DOI:
10.1039/D3RA07482J
(Paper)
RSC Adv., 2023,
13, 35926-35936
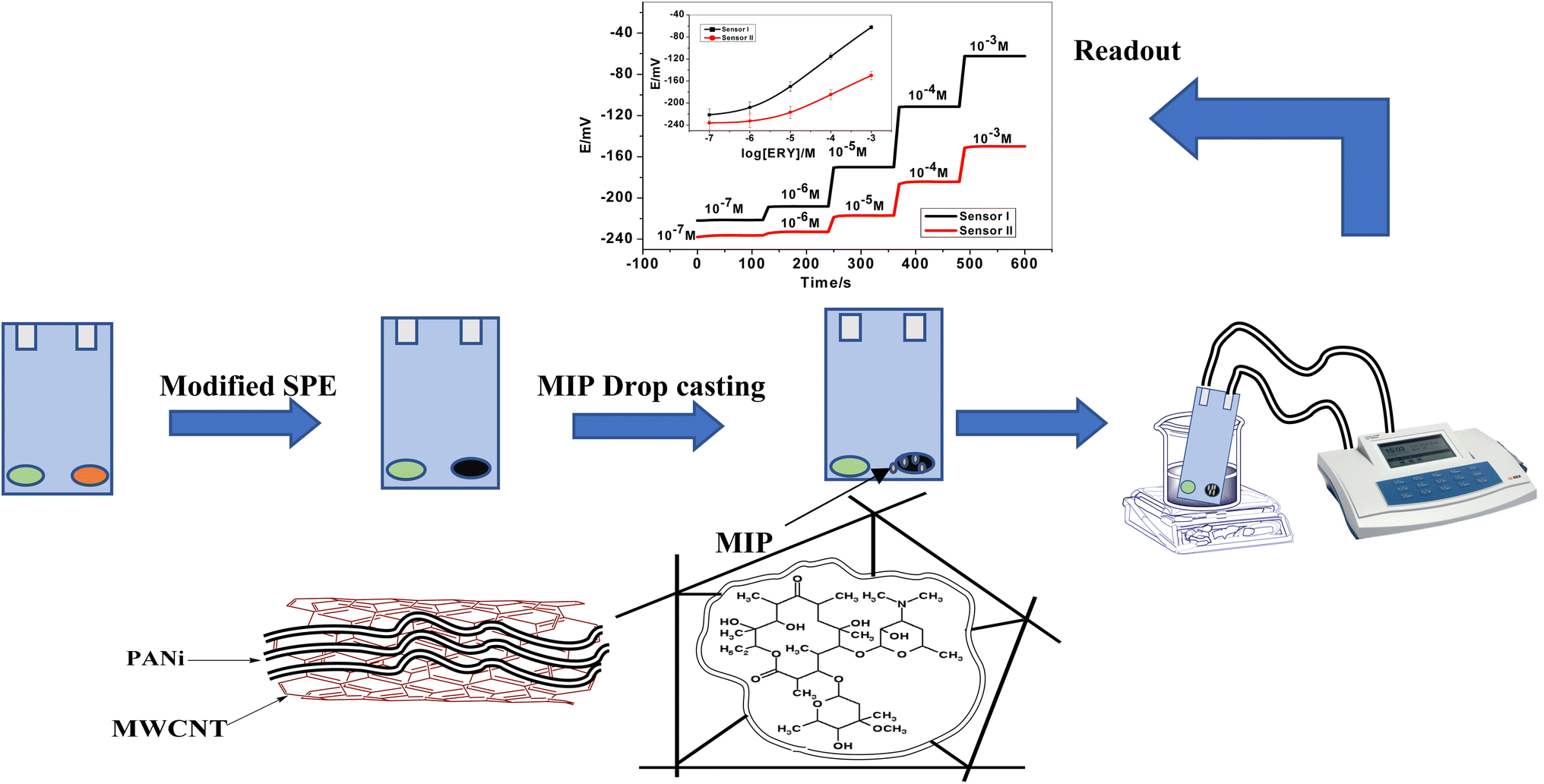
Potentiometric planar platforms modified with a multiwalled carbon nanotube/polyaniline nanocomposite and based on imprinted polymers for erythromycin assessment
Received
2nd November 2023
, Accepted 27th November 2023
First published on 11th December 2023
Abstract
A screen-printed potentiometric sensor for the erythromycin macrolide antibiotic (ERY) that is affordable, highly selective, and sensitive is made, described, and used for drug monitoring. Two circular carbon dots with a diameter of 4 mm make up the sensor. Multiwalled carbon nanotubes and polyaniline (f-MWCNTs/PANi) nanocomposites are used to change one carbon spot, which is then used as an ion-to-electron transfer material. Ag/AgCl is applied to the other spot, which is then used as a reference electrode. A solid-state polyvinyl butyral (PVB) is placed onto the second carbon spot to work as a reference electrode, and an ERY molecularly imprinted drug polymer (MIP) is coated onto the f-MWCNTs/PANi-containing strip to serve as a drug identification sensing material. Chronopotentiometry (CP) is used to analyze the integrated sensor's performance characteristics. It is confirmed that f-MWCNTs/PANi has an increased impact on the potential stability as well as the sensing membrane's interfacial double-layer capacitance. At a detection limit of 9.6 ± 0.4 × 10−7 M, the developed sensor exhibits a Nernstian slope of 54.0 ± 0.5 mV per decade (R2 = 0.9994) over the linear range of 4.6 × 10−6 to 1.0 × 10−3 M. When exposed to different related substances such azithromycin, clarithromycin, dirithromycin, paracetamol, and ascorbic acid, the sensor exhibits excellent selectivity. For the direct potentiometric determination of ERY in some pharmaceutical formulations and in samples of spiked human urine, the assay method has been validated and shown to be adequate. The obtained recovery ranges from 93.0 ± 0.5 to 104.3 ± 0.7 of the nominal or spiked concentration, with a mean relative standard deviation of ±0.6%. Due to the near closeness of the responsive membrane and the liquid junction, the use of all-solid-state electrodes coupled with a planar disposable platform enables applications with a minimum sample volume. The effectiveness of the suggested sensor in a complex urine matrix points to its use in hospitals for quick overdose patient detection as well as for quality control/quality assurance tests in the pharmaceutical sector.
1. Introduction
The first macrolide antibiotic that has been therapeutically utilized to treat human infections is erythromycin (ERY), which exhibits excellent action against both Gram-positive and Gram-negative bacteria.1 This medication is frequently utilized to advance the animal breeding sector in addition to preventing and treating several ailments.2–4 The overuse of this medication has led to a variety of residual issues in the environment and animal-derived foods. Due to antibiotics' ability to alter microbial structure and functions and have an impact on the emergence of antimicrobial resistance, the Rapid Alert System for Food and Feed (RASFF) of the European Union warns that even a small amount of antibiotic residue in edible animal parts may be harmful to the public's health.5
For the determination of erythromycin, numerous analytical techniques with low detection limits have been developed. They include surface plasmon resonance methods,6 Fourier transform infrared spectroscopy (FT-IR),7 capillary electrophoresis (CE),8 high performance liquid chromatography (HPLC),9 liquid chromatography/mass spectroscopy (LC/MS),10 liquid chromatography-mass/mass spectroscopy (LCMS/MS),11,12 voltammetry5,13–19 and potentiometry.20
Due to their excellent selectivity for target molecules, molecular imprinting polymers (MIPs)-based separation and sensing systems have been used in a variety of different fields.21–23 MIPs have several appealing qualities in addition to their antibody-like molecular selectivity. These features include robustness, stability at high temperatures and pressures, resistance to many chemical environments, use in both aqueous and non-aqueous media, low preparation costs, good shelf-lives, and repeated use without noticeably degrading their properties.24,25 For several decades, the integration of MIPs in chemical sensors has been steadily expanding.26–28 Since the beginning, the need for straightforward tools with the best selectivity for the detection of various compounds in industries like medical diagnosis, environmental and industrial monitoring, food, and toxicological analysis, and, more recently, the detection of explosives or their precursors, has driven their ongoing development.29,30
Sensors based on potentiometric transduction “ion-selective-electrodes (ISEs)” incorporating MIPs was a step forward towards the detection of organic ions.21–23 At zero-current trans-membrane ion fluxes, trace-level detection in potentiometric ion-sensors can be accomplished.31 To eliminate the ill-defined interface between the ion-sensing membrane (ISM) and the conductive substrate, an electron-to-ion transducing material was inserted into solid-contact potentiometric ISEs (SC/ISEs), which were used to overcome this limitation.32,33 Several reported sensors have been combined with various solid-contact materials to enhance their electrochemical characteristics. Some of these materials are carbon nanotubes (CNTs),34,35 conductive polymers,36,37 and others.38,39 Depending on the type of transducer utilized, each of these has a unique transduction mechanism, even though some of them demonstrate a significant improvement in potential stability and repeatability.40
In the current study, we sought to create a solid-contact potentiometric sensor that was highly stable, miniaturized, small, long-lasting, economical, and capable of detecting the erythromycin (ERY) macrolide antibiotic in a variety of pharmaceutical formulations and biological fluid matrices. We propose a screen-printed electrode (SPE) with molecularly imprinted erythromycin polymer (MIP) as recognition elements and f-MWCNTs/PANi nanocomposite as an ion-to-electron transducer material. Impedance spectroscopy and chronopotentiometry are used to examine the electrochemical properties of f-MWCNTs/PANi nanocomposite as an ion-to-electron transducer material in the constructed sensors. For measuring erythromycin (ERY) in pharmaceutical formulations and biological fluids, the SPE displays a good potential response with high potential stability, durability, sensitivity, and selectivity.
2. Experimental
2.1. Apparatus
A digital pH/mV meter (PXSJ-216 INESA, Scientific Instrument Co., Ltd., Shangahi, China) was used for all potentiometric measurements at room temperature. Ag/AgCl double-junction reference electrode (6.0729.100, Metrohm AG CH-9101 HERISAU, Switzerland) filled with 10% (w/v) KNO3. A Ross combined glass pH electrode was used for all pH solution adjustment. All polymeric beads were characterized and investigated using field-emission scanning electron microscope (FE-SEM) [ZEISS Sigma 300VP electron microscope instrument, Germany] and Fourier-transform infrared spectrometer (FT-IR) [Alpha II, Bruker ABCO, Germany].
2.2. Chemicals and reagents
All chemicals used in this work were of analytical reagent grade and used without any further purification. High molecular weight polyvinyl chloride (PVC), methacrylic acid (MAA), clarithromycin (purity, 96.0–102.0%), dirithromycin, azithromycin, and 2-nitrophenyl octyl ether (o-NPOE) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Ammonium persulfate (APS), polyvinyl butyral (PVB), ethylene glycol dimethacrylate (EGDMA), sodium tetrakis [3,5-bis(trifluoromethyl)phenyl] borate (NaTFPB) and tetrahydrofuran (THF) were purchased from Fluka. Ag/AgCl ink (E2414) was purchased from Ercon (Wareham, MA). Erythromycin (ERY) (<98.0% purity) was purchased from Cayman Chemical (Ann Arbor, Michigan 48108 USA). The chemicals and reagents were of analytical grade unless otherwise stated, and they were utilized without further purification. A Millipore water system made up of Milli-Q SP and Milli-RO 60 produced 18.25 M of de-ionized water.
A stock Erythromycine solution, 10−2 M was prepared in 50 mM phosphate buffer, pH 5.5 and 30 mM NaCl. Working ERY solutions (10−7 to 10−3 M) were prepared by accurate dilutions using the phosphate buffer.
2.3. Synthesis of imprinted beads (MIPs)
The imprinted polymeric beads were synthesized using the precipitation method. A 0.5 mmol of the template ERY, 3.0 mmol of MAA monomer, 3.0 mmol of the cross-linker EGDMA and 80 mg of the free-radical initiator APS were dissolved in 20 mL of acetonitrile (ACN) in a glass-capped bottle and the mixture was subjected to sonication for 10 min till a clear solution is obtained. De-oxygenation is carried out by passing a flow on N2 gas through the solution for 10 min and then it is placed in an oil bath at 70 °C for 17 h for complete polymerization. The polymer beads were precipitated from the solution, and the template was removed by batch-mode solvent extraction using methanol/acetic acid (8/2, v/v) and methanol. The MIP particles were left to dry at 40 °C overnight. For non-imprinted polymers (NIPs), they were prepared as mentioned above in absence of the template molecule.
2.4. Adsorption experiments
Adsorption capacity of the synthesized polymers towards ERY was evaluated using 20 mg of MIP in 10 mL of ERY solution with concentration [5.0–150.0 μg mL−1]. All were placed in an Erlenmeyer 25 mL closed bottle. At room temperature, the bottles were shaken for 6 hours, and then the MIP beads were separated via filtration through a membrane filter of 0.22 μm. The remaining ERY concentration was determined using UV visible absorption spectra at a λmax = 285 nm. After three repeated binding processes, the equilibrium adsorption quantity (Q, mmol g−1) was calculated according to eqn (1) and the corresponding adsorption isotherms and kinetics curves were drawn.where C0 (mg L−1) and C (mg L−1) are the initial and equilibrium concentration of Cit, respectively. V (mL) is the solution total volume and m (mg) is the weight of either MIPs or NIPs.
2.5. Synthesis of f-MWCNTs/PANI nanocomposite
f-MWCNTs/PANI is prepared by our group as reported before.41 In brief, f-MWCNTs weighing 20 mg were sonicated for 1 hour in 25 mL of 0.5 mol L−1 HCl. Like this, 3 mL of freshly distilled aniline was dissolved in 25 mL of 0.5 mol L−1 HCl and stirred for 30 minutes. The well-dispersed f-MWCNTs were then mixed with aniline hydrochloride solution and placed in an ultrasonic bath for 30 minutes at room temperature. As a polymerization initiator, ammonium persulfate (APS) (0.5 g) was added dropwise to the mixture while the mixture was continuously stirred for 1 hour. Overnight, the mixture was continuously stirred. The product was gathered, thoroughly cleaned with deionized water, and then given an ethanol rinse. The substance was then milled into a fine blackish green powder after being dried at 60 °C.
2.6. Sensor preparation and potential measurements
The electrochemical cell is constructed using screen-printed electrodes (SPEs). The design of the ceramic screen-printed electrode (SPE) is shown in Fig. 1. It contains two screens; one is made from carbon and the other from Ag/AgCl. The two screens were printed on alumina substrate of 0.1 mm thickness and 35 mm length. The screen for either carbon or Ag/AgCl ink printing was of 2 mm width. The sensing PVC membranes for either SPE/f-MWCNTs/PANi/MIP/ERY-ISE or SPE/f-MWCNTs/PANi/NIP/ERY-ISE were prepared by mixing NaTFPB (1.6 mg), PVC (31.2 mg), o-NPOE (58.5 mg), ETH 500 (2 mg) and MIP or NIP (6.7 mg). The membrane contents (100 mg in total) were dissolved in THF (2.0 mL). 10 μL of the membrane cocktail were dropped-casted onto the carbonic orifice in the SPE. The membrane was left over night for dryness. For the reference membrane, it was made by dissolving 70 mg of NaCl and 78.1 mg of polyvinyl butyral (PVB) in 1 mL of methanol. 20 μL of this membrane is drop-casted on the Ag/AgCl ink electrode surface.
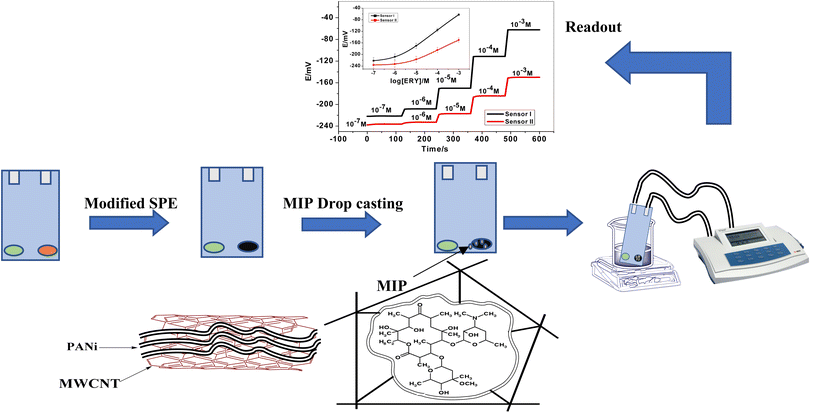 |
| | Fig. 1 A representation for the presented ERY membrane-based sensor. | |
The sensors were soaked in 1.0 × 10−3 M ERY and left for 24 h to be conditioned and stored in this solution when not in use.
Sensors' calibration was carried out when the integrated SPEs were inserted into a 25 mL beaker containing 9 mL of 50 mM phosphate buffer solution of pH 5.5. Different aliquots (0.5–1.0 mL) of 10−6 to 10−3 M standard ERY solutions were added and the potential response after each subsequent addition is recorded. The calibration plot [e.g., E (mV vs. log[ERY])] was constructed and used for all subsequent determination of unknown erythromycin samples.
2.7. Erythromycin assessment
Erythromycin is assessed in different pharmaceutical formulations collected from the local market using the proposed sensors. These samples include tablets (Erythryin 500, 500 mg per tablet, Misr Pharm., Egypt), gel (Benzamycin, 4 g/100 g, Sanofi, Egypt) and syrup solutions (200 mg/5 mL PHARCO Co., Egypt). In 5.0 mL of 50 mM phosphate buffer (pH 5.5), three tablets were thoroughly ground before being sonicated and filtered off. Using the proposed all-solid-state drug sensor, an aliquot comparable to one tablet was employed for direct potentiometric readings. A blank experiment was run in the same manner and the recorded potential was compared to the calibration plot.
A human urine samples were obtained from an adult male and diluted with phosphate-buffered solution (50 mM, pH 5.5) at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to assess the viability of the proposed SPE in complex matrices. These diluted urine samples underwent direct potentiometric measurements after being spiked with various ERY concentrations.
:10 to assess the viability of the proposed SPE in complex matrices. These diluted urine samples underwent direct potentiometric measurements after being spiked with various ERY concentrations.
All urine samples were collected in accordance with the Guidelines for Care and Use of National research Centre (NRC), Egypt, and approved by the Scientific Research Ethics Committee of The National Research Centre (NRC, Dokki, Egypt).
3. Results and discussion
3.1. Characterization of erythromycin imprinted polymers
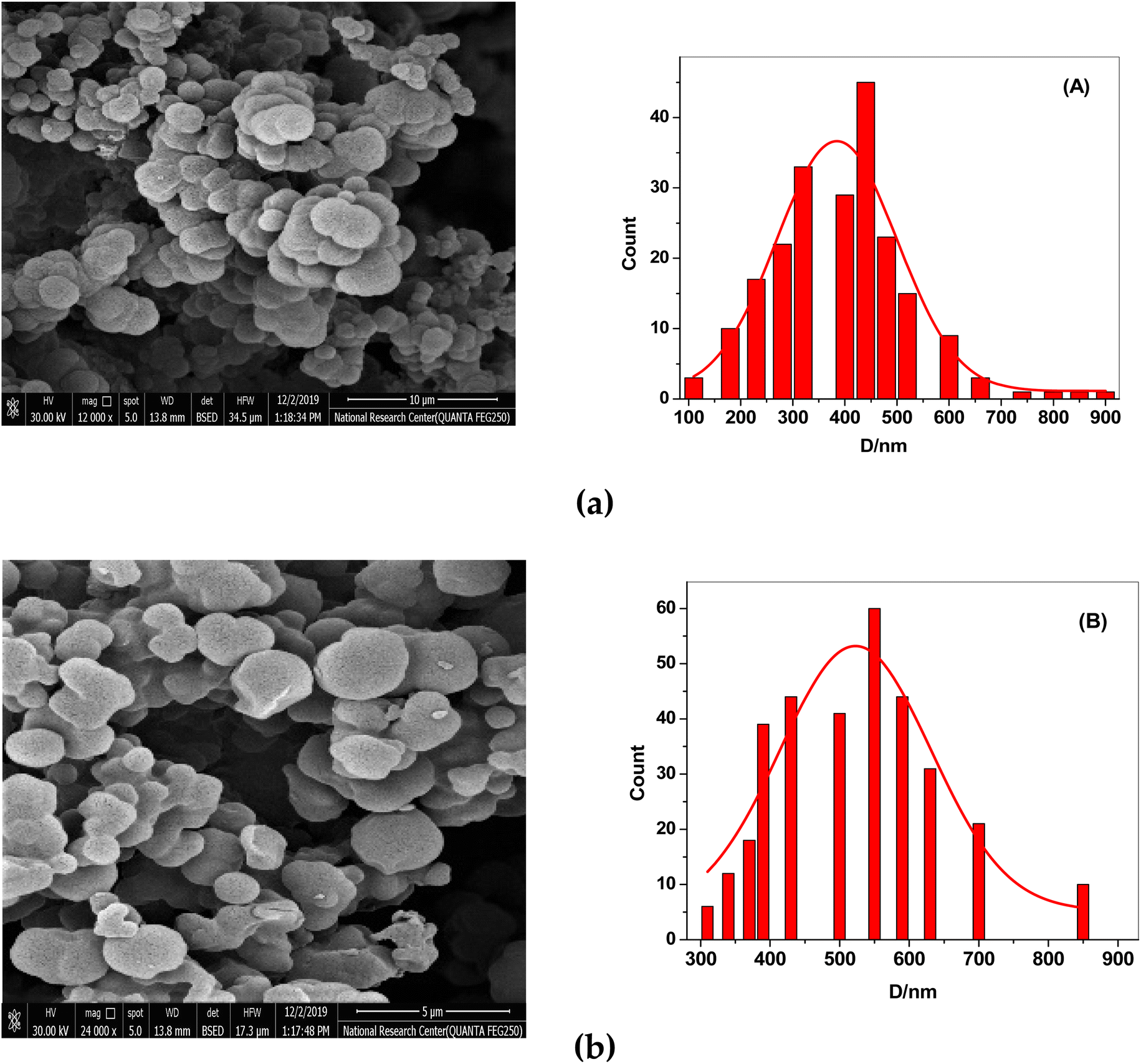
After the template (drug) was removed, the surface morphologies of both NIP and MIP were examined using scanning electron microscopy (SEM). SEM images show that the synthesized MIP and NIP are micro-spherical shape particles with different diameters of 0.1–0.9 μm. The imprinted NIP particles (average size 0.55 μm, Fig. 2B) were less aggregated than MIP particles (average size 0.42 μm, Fig. 2A). These morphological variations resulted from the absence of binding sites in the NIP beads, supporting the idea that the imprinting process took place in the MIP beads. The small and varied sizes of the MIP beads enhanced the dispersion and the distribution of these beads in the polymeric poly (vinyl chloride) matrix membrane of the developed sensor.
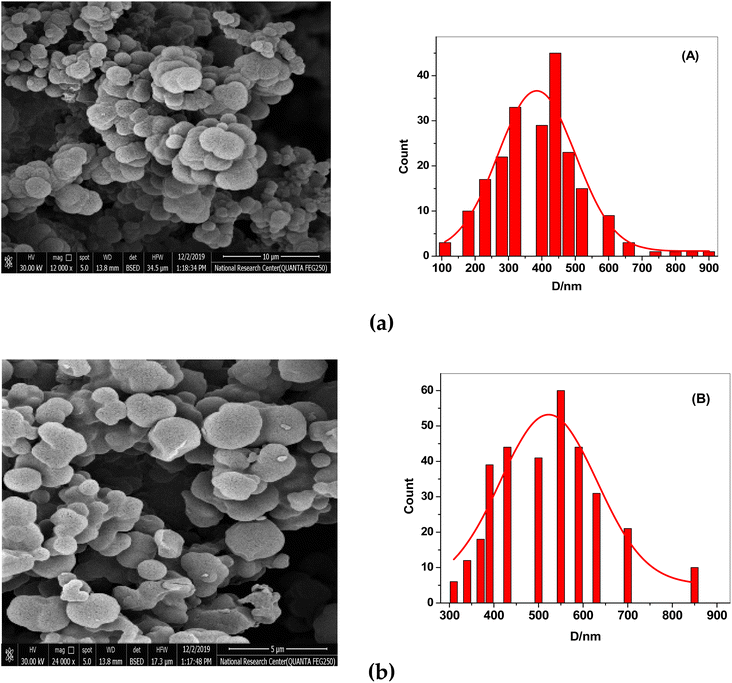 |
| | Fig. 2 SEM images of (a) MIPs and (b) NIPs. (A) The particle size distribution graph for MIPs and (B) for NIPs. | |
3.2. Binding affinity of MIP
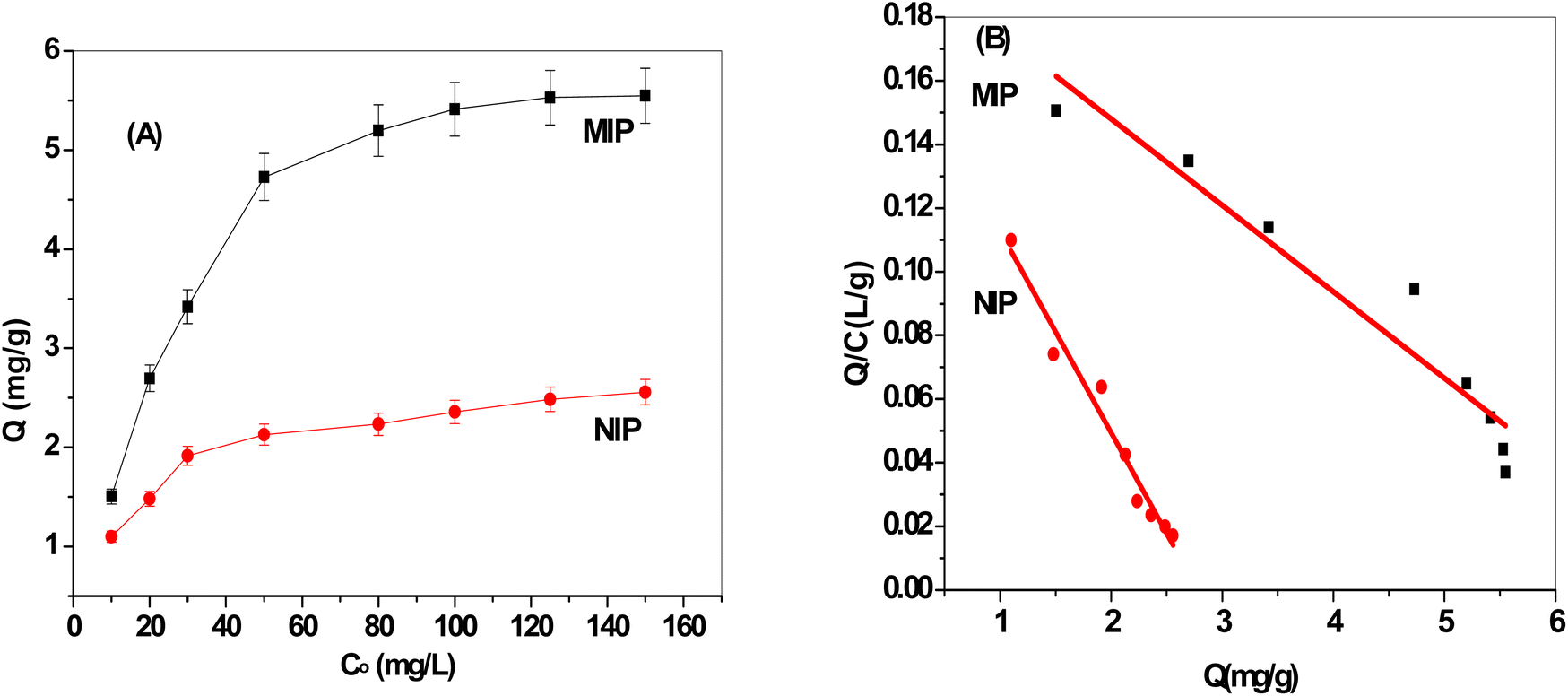
More ERY is absorbed by the MIP particles when the starting concentration of ERY increases, as seen in Fig. 3A. The plateau, which corresponds to the saturation adsorption capacity, reaches 5.5 and 2.48 mg g−1 for MIP and NIP beads, respectively. This demonstrates that compared to NIPs beads, MIPs exhibited a higher specific adsorption capability. The existence of template molecules and their involvement in the creation of MIPs can be used to explain this. The MIPs now possess active functional groups that play a complementary role in strongly recognizing the template molecules as well as active cavities that are compatible with the template ERY. As seen in Fig. 3A, NIP particles have a lesser adsorption capability because they lack these specific holes with the spatial structure and functional groups that would match the template molecules.
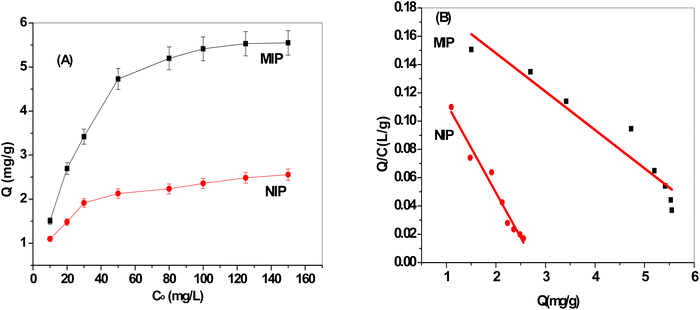 |
| | Fig. 3 (A) Adsorption isotherms and (B) Scatchard plots for both MIP and NIP beads. | |
A Scatchard plot created by eqn (2) was used in the Scatchard analysis to determine the maximal binding capacity Qmax.
where
Q (mg g
−1) is the number of MIPs that bind to ERY,
Qmax (mg g
−1) is the apparent maximum adsorption capacity,
Kd (mg L
−1) is the dissociation constant, and
C (mg L
−1) is the starting concentration of ERY.
As presented in Fig. 3B, the findings showed that the Scatchard model has a Q/C value of 0.202–0.0272 Q (r2 = 0.9865) and that the MIPs' adsorption isotherms toward the ERY are in good agreement with linearity. The equation showed that within the examined concentration range, MIPs have equal class binding sites for ERY, and their respective Kd and Qmax values for MIPs and NIPs were 15.79 mg L−1 and 7.45 ± 0.2 mg g−1 and 36.8 mg L−1 and 2.78 ± 0.4 mg g−1, respectively. Our results showed that MIPs had greater binding association constants than NIPs do.
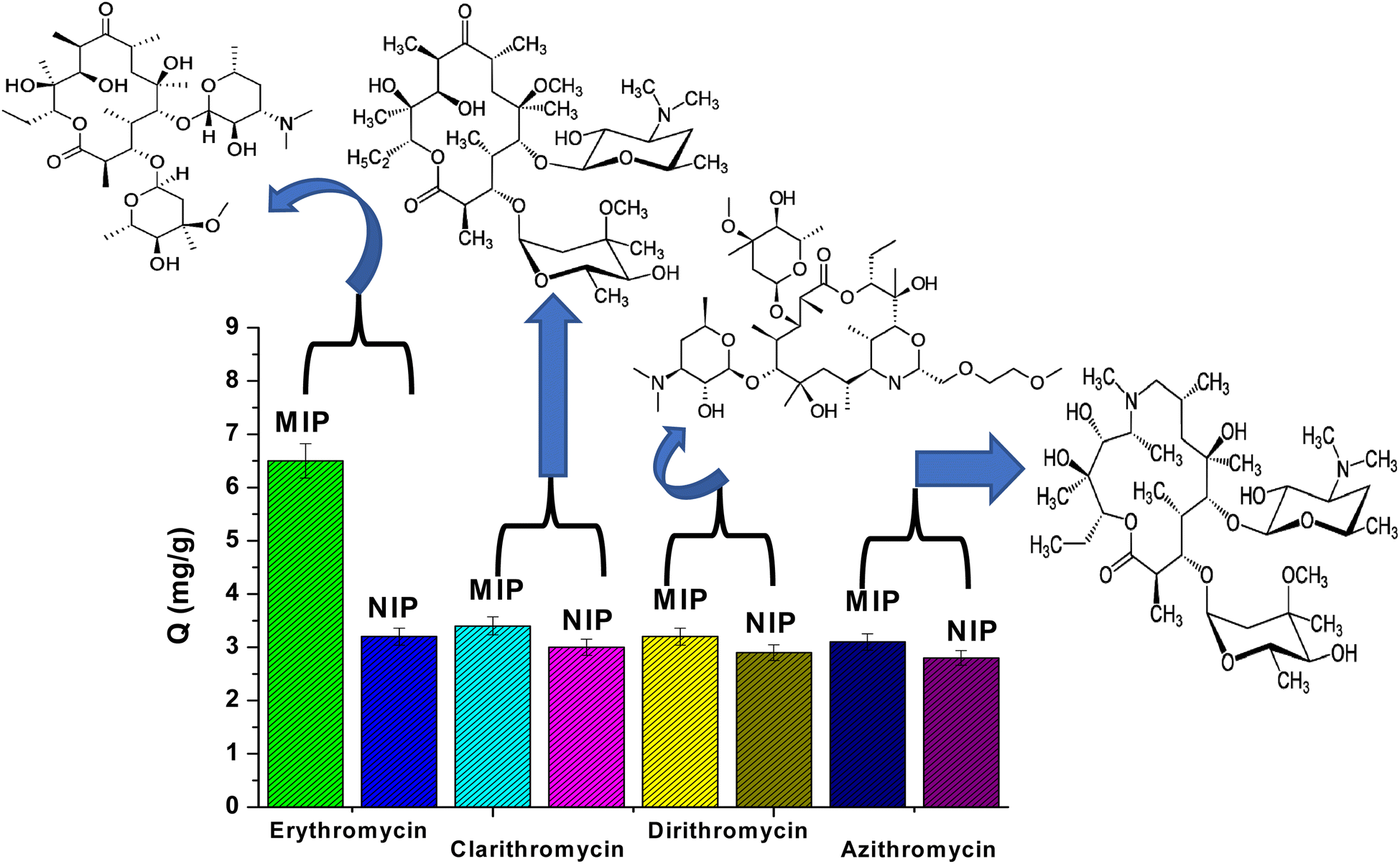
The selectivity of MIP and NIP particles was also studied for erythromycin (ERY), azithromycin (AZI), clarithromycin (CLA), and dirithromycin (DIR). According to Fig. 4, it was noticed that MIP beads had a higher ability for adsorption toward ERY than AZI, CLA and DIR. This can be explained by the fact that the active sites, cavity size, and shape of MIPs may not perfectly match those of AZI, CLA, and DIR. Furthermore, the NIPs are a general adsorbent to AZI, CLA, and DIR since they lack binding properties.
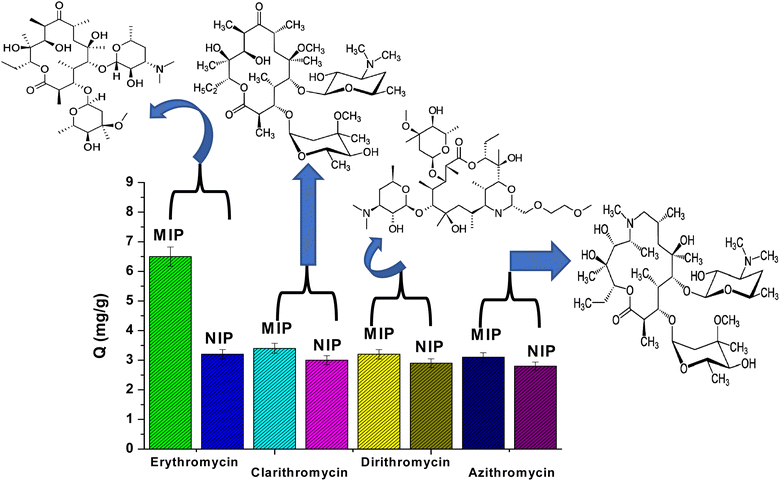 |
| | Fig. 4 Adsorption selectivity of MIPs and NIPs towards erythromycin, clarithromycin, dirithromycin, and azithromycin. | |
3.3. Potentiometric characteristics
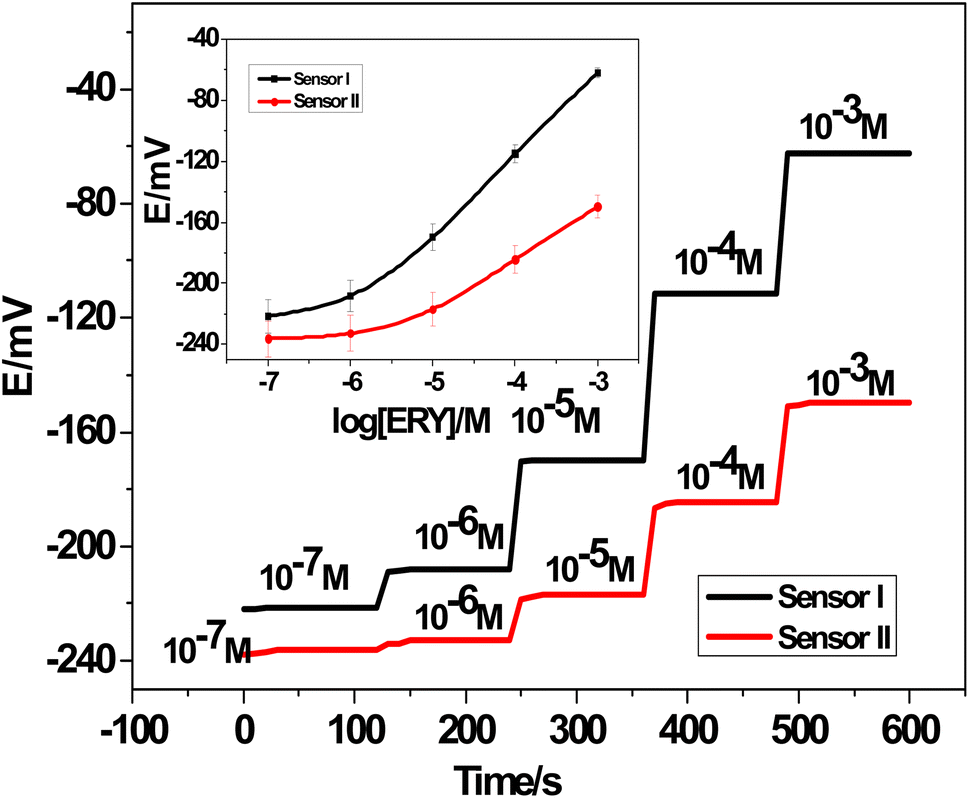
3.3.1. Potentiometric response. Using 1.0 × 10−7 to 1.0 × 10−2 M ERY drug test solutions at pH 5.5, the potentiometric response of the presented all-solid-state ERY sensors based on either MIPs or NIPs was assessed and compared (Fig. 5). For the modified MIPs sensor (SPE/f-MWCNTs/PANi/MIP/ERY-ISE), a Nernstian response was found with a calibration slope of 54.0 ± 0.5 mV per decade (n = 5, R2 = 0.9998) in a linear range of 4.6 × 10−6 to 1.0 × 10−3 M and a detection limit of 9.6 ± 0.4 × 10−7 M. It took about <15 s to get to the equilibrium state. On the other hand, a sensor based on NIP (MWCNTs/PANi/NIP/ERY-ISE) demonstrated a non-Nernstian response with a slope of 35.3 ± 0.6 mV per decade (n = 5, R2 = 0.9996) within a restricted linear range of 2.5 × 10−5 to 1.0 × 10−3 M, a detection limit of 4.0 × 10−6 M, and a response time The particular interaction between ERY molecules and MIP beads attached to the ERY-selective membrane was validated by these results.
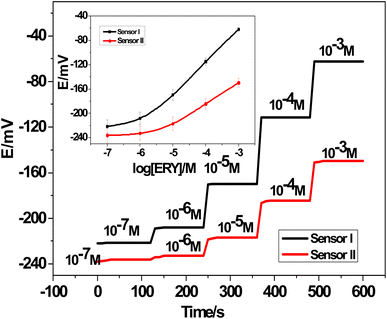 |
| | Fig. 5 Time/potential plot for both SPE/f-MWCNTs/PANi/MIP/ERY-ISE (sensor I) and MWCNTs/PANi/NIP/ERY-ISE (sensor II). [The inset plots: calibration plots for both SPE/f-MWCNTs/PANi/MIP/ERY-ISE (sensor I) and MWCNTs/PANi/NIP/ERY-ISE (sensor II)]. | |
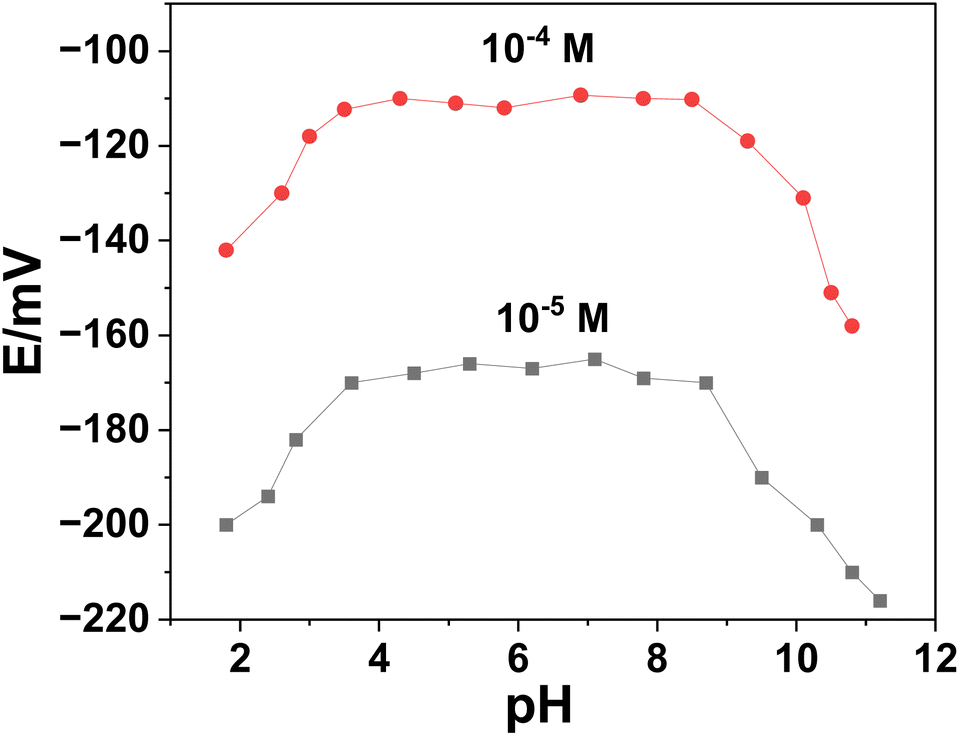
3.3.2. Effect of pH on the potentiometric response. Using two concentrations of ERY (1.0 × 10−4 and 1.0 × 10−5 M), the potential stability of SPE/f-MWCNTs/PANi/MIP/ERY-ISE over a range of pH values was examined (Fig. 6). With the use of HCl and/or NaOH, the test solution's pH was changed. Over the pH range of 3.5–8.5, high and steady potential measurements were recorded. The formation of the non-protonated ERY (pKa = 8.8), which caused negative potential drift, was observed at pH levels >8.5.42 A considerable drop in potential was seen at <pH 3 and was most likely caused by the macrolide drug's breakdown.42 In a (50 mM) phosphate buffer with a pH of 5.5, all following measurements with the all-solid-state ERY sensors were conducted.
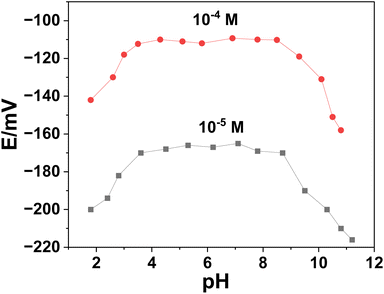 |
| | Fig. 6 Effect of pH on the potentiometric response of the presented sensor. | |
3.4. Sensor's selectivity
It investigated how selective various macrolides, organic compounds, and inorganic ions were toward SPE/f-MWCNTs/PANi/MIP/ERY-ISE. These interfering ions were anticipated to be present in human urine samples, as well as the fact that they are used in the production of various pharmaceutical formulations. The modified separate solution method (MSSM) was used to evaluate the selectivity coefficient values (Kpoti,j).43
At concentrations ranging from 1.0 × 10−5 to 1.0 × 10−2 M, calibration curves were constructed for each of the interfering species, starting with the most discriminated and ending with ERY. Eqn (3) was used to calculate the selectivity coefficient values from the extrapolated potentials at 1.0 M solutions.where: Eoi, Eoj, and S are the extrapolated potential readings of the primary (ERY) and interfering species, as well as the slope of the suggested sensor, respectively.
| | |
LogKpoti,j = (Eoi − Eoj)/S
| (3) |
Table 1 displays the selectivity coefficient values for SPE/f-MWCNTs/PANi/MIP/ERY-ISE. In the presence of several inorganic and organic species frequently found in pharmaceutical formulations and biological fluids, the sensor showed a high selectivity towards ERY over the entire test interfering ions. Although the selectivity of the sensor improved little because of the f-MWCNTs/PANi modification, the sensor's stability and endurance were greatly improved.
Table 1 The selectivity coefficient values for SPE/f MWCNTs/PANi/MIP/ERY-ISEa
| Interfering ion, j |
LogKpoti,j |
| ±SD (average of 3 measurements). |
| Clarithromycin |
−1.9 ± 0.3 |
| Dirithromycin |
−2.2 ± 0.4 |
| Azithromycin |
−2.5 ± 0.2 |
| Caffein |
−3.5 ± 0.6 |
| Paracetamol |
−3.7 ± 0.1 |
| Ascorbic acid |
−4.7 ± 0.4 |
| Urea |
−4.4 ± 0.3 |
| Creatinine |
−4.1 ± 0.2 |
| Glucose |
−4.6 ± 0.4 |
| Fructose |
−5.3 ± 0.7 |
| Ca2+ |
−6.2 ± 0.3 |
| NH4+ |
−4.5 ± 0.3 |
| K+ |
−5.1 ± 0.5 |
3.5. Sensor's durability
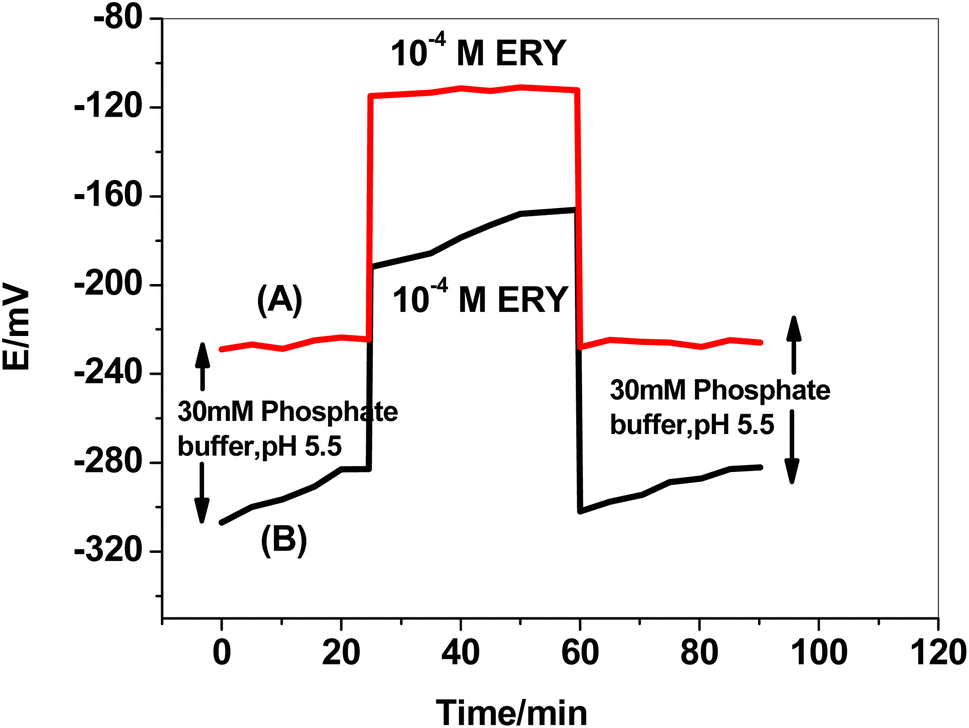
The formation of a water layer between the solid conducting substrate base in the SPE and the sensing polymeric membrane and the delamination of the electroactive polymeric film are two of the main issues with planar sensors' configuration. This consequence resulted in mechanical failure, potential fluctuation, hysteresis, instability, and a shorter sensor's life.44 A test was carried out to test the effect of utilizing f-MWCNTs/PANi as a hydrophobic layer. The sensor was immersed for 30 minutes in 50 mM phosphate buffer, then for another 30 minutes in 1.0 × 10−4 M ERY drug solution, before being returned to the phosphate buffer for another 30 minutes (Fig. 7). The formation of a thin water layer resulted in a continuous potential drift in the absence of f-MWCNTs/PANi. The water layer drastically minimized after utilizing f-MWCNTs/PANi, and the potential drift in the buffer test solution decreased from 29 to 4.6 mV/30 min. The potential drift was minimized from 26 to 2.6 mV/30 min in the second 30 minute cycle (ERY solution) and from 20 to 3.5 mV/30 min in the third 30 minute cycle (buffer solution).
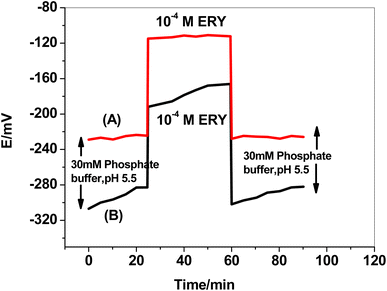 |
| | Fig. 7 Water layer test for (A) SPE/f-MWCNTs/PANi/MIP/ERY-ISE, and (B) SPE/MIP/ERY-ISE. | |
3.6. Method validation
The proposed SPE/f-MWCNTs/PANi/MIP/ERY-ISE approach for ERY measurement was thoroughly validated. Prior descriptions of the words and parameters were provided.45 These parameters include accuracy, trueness, bias, within-day repeatability, between-days reproducibility, standard deviations, precision, sensitivity, robustness, range, stability, durability, and limit of detection. According to ISO/IEC 17025, AOAC, USP, USEPA, and USFDA,45 a validation process was created, and the outcomes are shown in Table 2.
Table 2 Method validation data obtained with SPE/f-MWCNTs/PANi/MIP/ERY-ISE
| Parameter |
Value |
| Slope (mV per decade) |
54.0 ± 0.5 |
| Correlation coefficient (r2) |
0.9998 |
| Linear range (M) |
4.6 × 10−6 to 1.0 × 10−3 |
| Limit of detection, LOD (M) |
9.6 ± 0.4 × 10−7 |
| Working pH range (pH) |
3.5–8.5 |
| Response time for 10−4 M (s) |
<10 |
| Precision (%) |
0.4 ± 0.07 |
| Accuracy (%) |
99.1 ± 1.3 |
| Trueness, (%) |
98.6 ± 0.9 |
| Within-day reproducibility, Cvw (%) |
1.1 ± 0.2 |
| Between-days variability, Cvb (%) |
0.9 ± 0.07 |
| Relative standard deviation (%) |
0.8 ± 0.05 |
| Lifespan (days) |
15 |
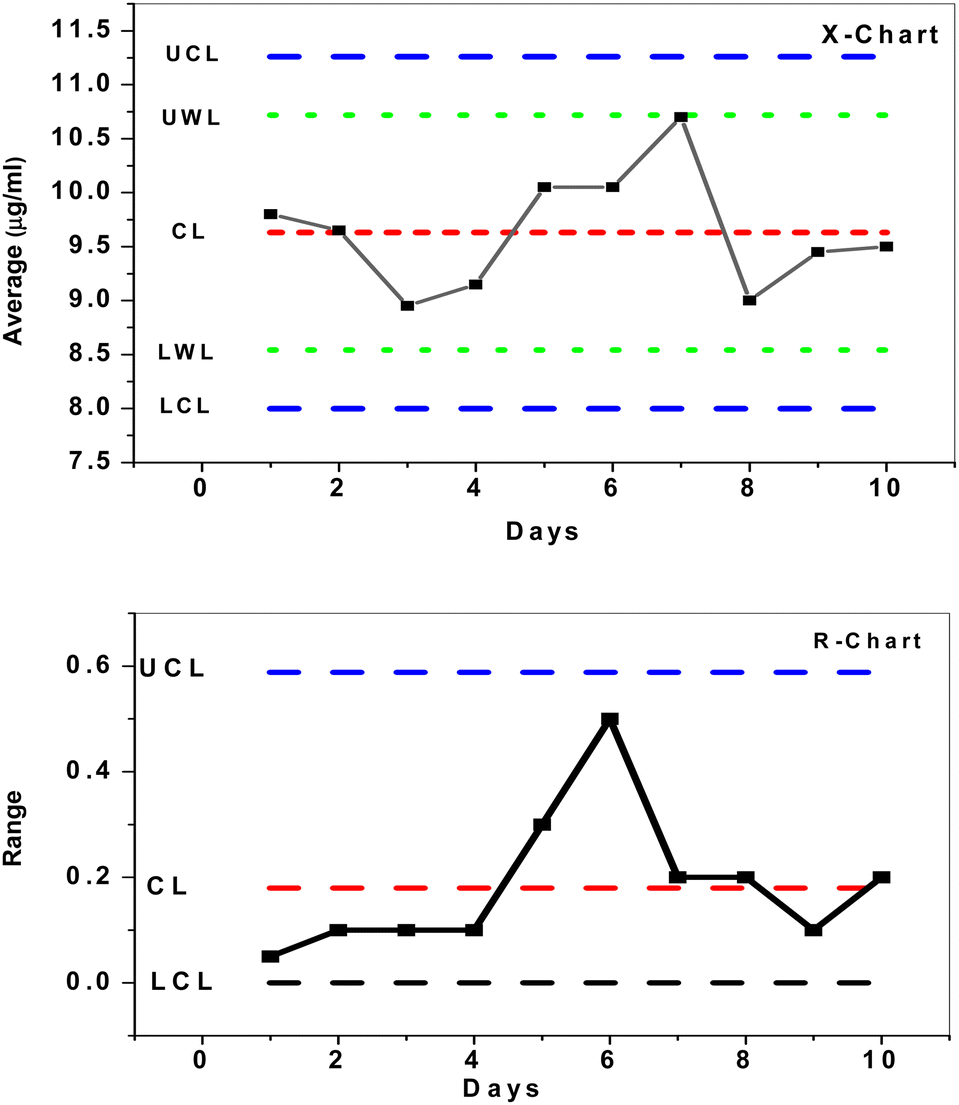
To investigate the mean and changes of duplicate measurements over time, quality X-bar and R-control charts of the data acquired by SPE/f-MWCNTs/PANi/MIP/ERY-ISE with a buffer solution of pH 5.5 were used.46 The results (Fig. 8) demonstrated that all measured concentrations [internal quality control ERY solution (IQC) = 10 μg mL−1] were statistically controlled, placed outside of the control (action) limit (UCL/LCL), and within the upper and lower warrant limits (UWL/LWL). This demonstrates that the sensor that has been provided may be successfully utilized for evaluating the quality of data as well as for quality control and assurance purposes.
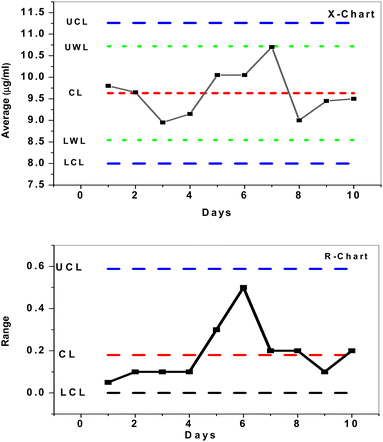 |
| | Fig. 8 X- and R control charts for 10 μM ERY solution using SPE/f-MWCNTs/PANi/MIP/ERY-ISE. | |
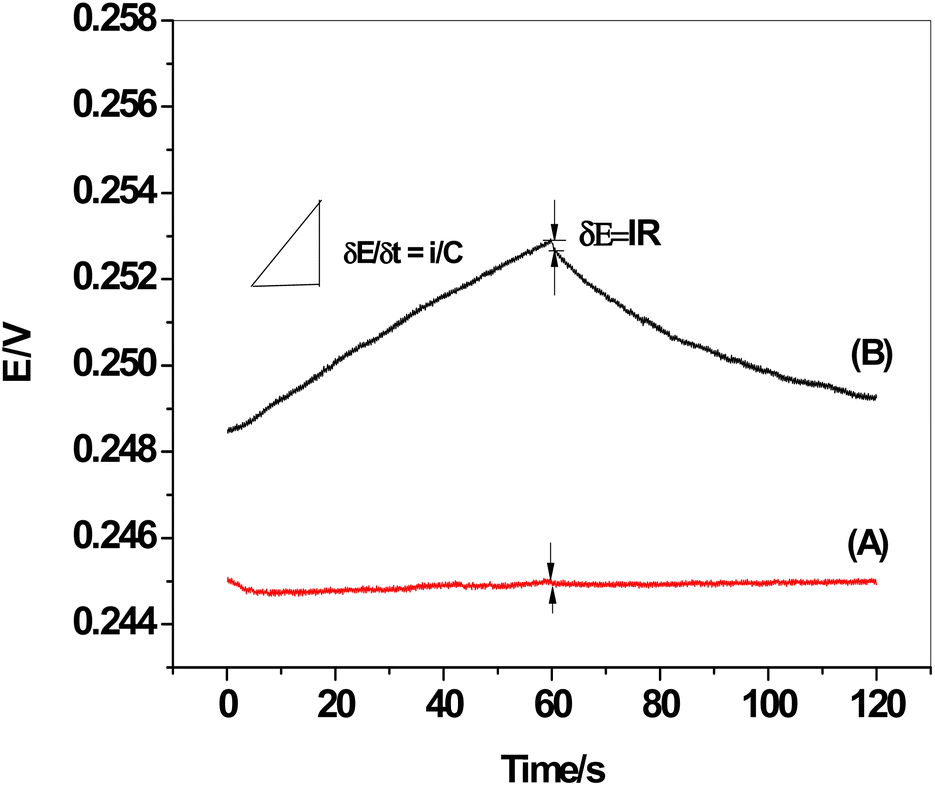
3.7. Chronopotentiometric measurements
The chronopotentiograms of the two sensors, SPE/f-MWCNTs/PANi/MIP/ERY-ISE and SPE/MIP/ERY-ISE (inserted in Fig. 9), were carried out by continuously applying cathodic and anodic currents of 1 nA for 60 seconds in a solution containing 1 × 10−5 M ERY. Eqn (4) and (5) were used to calculate the potential drift (ΔE/Δt), bulk membrane resistance (Rb), and interfacial double-layer capacitance (CL) using the plot of the potential values versus time.47where: ΔE, I, R, Δt, C, and R stand for the interfacial double-layer capacitance, applied current (1 nA), bulk membrane resistance, and time variation, respectively.
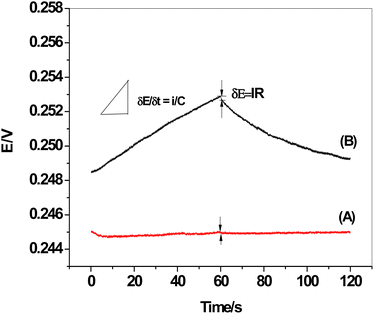 |
| | Fig. 9 Chronopotentiograms: (A) SPE/f-MWCNTs/PANi/MIP/ERY-ISE; and (B) SPE/MIP/ERY-ISE. Applied current: ±1 nA for 60 s. | |
The observed potential drift (ΔE/Δt), which decreased from 75.9 to 3.1 μV s−1 with a decrease in bulk membrane resistance from 0.51 to 0.4 MΩ, showed a considerable increase in the potential stability following modification with f-MWCNTs/PANi. The interfacial double-layer capacitance, however, changed dramatically from 13.2 to 324.7 μF. Our findings (Table 3) demonstrated the effectiveness of f-MWCNTs/PANi in enhancing the sensor's interfacial double-layer capacitance and short-time potential stability.
Table 3 Calculated information was gathered from the chromatograms of SPE/f-MWCNTs/PANi/MIP/ERY-ISE and SPE/MIP/ERY-ISE under a constant current of ±1 nA for 60 s in a solution of 1 × 10−5 M ERY
| Parameter |
SPE/f-MWCNTs/PANi/MIP/ERY-ISE |
SPE/MIP/ERY-ISE |
| ΔE (mV) |
0.40 |
0.51 |
| Resistance, Rb (MΩ) |
0.40 |
0.51 |
| Potential drift, ΔE/Δt (μV s−1) |
3.10 |
75.9 |
| Double-layer capacitance, CL (μF) |
324.7 |
13.2 |
3.8. Analytical applications
3.8.1 Determination of ERY in pharmaceutical formulations. SPE/f-MWCNTs/PANi/MIP/ERY-ISE was used to test the ERY content in some pharmaceutical samples to assess the suitability of the suggested sensor for drug analysis. The drug samples underwent various processing steps, dissolved in a phosphate buffer solution with a pH of 5.5, and assessed potentiometrically. The findings revealed that, on average, 99.8 ± 1.4% of the nominal values were recovered (Table 4). To verify the applicability of the suggested method, data collected using the conventional liquid chromatographic method (HPLC)48 were also included.
Table 4 Potentiometric determination of ERY in some pharmaceutical products using SPE/f-MWCNTs/PANi/MIP/ERY-ISE
| Pharmaceutical product |
Labeled content |
Found, as in the labeled contenta |
| Reference method [HPLC],48 |
Recovery, % |
ERY sensor |
Recovery, % |
| Average of 3 measurements (n = 3). |
| ERYTHRYIN 500 ®, Misr Pharm., Egypt |
500 mg per tablet |
495.3 ± 1.7 |
99.0 ± 0.5 |
481.2 ± 11.0 |
96.3 ± 1.2 |
| BENZAMYCIN®, Sanofi, Egypt |
4.0 g/100 g (gel) |
3.9 ± 0.4 |
97.5 ± 0.6 |
4.2 ± 0.8 |
105.0 ± 2.3 |
| ERYTHROMYCIN® PHARCO Co., Egypt |
200 mg/5 mL (syrup) |
197.6 ± 0.3 |
98.8 ± 0.3 |
196.4 ± 8.0 |
98.2 ± 0.9 |
3.8.2 Determination of ERY in spiked urine samples. To employ this sensor in detecting overdose patients, particularly in situations where a quick and accurate evaluative diagnosis is necessary, SPE/f-MWCNTs/PANi/MIP/ERY-ISE was also used to monitor ERY in human urine. Following the addition of known ERY concentrations to various aliquots of a human urine sample, potentiometric measurements were taken using the suggested sensor. As shown in Table 5, the average recovery was 97.7 ± 0.5% of the ERY concentration that had been spiked without any appreciable interference from the species that are often found in samples of natural human urine. This provided evidence that the suggested sensor may be used to measure ERY content in real samples while having little to no interference from these interfering species.
Table 5 Recovery values for determination of ERY in spiked human urine samples
| Sample |
ERY content, μM |
Recovery, (%) |
| Spiked |
Found |
| 1 |
10.0 |
9.8 ± 0.6 |
98.0 ± 0.6 |
| 2 |
20.0 |
18.6 ± 0.3 |
93.0 ± 0.5 |
| 3 |
50.0 |
47.7 ± 0.3 |
95.4 ± 0.3 |
| 4 |
100.0 |
104.3 ± 0.1 |
104.3 ± 0.7 |
3.9. Evaluation of the presented sensor relating to other electrochemical sensors
The presented all-solid-state potentiometric device was compared to other reported electrochemical sensors. As shown in Table 6, the proposed electrochemical device revealed high stability, long-term durability, and was easily produced for mass production of disposable devices for ERY determination. Using small sample volumes is made possible by miniaturization, which is the approach taken in this work.
Table 6 A summary of published electrochemical techniques for ERY assessmenta
| Sensing material |
Technique |
Type of electrode |
Slope, mV per decade |
Linearity range, M |
LOD, M |
Application |
Ref. |
| GR–AuNPs/CS–PtNPs/gold/MIP: chitosan–platinum nanoparticles (CS–PtNPs) and graphene–gold nanoparticles (GR–AuNPs) nanocomposites. mPD: m-phenylenediamine. PEDOT: poly(3,4-ethylenedioxythiophene). |
| MIPs modified carbon paste |
Voltammetry |
Macro-electrodes |
—, pH 7 |
5 × 10−8 to 1 × 10−5 |
1.9 × 10−8 |
Honey and dairy products |
15 |
| GR–AuNPs/CS–PtNPs/gold/MIP |
Voltammetry |
Macro-electrodes |
—, pH 4 |
7 × 10−8 to 9 × 10−5 |
2.3 × 10−8 |
Honey and milk |
16 |
| SPE/ERY-MIP (mPD) |
Voltammetry |
Micro-electrodes |
— |
2 × 10−9 to 1.6 × 10−8 |
1.2 × 10−10 |
Tap water |
17 |
| Erythromycin-tetraphenylborate |
Potentiometry |
Macro-electrodes |
58.7, pH 4–6 |
1 × 10−5 to 1.0 × 10−2 |
1.0 × 10−5 |
Pharmaceutical formulations |
20 |
| SPE/MWCNTs/PEDOT/MIP |
Potentiometry |
Micro-electrodes |
51, pH 4.5 |
1 × 10−7 to 1 × 10−3 |
6.6 × 10−8 |
Milk |
49 |
| SPE/f-MWCNTs/PANi/MIP |
Potentiometry |
Micro-electrodes |
54, pH 5.5 |
4 × 10−6 to 1 × 10−3 |
9.6 × 10−7 |
Pharmaceutical formulations and urine |
This work |
4. Conclusion
A highly sensitive and selective all-solid state screen-printed based sensor with low cost is presented and utilized to monitor the presence of the antibiotic erythromycin in some pharmaceutical products and spiked human urine samples. The sensor works by using MIP beads dispersed in a PVC membrane as a sensory element for detecting the drug. As a solid contact material, multi-walled carbon nanotube/polyaniline composite (f-MWCNTs/PANi) is utilized to enhance the ion-to electron transfer process. It greatly enhanced the sensor's electrical characteristics and removed the influence of the water layer that was present between the conductive substrate and the ion-sensing polymeric membrane. The sensor-based MIP exhibits enhanced selectivity towards the drug over different species and similar compounds and is applicable to 4.6 × 10−6 to 1.0 × 10−3 M ERY with a detection limit of 9.6 ± 0.4 × 10−7 M. These results enabled the proposed strip cell design to be readily applicable in the pharmaceutical sector for quick and accurate assessment as well as possible integration into automated and wearable devices. A comparison of the potentiometric characteristics of proposed SPE/f-MWCNTs/PANi/MIP/ERY-ISE with some of the earlier reported assay methods15–17,20,49 revealed some notable advantages in terms of sensitivity, accuracy, stability, and selectivity. The presented sensor consists of a reference electrode integrated with the ion-sensing electrode without internal filling solutions on the same planar substrate as a combined tiny planar (5 × 20 mm). It has the advantages of being miniaturized, affordable, stable, long-term durability, and easily produced for mass production of disposable devices.
Author contributions
The listed authors contributed to this work as follows: A. E. A. and A. H. K. provided the concepts of the work; A. A. A. and A. M. N. interpreted the results. A. H. K., L. S. A. and M. G. A. performed the experimental part. A. H. K. and A. E. A. prepared the manuscript; A. H. K. performed the revision before submission; A. A. A. and A. M. N. obtained the financial support for the work. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research (IFKSURC-1-0108).
References
- B. H. Schafhauser, L. A. Kristofco, C. M. R. de Oliveira and B. W. Brooks, Environ. Pollut., 2018, 238, 440–451 CrossRef CAS PubMed.
- S. Sun, X. Xie and E. Liu, J. Pediatr. Pharm., 2019, 25, 54–58 CAS.
- L. Wei, F. Li and N. He, Chin. J. Clin Ration. Drug Use, 2019, 12, 175–178 Search PubMed.
- T. S. Thompson and J. P. Van den Heever, Food Chem., 2012, 133, 1510–1520 CrossRef CAS.
- A. Veseli, F. Mullallari, F. Balidemaj, L. Berisha, L. Švorc and T. Arbneshi, Microchem. J., 2019, 148, 412–418 CrossRef CAS.
- E. Sari, R. Üzek, M. Duman and A. Denizli, Talanta, 2016, 150, 607–614 CrossRef CAS PubMed.
- M. Ali, S. T. H. Sherazi and S. A. Mahesar, Arabian J. Chem., 2014, 7, 1104–1109 CrossRef CAS.
- Y. M. Liu, Y. M. Shi, Z. L. Liu and W. Tian, Electrophoresis, 2010, 31, 364–370 CrossRef CAS.
- M. A. García-Mayor, G. Paniagua-González, B. Soledad-Rodríguez, R. M. Garcinuno- Martínez, P. Fernández-Hernando and J. S. Durand-Alegría, Food Chem. Toxicol., 2015, 78, 26–32 CrossRef.
- C. Benetti, R. Piro, G. Binato, R. Angeletti and G. Biancotto, Food Addit. Contam., 2006, 23, 1099–1108 CrossRef CAS.
- V. Licul-Kucera, M. Ladányi, G. Hizsnyik, G. Záray and V. G. Mihucz, Microchem. J., 2019, 148, 480–492 CrossRef CAS.
- X. Y. Zhang, Y. T. Bai, D. C. Hao, Y. Yu, Y. Q. Wang, W. Mao, W. Song, H. Sun and P. F. Li, Chin. J. Pharm. Anal., 2019, 39, 2184–2190 Search PubMed.
- H. Wang, A. Zhang, H. Cui, D. Liu and R. Liu, Microchem. J., 2000, 64, 67–71 CrossRef CAS.
- X. Z. Hu, P. Wang, J. Q. Yang, B. Zhang, J. Li, J. Luo and K. B. Wu, Colloids Surf., B, 2010, 81, 27–31 CrossRef CAS PubMed.
- B. Song, Y. Zhou, H. Jin, T. Jing, T. Zhou, Q. Hao, Y. Zhou, S. Mei and Y. I. Lee, Microchem. J., 2014, 116, 183–190 CrossRef CAS.
- W. J. Lian, S. Liu, J. H. Yu, X. R. Xing and J. D. Huang, Biosens. Bioelectron., 2012, 38, 163–169 CrossRef CAS PubMed.
- A. G. Ayankojo, J. Reut, V. Ciocan, A. Öpik and V. Syritski, Talanta, 2020, 209, 120502 CrossRef CAS PubMed.
- N. Li, D. Xie, H. Zhao, C. Yu, Z. Li, F. Li and Q. Cao, Microchem. J., 2022, 181, 107728 CrossRef CAS.
- T. Lai, H. Shu, X. Tian, J. Ren, X. Cui, H. Bai, X. C. Xiao and Y. D. Wang, Electrochemical sensor based on molecularly imprinted poly-arginine for highly sensitive and selective erythromycin determination, J. Mater. Sci.: Mater. Electron., 2023, 34(6), 1–13 Search PubMed.
- M. R. Ganjali, P. Shirin, F. Farnoush, A. Hosein, H. Morteza and N. Parviz, Int. J. Electrochem. Sci., 2011, 6, 1968–1980 CrossRef CAS.
- A. H. Kamel and H. R. Galal, Int. J. Electrochem. Sci., 2014, 9, 4361–4373 CrossRef.
- A. H. Kamel, H. R. Galal and N. S. Awaad, Anal. Methods, 2018, 10, 5406–5415 RSC.
- A. H. Kamel, A. E.-G. E. Amr, N. S. Abdalla, M. El-Naggar, A. M. Al-Omar, H. M. Alkahtani and A. Y. A. Sayed, Polymers, 2019, 11, 1796 CrossRef CAS.
- A. G. Ayankojo, J. Reut, V. B. C. Nguyen, R. Boroznjak and V. Syritski, Biosensors, 2022, 12, 441 CrossRef CAS PubMed.
- T. Karasu, E. Özgür and L. J. Uzun, Pharm. Biomed. Anal., 2023, 226, 115257 CrossRef CAS.
- N. Özcan, C. Karaman, N. Atar, O. Karaman and M. L. Yola, ECS J. Solid State Sci. Technol., 2020, 9, 121010 CrossRef.
- H. Karimi-Maleh, M. L. Yola, N. Atar, Y. Orooji, F. Karimi, P. S. Kumar, J. Rouhi and M. Baghayeri, J. Colloid Interface Sci., 2021, 592, 174–185 CrossRef CAS PubMed.
- C. Karaman, O. Karaman, N. Atar and M. L. Yola, Microchim. Acta, 2022, 189, 24–34 CrossRef CAS.
- A. Özcan, N. Atar and M. L. Yola, Biosens. Bioelectron., 2019, 130, 293–298 CrossRef PubMed.
- M. L. Yola and N. Atar, Composites, Part B, 2019, 175, 107113 CrossRef CAS.
- N. Rubinova, K. Y. Chumbimuni-Torres and E. Bakker, Sens. Actuators, B, 2007, 12, 135–141 CrossRef.
- M. Parrilla, M. Cuartero and G. A. Crespo, Trends Anal. Chem., 2019, 110, 303–320 CrossRef CAS.
- J. Hu, A. Stein and P. Bühlmann, Trends Anal. Chem., 2016, 76, 102–114 CrossRef CAS.
- A. M. Mahmoud, M. T. Ragab, N. K. Ramadan, N. A. El-Ragehy and B. El-Zeany, Electroanalysis, 2020, 32, 2803–2811 CrossRef CAS.
- H. S. Abd-Rabboh, A. E.-G. E. Amr, A. A. Almehizia and A. H. Kamel, Polymers, 2021, 13, 1192 CrossRef CAS.
- A. E.-G. E. Amr, A. H. Kamel, A. A. Almehizia, A. Y. Sayed, E. A. Elsayed and H. S. Abd Rabboh, ACS Omega, 2021, 6, 11340–11347 CrossRef CAS.
- S. S. M. Hassan, A. H. Kamel, A. E.-G. E. Amr, M. F. Abdelwahab and M. A. Al-Omar, Molecules, 2020, 25, 629 CrossRef CAS.
- N. Lenar, B. Paczosa-Bator and R. Piech, Membranes, 2020, 10, 182 CrossRef CAS PubMed.
- J. Xu, F. Li, C. Tian, Z. Song, Q. An, J. Wang, D. Han and L. Niu, Electrochim. Acta, 2019, 322, 134683 CrossRef CAS.
- Y. Shao, Y. Ying and J. Ping, Chem. Soc. Rev., 2020, 49, 4405–4465 RSC.
- S. S. M. Hassan, A. H. Kamel and M. A. Fathy, Anal. Chim. Acta, 2022, 1227, 340239 CrossRef CAS.
- https://pubchem.ncbi.nlm.nih.gov/compound/Erythromycin.
- E. Bakker, J. Electrochem. Soc., 1996, 143, L83 CrossRef CAS.
- M. Fibbioli, W. E. Morf, M. Badertscher, N. F. de Rooij and E. Pretsch, Electroanal, 2000, 12, 1286–1292 CrossRef CAS.
- U.D.O. Health, H. Services, Analytical Procedures and Methods Validation for Drugs and Biologics-Guidance for Industry, US Department of Health and Human Services, Washington, 2015 Search PubMed.
- J. C. Benneyan, Int. J. Qual. Health Care, 1998, 10, 69–73 CrossRef CAS PubMed.
- J. Bobacka, Anal. Chem., 1999, 71, 4932–4937 CrossRef CAS PubMed.
- British Pharmacopeia (BP), Version 17.0, Index+ © System Simulation Ltd, TSO, 2013 Search PubMed.
- M. A. Tantawy, A. M. Yehia and H. T. Elbalkiny, Microchim. Acta, 2023, 190, 408 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ahmed M. Naglaha,
Mashael G. Alanazib,
Abdel El-Galil E. Amrc and
Ayman H. Kamel
a,
Ahmed M. Naglaha,
Mashael G. Alanazib,
Abdel El-Galil E. Amrc and
Ayman H. Kamel