
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInstallation of electrophiles onto the C-terminus of recombinant ubiquitin and ubiquitin-like proteins†
Jakob
Farnung
 ,
Kateryna A.
Tolmachova
and
Jeffrey W.
Bode
*
,
Kateryna A.
Tolmachova
and
Jeffrey W.
Bode
*
Laboratorium für Organische Chemie, Department of Chemistry and Applied Biosciences, ETH Zürich, Zürich 8093, Switzerland. E-mail: bode@org.chem.ethz.ch
First published on 15th November 2022
Abstract
Ubiquitin and related ubiquitin-like proteins (Ubls) influence a variety of cellular pathways including protein degradation and response to viral infections. The chemical interrogation of these complex enzymatic cascades relies on the use of tailored activity-based probes (ABPs). Herein, we report the preparation of ABPs for ubiquitin, NEDD8, SUMO2 and ISG15 by selective acyl hydrazide modification. Acyl hydrazides of Ubls are readily accessible by direct hydrazinolysis of Ubl-intein fusions. The suppressed pKa and superior nucleophilicity of the acyl hydrazides enables their selective modification at acidic pH with carboxylic acid anhydrides. The modification proceeds rapidly and efficiently, and does not require chromatographic purification or refolding of the probes. We modified Ubl–NHNH2 with various thiol-reactive electrophiles that couple selectively with E2s and DUBs. The ease of modification enables the rapid generation and screening of ubiquitin probes with various C-terminal truncations and warheads for the selection of the most suitable combination for a given E2 or DUB.
Introduction
Ubiquitin and related ubiquitin-like modifiers (Ubl) are a wide-spread group of protein-based post-translational modifications involved in the regulation of various cellular pathways. Ubls affect protein stability,1 homeostasis,2 localization,3 and protein–protein interactions,4 among other functions. The essential role of ubiquitin and Ubls in cellular regulation and disease states has made them a focus of biological and chemical research.Ubls are typically attached to a lysine residue of protein substrates by an isopeptide-bond. Their conjugation is controlled by an intricate enzymatic cascade consisting of activating (E1), conjugating (E2) and ligating (E3) enzymes. The E1 enzymes activate the conserved C-terminal glycine and transfer Ubls to the corresponding E2s in a trans-thioesterification step. Substrate attachment is effected by an E2 conjugating enzyme, usually in conjunction with an E3 ligase. Ubl attachment is counter-balanced by the action of deubiquitylases (DUB), which cleave specific isopeptide bonds. Ubiquitin and related Ubls share a common beta-grasp structure, also known as the ubiquitin fold, despite little sequence similarity.5 The divergent sequences between Ubls confers specificity to the enzymatic cascades, allowing structurally similar but sequentially distinct Ubls to be processed orthogonally to one another.
Elucidation of the complex Ubl system requires the development of biochemical and chemical tools for its study and manipulation. Of particular utility are activity-based probes that provide precise information about Ubl attachment and removal.6,7 Activity-based probes covalently modify enzymes in their active site,8,9 and can be used to identify unknown enzymes in a specific cascade or, alternatively, as a chemical genetics approach to block pathways through enzyme inhibition.10,11 Cysteine-reactive probes are particularly useful for the interrogation of Ubl pathways because the majority of enzymes involved rely on cysteines for their function. E1s, E2s and certain E3s, which form covalent thioester intermediates with the Ubl C-terminus, are ideally suited for interrogation by these methods. Likewise, the majority of DUBs are cysteine-dependent proteases targetable by activity-based probes.12
Activity-based probes for the ubiquitin-like pathways are typically constructed by the installation of thiol-specific electrophiles on to the C-terminus of Ubls. This approach was pioneered by Pickart and Rose, who reported Ub–aldehyde as the first known Ub-ABP.13 Since this initial report two leading strategies have emerged for probe synthesis. Ploegh and Ovaa have generated ubiquitin electrophiles by direct aminolysis of expressed protein–thioesters or chemically synthesized proteins (Fig. 1a).14–16 In an alternative approach developed by Brik, chemically synthesized dehydroalanine (Dha) containing ubiquitin probes are accessed by native chemical ligation to investigate chain specific activity of DUBs.17–19 All of these methods generally require either HPLC purification or refolding of the Ubl-probe, in part due to long reaction times and poor conversions. A common feature of these strategies is the replacement of C-terminal Ubl residues with the probe to conserve the atomic register at the C-terminus, which is crucial for the productive recognition of a probe by its target enzyme. Probes such as Ub–propargylamine or Ub–Dha have been instrumental in the investigation of Ubl pathways and have contributed to our understanding of this complex biological network.20
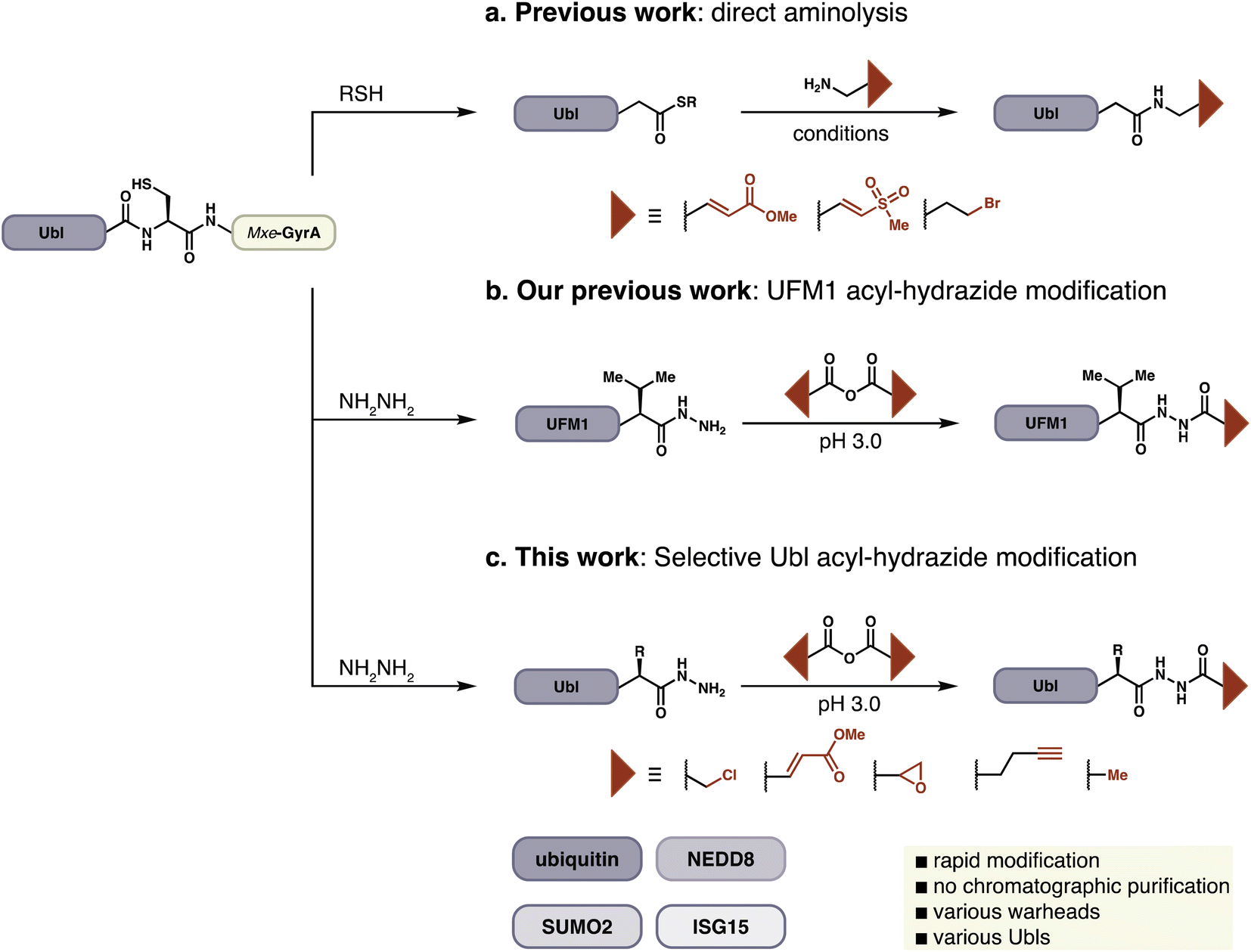 | ||
| Fig. 1 Preparation of Ubls with C-terminal warheads from Ubl-intein fusion proteins. (a) Previously reported approach by direct aminolysis of C-terminal thioesters by amine-functionalized electrophiles. Numerous conditions have been reported. (b) Previously reported modification of UFM1 acyl hydrazides by selective hydrazide acylation. (c) Selective modification of Ubl acyl hydrazides with various thiol-reactive electrophiles by acylation. | ||
In the course of preparing ABPs for UFMylation we faced difficulties with established approaches due to the presence of a sterically-demanding Val as the penultimate C-terminal residue of UFM1.21 We could, however, readily prepare the corresponding C-terminal acyl hydrazide from a UFM1-Mxe GyrA intein fusion and identified facile conditions for the site-specific attachment of electrophiles to the distal N-atom of the hydrazide, a process that could be conducted on the folded protein without the need for purification other than buffer exchange (Fig. 1b). The convenience of this protocol inspired us to extend it to other ABPs derived from Ub and Ubls, and we now report the preparation of >25 probes with a variety of electrophilic warheads from recombinantly expressed protein modifiers including Ub, SUMO2, NEDD8, and ISG15.
Results and discussion
Peptide hydrazides are commonly used as precursors for peptide-thioesters in the chemical synthesis of proteins by native chemical ligation.22 Hydrazides can be selectively activated by oxidation in aqueous acidic solution and displaced by thiols to form the corresponding thioester. Alternatively, acyl hydrazides have been used as key functional groups in hydrazone formation for protein modification.23,24 Likewise, hydrazine and hydrazone linkages have been used to introduce and study post-translational modifications.23,25 Our approach relies on the unique pKa of acyl hydrazides, enabling them to act as nucleophiles at a pH at which other nucleophiles including thiols, amines, and alcohols remain unreactive.26 We envisioned extension of this approach to full-length protein-derived activity-based probes beyond UFM1, by employing a chemoselective hydrazide modification platform (Fig. 1c).In order to extend this convenient method for the installation of electrophilic warheads onto the C-terminus of recombinant proteins other than UFM1, we first evaluated a variety of reaction conditions and activated carboxylic acids on model peptides. Carboxylic acid anhydrides displayed better reactivity and selectivity compared to NHS-esters, and various electrophilic amino acids were suitable for these reaction conditions (Fig. S1†).
With these results in hand, Ub(ΔGG) and Ub(ΔG76) were obtained in milligram quantities by hydrazinolysis of ubiquitin-Mxe GyrA intein fusion.27 The intein fusions were directly displaced by treatment with excess hydrazine to obtain the corresponding acyl hydrazides. By expressing ubiquitin as a C-terminal glycine deletion, ΔGG or ΔG76, the acyl hydrazide can function as an isostere of glycine and conserve the atomic register of the native C-terminus. Treatment of 100 μM Ub(ΔG76)–NHNH2 with 200-fold excess α-chloroacetic anhydride at pH 3.0 provided mono-acylated ubiquitin 2, as determined by LC-MS analysis (Fig. 2a and b). Complete conversion was observed within minutes, and prolonged reaction times did not affect the outcome. Modification with the corresponding NHS-ester led to only 60% conversion as judged by LC-MS analysis. No acylation was observed when Ub(ΔG76) with a C-terminal carboxylic acid was treated with α-chloroacetic anhydride, highlighting the selectivity of the reaction (Fig. 2b). As expected from previous work on hydrazide modification, the selectivity of the reaction was strictly dependent on pH; raising the pH from 3.0 to 6.0 leads to double and triple acylation products (Fig. S2†).28 This highlights again the superior nucleophilicity of acyl hydrazides at acidic pH even in the context of a full-length protein. We further confirmed the site-selectivity by digestion of acylated ubiquitin 2 with Asp-N endoprotease and tandem mass spectrometry (Fig. 2c).
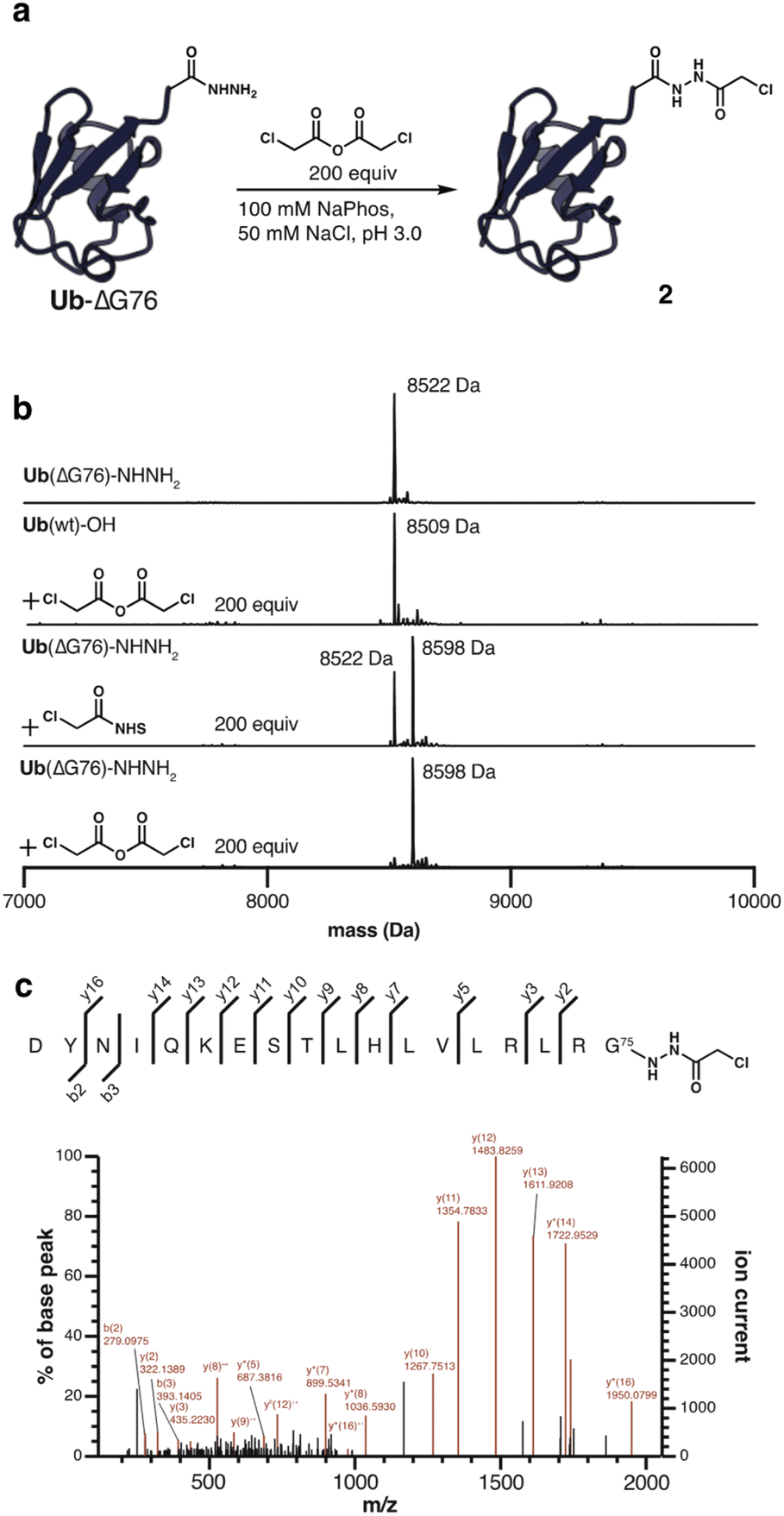 | ||
| Fig. 2 Ubiquitin(ΔG76) hydrazide is selectively modified by α-chloroacetic anhydride. (a) Ub(ΔG76) hydrazide (100 μM) was treated with α-chloroacetic anhydride at pH 3.0. (b) Deconvoluted MS spectra (ESI) of untreated Ub(ΔG76)–NHNH2, Ub(ΔG76)–OH, Ub(ΔG76)–NHNH2 treated with NHS ester, Ub(ΔG76)–NHNH2 treated with carboxylic acid anhydride. Complete conversion of Ub(ΔG76)–NHNH2 to the mono-acylated product 2 was observed upon treatment with anhydride of α-chloroacetic acid. Wild-type ubiquitin with a C-terminal carboxylic acid shows no reaction with α-chloroacetic anhydride under identical conditions. Calculated mass for Ub(ΔG76)–NHNH2 8522 Da, Ub(ΔG76)–OH 8508 Da, Ub(ΔG76)–NHNH α-chloroacetyl 8598 Da. (c) Tandem MS/MS spectrum of modified ubiquitin 2 after Asp-N digestion. Measured parent ion mass 2244.1771 Da, expected parent ion mass 2244.1894 Da. y2 measured m/z 322.1454, expected m/z 322.1389. | ||
Having established the utility of hydrazide acylation for electrophile attachment we investigated additional electrophiles that could be attached to Ub(ΔG76) hydrazide. Beyond α-chloroacetic acid we could attach methyl fumarate, glycidic acid29 and pentynoic acid. The pentynoic moiety was attached to mimic the reactivity of commonly employed propargylamine DUB-probes.30,31 Treatment with acetic anhydride provided the unreactive acetylated hydrazide, which functions as a negative control (Fig. 3a). The amount of anhydride required for each anhydride to achieve full conversion varied (Table S2†). For example, acetylation could be achieved with as little as 10 equivalents of acetic anhydride.
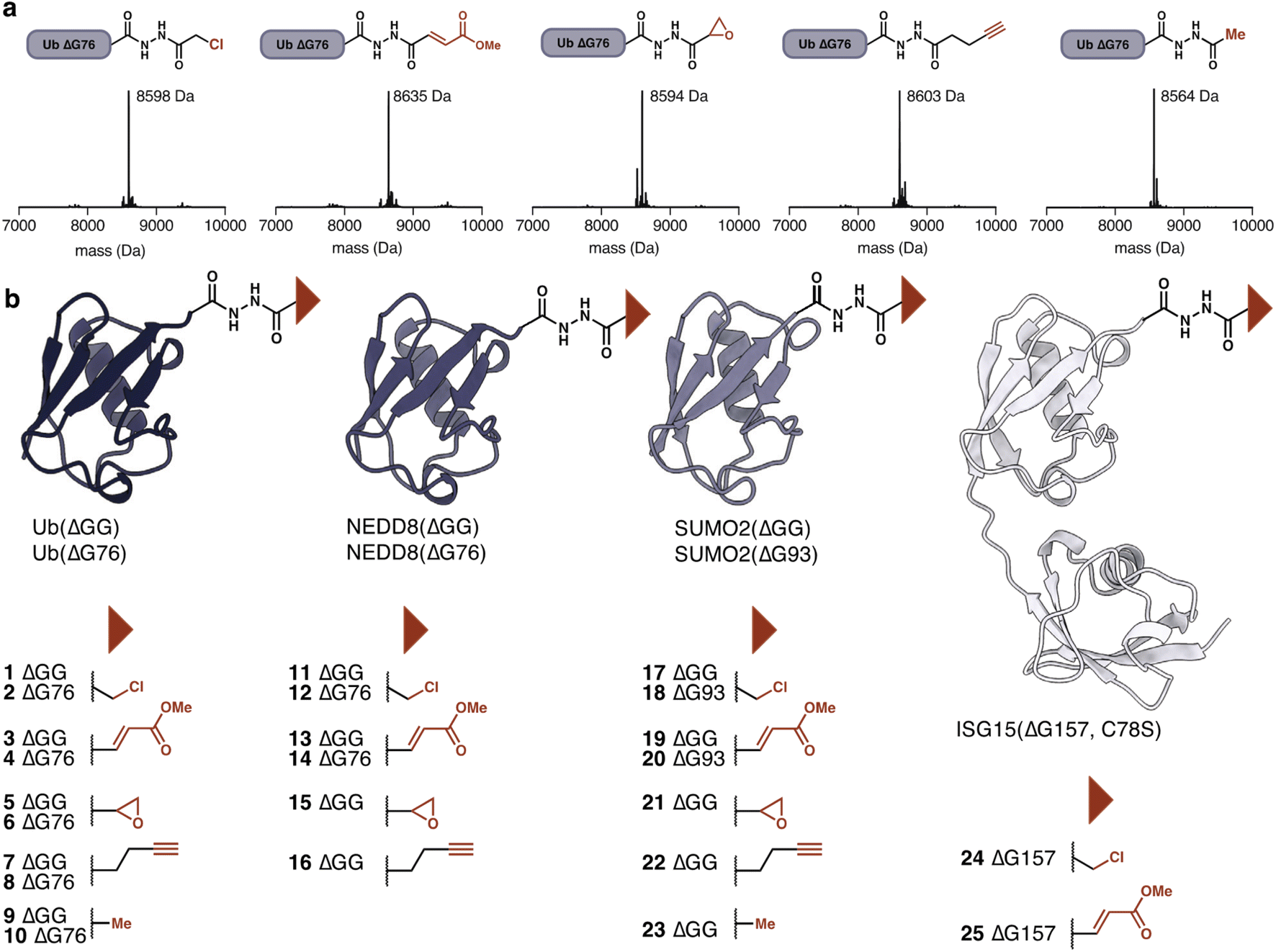 | ||
| Fig. 3 (a) Ubiquitin (ΔG76)–NHNH2 was modified with thiol-reactive warheads. Deconvoluted MS (ESI) spectra are shown. Calculated mass for Ub(ΔG76)–NHNH α-chloroacetyl; 8598 Da, Ub(ΔG76)–NHNH methyl fumarate; 8634 Da, Ub(ΔG76)–NHNH glycidate; 8592 Da, Ub(ΔG76)–NHNH pentynoate; 8602 Da, Ub(ΔG76)–NHNH acetyl; 8564 Da. (b) Ubls modified by hydrazide acylation are shown. Different C-terminal truncations were used for ubiquitin, NEDD8 and SUMO2. For ISG15 the C78S variant was employed. Electrophiles attached to each Ubl variant are shown below each Ubl. | ||
The ease of modification suggested that other ubiquitin-like modifiers should also be amenable to hydrazide modification to provide access to Ubl-probes. Beyond Ub(ΔG76, ΔGG), we prepared acyl hydrazides for NEDD8(ΔG76, ΔGG), SUMO2(ΔG93, ΔGG), and ISG15(ΔG157) (Fig. 3b). NEDD8 and SUMO2 probes were prepared using the general procedure developed for ubiquitin. The hydrazinolysis condition for ISG15(ΔG157) required optimization as ISG15 precipitated during intein cleavage. To circumvent this problem, we reduced the amount of hydrazine monohydrate to 25 mM and added 5 mM of MESNa to facilitate intein cleavage. Using these conditions ISG15(ΔG157)–NHNH2 was obtained. With the conditions optimized for Ub(ΔG76), we selectively modified these additional Ubl-hydrazides. The modifications proceeded efficiently and independently of the C-terminal residue, highlighting the generality of our acylation approach. In contrast, the success of aminolysis protocols varies based on the nature of the C-terminal residue.32 Following completion of the reaction, excess anhydride and carboxylic acid were removed by buffer exchange using dialysis or size exclusion chromatography. No further purification was required. The modified proteins remained stable and folded throughout the workflow, as determined by CD spectroscopy of the Ubl-hydrazide and the modified Ubls (Fig. S3–S6†). The probes were stored at −80 °C and were stable for weeks.
To test the activity of these probes with nucleophilic enzymes, we performed cross-linking experiments of the Ub, NEDD8 and SUMO2 probes with DUB and E2 enzymes. Activity-based probes have garnered special attention in the study of thiol-dependent DUBs because of their enhanced reactivity and use in structural33 and proteomic studies.34 SUMO2(ΔGG) probes (60 μM) were allowed to react with SENP1 (15 μM, catalytic domain) at 37 °C for 1 h. Probes 17 and 19 displayed excellent reactivity and selectivity with SENP1. 21 and 22 reacted less efficiently but are nevertheless selective for the catalytic cysteine of SENP1; no reaction was observed with catalytically incompetent SENP1 variant C603A. As expected, control probe SUMO2(ΔGG) acetate 23 did not react (Fig. 4a). We detected no cross-reactivity of SUMO2 probes with either ubiquitin, USP21, or NEDD8, SENP8, DUBs (Fig. 4b). SUMO2(ΔG93) probes 18 and 20 showed similar behavior to the SUMO2(ΔGG) probes with DUBs (Fig. S7†). Methyl fumarate probe 20 showed reduced reactivity, likely due to increased flexibility of the longer C-terminal tail upon SENP1 binding and protrusion from the SENP1 binding pocket.35
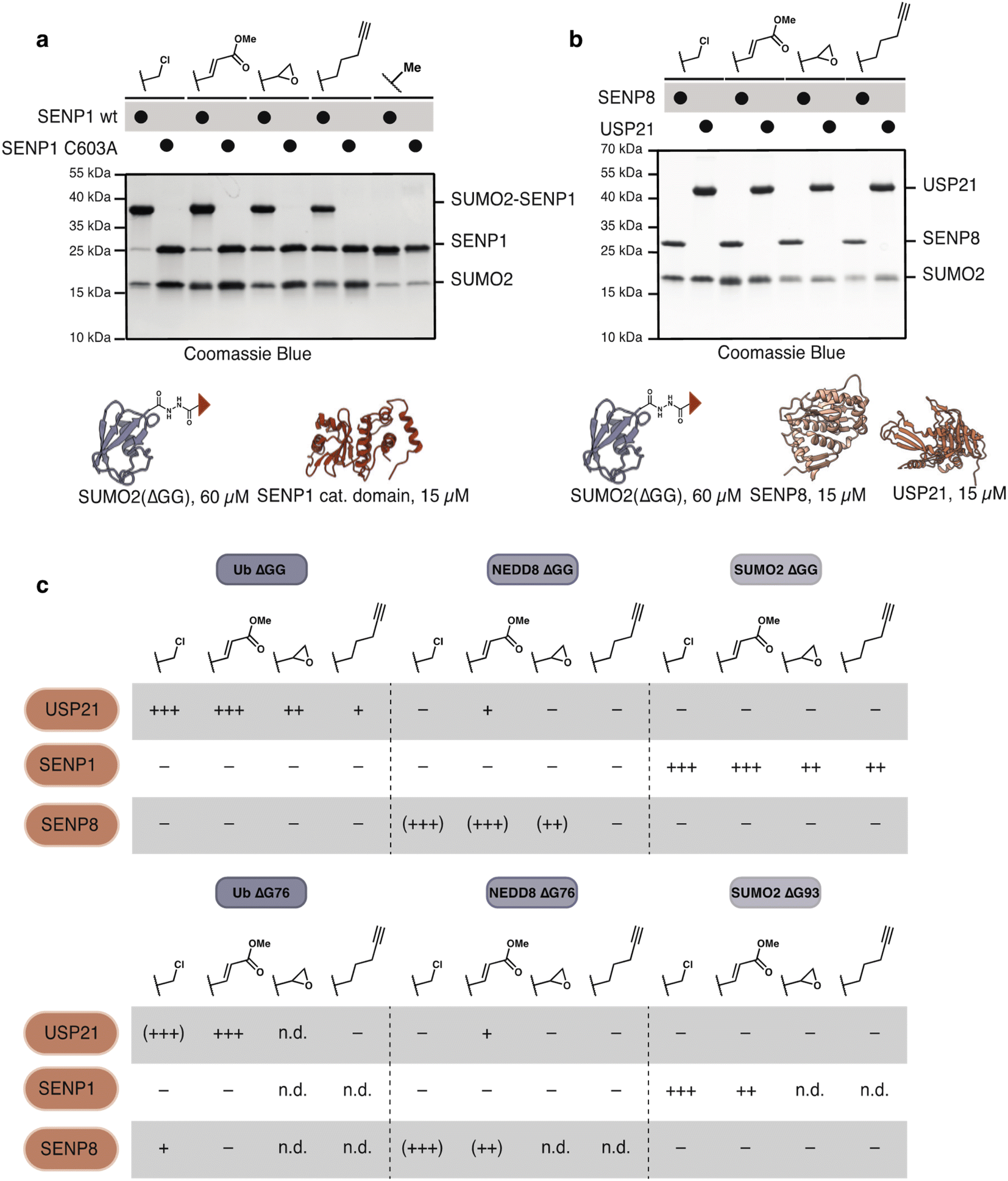 | ||
| Fig. 4 Ubl-hydrazide derived probes show selective modification of DUBs (a) and (b) SUMO2(ΔGG) probes (60 μM) react with (a) SENP1 catalytic domain (15 μM) in a catalytic-cysteine dependent manner and show no reaction with (b) SENP8 (15 μM) or USP21 (15 μM). (c) Table illustrating the reactivity of Ub(ΔGG, ΔG76), NEDD8(ΔGG, ΔG76) and SUMO2(ΔGG, ΔG93) probes with USP21, SENP1 and SENP8. Reactivity table derived from gels shown in (a, b) and Fig. S7–S11.† − indicates no reaction. + indicates degree of reactivity with ++ and +++ indicating higher reactivity. Parentheses indicate non-specific reactivity with Cys–Ala mutants. n.d. stands for not-determined. | ||
Likewise, Ub(ΔGG) probes reacted cleanly with a DUB, USP21, but not with the SUMO specific proteases SENP1 or NEDD8 specific protease SENP8 (Fig. S8†). Reaction of Ub(ΔG76) probes 2, 4 showed more unspecific reactivity with USP21 compared to Ub(ΔGG) α-chloroacetyl and methyl fumarate probes as cross-linking with USP21 C221A and SENP8 was observed. Ub(ΔG76) pentynoate probe 8 did not react with USP21 (Fig. S9†). This finding is in agreement with studies on Ub–propargylamine probes in which homologation of the alkyl chain between the propargyl electrophile and the ubiquitin C-terminus decreased probe reactivity.31 In contrast, most NEDD8-derived probes labelled SENP8 independently of the catalytic cysteine while the NEDD8(ΔGG) alkyne probe 16 did not react with SENP8 at all. This unspecific labelling is likely due to the presence of other cysteines close to the binding site of NEDD8 on SENP8.36
Despite the unselective modification of SENP8, NEDD8(ΔGG) probes showed no reaction with SENP1 (Fig. S10†). Minor cross-linking with USP21 was observed, likely due to USP21's known weak cross-reactivity with NEDD8.37,38 Similar to our observation for SUMO2 probes, NEDD8(ΔG76) probes 12 and 14 showed little difference in their reactivity compared to NEDD8(ΔGG) probes with 14 showing slightly depressed reactivity (Fig. S11†), mimicking the observation made for SUMO2(ΔG93) probe 20. Overall, our probes show excellent specificity for their cognate DUBs (Fig. 4c).
DUBs and related proteases are known to be far more active than E2 conjugating enzymes, and we expected that our probes would be less reactive towards these enzymes. We envisioned that modulation of the C-terminal atomic register and variation of the warhead could tune the reactivity to provide selective probes for specific E2s (Fig. 5a).
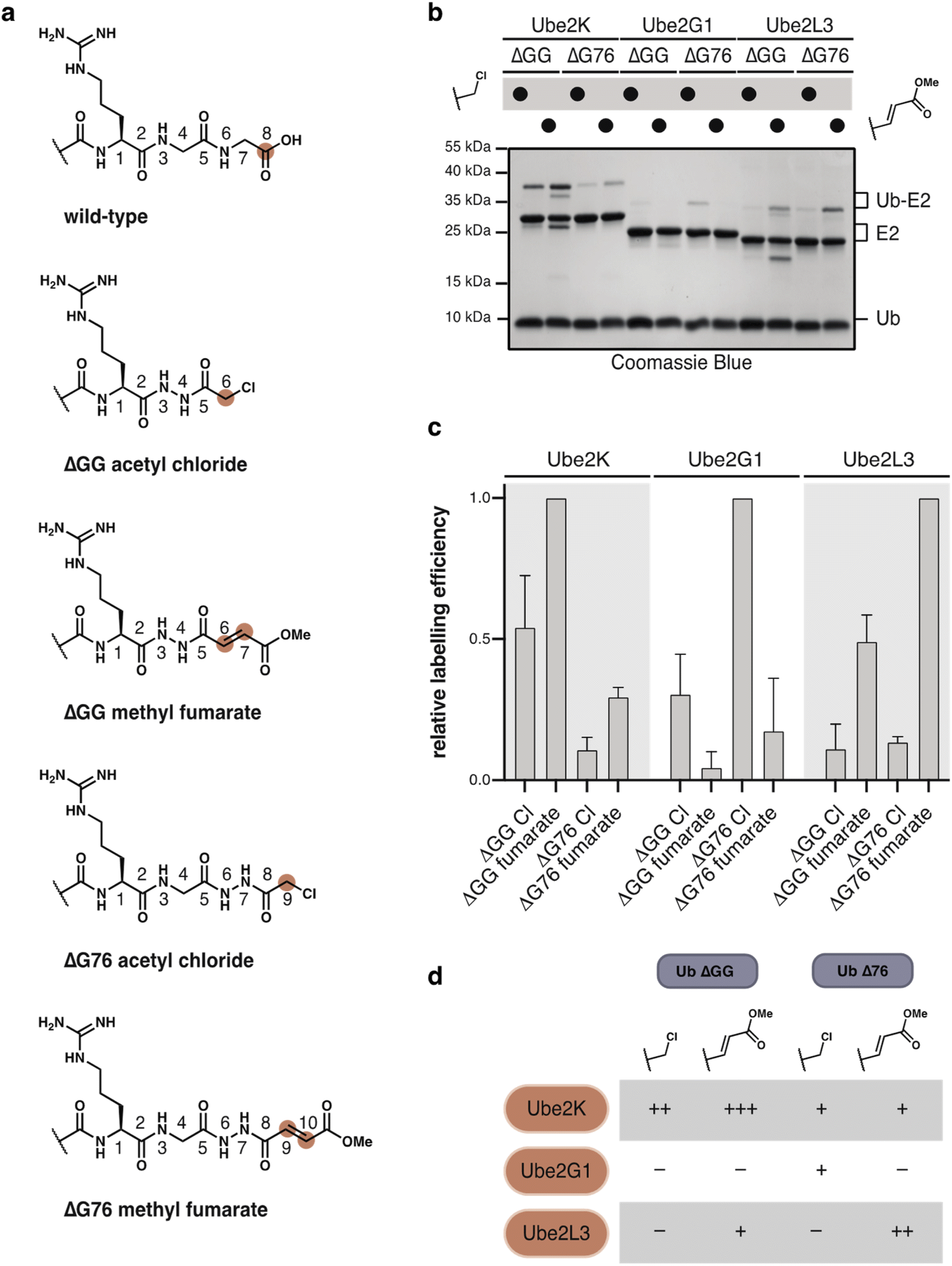 | ||
| Fig. 5 Probe reactivity is tuned by choice of ubiquitin truncation and warhead. (a) The C-terminal atomic register of ubiquitin probes can be altered by choice of ubiquitin deletion and probe. The carbon atom reacting with thiols is highlighted. For methyl fumarate two sites can potentially be attacked. (b) and (c) E2s show distinct reactivity with ubiquitin probes. Ubiquitin probes (100 μM) were allowed to react with E2s (15 μM). Relative labelling efficiency was determined by gel-densitometry of the Ub–E2 band. Efficiency is shown relative to most intense band for each E2. Results are shown as average of three measurements. (d) Table illustrating the comparative reactivity of each probe–E2 pair. Reactivity is indicated relative to Ub(ΔGG) methyl fumarate–Ube2K reaction. − indicates no reaction. + indicates degree of reactivity. | ||
E2 enzymes select specific nucleophiles such as lysine, cysteine or threonine, on their substrate.39–42 As a result the active-site of each E2 adopts a unique geometry, which will likely be reflected in its preference for a specific combination of C-terminal truncation and electrophile.
We first tested Ub(ΔGG) probes 1, 3 and 5 (100 μM) with Ube2K wt and Ube2K C92A (15 μM) at 37 °C. Both α-chloroacetyl 1 and fumarate 3 probes reacted with wild-type Ube2K but not with catalytically inactive Ube2K (Fig. S12a†). Glycidate probe 5 did not react with Ube2K. Ub(ΔGG) probes reacted selectively with Ube2K as no reaction was observed with either Ube2M, or Ubc9 (Fig. S12b†). Inspired by these results we also prepared Ub(ΔG76) α-chloroacetyl 2 and fumarate 4 probes. To test our hypothesis that E2s prefer distinct combinations of probe-type and atomic register we chose model E2s Ube2K, Ube2G1 and Ube2L3. Incubation of excess Ub–probes 1–4 (100 μM) with E2s (15 μM) at 37 °C revealed a distinct labelling profile for each E2 (Fig. 5b, c and S13†). Labelling efficiency and probe preference differed between each E2. Ube2K showed efficient crosslinking with Ub(ΔGG), with methyl fumarate reacting more efficiently than the α-chloroacetyl. In contrast, Ube2G1 showed generally weak labelling but uniquely prefers Ub(ΔG76) α-chloroacetyl probe 2. Ube2L3 preferentially reacted with the methyl fumarate probe 4 but with a distinct selectivity for Ub(ΔG76). This finding highlights that probe design is crucial to labelling efficiency and selectivity (Fig. 5d). Using hydrazide acylation, the ideal combination of deletion and probe type can be quickly screened to identify the most reactive probe design.
As anticipated, SUMO2(ΔGG) and NEDD8(ΔGG) probes also displayed poorer reactivity and diminished selectivity for their respective E2s, Ubc9 and Ube2M. We tested the more reactive α-chloroacetyl, methyl fumarate and glycidate electrophiles for E2 labelling. Higher concentrations of probe (150 μM) and E2 (30 μM) were required to observe any cross-linking. SUMO2(ΔGG) methyl fumarate was the only SUMO2 probe to react with Ubc9, albeit not specifically with C93 (Fig. S14†). NEDD8(ΔGG) α-chloroacetyl and methyl fumarate reacted with Ube2M. The α-chloroacetyl probe showed some unspecific reactivity with Ube2M C111A. However, both probes reacted unspecifically with Ube2K (Fig. S15†), likely due to high probe and enzyme concentration which can enhance probe reactivity. In addition, the high sequence similarity between ubiquitin and NEDD8 can lead to non-cognate interactions and facilitate thiol modification.43
To compare the hydrazide-derived ABPs with previously reported probes accessed by other approaches, we tested the reactivity of Ub(ΔG76) probe 2 and 4 with Ub vinyl methyl ester (VME) and dehydroalanine (Dha) probes. For UbVME we deleted G76 and substituted it with the desired electrophile. For the dehydroalanine probe G76 was mutated to Dha. To evaluate differences in the reactivity of the probes, we incubated them at equimolar concentration (15 μM) with both E2s and DUBs. All three probes reacted with USP21 with Ub G76Dha showing markedly reduced reactivity (Fig. 6a). Probes 2 and 4 also reacted with Ube2K even at these lower concentrations. However, neither Ub–VME nor Ub–Dha cross-linked with Ube2K (Fig. S16†). Cross-linking was observed for Ub–VME and Ub–Dha upon increasing probe concentrations to 60 μM. Nevertheless, even under these conditions the hydrazide probes reacted more efficiently with Ube2K (Fig. 6b). These findings highlight the superior reactivity of hydrazide-probes with even poorly electrophilic substrates such as E2s.
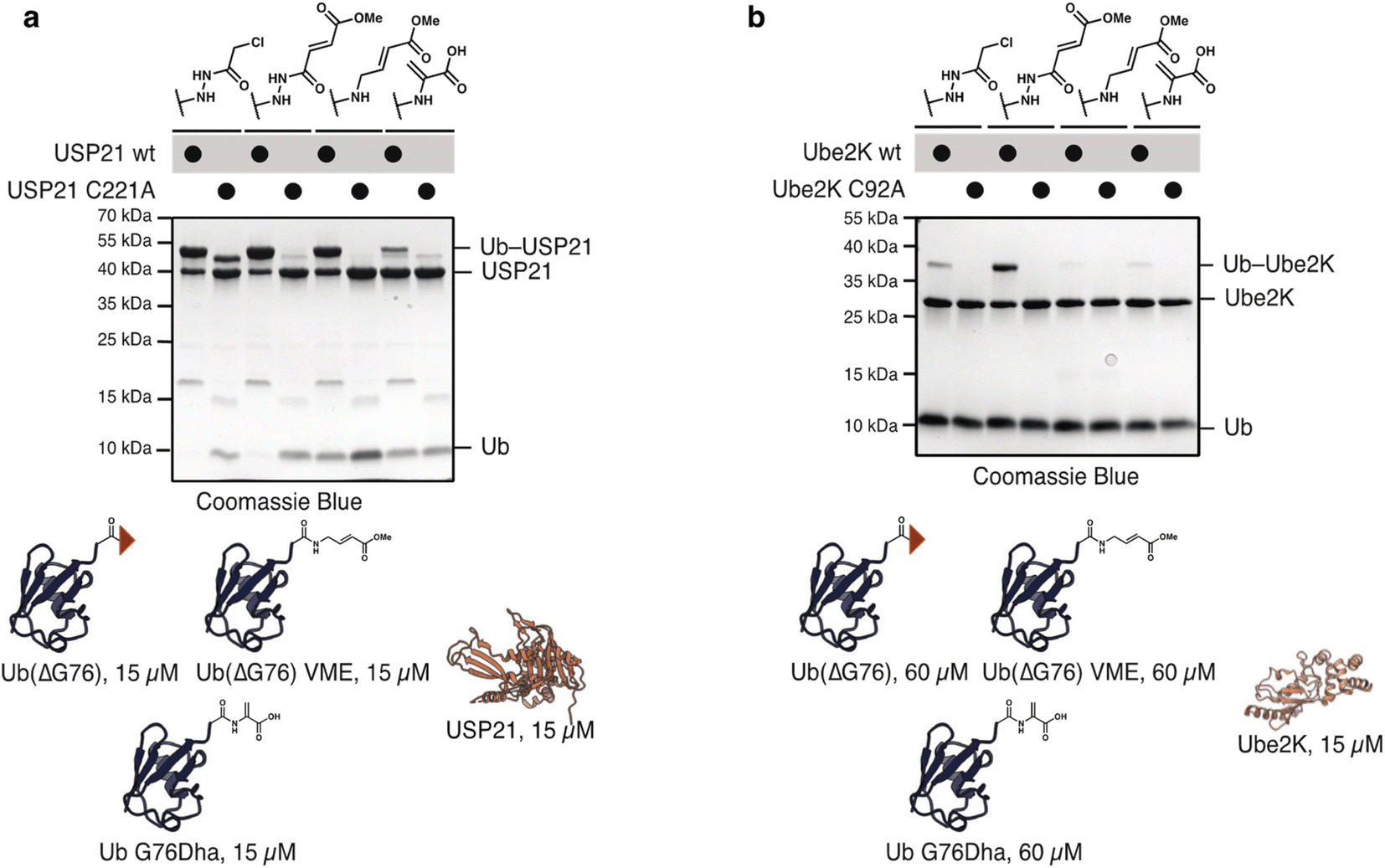 | ||
| Fig. 6 Comparison of hydrazide derived ABPs with state-of-the-art Ub–vinyl methyl ester (VME) probe and Ub dehydroalanine (Dha) probe. (a) Reaction of Ub(ΔG76) α-chloroacetyl 2, methyl fumarate 4, Ub(ΔG76)–VME and Ub G76Dha with USP21. All probes (15 μM) reacted with USP21. 1, 3 and Ub Dha showed some unspecific reactivity with USP21 (b) reaction of Ub(ΔG76) α-chloroacetyl, methyl fumarate, Ub(ΔG76)–VME and Ub G76Dha (15 μM) with Ube2K (15 μM). Only hydrazide derived probes react well with Ube2K. | ||
To confirm the utility of these probes in a more complex setting, we also tested the selective modification of recombinant SENP1 (1 μM) in HEK293T cell lysate with FLAG-SUMO2(ΔGG) probes. The reactivity profile of SUMO2(ΔGG) probes in vitro (Fig. 4a) was also observed in lysates. The α-chloroacetyl and methyl fumarate showed higher reactivity than glycidate and pentynoate (Fig. S17†).
Endogenous SENP1 was also labelled by α-chloroacetyl, methyl fumarate and glycidate probes (Fig. S18†). Visualization of probe reactivity using anti-FLAG immunoblotting showed that these probes displayed pronounced, but disparate reactivity, as evidenced by the appearance of distinct higher molecular weight bands for each probe. This difference in reactivity highlights again how the unique chemical properties of our probes lead to unique labelling profiles.
Conclusions
In summary, we demonstrated the chemoselective, site-specific attachment of electrophilic warheads to recombinantly produced C-terminal protein acyl hydrazides. The selective modification of the protein hydrazides is enabled by rapid (within seconds), selective coupling of anhydrides at acidic pH; the resulting probes require no further purification or refolding and offer a straightforward approach for the preparation of Ub and Ubl-derived ABPs for covalent crosslinking with E2s and DUBs involved in key biological pathways. For example, SUMO2(ΔGG)–NHNH2 derived probes cross-link with deSUMOylase SENP1 with excellent reactivity and no cross-reactivity with a DUB or deNEDDylase in vitro and in complex environments such as lysates. E2s generally show poor reactivity with ABPs. However, using a screen of C-terminal truncations and electrophiles we identified combinations with enhanced reactivity for specific E2s. For example, Ub(ΔG76) α-chloroacetyl, is the single combination which shows crosslinking with Ube2G1. This approach to C-terminal protein modification will facilitate access to activity-based probes beyond ubiquitin and expand the repertoire of expressed protein modification for applications in chemical biology.Data availability
All data is available in the manuscript and ESI.† Uncropped gel and blot images are shown in the ESI.† Further information is available from the authors upon reasonable request.Author contributions
J. F., K. A. T. and J. W. B. conceived the project. J. F. and K. A. T. performed all experiments. J. F., K. A. T. and J. W. B. analyzed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation (No. 188634) and a European Research Council Synergy Grant under the European Union's Horizon 2020 Research and Innovation Programme (No. 856581-CHUbVi). We acknowledge the Molecular and Biomolecular Analysis Service (MoBiAS) of the Department of Chemistry and Applied Biosciences at ETH Zürich for mass spectrometry. We thank B. Schulman for helpful discussions.References
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef CAS PubMed.
- N. Chondrogianni, I. Petropoulos, S. Grimm, K. Georgila, B. Catalgol, B. Friguet, T. Grune and E. S. Gonos, Mol Aspects Med, 2014, 35, 1–71 CrossRef CAS PubMed.
- Z. J. J. Chen and L. J. J. Sun, Mol. Cell, 2009, 33, 275–286 CrossRef CAS PubMed.
- E. J. Worden, X. Zhang and C. Wolberger, Elife, 2020, 9, e53199 CrossRef CAS PubMed.
- L. Cappadocia and C. D. Lima, Chem. Rev., 2018, 118, 889–918 CrossRef CAS PubMed.
- L. T. Henneberg and B. A. Schulman, Cell Chem. Biol., 2021, 28, 889–902 CrossRef CAS PubMed.
- X. Sui, Y. Wang, Y. X. Du, L. J. Liang, Q. Zheng, Y. M. Li and L. Liu, Chem. Sci., 2020, 11, 12633–12646 RSC.
- M. Bogyo, S. Shin, J. S. McMaster and H. L. Ploegh, Chem. Biol., 1998, 5, 307–320 CrossRef CAS PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS PubMed.
- S. Arastu-Kapur, E. L. Ponder, U. P. Fonovic, S. Yeoh, F. Yuan, M. Fonovic, M. Grainger, C. I. Phillips, J. C. Powers and M. Bogyo, Nat. Chem. Biol., 2008, 4, 203–213 CrossRef CAS PubMed.
- K. Shah, Y. Liu, C. Deirmengian and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3565–3570 CrossRef CAS PubMed.
- T. E. T. Mevissen and D. Komander, Annu. Rev. Biochem., 2017, 86, 159–192 CrossRef CAS PubMed.
- C. M. Pickart and I. A. Rose, J. Biol. Chem., 1986, 261, 210–217 CrossRef.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS PubMed.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed.
- A. Borodovsky, B. M. Kessler, R. Casagrande, H. S. Overkleeft, K. D. Wilkinson and H. L. Ploegh, EMBO J., 2001, 20, 5187–5196 CrossRef CAS PubMed.
- N. Haj-Yahya, H. P. Hemantha, R. Meledin, S. Bondalapati, M. Seenaiah and A. Brik, Org. Lett., 2014, 16, 540–543 CrossRef CAS PubMed.
- R. Meledin, S. M. Mali, O. Kleifeld and A. Brik, Angew. Chem., Int. Ed. Engl., 2018, 57, 5645–5649 CrossRef CAS PubMed.
- S. D. Whedon, N. Markandeya, A. S. J. B. Rana, N. A. Senger, C. E. Weller, F. Turecek, E. R. Strieter and C. Chatterjee, J. Am. Chem. Soc., 2016, 138, 13774–13777 CrossRef CAS PubMed.
- M. P. C. Mulder, K. Witting, I. Berlin, J. N. Pruneda, K. P. Wu, J. G. Chang, R. Merkx, J. Bialas, M. Groettrup, A. C. O. Vertegaal, B. A. Schulman, D. Komander, J. Neefjes, F. El Oualid and H. Ovaa, Nat. Chem. Biol., 2016, 12, 523–530 CrossRef CAS PubMed.
- K. A. Tolmachova, J. Farnung, J. R. Liang, J. E. Corn and J. W. Bode, ACS Cent. Sci., 2022, 8(6), 756–762 CrossRef CAS PubMed.
- G. M. Fang, J. X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS PubMed.
- D. K. Kolmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef CAS PubMed.
- K. Zuberbuhler, G. Casi, G. J. Bernardes and D. Neri, Chem. Commun., 2012, 48, 7100–7102 RSC.
- B. C. R. Dancy, S. A. Ming, R. Papazyan, C. A. Jelinek, A. Majumdar, Y. Sun, B. M. Dancy, W. J. Drury, R. J. Cotter, S. D. Taverna and P. A. Cole, J. Am. Chem. Soc., 2012, 134, 5138–5148 CrossRef CAS PubMed.
- C. R. Lindegren and C. Niemann, J. Am. Chem. Soc., 1949, 71, 1504 CrossRef CAS.
- J. Kalia and R. T. Raines, Chembiochem, 2006, 7, 1375–1383 CrossRef CAS PubMed.
- D. Bonnet, C. Rommens, H. Gras-Masse and O. Melnyk, Tetrahedron Lett., 2000, 41, 45–48 CrossRef CAS.
- D. Greenbaum, K. F. Medzihradszky, A. Burlingame and M. Bogyo, Chem. Biol., 2000, 7, 569–581 CrossRef CAS PubMed.
- E. Mons, R. Q. Kim, B. R. van Doodewaerd, P. A. van Veelen, M. P. C. Mulder and H. Ovaa, J. Am. Chem. Soc., 2021, 143, 6423–6433 CrossRef CAS PubMed.
- R. Ekkebus, S. I. van Kasteren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. R. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 CrossRef CAS PubMed.
- R. Y. P. Lue, G. Y. J. Chen, Y. Hu, Q. Zhu and S. Q. Yao, J. Am. Chem. Soc., 2004, 126, 1055–1062 CrossRef CAS PubMed.
- D. A. Boudreaux, T. K. Maiti, C. W. Davies and C. Das, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9117–9122 CrossRef CAS PubMed.
- W. J. Gui, C. A. Ott, K. Yang, J. S. Chung, S. Q. Shen and Z. H. Zhuang, J. Am. Chem. Soc., 2018, 140, 12424–12433 CrossRef CAS PubMed.
- L. Shen, M. H. Tatham, C. Dong, A. Zagorska, J. H. Naismith and R. T. Hay, Nat. Struct. Mol. Biol., 2006, 13, 1069–1077 CrossRef CAS PubMed.
- D. Reverter, K. Wu, T. G. Erdene, Z. Q. Pan, K. D. Wilkinson and C. D. Lima, J. Mol. Biol., 2005, 345, 141–151 CrossRef CAS PubMed.
- Y. Ye, M. Akutsu, F. Reyes-Turcu, R. I. Enchev, K. D. Wilkinson and D. Komander, EMBO Rep., 2011, 12, 350–357 CrossRef CAS PubMed.
- L. M. Gong, T. Kamitani, S. Millas and E. T. H. Yeh, J. Biol. Chem., 2000, 275, 14212–14216 CrossRef CAS PubMed.
- J. Liwocha, D. T. Krist, G. J. V. van Noort, F. M. Hansen, V. H. Truong, O. Karayel, N. Purser, D. Houston, N. Burton, M. J. Bostock, M. Sattler, M. Mann, J. S. Harrison, G. Kleiger, H. Ovaa and B. A. Schulman, Nat. Chem. Biol., 2021, 17, 272–279 CrossRef CAS PubMed.
- M. C. Rodrigo-Brenni, S. A. Foster and D. O. Morgan, Mol. Cell, 2010, 39, 548–559 CrossRef CAS PubMed.
- D. M. Wenzel, A. Lissounov, P. S. Brzovic and R. E. Klevit, Nature, 2011, 474, 105–U136 CrossRef CAS PubMed.
- X. Wang, R. A. Herr, M. Rabelink, R. C. Hoeben, E. J. Wiertz and T. H. Hansen, J. Cell Biol., 2009, 187, 655–668 CrossRef CAS PubMed.
- O. Leidecker, I. Matic, B. Mahata, E. Pion and D. P. Xirodimas, Cell Cycle, 2012, 11, 1142–1150 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04279g |
| This journal is © The Royal Society of Chemistry 2023 |