material–perborate systems was mineralized under identical conditions. Thus, the oxidation of phenol by perborate, using iron materials may generate more oxygen-containing radicals than the reaction with carbonaceous materials, which is consistent with the EPR results (Fig. 5).
3.5. Environmental implications
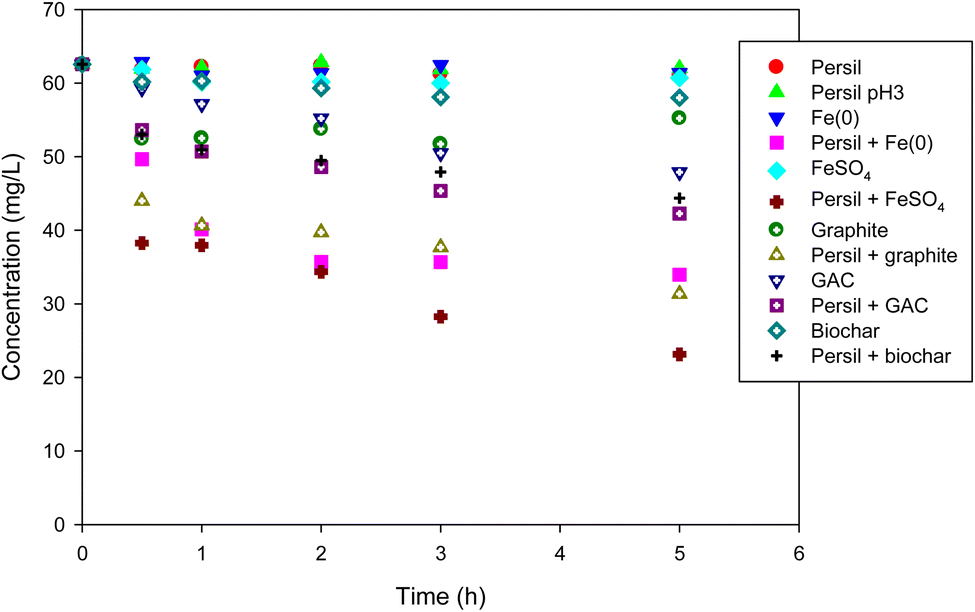
Based on the results of perborate oxidation in the presence of iron-bearing or carbonaceous materials, Persil®, a commercial detergent in which perborate is one of the primary ingredients, was tested as an oxidant under identical conditions. Compared with the direct oxidation by Persil®, the addition of iron-bearing or carbonaceous materials substantially enhanced the oxidation of phenol by the detergent (Fig. 6), which is consistent with the results obtained for perborate in the presence of iron or carbonaceous materials (Fig. 1 and 2). Phenol oxidation by Persil®, in the presence of iron materials, exhibited greater enhancement than that involving carbonaceous materials (Fig. 6), which is also consistent with the perborate systems (Fig. 1 and 2). In the case of iron-rich soils and sediments, the direct addition of Persil® may promote the oxidation of organic pollutants in field applications. The results revealed that Persil®, a readily available commercial detergent, can be used as an oxidant for the practical application of AOPs for environmental remediation of soils and sediments contaminated with refractory organic pollutants. Using perborate or Persil® as oxidants for AOPs would help maintain the reactivity of oxidants in field applications. The rapid consumption of hydrogen peroxide generated from perborate can be controlled to some degree because perborate is transformed into hydrogen peroxide in water in a stepwise process. The oxidation capability of perborate is similar to that of hydrogen peroxide and this compound is also easy to handle. Thus, perborate is a promising oxidant for AOPs in environmental remediation.
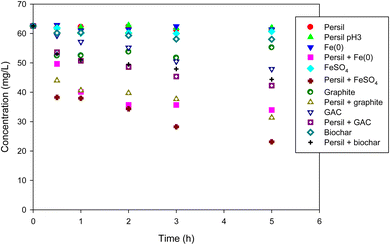 |
| | Fig. 6 Degradation of phenol with Persil® in the presence of iron-bearing and carbonaceous materials. The concentration of Persil®, the initial concentration of phenol, the amount of iron-bearing and carbonaceous materials, the volume of solution, and initial pH are 144 mg L−1, 1 mM, 1 g (except for GAC: 0.3 g), 250 mL, and pH 3, respectively. | |
4. Conclusions
Our results showed that the addition of iron or carbonaceous materials (FeSO4, Fe(0), graphite, ACM, GAC, and biochar) substantially enhanced the oxidation of phenol by perborate. Hydrogen peroxide generated by the dissociation of perborate was activated by the iron-bearing or carbonaceous materials. Hydroxyl and superoxide radicals were responsible for the enhancement of phenol oxidation in the iron– or carbonaceous–perborate systems. Carbonaceous materials also played a role as electron transfer mediators between phenol and hydrogen peroxide to enhance the oxidation of phenol by perborate. The applicability of Persil® (a commercial detergent) as an oxidant in combination with iron-bearing or carbonaceous materials was confirmed. Our results suggest that perborate may be a promising oxidant for use in AOPs for environmental remediation of contaminated areas.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Regional Innovation Cluster Development (R&D) by the Ministry of Trade, Industry and Energy (MOTIE, South Korea) [Project Name: Open Innovation Project for Convergence Industry of Battery/Fuel Cell for Mobility Electrification and Energy Production/Storage (P0025406)]. It was also in part supported by a National Research Foundation (NRF) of Korea grant funded by the Korean government (MSIT) (2020R1A2C1010855) and by the Korea Institute for Advancement of Technology (KIAT) grant funded by MOTIE (P0008421).
References
- M. Manna and S. Sen, Advanced oxidation process: a sustainable technology for treating refractory organic compounds present in industrial wastewater, Environ. Sci. Pollut. Res., 2023, 30, 25477–25505 CrossRef CAS PubMed.
- M. P. Rayaroth, M. Marchel and G. Boczkaj, Advanced oxidation processes for the removal of mono and polycyclic aromatic hydrocarbons - a review, Sci. Total Environ., 2023, 857(2), 159043 CrossRef CAS PubMed.
- Z. Liu, X. Ren, X. Duan, A. K. Sarmah and X. Zhao, Remediation of environmentally persistent organic pollutants (POPs) by persulfates oxidation system (PS): a review, Sci. Total Environ., 2023, 863, 160818 CrossRef CAS PubMed.
- H. Yan, C. Lai, S. Liu, D. Wang, X. Zhou, M. Zhang, L. Li, X. Li, F. Xu and J. Nie, Metal-carbon hybrid materials induced persulfate activation: application, mechanism, and tunable reaction pathways, Water Res., 2023, 234, 119808 CrossRef CAS PubMed.
- S. Y. Oh, H. W. Kim, J. M. Park, H. S. Park and C. Yoon, Oxidation of polyvinyl alcohol by persulfate activated with heat, Fe2+, and zero-valent iron, J. Hazard. Mater., 2009, 168, 346–351 CrossRef CAS PubMed.
- S. Y. Oh, S. G. Kang, D. W. Kim and P. C. Chiu, Degradation of 2,4-dinitrotoluene by persulfate activated with iron sulfides, Chem. Eng. J., 2011, 172, 641–646 CrossRef CAS.
- X. Hou, H. Dong, Y. Li, J. Xiao, Q. Dong, S. Xiang and D. Chu, Activation of persulfate by graphene/biochar composites for phenol degradation: performance and nonradical dominated reaction mechanism, J. Environ. Chem. Eng., 2023, 11, 109348 CrossRef CAS.
- X. Huang, J. Yuan, X. Zhao, S. Li, L. Yang, W. Li, K. Peng, C. Cai and J. Liu, Effects of adsorption characteristics of carbocatalysts on persulfate-based advanced oxidation processes: organic removal mechanisms and optimization strategies, Chem. Eng. J., 2023, 465, 142801 CrossRef CAS.
- S. Li, R. Guo, B. Li, Y. Liang, Z. Wang and R. Qu, Kinetics and mechanism studies of efficient degradation of 1-hexyl-3-methylimidazolium by zero-valent iron activated persulfate, Chem. Eng. J., 2023, 460, 141575 CrossRef CAS.
- Y. Shen, X. Mao, F. Liu, W. Yin, W. Shi and B. Zhang, A comparison study of heat-assisted Fe2+/persulfate and powdered activated carbon/persulfate wastewater pre-treatment for membrane fouling alleviation, J. Cleaner Prod., 2023, 406, 137127 CrossRef CAS.
- C. Shi, L. Nie, K. Hu, C. Zheng, C. Xu, H. Song and G. Wang, New insights into peroxydisulfate activation by nanostructured and bulky carbons, Appl. Catal., B, 2023, 325, 122371 CrossRef CAS.
- D. Guo, S. You, F. Li and Y. Liu, Engineering carbon nanocatalysts towards efficient degradation of emerging organic contaminants via persulfate activation: a review, Chin. Chem. Lett., 2022, 33, 1–10 CrossRef CAS.
- G. W. Kabalka, T. M. Shoup and N. M. Goudgaon, Sodium perborate: a mild and convenient reagent for efficiently oxidizing organoboranes, J. Org. Chem., 1989, 54, 5930–5933 CrossRef CAS.
- C. Karunakaran and B. Muthukumaran, Molybdenum(VI) catalysis of perborate or hydrogen peroxide oxidation of iodide ion, Transition Met. Chem., 1995, 20, 460–462 CrossRef CAS.
- C. Karunakaran and B. Muthukumaran, Zirconium(IV) catalysis in perborate oxidation of iodide, React. Kinet. Catal. Lett., 1997, 60, 387–394 CrossRef CAS.
- M. V. Gómez, R. Caballero, E. Vázquez, A. Moreno, A. de la Hoz and Á. Díaz-Ortiz, Green and chemoselective oxidation of sulfides with sodium perborate and sodium percarbonate: nucleophilic and electrophilic character of the oxidation system, Green Chem., 2007, 9, 331–336 RSC.
- E. Pesman, S. Imamoglu, E. E. Kalyoncu and H. Kırci, The effects of sodium percarbonate and perborate usage on pulping and flotation deinking instead of hydrogen peroxide, BioResources, 2014, 9, 523–536 CAS.
- K. Kurin-Csörgei, E. Poros-Tarcali, I. Molnár, M. Orbán and I. Szalai, Chemical oscillations with sodium perborate as oxidant, Front. Chem., 2020, 8, 561788 CrossRef PubMed.
- S. S. Devi, B. Muthukumaran and P. Krishnamoorthy, Vanadium(V) catalysis of perborate oxidation of substituted 5-oxo acids: a kinetic and mechanistic study, Ionics, 2014, 20, 1783–1794 CrossRef.
- H. R. Sindelar, M. T. Brown and T. H. Boyer, Evaluating UV/H2O2, UV/percarbonate, and UV/perborate for natural organic matter reduction from alternative water sources, Chemosphere, 2014, 105, 112–118 CrossRef CAS PubMed.
- H. H. Rump and H. Krist, Laboratory Manual for the Examination of Water, Wastewater and Soil, VCH, New York, 1988 Search PubMed.
- P. R. Hesse, A Textbook of Soil Chemical Analysis, John Murray, London, UK, 1971 Search PubMed.
- P. Faria, J. Órfão and M. Pereira, Adsorption of anionic and cationic dyes on activated carbons with different surface chemistries, Water Res., 2004, 38, 2043–2052 CrossRef CAS PubMed.
- G. Fang, C. Liu, Y. Wang, D. D. Dionysiou and D. Zhou, Photogeneration of reactive oxygen species from biochar suspension for diethyl phthalate degradation, Appl. Catal., B, 2017, 214, 34–45 CrossRef CAS.
- J. Al-Nu'airat, B. Z. Dlugogorski, X. Gao, N. Zeinali, J. Skut, P. R. Westmoreland, I. Oluwoye and M. Altarawneh, Reaction of phenol with singlet oxygen, Phys. Chem. Chem. Phys., 2019, 21, 171–183 RSC.
- X. Cheng, H. Guo, Y. Zhang, G. V. Korshin and B. Yang, Insights into the mechanism of nonradical reactions of persulfate activated by carbon nanotubes: activation performance and structure-function relationship, Water Res., 2019, 157, 406–414 CrossRef CAS PubMed.
- S. H. Ho, Y. D. Chen, R. Li, C. Zhang, Y. Ge, G. Cao, M. Ma, X. Duan, S. Wang and N. Q. Ren, N-doped graphitic biochars from C-phycocyanin extracted Spirulina residue for catalytic persulfate activation toward nonradical disinfection and organic oxidation, Water Res., 2019, 159, 77–86 CrossRef CAS PubMed.
- A. Mckillop and W. R. Sanderson, Sodium perborate and sodium percarbonate: cheap, safe and versatile oxidising agents for organic synthesis, Tetrahedron, 1995, 51, 6145–6166 CrossRef CAS.
- A. Mckillop and D. Kemp, Further functional group oxidation using sodium perborate, Tetrahedron, 1989, 45, 3299–3306 CrossRef CAS.
- M. Wang, S. Qiu, H. Yang, Y. Huang, L. Dai, B. Zhang and J. Zou, Spectrophotometric determination of hydrogen peroxide in water with peroxidase-catalyzed oxidation of potassium iodide and its applications to hydroxylamine-involved Fenton and Fenton-like systems, Chemosphere, 2021, 270, 129448 CrossRef CAS PubMed.
- C. Walling, Fenton's reagent revisited, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- R. W. Coughlin and F. S. Ezra, Role of surface acidity in the adsorption of organic pollutants on the surface of carbon, Environ. Sci. Technol., 1968, 2, 291–297 CrossRef CAS.
- J. A. Mattson, H. B. Mark, M. D. Malbin, W. J. Weber and J. C. Crittenden, Surface chemistry of active carbon: specific adsorption of phenols, J. Colloid Interface Sci., 1969, 31, 116–130 CrossRef CAS.
- M. Pu, Y. Ma, J. Wan, Y. Wang, M. Huang and Y. Chen, Fe/S doped granular activated carbon as a highly active heterogeneous persulfate catalyst toward the degradation of Orange G and diethyl phthalate, J. Am. Chem. Soc., 2014, 418, 330–337 CAS.
- W. D. Oh, S. K. Lua, Z. Dong and T. T. Lim, Performance of magnetic activated carbon composite as peroxymonosulfate
activator and regenerable adsorbent via sulfate radical-mediated oxidation processes, Appl. Catal., B, 2015, 284, 1–9 CAS.
- J. Yan, L. Han, W. Gao, S. Xue and M. Chen, Biochar supported nanoscale zerovalent iron composite used as persulfate activator for removing trichloroethylene, Bioresour. Technol., 2015, 175, 269–274 CrossRef CAS PubMed.
- T. H. A. Nguyen and S. Y. Oh, Anode carbonaceous material recovered from spent lithium-ion batteries in electric vehicles for environmental application, Waste Manage., 2021, 120, 755–761 CrossRef PubMed.
- J. M. Ducéré, C. Lepetit and R. Chauvin, Carbo-graphite: structural, mechanical, and electronic properties, J. Phys. Chem. C, 2013, 117, 21671–21681 CrossRef.
- K. Masaru and M. Ikuko, Discovery of the activated-carbon radical AC+ and the novel oxidation-reactions comprising the AC/AC+ cycle as a catalyst in an aqueous solution, Bull. Chem. Soc. Jpn., 1994, 67, 2357–2360 CrossRef.
- E. Saputra, S. Muhammad, H. Sun and S. Wang, Activated carbons as green and effective catalysts for generation of reactive radicals in degradation of aqueous phenol, RSC Adv., 2013, 3, 21905–21910 RSC.
- S. Y. Oh and T. H. A. Nguyen, Ozonation of phenol in the presence of biochar and carbonaceous materials: the effect of surface functional groups and graphitic structure on the formation of reactive oxygen species, J. Environ. Chem. Eng., 2022, 10, 107386 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Jun-Hwan Kim
* and
Jun-Hwan Kim