
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of the conversion of ethanol to 1,3-butadiene: effects of chemical and structural heterogeneity on the activity of MgO–SiO2 mixed oxide catalysts†
Blanka
Szabó
 a,
Virág
Hutkai
b,
Gyula
Novodárszki
a,
Ferenc
Lónyi
a,
Zoltán
Pászti
a,
Zsolt
Fogarassy
c,
József
Valyon
a and
Róbert
Barthos
*a
a,
Virág
Hutkai
b,
Gyula
Novodárszki
a,
Ferenc
Lónyi
a,
Zoltán
Pászti
a,
Zsolt
Fogarassy
c,
József
Valyon
a and
Róbert
Barthos
*a
aInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Magyar tudósok körútja 2, Budapest 1117, Hungary. E-mail: barthos.robert@ttk.hu; Tel: +36 1 3628842
bErhvervsakademi Aarhus, Sønderhøj 30, 8260 Viby J, Denmark
cCentre for Energy Research, Institute for Technical Physics and Materials Science, Konkoly-Thege M. út 29-33, Budapest 1121, Hungary
First published on 8th December 2022
Abstract
The ethanol-to-butadiene (ETB) transformation proceeds through consecutive reactions, involving hydrogenation/dehydrogenation, C–C coupling, and dehydration. Uniform active sites are needed to attain high catalytic selectivity. It is a challenge to generate a catalyst containing three kinds of co-operating active sites in high homogeneity. Lacking dehydration activity, basic MgO is active in converting ethanol mainly to acetaldehyde and butanol, whereas the main products obtained over SiO2 catalysts are dehydration products ethylene and diethyl ether. 1,3-Butadiene could be obtained over MgO–SiO2 mixed oxide catalysts, having acidic and basic sites of strength and concentration favoring all three reactions. Silica was either precipitated over the surface of MgO, or wet-kneaded with MgO to get mixed oxide catalysts. More active ETB catalysts were obtained if the MgO component has a higher specific surface area. XRD, EDS, XPS and acidity/basicity examinations showed that Mg atoms got incorporated into the silica phase, generating new Lewis acid surface sites. An amorphous MgO–SiO2 mixed oxide preparation, having the highest surface Mg/Si ratio and atomic homogeneity, had the highest activity and 1,3-butadiene selectivity. The catalyst was obtained by hydrolyzation/condensation/precipitation of an Si,Mg–alkoxide solution within carbon mesopores and burning out the carbon/precipitate material. The catalytic ETB mechanism is discussed.
1. Introduction
Bioethanol, which can be economically produced at the industrial scale, is one of the most widely used renewable, carbon neutral feedstocks in the chemical industry. 1,3-Butadiene (BD) which can be prepared also from ethanol is a constituent of many common polymers (synthetic rubbers, polymer resins, elastomers, etc.). Nowadays, BD is produced almost exclusively from petroleum; however, due to environmental concerns and the uncertain availability of crude oil and natural gas, ethanol conversion to butadiene (the ETB reaction) is receiving considerable industrial and academic attention.1 The potential of the heterogeneous catalytic ETB process has even been demonstrated at the industrial level.2The most commonly used supports/catalysts for the reaction are mixed oxides, such as MgO–SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3–29 or ZrO2–SiO2,30–34 and their derivatives, obtained through modification by metals, such as Ag,4,27,31,35,36 Cu,16,27,33 and Au,10 or by metal oxides ZrO2,9,36,37 ZnO,9,16,19,23,32,37 In2O3,23 and Ga2O3.23,38 Previous studies have shown that neat mixed oxides have significant catalytic activity in the ETB reaction and that additives can increase the activity by promoting ethanol dehydrogenation and C–C coupling reactions. It has also been shown that the catalytic properties of the modified mixed oxides were still determined mainly by the properties of the mixed oxide components.
3–29 or ZrO2–SiO2,30–34 and their derivatives, obtained through modification by metals, such as Ag,4,27,31,35,36 Cu,16,27,33 and Au,10 or by metal oxides ZrO2,9,36,37 ZnO,9,16,19,23,32,37 In2O3,23 and Ga2O3.23,38 Previous studies have shown that neat mixed oxides have significant catalytic activity in the ETB reaction and that additives can increase the activity by promoting ethanol dehydrogenation and C–C coupling reactions. It has also been shown that the catalytic properties of the modified mixed oxides were still determined mainly by the properties of the mixed oxide components.
The modification could increase the specific surface area (SSA) of the catalyst. The increase in the ETB activity was often higher than what was justified by the increased SSA.31,39,40 Different types of high-SSA silica materials, such as SBA-15,18,24,31,41–45 SBA-16,31,33,36 MCM-41,4,41 MCM-48,41 TUD-1,39,40,46,47 COK-12,4 Q-6, and KSMG,31 as well as, zeolites, having the framework type LTA48 and BEA,39,41,48–55 which are zeolites of a high Si-to-Al ratio or dealuminated, have been studied in the ETB reaction. The combination of high-SSA silica or silica-containing materials with magnesia gave catalysts of significant activity.
Hardly any study has been reported about the ETB reaction over mixed oxide catalysts containing a high-SSA MgO component. Men et al.17,21 reported about catalysts made by calcining a MgO precursor and impregnating the obtained MgO with a silica sol. In order to get a high-SSA MgO catalyst component, the MgO precursor was synthetized by a hydrothermal process with the aid of surfactants. Recently, Reschetilowski et al.20 published a paper about mesoporous MgO-supported SiO2 catalysts. The high-SSA support was prepared by hydrothermal treatment of precipitated MgCO3.
It is known that thermal decomposition of magnesium salts results in non-porous MgO having a SSA smaller than 5 m2 g−1.56,57 The present study is concerned with new MgO–SiO2 ETB catalysts, containing a high-SSA, microporous and/or mesoporous MgO (HSM) component. HSM materials can be produced relatively simply, for instance, by urea-assisted, hydrothermal, and homogeneous precipitation methods,56,58,59 solvothermal transformation of magnesium acetate,21,60–63 or by sol–gel synthesis.64 A complexation–combustion method was described by Hiremath et al.65 It was shown that combustion of magnesium nitrate with a fuel, which is a good complexing agent of magnesium, like glycine, gave a HSM material.
A particular synthesis method for the production of HSM materials is the so-called hard-templating (HT) method. Accordingly, an MgO precursor compound is introduced into a porous template, and by converting the compound to an oxide and removing the template, a porous HSM material is obtained that is a replica of the template porosity. For template materials, carbon has been proven to be the most suitable because it can be removed by simple combustion while the magnesia precursor is converted to MgO. Recently, waste cotton was described as a carbon source for the preparation of HSM templates.66 However, the most widely used HSM templates are carbon CMK-3, which is a replica of the HT SBA-15 material,67 and porous carbon obtained by carbonization of a resorcinol–formaldehyde (RF) aerogel.68–72
HSM materials are active in reactions that require strong base active sites, such as the Claisen–Schmidt condensation.58,73 In combination with redox catalytic function, they also show activity in the oxidative ethylbenzene dehydrogenation reaction.67 Moreover, these materials are commonly used as a basic adsorbent for the sequestration of carbon dioxide,56,59,65 and as a scavenger of organic dyes61,66,74 or various metal cations, such as Pb2+,61 As3+,64,72 As5+ (ref. 62) and Cr6+.64
To increase its mechanical strength, the HSM adsorbent is often coated with a silica shell.57,60,61,63 The coating is usually generated by hydrolyzing tetraethyl ortosilicate (TEOS) with ammonium hydroxide either in the presence60,61 or in the absence57 of a cetyltrimethyl ammonium bromide (CTABr) template. In addition to the structure-strengthening effect, the silica coating provides acidic properties and, thereby, ETB activity to the material. Recently, Larina et al.41,45 established that the porous structure (SSA, pore size distribution) of the silica component of MgO–SiO2 catalysts is not an essential factor from the point of view of achieving high BD yields. The present study deals with three novel MgO–SiO2 preparations, containing a HSM component. Our main objective was to clarify the influence of MgO distribution on the ETB activity of the catalyst preparations. The preparations were characterized by structure, texture, morphology and ETB activity. For comparison, similarly prepared catalysts, based on low-SSA MgO (LSM), were also examined. In light of the results, the possible reaction mechanisms are reviewed.
2. Experimental
2.1 Preparation of catalysts
A mesoporous carbon aerogel (MCA) was saturated with a Mg(NO3)2 solution and calcined to combust carbon and obtain high-SSA mesoporous MgO (HSM).Using TEOS as a silica source, mixed oxide MgO–SiO2 catalysts were prepared. TEOS either was hydrolyzed to silica in the presence of HSM with the intention to form a silica coating (SC) on the HSM surface or was first hydrolyzed and the obtained silica was compounded with the HSM by wet kneading (WK). The catalysts were designated as SCH and WKH, respectively. A third catalyst was prepared by hydrolyzing a TEOS/magnesium methoxide solution in the internal voids of the carbon template, referred to as internal hydrolysis (IH), and calcination to get sample IHH. Catalysts were also prepared by the same methods using LSM, obtained by Mg(NO3)2 calcination omitting the use of carbon. The latter preparations were identified as SCL, WKL, and IHL. The catalysts, prepared using low and high-SSA MgO or with MCA assistance, are referred to as L and H catalysts, respectively. The WK, SC or IH catalyst designations apply to the L and H catalyst versions together. An attached list helps the reader to match the abbreviation and its meaning. The preparation procedures are described in detail below.
The MCA was produced by adding 194 g of resorcinol to 286.75 g of formaldehyde under continuous stirring. In a separate step, 0.375 g of sodium carbonate was dissolved in 519.75 g deionized water and then added to the previous solution as a polymerization catalyst. The product mixture was aged for 24 hours at room temperature, and then sealed in a PET reactor and kept at 50 °C for 24 hours and at 90 °C for 72 hours. The wet resorcinol–formaldehyde polymer gel was washed with acetone. To remove the acetone, the sample was dried at room temperature for 24 hours. The obtained RF aerogel was pyrolyzed to get MCA. The pyrolysis was carried out by heating up the RF aerogel in a N2 flow (200 mL min−1) at a rate of 1 °C min−1 to 800 °C and maintaining this temperature for 5 hours.
Mesoporous HSM was produced by soaking the air-dried MCA in saturated Mg(NO3)2 solution for 24 h at room temperature. Thereafter, the saturated carbon was separated from the Mg(NO3)2 solution by filtration, and dried at 110 °C for 24 hours. To remove the carbon template and obtain mesoporous HSM, the sample was heated up to 600 °C at a rate of 1 °C min−1 in air and calcined for an additional 8 hours at this temperature. To get LSM reference MgO, Mg(NO3)2·6H2O was decomposed using similar calcination conditions.
The SC catalysts were prepared by suspending 1 g of HSM or LSM type MgO in a mixture of 180 g deionized water and 150 g ethanol. The pH of the mixture was adjusted to 11.7 with 25 wt% NH3 solution, and then 0.775 g CTABr was added to the mixture during continuous stirring. After 30 min, 2.583 g TEOS was added to the mixture dropwise and the stirring was continued for an additional 2 hours. Then, the suspension was centrifuged. The thus separated solid was washed with 200 mL water and then with 200 mL methanol. The solid was dried at 100 °C overnight and then its temperature was ramped up at a rate of 1 °C min−1 to 550 °C. The solid was calcined at this temperature for 5 hours.
The WK catalysts were synthesized by mixing pre-hydrolyzed TEOS with HSM or LSM as described earlier.29
The internal hydrolysis method, developed by Ogura et al.,75 was applied in a slightly modified form to get catalyst IHH. As the first step, the MCA was soaked in a mixture of magnesium methoxide and TEOS (Mg/Si = 2) for four days. After filtration separation, the impregnated MCA was placed in a desiccator, wherein hydrolysis of the alkoxides was initiated by making contact with the vapor from a 25 wt% NH3 solution for three days. The material was then dried at 120 °C overnight and calcined at 600 °C for 8 h (the rate of heating up was 1 °C min−1). The same procedure was followed to get the IHL catalyst but without the use of the MCA.
As a reference material, pure silica was prepared by adding TEOS to a 1.5 M NH3 solution dropwise at 70 °C during continuous stirring. The liquid phase was evaporated. The solid residue was dried at 120 °C for 24 h and then calcined at 550 °C to get a neat SiO2 sample.
2.2 Catalyst characterization
The elemental composition of the preparations was determined by the inductively coupled plasma optical emission spectrometry (ICP-OES) method with axial plasma observation (Spectro Genesis).Nitrogen adsorption/desorption isotherms were measured using a Thermo Scientific Surfer automatic volumetric adsorption analyzer at −196 °C. Prior to the measurements, the samples were evacuated for 2 h at 250 °C.
Powder X-ray diffractograms were recorded with a Philips PW 1810/3710 diffractometer applying monochromatized CuKα (λ = 0.15418 nm) radiation (40 kV, 35 mA) and a proportional counter. Data were collected between 1.5° and 70° 2θ, in 0.04° steps with 0.5 s each step.
The morphology of the samples was examined using a Cs-corrected ThermoFisher Themis 200 scanning transmission electron microscope (STEM) with an accelerating voltage of 200 keV. Energy dispersive spectroscopic (EDS) elemental maps were recorded using Super-X EDX detectors in STEM mode. The crystalline structure of selected sample areas was analyzed by electron diffraction (SAED).
X-ray photoelectron spectroscopic measurements were carried out using a spectrometer manufactured by OMICRON nanotechnology GmbH (Germany). The photoelectrons were excited by both MgKα (1253.6 eV) and AlKα (1486.6 eV) radiation. Spectra were recorded in the Constant Analyser Energy mode of an EA125 energy analyser with a 30 eV pass energy resulting in a spectral resolution of 1.0 eV. Calibration of the energy scale of the instrument was performed according to the ISO 15472 standard. The estimated accuracy of the reported binding energy (BE) values is ±0.2 eV. As the samples are insulating materials, considerable charging was observed during the measurements. For silicates, generally the C 1s line is used to account for the charging effect.76 Accordingly, in the present study, the hydrocarbon component of the C 1s spectrum at a 285.0 eV BE was selected as a reference point. Data were processed using the CasaXPS software package77 by fitting the spectra with Gaussian–Lorentzian product peaks after removing the Shirley or linear background. Nominal surface compositions were calculated using the XPS MultiQuant software package78,79 with the assumption of a homogeneous depth distribution for all components. The chemical states were identified using XPS databases.80,81
The acidity and basicity of the preparations were characterized by temperature-programmed desorption (TPD) of NH3 and CO2, as well as using the FT-IR spectra of adsorbed pyridine and CDCl3, respectively. The experimental details are described in our earlier papers.23,29
2.3 Catalytic ETB reaction
A fixed bed, continuous flow, quartz tube (l = 400 mm, i. d. = 8 mm) microreactor was used at atmospheric pressure. The microreactor was connected to an online gas chromatograph (GC) equipped with a flame ionization detector (FID) for analyzing the carbon-containing compounds and also with another FID for analyzing the oxygenates. The evaporation zone of the microreactor was heated to 120°C. Catalysts (∼1.00 g, particle size 0.315–0.65 mm) were activated in an O2-flow in situ in the reactor at 550 °C for 30 min. Ethanol was fed into the reactor to a He flow using a Gilson 307 type pump to get a 15 v/v% ethanol/He gas flow in the hot reactor. Temperature-dependence of the activity was determined in the range of 300–475 °C at a total flow rate of 30 ml min−1. This flow rate corresponds to weight hourly space velocity (WHSV) of 0.5 gethanol gcat−1 h−1. Further, these reaction conditions are referred to as the standard reaction conditions. Space–time dependence of the activity was determined at temperatures of 350 and 400 °C to compare the catalytic activities at 20 and 50% conversions and to identify reaction intermediates. The flow rate was varied between 10 and 180 ml min−1 (WHSV = 0.17–3.0 gethanol gcat−1 h−1). The concentration of the reactant and all major products were determined by GC. Calibration was made separately for each component of the product mixture.
Conversions/selectivities/formation rates were calculated based on the number of carbon atoms in the feed and in the products.
3. Results
3.1 Composition, structure, and texture
In the procedure of catalyst preparation, Mg and Si sources were mixed to get a Mg/Si molar ratio of 2; however, chemical analysis gave a ratio of 2.2–2.5 (Table 1).| Sample ID | Mg/Sia | SSA,b m2 g−1 | PV,c cm3 g−1 | PD,d nm | |
|---|---|---|---|---|---|
| ICP | XPS | ||||
| a Molar ratio of the bulk (ICP) and the surface (XPS). b Specific surface area, determined by the BET method. c Pore volume determined by the Gurvich method. d Most frequent pore diameter, calculated by the BJH method. | |||||
| LSM | — | — | 5 | 0.03 | 5.56 |
| HSM | — | — | 40 | 0.27 | 20.32 |
| SiO2 | — | — | 300 | 0.39 | 4.30 |
| SCH | 2.51 | 0.94 | 253 | 0.41 | 21.02 |
| SCL | 2.20 | 0.63 | 386 | 0.23 | — |
| WKH | 2.24 | 1.41 | 262 | 0.38 | 3.84 |
| WKL | 2.24 | 1.16 | 156 | 0.19 | — |
| IHH | 2.46 | 1.40 | 99 | 0.36 | 10.55 |
| IHL | 2.28 | 1.55 | 29 | 0.11 | — |
The difference in the expected and measured ratios may come from a minor analytical error related to the difficulty of rendering silica quantitatively soluble. Nonetheless, the XPS analysis (Table 1) showed Si enrichment on the surface of all the catalysts relative to its bulk concentration, i.e., 0.63 < Mg/Si < 1.55 (vide infra). The sample-to-sample variation of the Mg to Si surface ratio reflects structural differences as discussed below.
The N2 physisorption isotherms of the WK and IH catalysts are of type IV and, with the exception of that of the SCL sample, show an H3 type hysteresis loop (Fig. 1B). The isotherms of the SC samples seem to be a combination of the isotherm obtained for the MgO component (Fig. S1†) and an isotherm similar to those generally reported for MCM-41 silica materials. This is not surprising if we consider that the conditions of sample preparation were hardly different from the synthesis conditions of MCM-41 materials. It is also obvious that the SSA and pore volume (PV) of the samples, produced using a MCA template, are higher than those of the reference samples, made without a template (Table 1).
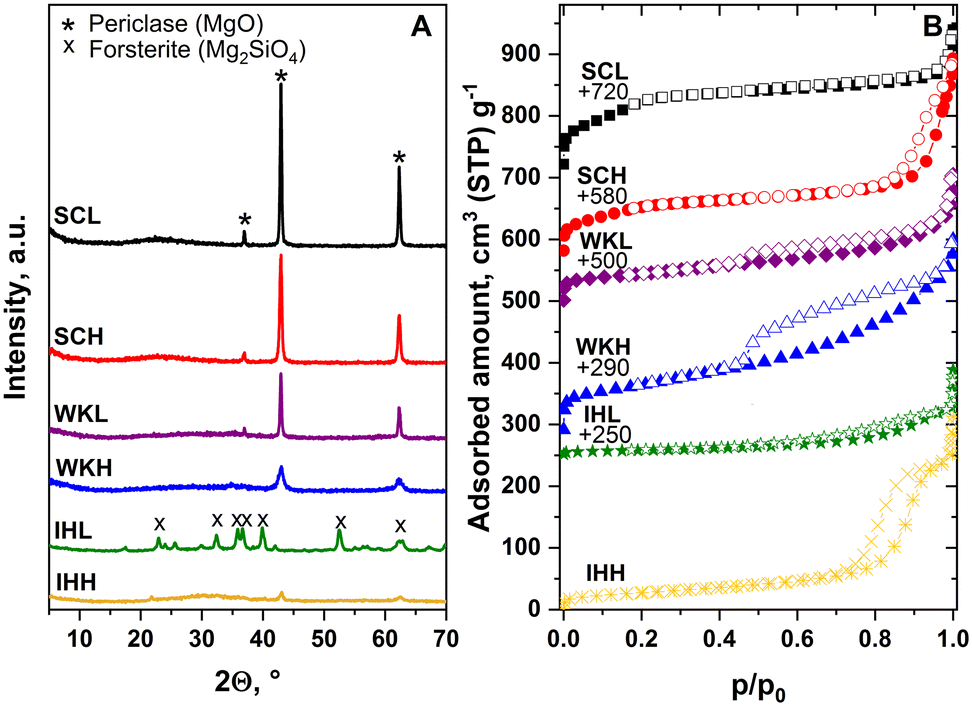 | ||
| Fig. 1 (A) XRD patterns and (B) N2 physisorption isotherms of activated MgO–SiO2 mixed oxide catalysts, prepared by coating silica over MgO (SC), by wet-kneading of the oxides (WK), and by hydrolyzation of MgO and SiO2 precursors inside the pores of porous carbon (IH). In the sample designation, the ending L denotes the preparation, which involved the use of low-SSA MgO. Samples designated with the ending H were made using high-SSA MgO or with the assistance of MCA. | ||
Fig. 2 shows the STEM micrographs and the EDS elemental maps of the samples. In the micrographs of the SC samples, amorphous SiO2 spheres (Ø ∼ 200 nm) appeared. Among the spheres, there is unordered material, containing both Mg–O and Si–O moieties. Mg–O and fine-grained Si–O areas are mostly separated; however, some areas can be also distinguished where these components are mixed.
 | ||
| Fig. 2 Elemental maps of the catalysts. HAADF: high-angle annular dark-field imaging. | ||
Electron diffraction analysis shows that the Mg–O regions are crystalline (periclase), whereas the Si–O regions have an amorphous structure. The WK samples have a sponge-like structure in which the MgO and SiO2 components overlap. The samples contain a crystalline MgO phase; however, no significant difference is found between samples made using LSM and HSM.
Due to the heterogeneity of the samples, the Mg/Si ratio could not be determined from the EDS elemental maps. However, it could be established that silica-rich particles contained a small amount of homogeneously distributed Mg (2–4 at.%) and vice versa, magnesia-rich particles contained some Si.
The IH samples have a lamellar structure and are visibly much more homogeneous than the sample pairs described above. With this in line, XPS detected a higher Mg concentration on the surface than on the surface of the other catalyst preparations.
3.2 Surface topology
XPS can provide information about the bonding environment of surface Mg and Si species. As described in the literature, the electron binding energies (BEs) of Si and O in silicate materials primarily carry information about the way the SiO4 tetrahedral building units are interlinked.79,82 For example, in crystalline silica, e.g., in quartz, each SiO44− unit is connected to four other units, and thus there is no formal charge on the tetrahedra, which results in high binding energies (BEs) for both the Si 2p (103.5–104.0 eV) and the O 1s (around 533 eV) electrons.81 If each tetrahedron shares three oxygen ions with adjacent tetrahedra in such a way that an infinite 2-dimensional sheet is formed (such as in talc), the formal charge of each tetrahedron is −1, which results in shifting of both the Si and O peaks towards lower binding energies by 0.5–0.6 eV. Indeed, studies mention Si 2p BEs in talc at around 103.4–103.5 eV (ref. 76) and O 1s peak positions at around 532.5 eV.82,83 At the other end of the range, in forsterite (Mg2SiO4), the SiO4 tetrahedra are surrounded by Mg2+ cations; thus, the charge on each SiO4 unit is −4 and the corresponding Si 2p and O 1s BEs are as low as 101.8 and 530.9 eV, respectively.84 The pronounced shift of the Si and O photoelectron lines is accompanied by a similar but smaller shift of the charge balancing cations. The Mg 2p line of talc is at 50.7 eV,83 whereas that of forsterite is at 50.2–50.4 eV.84In general, the photoelectron peaks of the studied catalysts were adequately modeled by a single symmetric but rather broad peak (except for the C 1s peaks, see below). It has to be noted that these broad but featureless peak shapes do not necessarily indicate the presence of a single chemical state: they probably arise as the envelope of a range of slightly different chemical environments. The measured binding energies of the Mg 2p, Si 2p and O 1s peaks of the mixed oxides are listed in Table 2 with respect to the hydrocarbon component of the C 1s band, fixed at 285.0 eV BE. For MgO, studies report BEs of around 50.0–50.5 eV,80,81,85,86 which corresponds to our results. The Mg 2p binding energies are quite similar for the different catalyst preparations (Table 2). The found values 50.5–50.7 eV seem to be a little high, although these agree well with the BE obtained for the WK material in our previous study.23 Similar Mg 2p BEs were found for Mg-silicates involving talc.76 The Mg 2p BE therefore suggests a mixed oxide that contains Si-related species in the environment of the Mg2+ cations, rather than a pure MgO-like arrangement.
| Sample | Composition | Binding energies (eV) | |||
|---|---|---|---|---|---|
| at% | Mg 2p | Si 2p | O 1s | C 1sa,b | |
| a The main component of the C 1s spectrum arising from hydrocarbons was used for charge compensation (fixed at 285.0 eV). b The smaller component of the C 1s spectrum is due to carbonate species at around 290.5 eV BE. The number in parentheses shows the relative strength of the carbonate contribution. | |||||
| WKL | Mg: 17.4 | 50.7 | 102.7 | 532.0 | 285.0 |
| Si: 15.0 | |||||
| O: 56.3 | 290.5 (30%) | ||||
| C: 11.3 | |||||
| WKH | Mg: 19.1 | 50.6 | 102.8 | 532.2 | 285.0 |
| Si: 13.5 | |||||
| O: 56.7 | 290.6 (38%) | ||||
| C: 10.7 | |||||
| SCL | Mg: 13.2 | 50.7 | 103.1 | 532.4 | 285.0 |
| Si: 21.1 | |||||
| O: 59.2 | 290.4 (38%) | ||||
| C: 6.5 | |||||
| SCH | Mg: 17.7 | 50.5 | 103.0 | 532.2 | 285.0 |
| Si: 18.9 | |||||
| O: 59.4 | 290.5 (40%) | ||||
| C: 4.0 | |||||
| IHL | Mg: 22.6 | 50.5 | 102.2 | 531.6 | 285.0 |
| Si: 14.6 | |||||
| O: 57.6 | 532.7 | 290.3 (44%) | |||
| C: 5.2 | |||||
| IHH | Mg: 20.7 | 50.5 | 102.2 | 531.4 | 285.0 |
| Si: 14.8 | |||||
| O: 58.3 | 532.6 | 290.6 (64%) | |||
| C: 5.7 | |||||
A clearer trend is seen for the Si 2p BEs of the catalysts. The smallest values were obtained for the IH materials (around 102.2 eV), significantly higher values were found for the WK samples (slightly below 103.0 eV) and still somewhat higher values were measured for the SC oxides. Considering that the Si 2p BE in silicate systems reflects the formal charge on the SiO4 building blocks (see above), which is directly connected to the arrangement of these blocks and their relation to the heteroatom (Mg) content, two conclusions can be drawn: (i) the Si 2p BEs are always lower than those in SiO2, suggesting some dilution of the silica network with Mg ions (i.e. formation of a mixed oxide) even in the case of the SC systems. (ii) The bonding arrangement of the Si4+ cations is the “most silica-like” for the SC systems, with the WK materials being similar to the SC materials but probably having higher Mg2+ incorporation, whereas the highest disruption of the silica network (and the highest Mg2+ penetration) occurs in the IH systems. This behavior is virtually independent from the surface area of the magnesia backbone, as the Si 2p BEs of the low and high-SSA variants are almost the same.
In general, the BE of the broad, symmetric, and unstructured O 1s envelope of the catalysts always remains significantly below the 533 eV BE of SiO2, which is again consistent with mixed oxide formation. In the case of the IH sample pair, the asymmetry of the O 1s peak shape pointed to the existence of low BE (around 531.5 eV) and higher BE (around 532.6 eV) components. Considering the parallel shift of the Si and O peaks upon changing the environment of the SiO4 units (see above), a possible interpretation for the low BE component is that it could arise from relatively Mg-rich regions, probably along with contributions from hydroxylated–carbonated surface Mg species,82 while the higher BE part may indicate the presence of more silica-like regions.
In all samples, carbon occurred in hydrocarbon form (contamination collected from ambient air) and carbonate form (with C 1s peaks at 285.0 eV and 290.5 eV BE,84,85,87 respectively). The carbonate signal always represents a significant fraction of the total carbon content and is generally higher for the HSM-based systems.
3.3 Acidity and basicity
The acidity and basicity characteristics, derived from NH3- and CO2-TPD and FT-IR spectra of adsorbed pyridine and CDCl3, are presented in the ESI† (Fig. S2 and S3) and are given in Table 3. The spectra show bands of Lewis acid site-bound pyridine only, suggesting that after the applied pre-treatment, the catalysts contain only Lewis acid sites. Due to strong surface carbonate bands in the frequency region of pyridine vibrations, no pyridine bands could be recorded for the IH samples. The acidity of these samples was characterized by NH3-TPD only. The amount of adsorbed base roughly paralleled the SSA of the samples. The MCA assisted mixed oxide preparations always had a higher SSA and NH3 adsorption capacity than those made using low-SSA MgO (LSM) as the magnesia component. The low and high-SSA catalysts, prepared in the same way, gave virtually the same weight specific adsorption capacity (Table S1†). An exception was the SC pair, where the total peak areas of adsorbed pyridine were nearly identical for the two samples.| Acidity | Basicity | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample ID | Ca weak | Ca medium | ΣCa | Pyb | Cc | CDCl3d weak | CDCl3e medium | ΣCDCl3 |
| a Adsorbed amount of NH3 (μmol g−1), determined by TPD measurement, applying a linear heating rate of 10 °C min−1. An isothermal step was inserted in the heating program at 150 °C for 30 min. The ammonia desorbed in this step was considered to be weakly bound NH3. The ammonia desorbed above 150 °C was considered as NH3 bound by medium strength. b Area of the FT-IR absorption band of adsorbed pyridine at 1448 cm−1, indicating the amount of Lewis acid sites. Experimental details: the pellets were pre-treated in a vacuum at 450 °C for 1 h. The spectra were recorded at room temperature after adsorption of pyridine at a 666 Pa pressure at 200 °C and evacuation at the same temperature for 30 min. c Adsorbed amount of CO2 (μmol g−1), determined by TPD measurement. Adsorption of CO2 at 13 kPa and room temperature, flushing for 15 min, evacuation, and then ramping up in a He flow at a rate of 10 °C min−1 to 500 °C and holding this temperature for 1 h. d Area of the FT-IR absorption band of CDCl3 at 2255 cm−1, indicating the amount of weakly adsorbed CDCl3. The pellets were pre-treated in a vacuum at 450 °C for 1 h. The spectra were recorded at room temperature in the presence of CDCl3 at about a 933 Pa pressure. e Area of the FT-IR absorption band of CDCl3 at 2227 cm−1. The area indicates the amount of CDCl3 adsorbed with medium strength. f Could not be measured due to strong carbonate bands overlapping with the possible pyridine bands. | ||||||||
| SiO2 | 64 | — | 64 | 0.06 | 1 | 0.24 | — | 0.24 |
| HSM | 17 | — | 17 | 0.02 | 22 | 0.64 | — | 0.64 |
| LSM | 4 | — | 4 | 0.05 | 67 | 0.71 | 0.05 | 0.76 |
| SCH | 127 | 35 | 162 | 0.45 | 8 | 0.55 | 0.56 | 1.11 |
| SCL | 91 | 16 | 108 | 0.48 | 2 | 1.16 | — | 1.16 |
| WKH | 254 | 175 | 430 | 1.14 | 41 | 1.09 | 1.74 | 2.83 |
| WKL | 166 | 138 | 304 | 0.34 | 20 | 0.93 | 0.47 | 1.40 |
| IHH | 155 | 57 | 212 | —f | 57 | 1.36 | 1.22 | 2.58 |
| IHL | 36 | 28 | 64 | —f | 28 | 1.27 | — | 1.27 |
Regarding the basicity of the catalysts, it can be concluded that the basicity of the samples prepared via the route using the MCA template also exceeds the basicity of the samples prepared without a template. The only exception was again the SC catalyst pair, where the total CDCl3 peak areas were nearly identical for the two samples. However, the SCL sample did not contain medium-strength base sites, whereas its SCH counterpart did.
3.4 Conversion and selectivity
The dependence of ethanol conversion activity on the reaction temperature is shown in Fig. 3 for the three catalyst pairs. The main reaction products were 1,3-butadiene (BD), ethylene (EE), diethyl ether (DEE), and acetaldehyde (AL). Crotyl alcohol (CAL) also appeared as major product in the product mixture obtained using the IHH catalyst. Minor products, appearing with a selectivity less than about 5%, were butanol (BOL), butenes (BUE), and propene (PE). Interestingly, regardless of the catalyst used, crotonaldehyde (CAD), which is generally considered as a possible intermediate in the consecutive reaction route of BD formation, was only found in traces or not at all. Higher BD selectivity was achieved over the catalysts prepared by the SC and WK methods using HSM, and by the IH method using the MCA template, than over the catalysts prepared using LSM.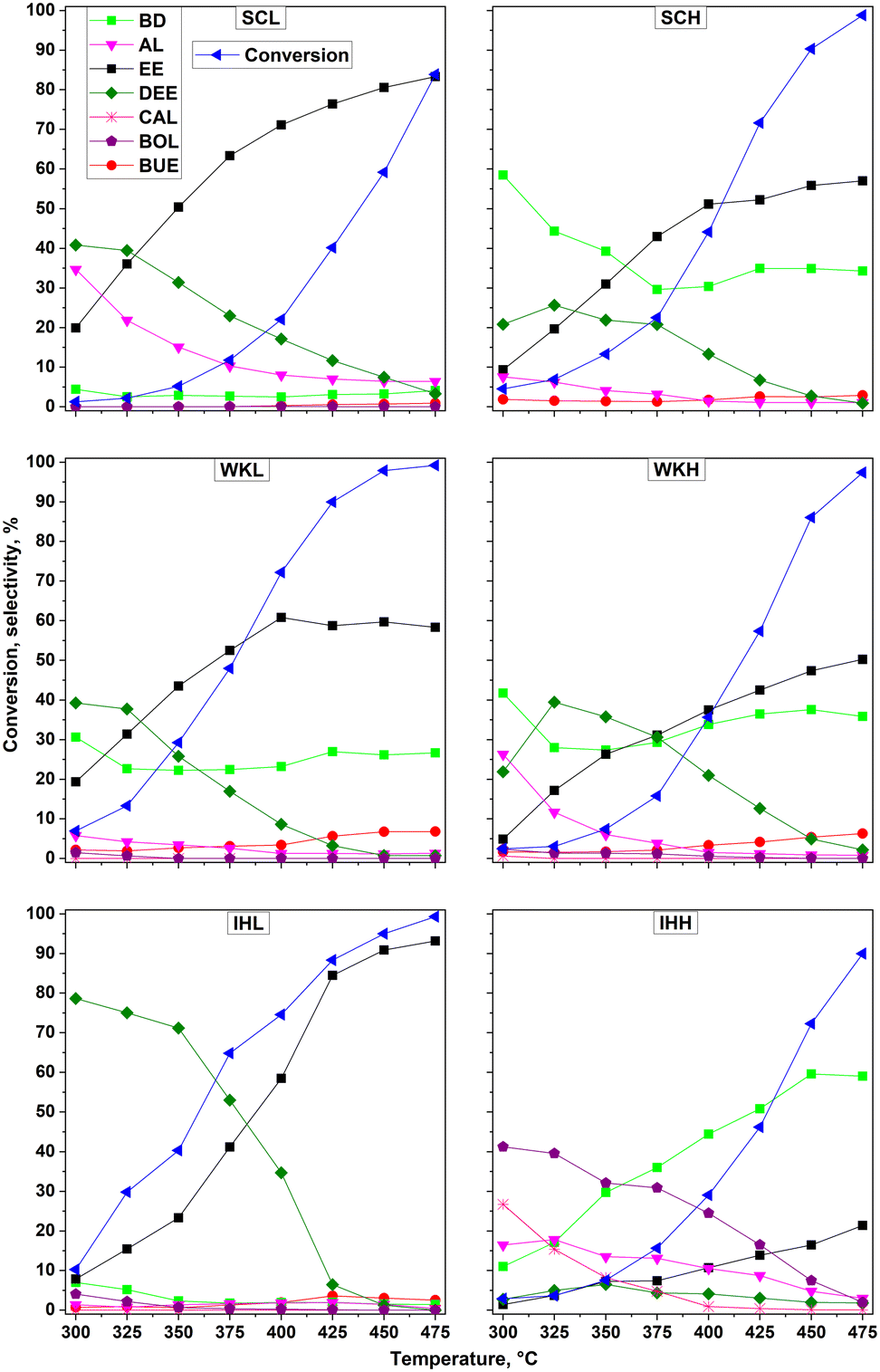 | ||
| Fig. 3 Catalytic conversion of ethanol at different temperatures over the MgO–SiO2 catalyst preparations, having a Mg to Si ratio of 2 to 1 under standard reaction conditions, such as atmospheric pressure, 15 kPa ethanol/He, 0.5 gethanol gcat−1 h−1, and a total gas flow rate of 30 ml min−1. | ||
A lower BD selectivity was always accompanied with a higher DEE and EE selectivity. In the case of the SCL and IHL catalysts, the BD selectivity remained below 10% almost in the entire applied temperature range. Over the SCL catalyst, in addition to the EE and DEE products, AL was also formed with a selectivity from 7 to 35%, depending on the reaction temperature. The catalytic activities of the WKL and WKH samples were very similar to each other; nevertheless, EE and DEE were formed with a somewhat higher selectivity over the WKL catalyst. Consequently, the BD selectivity of the WKL catalyst was about 5–10% lower than that of the WKH catalyst. In terms of BD yield, the best results were provided by the SCH and IHH catalysts. The dehydration selectivity of the SCH and IHH catalysts, i.e., the selectivity for EE plus DEE, was about 60% and less than 20%, respectively. It is also worth noting that the total selectivity for C4 products, such as BD, BOL, CAL, and BUE, was remarkably high (∼70%) over the IHH sample. As the amounts of undesirable by-products (hexadienes, hexatrienes, hexanol, 2-ethyl-1-butanol, etc.) increased significantly at higher temperatures (>400 °C), the carbon amount, fed in with ethanol, and that of the quantified main products became more unbalanced. CAD and/or CAL are necessary intermediates in the ETB route even if they do not appear in the product mixture. CAL showed up as a product, formed over the IHH catalyst. As the reaction temperature was raised, the BD selectivity of the catalyst increased at the expense of the CAL selectivity, substantiating that BD was formed from CAL by dehydration.
It is possible that the dehydrogenation of ethanol to AL is the rate-determining step in the ETB reaction. In further consecutive steps, AL can participate in reactions resulting in various C4 products, such as BD, CAD, CAL, BOL and BUE. It should be mentioned that AL can also be formed by the MPV reaction, in which ethanol hydrogenates CAD, forming AL and CAL. To characterize the activities of ethanol dehydrogenation, we summarized the formation rate of acetaldehyde, and twice the formation rate of the C4 products. This approximation assumes that no MPV reaction takes place and all the C4 products were formed in a transformation involving the coupling of two AL molecules. Although neither of these assumptions are justified, we believe that the rates obtained by this approximation characterizes the dehydrogenation activity of the catalysts (Fig. 4A). The dehydrogenation activity of the H-series catalysts, prepared using a mesoporous carbon template, is higher, whereas their dehydration activity (Fig. 4C) is lower than that of their corresponding L-series catalyst pair. Assessing the temperature-dependent formation rate of C4 products except BD, and the formation rate of BD (ΣC4 in Fig. 4B, and BD in 4D), it can be concluded that the formation rate of ΣC4 products roughly parallels the rate of BD formation. Only the IHL catalyst does not seem to follow this trend. This catalyst has an exceptionally high ethanol dehydration activity which makes the ETB route subordinate. It should be noted that the rate of the consecutive process steps must be the same as that of the rate determining step of the process. This reaction rate is nothing to do with the rate as the products appearing in the product mixture.
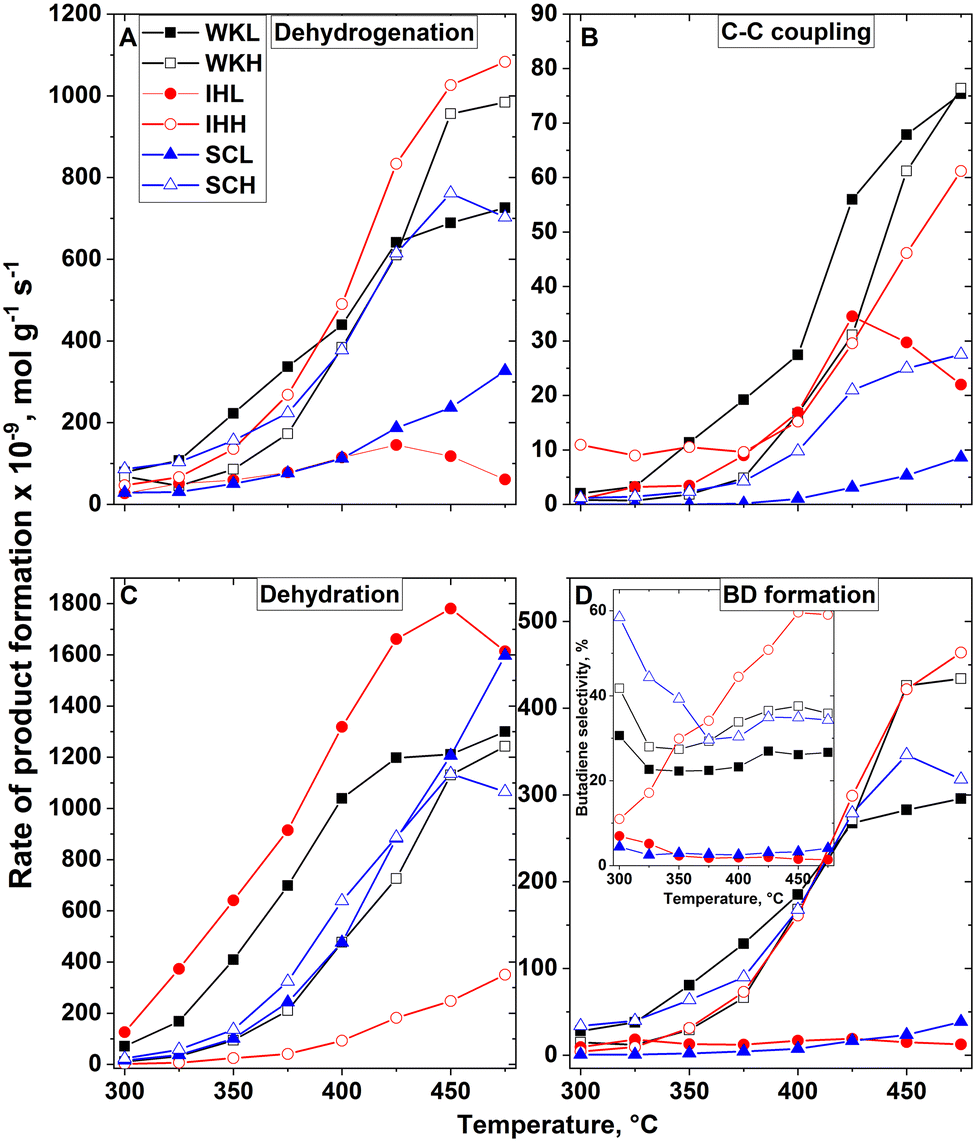 | ||
| Fig. 4 Formation rates of ETB reaction products obtained by (A) dehydrogenation to AL, (B) C–C coupling to ΣC4 products (BOL, BUE, CAL) but not to BD, (C) ethanol dehydration (EE plus DEE), and (D) C–C coupling to BD as a function of reaction temperature under standard reaction conditions. The insert in section (D) shows the butadiene selectivities. The rate of dehydrogenation was calculated as the rate of AL formation plus twice the rate of ΣC4 formation. | ||
To better understand the ETB activity of the mixed oxide catalysts, the activity of the individual components was examined (Fig. 5). Both the LSM and HSM preparations were active in the dehydrogenation of ethanol to AL and in the C–C coupling reaction (Fig. 5A and B). The HSM material, forming mainly BOL, showed significantly higher activity than the LSM material. BUE and CAD appeared in traces only. The LSM sample showed significant activity in the ethanol dehydration to EE (Fig. 5A).
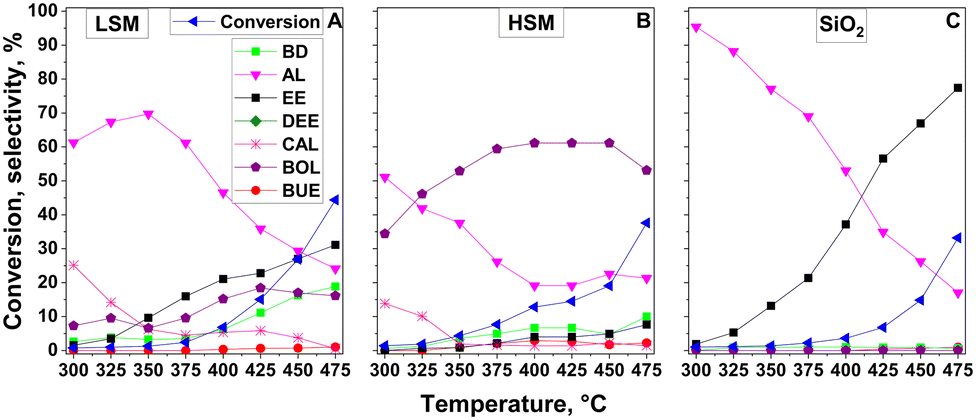 | ||
| Fig. 5 Catalytic conversion of ethanol as a function of temperature over (A) LSM, (B) HSM, and (C) SiO2 under standard reaction conditions. | ||
Over pure SiO2, dehydrogenation prevailed at lower temperatures, whereas dehydration became the dominating reaction at higher temperatures. The pure silica had no C–C coupling activity at all (Fig. 5C).
Fig. 6 shows the space time dependence of the ethanol conversion at 350 °C over the MgO (HSM and LSM) and MgO–SiO2 (IHH) catalysts. Over the LSM catalyst (Fig. 6A) the conversion remained low, and AL was the main product. Some CAL and BOL were also obtained.
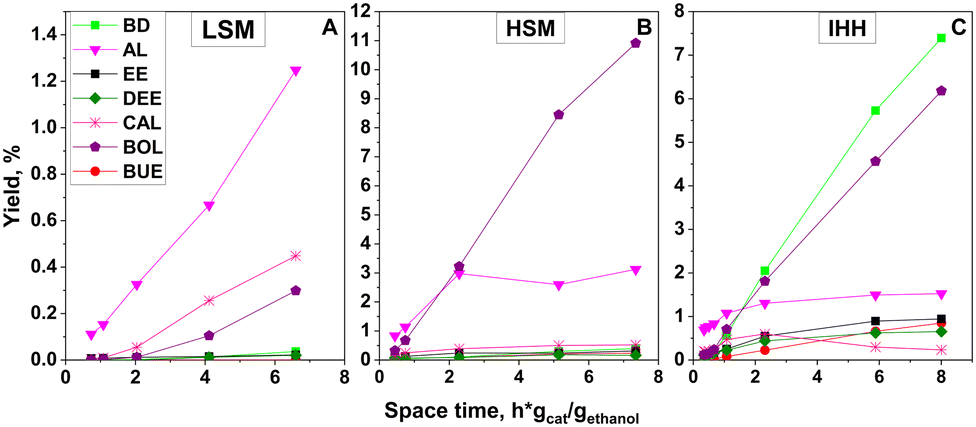 | ||
| Fig. 6 Effect of space time on the conversion of ethanol at 350 °C over the (A) LSM, (B) HSM and (C) IHH catalysts. The WHSV of ethanol was varied between 0.2 and 3.0 gethanol gcat−1 h−1. Otherwise, standard reaction conditions were used. | ||
AL must be an intermediate participating in the C–C coupling reaction with ethanol to CAL or in aldol condensation with another AL molecule to CAD. BOL could be formed by the hydrogenation of CAL or CAD. No doubt that the LSM catalyst has also dehydration and hydrogenation activity. The conversion over HSM (Fig. 6B) was significantly higher, and BOL was the main product besides AL and traces of CAD, CAL and BD. Traces of DEE were obtained over both magnesia catalysts but for clarity are not shown in the figure. Over the IHH mixed oxide catalyst (Fig. 6C), BD was the main product besides some dehydration products EE and DEE. Traces of PE were also detected. The yields of BOL and AL were low relative to their yields over the HSM catalyst. The product distribution suggests that the IHH mixed oxide catalyst has higher dehydration activity than the pure magnesia preparations. The enhanced dehydration activity must be related to the stronger acidity generated by intimate mixing of silica and magnesia components. CAL and a minor amount of CAD were also detected in the product mixture.
The activity of the WKH and IHH catalysts was examined at a 400 °C reaction temperature as a function of time on stream (Fig. S4†). The conversion decreased by 4–5% in the first half hour and then hardly changed for 32 hours, while the product selectivities also remained virtually unchanged.
The selectivities of the catalysts for the different products are compared in Fig. 7. Identical conversions were achieved using the catalysts of different activities by changing the space time of the reactant ethanol at the selected two reaction temperatures (the SCL catalyst is omitted from Fig. 7 because the desired conversion could not be attained with it). Over the mixed oxide catalysts, containing HSM, higher BD selectivities were found than those over the catalysts containing the LSM component. Accounting for all the C4 products, the IHH catalyst showed the highest selectivity at both 20 and 50% ethanol conversions. At 50% conversion, the IHH sample was the most BD selective catalyst, showing about a 55% BD selectivity.
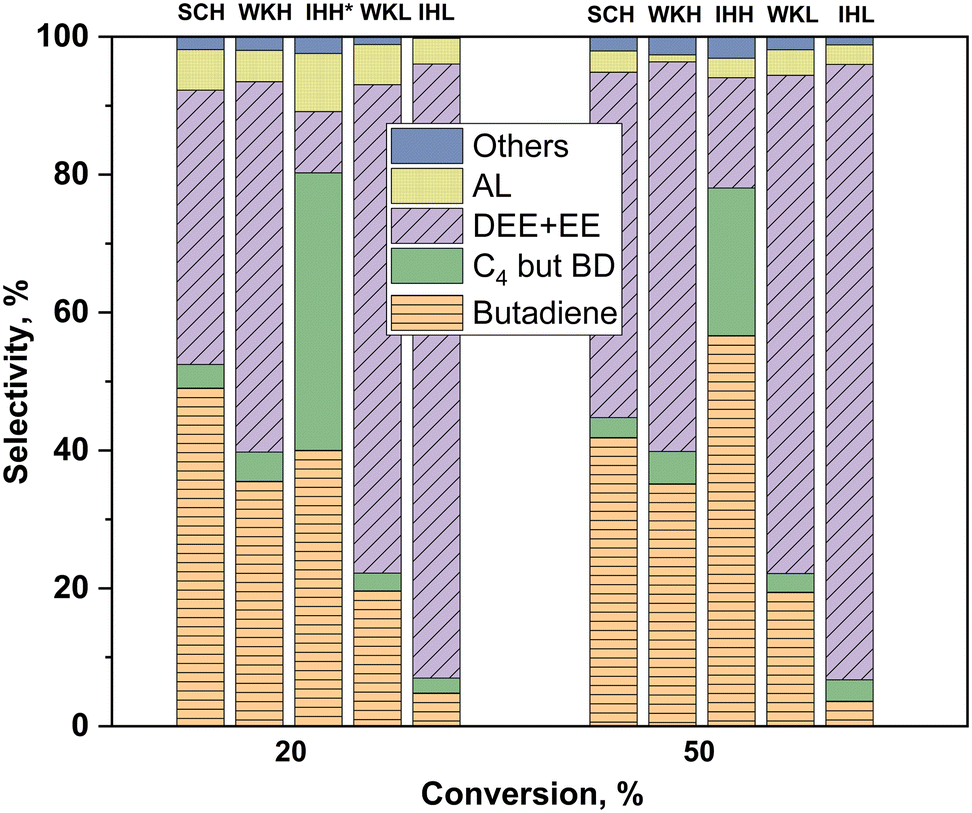 | ||
| Fig. 7 Product distribution at 20 and 50% conversion levels over different MgO–SiO2 catalysts. Identical conversions with the catalysts of different activities were achieved by adjusting the ethanol WHSV between 0.2 and 3.0 gethanol gcat−1 h−1 at about a 15 kPa partial pressure at 350 °C for 20% conversion and 400 °C for 50% conversion. *Over the IHH sample, the attained maximum conversion was only ∼18%. Others: methane, propylene, higher olefins. | ||
4. Discussion
In agreement with the STEM/EDS and XRD results, the observations based on XPS point to certain structural differences of the samples. The highest apparent Si content and its most silica-like state in the SC systems confirm the formation of thick Si-rich domains on the outer surface of the materials with the smallest Mg incorporation. The WK samples are similar but have more Mg-rich material on the surface and slightly higher Mg incorporation into the silica. Finally, the most disrupted silica network, having the highest extent of Mg incorporation, occurs in the IH systems. These results suggest that there are surface Si–O–Mg bonds in all catalysts. The Si–O–Mg surface moieties represent Lewis acid sites. The immediate environment and abundance of such bonds determine the strength and concentration, respectively, of the acid sites. It was shown that the surface acidity of these mixed oxide catalysts was higher, in which the catalysts had a lower surface Mg/Si ratio.The ETB activity of the MgO–SiO2 mixed oxide catalysts could be improved by applying a high-SSA MgO component for catalyst preparation (Fig. 3 and 6). This is in harmony with the findings of previous studies, showing that a MgO component of higher SSA and porosity brings about stronger basicity and, as a result, higher BD selectivity.17,20,88 Reschetilowski et al.20 pointed out that the activity could have been improved also by the higher amount of acidic Mg–O–Si moieties, generated over the higher-SSA MgO component. Angelici et al.6 concluded that the best performing catalysts were those containing some acid sites and small amounts of strong and weak base sites. Obviously, the role of acid/base sites in the ETB reaction is unclear yet.
Our acidity/basicity measurements gave rather controversial results. We found only trend-like relations between the quantified acid/base characteristics and the ETB activity. The acidity and basicity were characterized by the weight specific amount of adsorbed base (NH3, pyridine) and acid (CO2, CDCl3), respectively. The amount of adsorbate retained by the catalyst under the selected adsorption conditions depends not only on the chemical character of the catalyst surface but also on the SSA that changes from sample to sample (Table 1). The correlation between the measured weight-specific acidity/basicity and the weight-specific catalytic activity was investigated. A strong correlation could not be expected at all, considering that the acid/base properties of the adsorbates, used to probe the catalyst surface were completely different from those of the reactant ethanol. In addition, the interaction of the catalysts with ethanol and reaction products, including water, could chemically modify and even restructure the active surface.
We measured higher acidity and basicity for catalysts, except for the pure magnesia samples, prepared using the MCA template. These catalysts proved to be more efficient, regarding the BD yields, than those prepared without using the template. It was also observed that the catalysts, having similar acid/base site concentrations, give similar BD yields regardless of their different SSAs [cf.Fig. 3, C and D]. The acidity of the pure oxide components of the catalysts follows the order LSM < HSM < SiO2. As was expected for the basicity, the reverse trend prevails (Table 3, Fig. S5†). The acidity of the mixed oxides usually exceeded that of the individual components, indicating that incorporation of Mg into the silica generated acidity (Table 3). The admixing of silica to magnesia had a controversial effect on the measured basicity. We thought that diluting magnesia with silica will likely cause a basicity decrease. Surprisingly, the CDCl3 probe detected an increased total basicity. In contrast, against the acidic probe CO2, the basicity of the mixed oxides, in harmony with our expectation, was lower than that of their corresponding MgO component; however, two preparations (WKH, IHH) showed again unexpectedly high basicity (Table 2).
The stoichiometric equation of the ETB reaction (2CH3–CH2–OH → CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHCH2 + H2 + 2H2O) shows that the catalyst of the reaction must have C–C coupling, hydrogenation/dehydrogenation and dehydration activity. The LSM and HSM catalysts, having low Lewis acidity, virtually do not have dehydration activity. These pure MgO catalysts initiated dehydrogenation and were basic enough to initiate C–C coupling, as indicated by the formation of BOL (2CH3–CH2–OH → CH3–CH2–CH2–CH2–OH + H2O (Fig. 5 and 6, A and B)). In contrast, pure SiO2, being more acidic than MgO, has dehydrogenation and dehydration activity but, being less basic than MgO, has no C–C coupling activity (Fig. 5C). The pure silica provides mainly AL, EE, and DEE. BD was obtained only over mixed oxides.
CH–CHCH2 + H2 + 2H2O) shows that the catalyst of the reaction must have C–C coupling, hydrogenation/dehydrogenation and dehydration activity. The LSM and HSM catalysts, having low Lewis acidity, virtually do not have dehydration activity. These pure MgO catalysts initiated dehydrogenation and were basic enough to initiate C–C coupling, as indicated by the formation of BOL (2CH3–CH2–OH → CH3–CH2–CH2–CH2–OH + H2O (Fig. 5 and 6, A and B)). In contrast, pure SiO2, being more acidic than MgO, has dehydrogenation and dehydration activity but, being less basic than MgO, has no C–C coupling activity (Fig. 5C). The pure silica provides mainly AL, EE, and DEE. BD was obtained only over mixed oxides.
In the SCL mixed oxide catalyst, a large fraction of the MgO surface is coated by silica as evidenced by the low Mg/Si surface ratio (Table 1). Therefore, over the SCL catalyst, dehydration reactions, characteristic of pure silica, dominated, while C–C coupled products were hardly formed (Fig. 3A). In contrast, the SCH catalyst, having similar acidity to the SCL catalyst (Table 3), has significant (35–60%) BD selectivity. This can be explained by the lower silica coverage of the HSM component, leaving the basic MgO surface sites accessible for the reactant and intermediates to initiate C–C coupling (Fig. 3B). If anything, the medium strength basicity of the catalysts (Table 3) seems to show a correlation with the found difference in BD selectivity. Besides hydrogenation/dehydrogenation activity, due to the silica-generated acidity and the residual medium strength basicity, the SCH catalyst had both dehydration activity and C–C coupling activity, as indicated by the appearance of DEE and EE, and also BD. No BOL was formed over either of the silica-coated magnesia catalysts.
The WKL and WKH mixed oxide preparations showed a rather similar catalytic behavior, though the WKH catalyst was more basic and more acidic than its WKL counterpart. This difference most probably is related to the differences in the SSA of MgO, used for catalyst preparation. When the MgO and SiO2 components were forced by wet kneading to get into chemical interaction, the higher-SSA MgO allowed the formation of more Mg–O–Si bonds, providing surface species that are Lewis acid sites. Larina et al.5,26 have previously demonstrated that the formation of Mg–O–Si bonds results in Lewis acid sites, which play a decisive role in the ETB reaction. Indeed, the surface Mg/Si ratio of the mixed oxide WKH is higher than that of its WKL counterpart (Table 1). The higher basicity of the WKH catalyst is due to the higher MgO surface, remaining free after wet kneading. This free MgO surface is responsible for the higher dehydrogenation/coupling activity of the WKH sample, while the higher free SiO2 surface provides WKL with higher dehydration activity. The higher dehydrogenating and coupling activity of the sample prepared with HSM can be explained by the higher concentration of low-coordinated O2− anions present at corner or edge sites of high-SSA and mesoporous MgO.89 Comparing the activities of the catalyst pair at the same conversion levels, the WKH catalyst showed higher BD and C4 selectivities than its WKL counterpart (Fig. 7).
Interestingly, the crystallinity of the IH preparations depended on the co-precipitation method used. The IHL catalyst consisted of forsterite, while the IHH catalyst was X-ray amorphous (Fig. 1A). The forsterite was a weak acid, whereas the amorphous catalyst showed significant acidity and basicity (Table 3). In harmony with its dominating acidic character, only EE and DEE were formed over the IHL catalyst (Fig. 3E). The low ETB activity of forsterite has already been demonstrated.12 In contrast, the IHH catalyst had significant ETB activity (Fig. 3F). In the temperature range 300–350 °C, only AL and products of C–C coupling, such as BOL, CAL, and BD, appeared in the reactor effluent. It is interesting that CAL was formed with high selectivity, although it has been reported that the dehydration of CAL was very facile even at these temperatures.24 At higher temperatures, BD and EE became the main products at the expense of CAL and BOL formation, indicating that the dehydration activity was more strongly affected by the temperature rise than the C–C coupling activity (Fig. 3F).
Before getting to a deeper mechanistic understanding of the consecutive steps of the ETB process, first the surface intermediates must be substantiated. The rate of each consecutive process step must be the same as that of the rate determining slowest step, whereas the surface concentration of the intermediates is determined by the rate constants of the processes generating and consuming a given intermediate. The most convincing proof for a suggested reaction route is if the hypothesized key intermediates can be discerned in the product mixture. However, it can happen that some presumed intermediates do not appear as a detectable product because of their surface concentration being very low and/or the desorption of the activated surface intermediate being not favored. In this case, proving the reaction route can be attempted by debatable interpretation of results, provided by operando surface spectroscopy and theoretical calculations. AL always appeared as the main product (Fig. 3). It is not questioned that one intermediate of the ETB reaction is AL, formed from ethanol by dehydrogenation. Another one is CAL that can be dehydrated to get BD and also appeared as a significant reaction product over some catalysts (Fig. 6). These results make it likely that CAL-like activated surface species participate in the ETB reaction, even if CAL is not always detected as a product. In the scientific literature, there is a discussion about the way CAL or CAL-like surface intermediates are formed. It is argued that these species are formed either via direct surface reaction between AL and ethanol,11,90 or by Meerwein–Ponndorf–Verley (MPV) reduction of CAD, which is obtained from AL by aldol addition and condensation.13,55,91,92 Recently, Yang et al.28 successfully identified the aldol addition product 3-hydroxybutanal (i. e., acetaldol), the intermediate of CAD, but not under commonly used reaction conditions. We never detected acetaldol and never found CAD in the product mixture in concentration that could be considered as strong enough evidence for the role of CAD or CAD-like surface species as an intermediate of the ETB reaction. It should be noted that CAD may also be formed due to disproportionation of CAL,23 enhancing the doubts that detection of CAD traces could provide enough support for its participation in the ETB reaction as an intermediate.
Our results support the notion that the ethanol conversion to BOL and/or BD proceeds through a common intermediate CAL. We cannot rule out the fact that the route to CAL formation leads through intermediates AL and CAD. Sites of weak base strength were considered responsible for ethanol dehydrogenation to AL, whereas strongly basic sites were presumed to initiate aldol condensation of AL to CAD and MPV reduction of CAD to CAL. It was suggested that sites of strong acidity could initiate both CAD formation and CAL dehydration to BD, whereas sites of weak acid strength effected the undesired ethanol dehydration.15
The catalytic process of BOL formation over pure MgO raises similar mechanistic questions, as does the ETB process over MgO–SiO2 mixed oxides. The reaction is suggested to proceed through the CAD and CAL intermediates (aldol route, Guerbet mechanism)93,94 or through direct coupling of two ethanol molecules. According to the latter route, AL could have been also formed but did not have any role in the C–C coupling process.90,95 Based on kinetic analysis, diffuse-reflectance infrared spectroscopic investigation, and density functional theory calculations, Toniolo et al.96 substantiated that the dominating mechanism of BOL formation depends on the reaction temperature. It was suggested that below about 373 °C, the so-called Cβ–H mechanism is dominating, whereas above this temperature, up to 473 °C, the well-known Guerbet mechanism. The process according to latter mechanism proceeds through adsorbed CAD and the MPV reduction of CAD to CAL and finally to BOL. In contrast, according to the Cβ–H mechanism, surface ethoxy species are activated by donating a proton from their β-carbon atom to a basic surface oxide ion. The obtained ethoxy anion attacks then the α-carbon of an ethoxy group, coordinated to a neighboring a Lewis acid site. The result is surface-bound BOL that can be released as a product. The Cβ–H mechanism corresponds to that suggested by Chieregato et al.11,90 These studies describe a similar mechanism for CAL and BD formation. According to this idea, the activated ethoxy carbanion attacks not the α-carbon of an ethoxy group but that of AL, coordinated to a neighboring Lewis acid site. Climent et al.97 have pointed out that coordination of a carbonyl group to an acid site can polarize the C–O bond, increasing the density of positive charge on the carbon. This effect makes the carbon more susceptible to attack by the carbanion. Briefly, the latter mechanism assumes the formation of AL that directly couples with an ethanol molecule to give CAL and then BD by dehydration.
At low temperatures, when the conversion of ethanol is only a few percent, the surface of the catalysts must be predominantly covered by ethanol, and the surface concentration of AL must be low even at high AL selectivities. Nevertheless, we got BD with relatively high selectivity (Fig. 3). In the product mixture, formed over the WK and IH catalysts, even the intermediate CAL could be detected. The low AL coverage must make the probability of bimolecular surface aldol reaction low, substantiating that CAL and BD could have been formed by the Cβ–H mechanism.
At higher temperatures and conversions, the aldol route may become the prevailing ETB mechanism. The conversion test of an ethanol/CAD mixture was found to result in BD formation (not shown). However, we do not believe that this is strong enough evidence for the aldol ETB route. The absence of CAD in the product mixture may be a result of its low surface concentration due to the high rate constant of its MPV hydrogenation. However, no CAD could be detected in the product mixture even using very short or longer space times (Fig. 6).
The catalytic ETB selectivities of the H and L series catalysts were compared at the same temperature and conversion (Fig. 7). The comparison was made at 350 °C and 20% conversion, and also at 400 °C and 50% conversion. The BD selectivity was significantly higher for the H series catalysts at both conversions. The IHH catalyst showed the highest selectivity, about 80% ΣC4, at both temperatures. In contrast, under identical conditions, the EE plus DEE selectivity of the IHL catalyst was near 90%. The results obtained at the two temperatures/conversions demonstrates the strong effect of these reaction parameters on the ETB selectivity. For instance, at the two temperatures, the IHH catalyst has the same ΣC4 selectivity but a higher BD selectivity and much higher BD yield at the higher temperature.
5. Conclusions
The activities of MgO–SiO2 mixed oxide catalysts, having a Mg to Si molar ratio of 2, were compared in the ethanol-to-butadiene (ETB) reaction. The aim of the study was to clarify the influence of MgO distribution within the MgO–SiO2 catalysts on the ETB activity. Catalysts were prepared either by wet kneading of MgO, having a high specific surface area (SSA), and SiO2, or by coating the surface of high-SSA MgO with silica. The high-SSA MgO component was generated by combustion of mesoporous carbon, saturated by a Mg(NO3)2 solution. A third catalyst was prepared by hydrolyzation/condensation/precipitation of an Si,Mg–alkoxide solution within mesopores of carbon and burning out the carbon/precipitate material. The materials obtained allow the evaluation of the effect of Mg–Si atomic homogeneity on the ETB activity. For comparison, a corresponding set of catalysts was prepared by the abovementioned methods but the use of a carbon template was fully omitted. Low-SSA MgO was prepared by the thermal decomposition of Mg(NO3)2. XPS examination proved that the surface Mg/Si ratio was higher for the mixed oxide catalysts, obtained using high-SSA MgO, than that for those prepared using low-SSA MgO. The metal distribution in the catalysts, prepared by alkoxide hydrolysis, was the most homogeneous. Catalytic ETB tests showed that the pure, high-SSA MgO catalysts had significantly higher ethanol coupling and dehydrogenation activity than their low-SSA counterpart. This was attributed to the higher concentration of low-coordinated O2− anions present in corners or edge sites of high SSA and mesoporous MgO. Acetaldehyde (AL), butanol (BOL), and crotyl alcohol (CAL) were the main products over both catalysts. Over pure silica, virtually only products of ethanol dehydrogenation and dehydration, such as acetaldehyde (AL), ethylene (EE), and diethyl ether (DEE), were formed. The acidity of the MgO samples, probed using basic adsorbates NH3 and pyridine, increased upon introduction of a SiO2 component. The basicity measurements, using acidic adsorbate CO2 and CDCl3 to probe the surface, gave controversial results. Over the mixed oxides, having balanced acid–base properties, butadiene (BD) became the main C4 product besides the products AL, EE, and DEE. No substantial amount of BOL and CAL was formed. The BD selectivity of the MgO–SiO2 catalysts, made using high-SSA MgO, was higher than that of the catalysts made using low-SSA MgO both at the same reaction temperature and at the same conversion level. The silica-coated low-SSA MgO showed catalytic activity similar to that of pure SiO2. The high-SSA MgO, coated by the same amount of SiO2, showed activity, similar to that of the wet-kneaded catalysts. These results suggest that MgO can partly retain its original properties in the MgO–SiO2 catalysts. Alkoxide hydrolysis and calcination resulted in low-SSA forsterite (Mg2SiO4), while calcination, following hydrolysis within the pores of the carbon template, gave high-SSA amorphous MgO–SiO2 mixed oxides. Using the former preparation as an ETB catalyst, only EE and DEE were formed. Meanwhile, using the catalyst prepared by template assistance, a higher coupling activity could be attained than that with any other studied catalyst. For instance, at 50% ethanol conversion, the C4 selectivity was as high as 80%, while BD was formed at 55% selectivity. It was substantiated that, at least at low conversions, the BD-intermediate CAL could have been formed by direct acetaldehyde–ethanol coupling, bypassing crotonaldehyde (CAD) formation and Meerwein–Ponndorf–Verley (MPV) reduction of CAD.Abbreviations
| AL | Acetaldehyde |
| BD | 1,3-Butadiene |
| BUE | Butenes |
| BOL | Butanol |
| CAD | Crotonaldehyde |
| CAL | Crotyl alcohol |
| CTABr | Cetyltrimethyl ammonium bromide |
| DEE | Diethyl ether |
| EE | Ethylene |
| ETB | Ethanol-to-butadiene |
| H | Catalysts prepared using high-specific surface area MgO |
| HSM | High-specific surface area MgO |
| HT | Hard-templating |
| IH | Internal hydrolysis |
| IHH | Internally hydrolyzed sample containing high specific surface area MgO |
| IHL | Internally hydrolyzed sample containing low specific surface area MgO |
| L | Catalysts prepared using low-specific surface area MgO |
| LSM | Low-specific surface area MgO |
| MCA | Mesoporous carbon aerogel |
| PE | Propene |
| RF | Resorcinol–formadehyde |
| SC | Silica coating |
| SCH | Silica coated sample containing high specific surface area MgO |
| SCL | Silica coated sample containing low specific surface area MgO |
| SSA | Specific surface area |
| TEOS | Tetraethyl orthosilicate |
| WK | Wet kneading |
| WKH | Wet kneaded sample containing high specific surface area MgO |
| WKL | Wet kneaded sample containing low specific surface area MgO |
Author contributions
Blanka Szabó: conceptualization, validation, investigation, and writing – original visualization draft. Virág Hutkai: investigation. Gyula Novodárszki: investigation. Ferenc Lónyi: conceptualization and validation, writing – review & editing. Zoltán Pászti: investigation and writing – original draft. Zsolt Fogarassy: investigation and writing – original draft. József Valyon: conceptualization, validation, writing – review & editing, and supervision. Róbert Barthos: conceptualization, validation, investigation, writing – original draft, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank to the Ministry of Innovation and Technology of Hungary for financing this study from the National Research, Development and Innovation Fund, under the 2019-2.1.13-TÉT_IN funding scheme (Project No. 2019-2.1.13-TÉT_IN-2020-00043). Thanks are also given to the Interreg V-A Slovakia – Hungary Cooperation Program, SKHU/1902 (Project No: SKHU/1902/4.1/001), and the Competitive Central Hungary Operational Program, VEKOP, supported by the European Union, the State of Hungary, and co-financed by the European Regional Development Fund (Project No. VEKOP-2.3.2-16-2017-00013), and the European Structural and Investment Funds (Project No. VEKOP-2.3.3-15-2016-00002) for supporting this work. One of the authors (Zs. Fogarassy) says thanks also for the support from the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. One of the authors (B. Szabó) is supported by the ÚNKP-22-4-SZTE-520 New National Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund.References
- G. Pomalaza, P. Arango Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911 RSC.
- The Michelin Group, The Biobutterfly Project, https://www.ifpenergiesnouvelles.fr/article/michelin-ifpen-et-axens-donnent-ensemble-nouvelle-dimension-au-projet-biobutterfly, (accessed 2 October 2022) Search PubMed.
- S. Kvisle, A. Aguero and R. P. A. Sneeden, Appl. Catal., 1988, 43, 117–131 CrossRef CAS.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2015, 8, 994–1008 CrossRef CAS.
- O. V. Larina, P. I. Kyriienko and S. O. Soloviev, Catal. Lett., 2015, 145, 1162–1168 CrossRef CAS.
- C. Angelici, M. E. Z. Velthoen, B. M. Weckhuysen and P. C. A. Bruijnincx, Catal. Sci. Technol., 2015, 5, 2869–2879 RSC.
- M. Zhang, M. Gao, J. Chen and Y. Yu, RSC Adv., 2015, 5, 25959–25966 RSC.
- S.-H. Chung, C. Angelici, S. O. M. Hinterding, M. Weingarth, M. Baldus, K. Houben, B. M. Weckhuysen and P. C. A. Bruijnincx, ACS Catal., 2016, 6, 4034–4045 CrossRef CAS.
- S. Da Ros, M. D. Jones, D. Mattia, J. C. Pinto, M. Schwaab, F. B. Noronha, S. A. Kondrat, T. C. Clarke and S. H. Taylor, ChemCatChem, 2016, 8, 2376–2386 CrossRef CAS.
- S. Shylesh, A. A. Gokhale, C. D. Scown, D. Kim, C. R. Ho and A. T. Bell, ChemSusChem, 2016, 9, 1462–1472 CrossRef CAS.
- J. V. Ochoa, C. Bandinelli, O. Vozniuk, A. Chieregato, A. Malmusi, C. Recchi and F. Cavani, Green Chem., 2016, 18, 1653–1663 RSC.
- Q. Zhu, B. Wang and T. Tan, ACS Sustainable Chem. Eng., 2017, 5, 722–733 CrossRef CAS.
- W. E. Taifan, G. X. Yan and J. Baltrusaitis, Catal. Sci. Technol., 2017, 7, 4648–4668 RSC.
- S. Da Ros, M. D. Jones, D. Mattia, M. Schwaab, E. Barbosa-Coutinho, R. C. Rabelo-Neto, F. B. Noronha and J. C. Pinto, Chem. Eng. J., 2017, 308, 988–1000 CrossRef CAS.
- W. E. Taifan and J. Baltrusaitis, J. Phys. Chem. C, 2018, 122, 20894–20906 CrossRef CAS.
- W. E. Taifan, Y. Li, J. P. Baltrus, L. Zhang, A. I. Frenkel and J. Baltrusaitis, ACS Catal., 2019, 9, 269–285 CrossRef CAS.
- S. Li, Y. Men, J. Wang, S. Liu, X. Wang, F. Ji, S. Chai and Q. Song, Appl. Catal., A, 2019, 577, 1–9 CrossRef CAS.
- H. T. Abdulrazzaq, A. Rahmani Chokanlu, B. G. Frederick and T. J. Schwartz, ACS Catal., 2020, 10, 6318–6331 CrossRef CAS.
- X. Wang, Y. Men, J. Wang, S. Liu, Q. Song and M. Yang, Appl. Catal., A, 2020, 598, 117565 CrossRef CAS.
- W. Reschetilowski, M. Hauser, F. Alscher, M. Klauck and G. Kalies, Catalysts, 2020, 10, 854 CrossRef CAS.
- X. Huang, Y. Men, J. Wang, W. An and Y. Wang, Catal. Sci. Technol., 2017, 7, 168–180 RSC.
- M. Zhang, Y. Qin, X. Tan, L. Wang, Y. Yu and H. Jiang, Catal. Lett., 2020, 150, 1462–1470 CrossRef CAS.
- B. Szabó, G. Novodárszki, Z. Pászti, A. Domján, J. Valyon, J. Hancsók and R. Barthos, ChemCatChem, 2020, 12, 5686–5696 CrossRef.
- B. Szabó, G. Novodárszki, Z. May, J. Valyon, J. Hancsók and R. Barthos, Mol. Catal., 2020, 491, 110984 CrossRef.
- S. H. Chung, A. Ramirez, T. Shoinkhorova, I. Mukhambetov, E. Abou-Hamad, S. Telalovic, J. Gascon and J. Ruiz-Martínez, Nanomaterials, 2021, 11, 1–17 Search PubMed.
- P. I. Kyriienko, O. V. Larina, D. Y. Balakin, A. O. Stetsuk, Y. M. Nychiporuk, S. O. Soloviev and S. M. Orlyk, Appl. Catal., A, 2021, 616, 118081 CrossRef CAS.
- P. I. Kyriienko, O. V. Larina, D. Y. Balakin, S. O. Soloviev and S. M. Orlyk, Catal. Lett., 2022, 152, 921–930 CrossRef CAS.
- Y. Yang, X. Guo, Y. Pan and Y. Fang, Catal. Sci. Technol., 2022, 12, 1746–1750 RSC.
- B. Szabó, G. Novodárszki, F. Lónyi, L. Trif, Z. Fogarassy, J. Valyon and R. Barthos, J. Mol. Struct., 2022, 1259, 132764 CrossRef.
- Z. Han, X. Li, M. Zhang, Z. Liu and M. Gao, RSC Adv., 2015, 5, 103982–103988 RSC.
- V. L. Dagle, M. D. Flake, T. L. Lemmon, J. S. Lopez, L. Kovarik and R. A. Dagle, Appl. Catal., B, 2018, 236, 576–587 CrossRef CAS.
- T. Miyazawa, Y. Tanabe, I. Nakamura, Y. Shinke, M. Hiza, Y. K. Choe and T. Fujitani, Catal. Sci. Technol., 2020, 10, 7531–7541 RSC.
- S. A. Akhade, A. Winkelman, V. Lebarbier Dagle, L. Kovarik, S. F. Yuk, M.-S. Lee, J. Zhang, A. B. Padmaperuma, R. A. Dagle, V.-A. Glezakou, Y. Wang and R. Rousseau, J. Catal., 2020, 386, 30–38 CrossRef CAS.
- J. L. Vieira, P. Destro, L. O. Laier, C. M. P. Marques, J. M. R. Gallo and J. M. C. Bueno, Mol. Catal., 2022, 532, 112718 CrossRef CAS.
- Y. Shinke, T. Miyazawa, M. Hiza, I. Nakamura and T. Fujitani, React. Chem. Eng., 2021, 6, 1381–1385 RSC.
- F. Lin, V. L. Dagle, A. D. Winkelman, M. Engelhard, L. Kovarik, Y. Wang, Y. Wang, R. Dagle and H. Wang, ChemCatChem, 2021, 13, 999–1008 CrossRef CAS.
- K. Wang, W. Gao, F. Chen, G. Liu, J. Wu, N. Liu, Y. Kawabata, X. Guo, Y. He, P. Zhang, G. Yang and N. Tsubaki, Appl. Catal., B, 2022, 301, 120822 CrossRef CAS.
- J. V. Ochoa, A. Malmusi, C. Recchi and F. Cavani, ChemCatChem, 2017, 9, 2128–2135 CrossRef CAS.
- G. Pomalaza, G. Vofo, M. Capron and F. Dumeignil, Green Chem., 2018, 20, 3203–3209 RSC.
- G. Pomalaza, P. Simon, A. Addad, M. Capron and F. Dumeignil, Green Chem., 2020, 22, 2558–2574 RSC.
- O. V. Larina, N. D. Shcherban, P. I. Kyriienko, I. M. Remezovskyi, P. S. Yaremov, I. Khalakhan, G. Mali, S. O. Soloviev, S. M. Orlyk and S. Dzwigaj, ACS Sustainable Chem. Eng., 2020, 8, 16600–16611 CrossRef CAS.
- P. Tu, B. Xue, Y. Tong, J. Zhou, Y. He, Y. Cheng, J. Ni and X. Li, ChemistrySelect, 2020, 5, 7258–7266 CrossRef CAS.
- H. T. Abdulrazzaq, A. Rahmani Chokanlu, B. G. Frederick and T. J. Schwartz, ACS Catal., 2020, 10, 6318–6331 CrossRef CAS.
- K. Wang, X. Peng, X. Gao, Y. Araki, H. Zhao, J. Liang, L. Xiao, J. Chen, G. Liu, J. Wu, G. Yang and N. Tsubaki, React. Chem. Eng., 2021, 6, 548–558 RSC.
- P. I. Kyriienko, O. V. Larina, N. D. Scherban, I. M. Remezovskyi, L. M. Alekseenko, P. S. Yaremov, S. O. Soloviev and S. M. Orlyk, Theor. Exp. Chem., 2020, 56, 329–337 CrossRef.
- G. Pomalaza, M. Capron and F. Dumeignil, Appl. Catal., A, 2020, 591, 117386 CrossRef.
- K. Wang, L. Guo, W. Gao, B. Zhang, H. Zhao, J. Liang, N. Liu, Y. He, P. Zhang, G. Yang and N. Tsubaki, ACS Sustainable Chem. Eng., 2021, 9, 10569–10578 CrossRef CAS.
- A. Klein and R. Palkovits, Catal. Commun., 2017, 91, 72–75 CrossRef CAS.
- V. L. Sushkevich, D. Palagin and I. I. Ivanova, ACS Catal., 2015, 5, 4833–4836 CrossRef CAS.
- A. Klein, K. Keisers and R. Palkovits, Appl. Catal., A, 2016, 514, 192–202 CrossRef CAS.
- P. I. Kyriienko, O. V. Larina, S. O. Soloviev, S. M. Orlyk and S. Dzwigaj, Catal. Commun., 2016, 77, 123–126 CrossRef CAS.
- V. L. Sushkevich and I. I. Ivanova, ChemSusChem, 2016, 9, 2216–2225 CrossRef CAS PubMed.
- P. I. Kyriienko, O. V. Larina, S. O. Soloviev, S. M. Orlyk, C. Calers and S. Dzwigaj, ACS Sustainable Chem. Eng., 2017, 5, 2075–2083 CrossRef CAS.
- L. Qi, Y. Zhang, M. A. Conrad, C. K. Russell, J. Miller and A. T. Bell, J. Am. Chem. Soc., 2020, 142, 14674–14687 CrossRef CAS.
- M. Zhang, X. Guan, J. Zhuang and Y. Yu, Appl. Surf. Sci., 2022, 579, 152212 CrossRef CAS.
- A. Hanif, S. Dasgupta and A. Nanoti, Ind. Eng. Chem. Res., 2016, 55, 8070–8078 CrossRef CAS.
- H. T. Hoang, A. A. Sertsova, S. I. Marakulin, E. N. Subcheva, M. P. Zaitseva and E. V. Yurtov, Russ. J. Inorg. Chem., 2018, 63, 1414–1418 CrossRef CAS.
- W. Peng, J. Li, B. Chen, N. Wang, G. Luo and F. Wei, Catal. Commun., 2016, 74, 39–42 CrossRef CAS.
- X. Zhao, G. Ji, W. Liu, X. He, E. J. Anthony and M. Zhao, Chem. Eng. J., 2018, 332, 216–226 CrossRef CAS.
- Z. M. Cui, Z. Chen, C. Y. Cao, W. G. Song and L. Jiang, Chem. Commun., 2013, 49, 6093–6095 RSC.
- C. X. Gui, Q. J. Li, L. L. Lv, J. Qu, Q. Q. Wang, S. M. Hao and Z. Z. Yu, RSC Adv., 2015, 5, 20440–20445 RSC.
- S. Yang, P. Huang, L. Peng, C. Cao, Y. Zhu, F. Wei, Y. Sun and W. Song, J. Mater. Chem. A, 2015, 4, 400–406 RSC.
- Z. M. Cui, J. Hao, C. Y. Cao and W. Song, J. Porous Mater., 2017, 24, 103–108 CrossRef CAS.
- M. S. Hamdy, N. S. Awwad and A. M. Alshahrani, Mater. Des., 2016, 110, 503–509 CrossRef CAS.
- V. Hiremath, R. Shavi and J. Gil Seo, J. Colloid Interface Sci., 2017, 498, 55–63 CrossRef CAS.
- W. Chen, Y. Zong, Y. Zhou, W. Lu, Y. Zhang and J. Qian, Colloids Surf., A, 2019, 571, 160–167 CrossRef CAS.
- S. Jarczewski, M. Drozdek, P. Michorczyk, C. Cuadrado-Collados, J. Gandara-Loe, J. Silvestre-Albero, L. Lityńska-Dobrzyńska and P. Kuśtrowski, Appl. Surf. Sci., 2020, 504, 144336 CrossRef CAS.
- H. Tamon and H. Ishizaka, J. Colloid Interface Sci., 2000, 223, 305–307 CrossRef CAS.
- W.-C. Li, A.-H. Lu, C. Weidenthaler and F. Schüth, Chem. Mater., 2004, 16, 5676–5681 CrossRef CAS.
- M. Yoshimune, T. Yamamoto, M. Nakaiwa and K. Haraya, Carbon, 2008, 46, 1031–1036 CrossRef CAS.
- K. Z. Gaca and J. Sefcik, J. Colloid Interface Sci., 2013, 406, 51–59 CrossRef CAS.
- S. Purwajanti, H. Zhang, X. Huang, H. Song, Y. Yang, J. Zhang, Y. Niu, A. K. Meka, O. Noonan and C. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 25306–25312 CrossRef CAS.
- X. Qi, X. Yan, W. Peng, J. Zhang, Y. Tong, J. Li, D. Sun, G. Hui and J. Zhang, New J. Chem., 2019, 43, 4698–4705 RSC.
- M. Zare, J. Dispersion Sci. Technol., 2020, 0, 1–8 CAS.
- K. K. Cheralathan, T. Hayashi and M. Ogura, Microporous Mesoporous Mater., 2008, 116, 406–415 CrossRef CAS.
- C. Elmi, S. Guggenheim and R. Gieré, Clays Clay Miner., 2016, 64, 537–551 CrossRef CAS.
- N. Fairley, CasaXPS software package, https://www.casaxps.com Search PubMed.
- M. Mohai, Surf. Interface Anal., 2004, 36, 828–832 CrossRef CAS.
- M. Mohai, XPS MultiQuant: Multi-model X-ray photoelectron spectroscopy quantification program, Version 7.00.92, https://aki.ttk.hu/XMQpages/XMQhome.php Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp., Eden Prairie, Minnesota, USA, 1992 Search PubMed.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble Jr, NIST X-ray Photoelectron Spectroscopy Database, Version 3.4, National Institute of Standards and Technology, Gaithersburg, MD, 2003, https://srdata.nist.gov/xps/ Search PubMed.
- A. R. Gonzalez-Elipe, J. P. Espinos, G. Munuera, J. Sanz and J. M. Serratosa, J. Phys. Chem., 1988, 92, 3471–3476 CrossRef CAS.
- C. D. Wagner, D. E. Passoja, H. F. Hillery, T. G. Kinisky, H. A. Six, W. T. Jansen and J. A. Taylor, J. Vac. Sci. Technol., 1982, 21, 933–944 CrossRef CAS.
- J. H. Kwak, J. Z. Hu, R. V. F. Turcu, K. M. Rosso, E. S. Ilton, C. Wang, J. A. Sears, M. H. Engelhard, A. R. Felmy and D. W. Hoyt, Int. J. Greenhouse Gas Control, 2011, 5, 1081–1092 CrossRef CAS.
- S. Ardizzone, C. L. Bianchi, M. Fadoni and B. Vercelli, Appl. Surf. Sci., 1997, 119, 253–259 CrossRef CAS.
- V. Fournier, P. Marcus and I. Olefjord, Surf. Interface Anal., 2002, 34, 494–497 CrossRef CAS.
- A. Chakradhar and U. Burghaus, Surf. Sci., 2013, 616, 171–177 CrossRef CAS.
- X. Huang, Y. Men, J. Wang, W. An and Y. Wang, Catal. Sci. Technol., 2017, 7, 168–180 RSC.
- J. I. Di Cosimo, V. K. Díez, C. Ferretti and C. R. Apesteguía, in Catalysis, 2014, vol. 26, pp. 1–28 Search PubMed.
- A. Chieregato, J. V. Ochoa, C. Bandinelli, G. Fornasari, F. Cavani and M. Mella, ChemSusChem, 2015, 8, 377–388 CrossRef CAS.
- L. H. Chagas, P. C. Zonetti, C. R. V. Matheus, C. R. K. Rabello, O. C. Alves and L. G. Appel, ChemCatChem, 2019, 11, 5625–5632 CrossRef CAS.
- M. Zhang, R. Li, Y. Wu and Y. Yu, Ind. Eng. Chem. Res., 2021, 60, 2871–2880 CrossRef CAS.
- Z. D. Young and R. J. Davis, Catal. Sci. Technol., 2018, 8, 1722–1729 RSC.
- T. W. Birky, J. T. Kozlowski and R. J. Davis, J. Catal., 2013, 298, 130–137 CrossRef CAS.
- A. S. Ndou, N. Plint and N. J. Coville, Appl. Catal., A, 2003, 251, 337–345 CrossRef CAS.
- E. F. de Souza, H. P. Pacheco, N. Miyake, R. J. Davis and F. S. Toniolo, ACS Catal., 2020, 10, 15162–15177 CrossRef CAS.
- M. J. Climent, A. Corma, V. Fornés, R. Guil-Lopez and S. Iborra, Adv. Synth. Catal., 2002, 344, 1090–1096 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00450j |
| This journal is © The Royal Society of Chemistry 2023 |