
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEquilibrating parent aminomercaptocarbene and CO2 with 2-amino-2-thioxoacetic acid via heavy-atom quantum tunneling†
Bastian
Bernhardt‡
 ,
Markus
Schauermann‡
,
Ephrath
Solel
,
André K.
Eckhardt§
and
Peter R.
Schreiner
*
,
Markus
Schauermann‡
,
Ephrath
Solel
,
André K.
Eckhardt§
and
Peter R.
Schreiner
*
Institute of Organic Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: prs@uni-giessen.de
First published on 17th November 2022
Abstract
The search for methods to bind CO2 and use it synthetically as a C1-building block under mild conditions is an ongoing endeavor of great urgency. The formation of heterocyclic carbene–carbon dioxide adducts occurs rapidly when the carbene is generated in solution in the presence of CO2. Here we demonstrate the reversible formation of a complex of the hitherto unreported aminomercaptocarbene (H2N–![[C with combining umlaut]](https://www.rsc.org/images/entities/char_0043_0308.gif) –SH) with CO2 isolated in solid argon by photolysis of 2-amino-2-thioxoacetic acid. Remarkably, the complex disappears in the dark as deduced by time-dependent matrix infrared measurements, and equilibrates back to the covalently bound starting material. This kinetically excluded process below ca. 8 K is made possible through heavy-atom quantum mechanical tunneling, as also evident from density functional theory and ab initio computations at the CCSD(T)/cc-pVTZ level of theory. Our results provide insight into CO2 activation using a carbene and emphasize the role of quantum mechanical tunneling in organic processes, even involving heavy atoms.
–SH) with CO2 isolated in solid argon by photolysis of 2-amino-2-thioxoacetic acid. Remarkably, the complex disappears in the dark as deduced by time-dependent matrix infrared measurements, and equilibrates back to the covalently bound starting material. This kinetically excluded process below ca. 8 K is made possible through heavy-atom quantum mechanical tunneling, as also evident from density functional theory and ab initio computations at the CCSD(T)/cc-pVTZ level of theory. Our results provide insight into CO2 activation using a carbene and emphasize the role of quantum mechanical tunneling in organic processes, even involving heavy atoms.
Introduction
Incorporating CO2 in industrial synthesis for basic chemicals, drugs, or fuels is a key goal in sustainable chemistry1,2 and much effort has been devoted to develop CO2 activating systems: besides frustrated Lewis pairs,3 ionic liquids,4 superbases,5 poly-oxometallates,6 and phosphorus ylides,7 N-heterocyclic carbenes (NHCs) can be used to activate CO2.8,9 Such carbenes form carboxylates that can be further used for synthetic applications, e.g., for carbene catalyzed carba-, sulfa-, and phospha-Michael additions10 or as catalysts for other carbene-promoted CO2 fixation reactions,11–16e.g., as organic carbonates.17 Azolium carboxylates are remarkably stable,18 because intramolecular neutralization even through transfer of the R3-group does not occur (Scheme 1).19 The zwitterionic structure, however, also implies that charge separation has to occur in the attack of the carbene on the highly unreactive carbon of CO2. NHC-carboxylates can be used as carbene-precursors when CO2 is thermally extruded in situ. Some decarboxylation reactions can be achieved photochemically, e.g., acetic acid extrudes CO2 when irradiated with UV light.20 Photodecarboxylation has also been used for synthetic applications.21 The photolysis of thiazol-2-carboxylic acid and imidazole-2-carboxylic acid results in the formation of carbene–CO2 complexes, but the reverse reaction has not been reported.22,23 For various C(sp3) carboxylates 13CO2 exchange in solution was reported recently and carbon nucleophiles and enolates were postulated as intermediates.24 Our results suggest the presence of carbene–CO2 complexes in such reactions.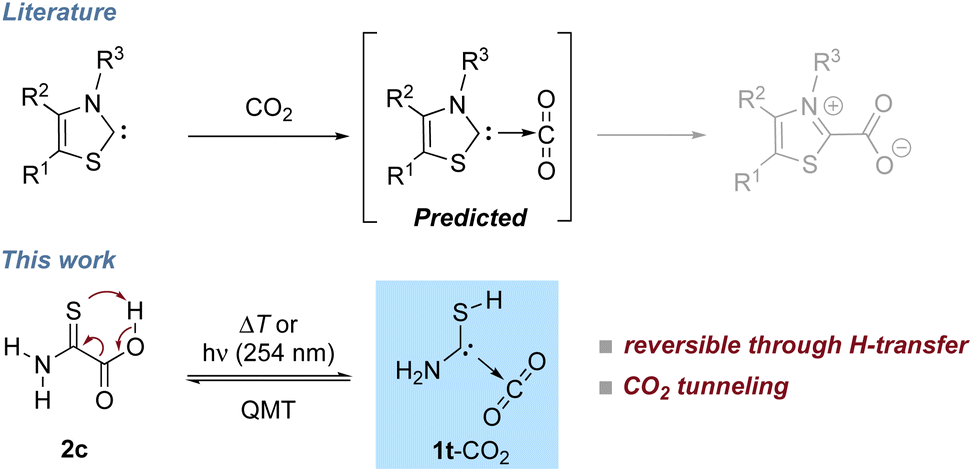 | ||
| Scheme 1 In solution NHCs add to CO2 to form carboxylate zwitterions (top, gray). In the present study 2c photodecarboxylates to give 1t-CO2. This hetero-ene-type reaction is reversible under cryogenic conditions and dominated by QMT below ca. 8 K indicating non-covalent bound complexes as intermediates of the carboxylate formation. | ||
Here we present the preparation and reaction of a novel carbene, namely aminomercaptomethylene (1, H2N––SH) in its complex with CO2 (1-CO2) that reacts back to 2-amino-2-thioxoacetic acid (2) under cryogenic conditions (Scheme 1). The concomitant transfer of a proton onto the CO2 moiety leads to a neutral system instead of a zwitterion as in case of the azolium carboxylates, and this process is associated with a very low barrier.
Additionally, we demonstrate that the association reaction is accelerated through heavy-atom quantum mechanical tunneling (QMT) that opens new possibilities for affecting the reactions of CO2 with carbenes or other nucleophiles. In this context we demonstrate the first evidence for 1, the parent structure of nature's thiazol-2-ylidene active site in, e.g., thiamine (vitamin B1) and pyruvate decarboxylase,25 in its complex with CO2. Coincidently, 1-CO2 resembles a rare example of a spectroscopically characterized member of the family of mercaptocarbenes (R––SH) of which hydroxymercaptomethylene was the first spectroscopically identified member.26 Spectrometric evidence has been reported for parent mercaptomethylene.27,28
Under cryogenic conditions the contribution of QMT to the overall reaction rate is larger compared to ambient conditions and sometimes completely determines the qualitative outcome of a reaction.29–31 While proton tunneling is a rather common feature, heavy-atom QMT is encountered less frequently.32–35 However, sometimes even larger groups are transferred as in the case of trifluoroacetyl nitrene, which reacts to trifluoromethyl isocyanate by transferring the CF3-group in a formal [1,2]-shift via QMT.36
High-vacuum flash pyrolysis (HVFP) of α-keto carboxylic acids gives rise to the corresponding hydroxycarbenes that have been investigated via matrix isolation spectroscopy.31,37–43 Besides their intriguing QMT behavior, some hydroxycarbenes add to carbonyls in nearly barrierless carbonyl–ene reactions.44 The reaction 1t-CO2 → 2c (Scheme 1) resembles another example of this reaction type. In analogy to these studies, we used 2 as the precursor for the generation of 1 complexed with CO2.
Results and discussion
We synthesized 2via saponification of commercially available ethyl thiooxamate (see the ESI and Fig. S1† for details). After deposition of 2 on a CsI window at 3 K together with a large excess of Ar, we exclusively observe its most stable anti-(E)-conformer 2c (Fig. 1). The anti-(Z)-conformer 2t is 4.3 kcal mol−1 higher in energy (CCSD(T)/cc-pVTZ) and, hence, not populated at 3 K. To isolate free aminomercaptomethylene 1 we performed multiple HVFP experiments exposing 2 to pyrolysis temperatures ranging from 300 up to 1150 °C. However, we could not detect even traces of 1 and only observed CO2 and thioformamide together with small amounts of its thiolimine tautomer45 as the pyrolysis products. This result is not completely unexpected as the corresponding carbonyl species (here a thioamide) has been the main pyrolysis product in all our previous studies on the matrix isolation of hydroxycarbenes, which follows similar strategies.29 Furthermore, the computed Gibbs free potential energy hypersurface (PES) of the pyrolysis explains the absence of 1 in such experiments (cf. ESI, Fig. S49†). Fortunately, we were able to isolate the desired carbene 1 complexed with CO2 photochemically as outlined in the following.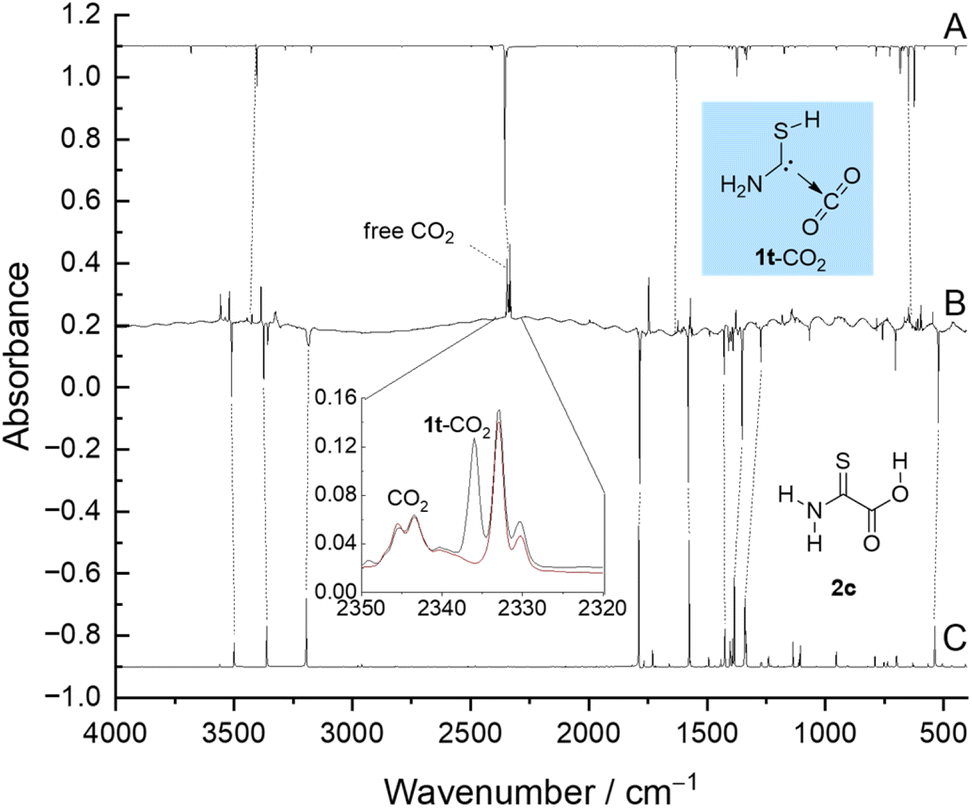 | ||
| Fig. 1 Experimental matrix-IR difference spectrum (B) of spectra measured before and after 4 min of irradiation at 254 nm compared with the anharmonic spectrum of 1t-CO2 (A) and 2c (C) computed at the B3LYP/6-311++G(3df,3pd) level of theory (anharmonic). Increasing bands not assigned here are discussed in the ESI.† (Inset) Spectra recorded before (black) and after (red) keeping the matrix in the dark for 70 h. Other time-dependent band profiles are shown in Fig. S14–S23.† | ||
Upon UV irradiation there are several conceivable reaction paths of 2c. Photoinduced rotamerizations of carboxylic acids46–50 and isomerizations of thioamides to thiolimines51–62 are well known under cryogenic conditions. As 2c contains both of these functionalities, many photoproducts can be envisaged, e.g., the higher lying conformer 2t and 16 conformationally distinct thiolimines, which are 11.4 to 34.4 kcal mol−1 higher in energy than 2c (B3LYP/6-311++G(3df,3pd), see the ESI† for an energetic ranking (Fig. S43†) and experimental data of the rotamerization (Fig. S24†) and tautomerization (Fig. S27†) of 2). Furthermore, we observed the formation of a complex of trans-1 and CO2 (1t-CO2) evidenced by a characteristic matrix infrared (IR) band with maxima at 2336.1, 2333.2, and 2330.4 cm−1 in good agreement with the computed antisymmetric CO2 stretching vibration at 2356.7 cm−1 (B3LYP/6-311++G(3df,3pd), anharmonic) in the complex. Weaker bands at 3444.9 (computed: 3404.6), 1634.8 (1633.0), and 641.8 (625.3) cm−1 can be assigned to 1t-CO2 as well. These bands reach their maximum intensity after 4 min of irradiation at 254 nm (Fig. 1). The assignment is further supported by comparing experimental and computed shifts of perdeuterated 1t-CO2-d3 (ESI, Table S2†).
Much to our surprise, once generated, 1t-CO2 converts back to 2c in the dark. The half-life (t1/2) of this process depends on the matrix site63 and can be derived by monitoring the time-dependent band profile of the antisymmetric CO2 stretching vibration of 1t-CO2. The decay of the maximum at 2336.1 cm−1 yields t1/2 = 26 min (3 K) while the maximum at 2333.2 cm−1 yields t1/2 = 3.8 d (20 K, no reaction at 3 K). The third maximum (2330.4 cm−1) cannot be reliably evaluated due to its small intensity and long half-life. Distinct matrix sites presumably lead to different distances between the two fragments in 1t-CO2, which result in different half-lives. The first value is in excellent agreement with CVT/SCT//B3LYP/6-311+G(d,p) computations yielding t1/2 = 55 min at 3 K for the 1t-CO2 → 2c reaction while the second value agrees well with CVT/SCT//B3LYP/6-311++G(3df,3pd) computations (t1/2 = 7.6 d). The C–C distance in 1t-CO2 is 2.971 Å at the first and 3.005 Å at the latter level of theory; the activation barriers towards 2c are reduced to 1.9 and 2.2 kcal mol−1, respectively. For details on the kinetic analyses see the ESI.†
Even though the computed barrier of 4.2 kcal mol−1 (CCSD(T)/cc-pVTZ) is low, the 1t-CO2 → 2c reaction cannot occur thermally at 3 K, and only QMT explains the experimental observation. To ensure that there is no activation by the spectrometer's light source we repeated the experiment measuring every 5 min while the matrix was not exposed to the spectrometer globar beam between measurements. We also prepared perdeuterated 1t-CO2-d3 whose half-life extends to t1/2 = 36 min (3 K) in the first and t1/2 = 5.1 d (20 K) in the second matrix site (see the ESI† for details). This is in good agreement with CVT/SCT//B3LYP/6-311++G(3df,3pd) computations (t1/2 = 259 min at 3 K, second matrix site: 7.6 d at 20 K). This effect is small (KIE = 1.4 at 3 K, computed: 4.7 at 3 K) owing to the minute movements of the H/D atoms in the QMT process (vide infra). Additionally, we performed kinetic measurements at temperatures between 3 K and 12 K to solidify the QMT mechanism of the reaction 1t-CO2 (2336.1 cm−1) → 2c (Fig. 2). Note that at 20 K we could not detect 1t-CO2 in this matrix site presumably due to its very fast reaction (t1/2 < 1 min).
 | ||
| Fig. 2 Arrhenius plot of the experimental rate constants (k) of the reaction of 1t-CO2 (2336.1 cm−1) to 2c at different temperatures (T; triangles) compared to computed rates (CVT/SCT//B3LYP/6-311+G(d,p); circles). Above ca. 20 K the classic thermal reactivity dominates this reaction (linear curve). Below ca. 8 K (constant values) this reaction can only occur via QMT. | ||
The logarithmic rate vs. inverse temperature plots (Arrhenius plots) of theory and experiment in Fig. 2 agree well with the regions of Arrhenius and non-Arrhenius behavior, underlining our QMT hypothesis. We conclude that at temperatures below ca. 8 K QMT dominates this reaction entirely. Above ca. 20 K the rate grows exponentially since the barrier of 4.2 kcal mol−1 (CCSD(T)/cc-pVTZ) can be overcome thermally. This system allows for measuring of the kinetics up to temperatures that are in the transition range between the QMT-dominated and the thermally-dominated regions.
Our result can be rationalized comparing the geometry of 1t-CO2 with that of TS_CO2 (point B; Fig. 3). In TS_CO2 the S–H bond does not elongate; the distance the hydrogen atom moves is the result of the H–S–C angle decreasing. Instead, the main movement in TS_CO2 is the two carbon atoms approaching each other by about 0.7 Å. Two hydrogen bonds form between the thiol- and the amino-group facilitating the bonding and activation of CO2. Upon C–C-bond formation the curve flattens and reaches point C (Fig. 3B) corresponding to a zwitterionic structure similar to the carboxylate products in reactions of NHCs with CO2 (cf.Scheme 1). However, point C is not a minimum on the PES (υi = 164.5 cm−1, B3LYP/6-311++G(3df,3pd)) and a hydrogen shift immediately occurs yielding an uncharged species. The potential of this hydrogen transfer is steep and this step does not contribute to the observable kinetics of the reaction. Hence, the measured kinetics (Fig. 2) are due to the two fragments 1 and CO2 approaching each other and not the subsequent hydrogen transfer. Therefore, below ca. 8 K the reaction mechanism can be best described as heavy-atom QMT.
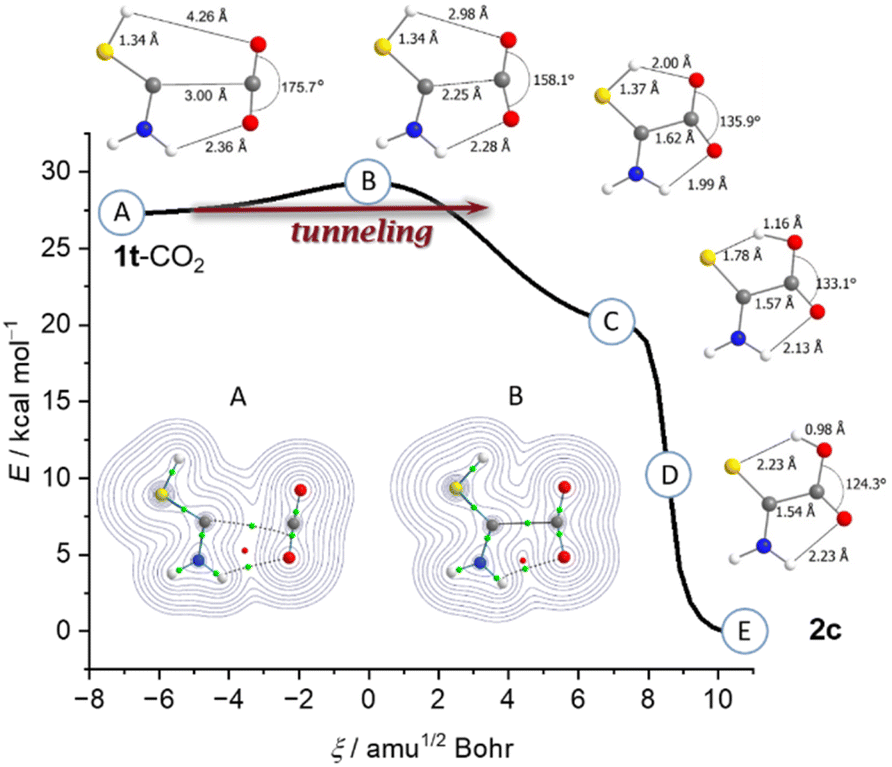 | ||
| Fig. 3 Intrinsic reaction coordinate (B3LYP/6-311++G(3df,3pd)) of the reaction of 1t-CO2 to 2c. Bond critical points (green) and a ring critical point (red) in 1t-CO2 indicate bonding interactions between the carbon atoms and between oxygen and the amino group. This is also visualized by the Laplacian (inset). Once the C–C-bond formed, the H-shift occurs without further activation. | ||
Carbene 1 possesses a singlet ground state and the vertical (adiabatic) singlet/triplet energy separation amounts to 59.4 (38.1) kcal mol−1 at the B3LYP/6-311++G(3df,3pd) level of theory. In 1t-CO2 these values are 61.8 (57.2) kcal mol−1. Complex 1t-CO2 is stabilized by 4.2 kcal mol−1 (CCSD(T)/cc-pVTZ) compared to the free fragments. A bond critical point analysis (Fig. 3, inset) of 1t-CO2 suggests hydrogen bonding interactions (green) between the amino group and CO2 as well as an onset of interactions between the carbon atoms, even at a distance of nearly 3 Å. This leads to a circular arrangement of bonding interactions indicated by a ring critical point (red). The attractive interaction can be interpreted by electron donation from the carbene lone pair to the π*-CO2 orbital (Fig. S38†).
Complexes of carbenes with CO2 might represent transient intermediates in carbene mediated CO2 activation in general. We theoretically found complexes of aminomethylene,64 dihydroxymethylene,38 and aminohydroxymethylene65 with CO2 to be minimum structures on their PES. The carbonyl–ene reactions of aminomethylene and dihydroxymethylene are barrierless while the CO2 addition of aminohydroxymethylene is associated with an activation barrier of 3.9 kcal mol−1 (CCSD(T)/cc-pVTZ). However, these complexes have not been observed experimentally since the mentioned carbenes have been generated under HVFP conditions in the gas phase, when entropy precludes their formation.
As noted above, a CO2 complex of thiazolylidene has been spectroscopically identified earlier, but the back reaction, i.e., CO2 activation has not been reported. We reproduced these results and also found no evidence for the reverse reaction to take place even upon annealing the matrix to 32 K. Note that in thiazolylidene the proton has to be transferred from the NH group (and not from the SH moiety as in 1t-CO2). This possibility is in principle also given in 1t-CO2viaTS15 (Fig. 4).
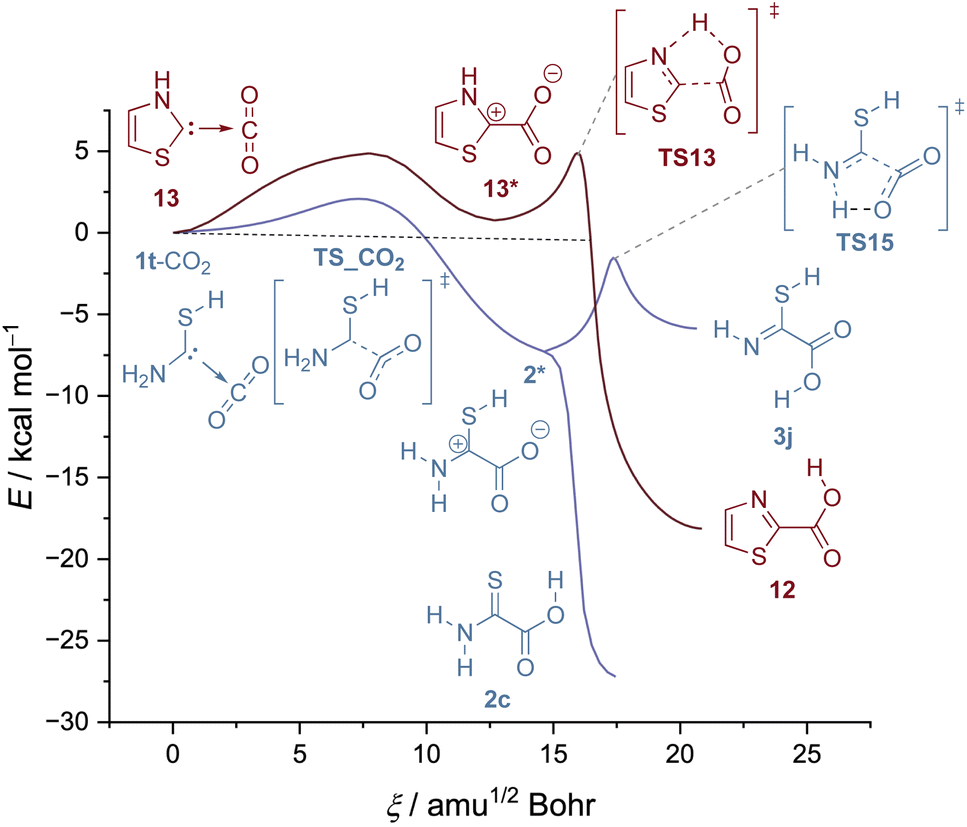 | ||
| Fig. 4 IRC curves for the H-transfer in 1t-CO2 from the SH group and the NH2 group (blue) compared to the reaction profile of 13 (red). All IRC curves computed at B3LYP/6-311++G(3df,3pd). | ||
While the H-transfer from SH is barrierless (Fig. 4, blue), in both cases transfers from NH feature a second barrier after the formation of the zwitterion (2* and 13*). In the case of thiazolylidene the formation of the zwitterion itself is even endothermic. This leads to a large barrier integral and QMT cannot take place. Accordingly, only for the reaction of 1t-CO2 to 2c tunneling was observed.
Conclusions
We isolated a complex of aminomercaptomethylene with CO2 in solid argon by photolysis of 2-amino-2-thioxoacetic acid. The carbene itself is a rare example of spectroscopically examined aminocarbenes as well as mercaptocarbenes. Once generated, the complex reacts back to the precursor in a heavy-atom quantum tunneling process at 3 K.While NHCs readily react with CO2 in solution to form stable carboxylates, this is not possible in the gas phase or in inert gas matrices due to the charge separation. In 1t-CO2 the thiol group facilitates the formation of a covalent bond by avoiding charge separation through an H shift. This, together with the heavy-atom tunneling uncovered here, opens new avenues for reactions for the activation of small molecules, in particular, CO2.
A related mechanism, albeit thus far not considered may also operate in the initial steps of the conversion of CO2 to formic acid, catalyzed by 1,2,3-triazole.66
Data availability
All experimental and computed data are collected in the ESI.†Author contributions
B. B. and M. S. performed the matrix isolation experiments and evaluated the experimental data. B. B., M. S., E. S., and A. K. E. conducted the computational work. P. R. S. supervised the project. All authors interpreted the results and contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Dennis Gerbig and Marcel Ruth (both JLU Giessen) for fruitful discussions. B. B. thanks the Fonds der Chemischen Industrie for a doctoral scholarship. E. S. thanks funding provided by the Alexander von Humboldt Foundation and VATAT – the Israeli Council for Higher Education.Notes and references
- Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed.
- A. Gulzar, A. Gulzar, M. B. Ansari, F. He, S. Gai and P. Yang, Chem. Eng. J., 2020, 100013 CAS.
- C. M. Momming, E. Otten, G. Kehr, R. Frohlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643 CrossRef PubMed.
- C. Wang, H. Luo, D. E. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978 CrossRef CAS.
- C. Villiers, J. P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465 CrossRef CAS.
- M.-Y. Wang, Q.-W. Song, R. Ma, J.-N. Xie and L.-N. He, Green Chem., 2016, 18, 282 RSC.
- H. Zhou, G.-X. Wang, W.-Z. Zhang and X.-B. Lu, ACS Catal., 2015, 5, 6773 CrossRef CAS.
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962 CrossRef CAS PubMed.
- A. Tudose, A. Demonceau and L. Delaude, J. Organomet. Chem., 2006, 691, 5356 CrossRef CAS.
- M. Hans, L. Delaude, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2014, 79, 2758 CrossRef CAS.
- L. Gu and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 914 CrossRef CAS.
- J. D. Holbrey, W. M. Reichert, I. Tkatchenko, E. Bouajila, O. Walter, I. Tommasi and R. D. Rogers, Chem. Commun., 2003, 28 RSC.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 40, 112 RSC.
- I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2006, 47, 6453 CrossRef CAS.
- I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2005, 46, 2141 CrossRef CAS.
- H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039 CrossRef CAS PubMed.
- Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 121, 4258 CrossRef.
- D. M. Denning and D. E. Falvey, J. Org. Chem., 2014, 79, 4293 CrossRef CAS PubMed.
- L. Delaude, Adv. Synth. Catal., 2020, 362, 3259 CrossRef CAS.
- L. Farkas and O. H. Wansbrough-Jones, Z. Phys. Chem B, 1932, 18, 124 Search PubMed.
- M. K. R. Noyori, M. Kawanisi and H. Nozaki, Tetrahedron, 1969, 25, 1125 CrossRef.
- G. Maier and J. Endres, Eur. J. Org. Chem., 1998, 1998, 1517 CrossRef.
- G. Maier, J. Endres and H. P. Reisenauer, Angew. Chem., Int. Ed., 1997, 36, 1709 CrossRef CAS.
- P. J. M. D. Kong, E. K. J. Lui, O. Bsharat and R. J. Lundgren, Science, 2020, 369, 557 CrossRef PubMed.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- M. Schauermann and P. R. Schreiner, J. Phys. Chem. Lett., 2022, 13, 3138 CrossRef CAS PubMed.
- E. E. Gard, M. J. Kleeman, D. S. Gross, L. S. Hughes, J. O. Allen, B. D. Morrical, D. P. Fergenson, T. Dienes, M. E. Gälli and R. J. Johnson, Science, 1998, 279, 1184 CrossRef CAS PubMed.
- S. Doddipatla, C. He, R. I. Kaiser, Y. Luo, R. Sun, G. R. Galimova, A. M. Mebel and T. J. Millar, Proc. Natl. Acad. Sci., 2020, 117, 22712 CrossRef CAS.
- P. R. Schreiner, J. Am. Chem. Soc., 2017, 139, 15276 CrossRef CAS PubMed.
- P. R. Schreiner, Trends Chem., 2020, 2, 980 CrossRef CAS.
- P. R. Schreiner, H. P. Reisenauer, D. Ley, D. Gerbig, C.-H. Wu and W. D. Allen, Science, 2011, 332, 1300 CrossRef CAS PubMed.
- W. T. Borden, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 20 CAS.
- R. S. Sheridan, in Review Reactive Intermediate Chemistry, 2007, p. 415 Search PubMed.
- C. Castro and W. L. Karney, Angew. Chem., Int. Ed., 2020, 59, 8355 CrossRef CAS PubMed.
- C. M. Nunes, A. K. Eckhardt, I. Reva, R. Fausto and P. R. Schreiner, J. Am. Chem. Soc., 2019, 141, 14340 CrossRef CAS.
- Z. Wu, R. Feng, H. Li, J. Xu, G. Deng, M. Abe, D. Bégué, K. Liu and X. Zeng, Angew. Chem., Int. Ed., 2017, 129, 15878 CrossRef.
- P. R. Schreiner, H. P. Reisenauer, F. C. Pickard IV, A. C. Simmonett, W. D. Allen, E. Mátyus and A. G. Császár, Nature, 2008, 453, 906 CrossRef CAS PubMed.
- P. R. Schreiner and H. P. Reisenauer, Angew. Chem., Int. Ed., 2008, 47, 7071 CrossRef CAS.
- D. Ley, D. Gerbig, J. P. Wagner, H. P. Reisenauer and P. R. Schreiner, J. Am. Chem. Soc., 2011, 133, 13614 CrossRef CAS.
- D. Ley, D. Gerbig and P. R. Schreiner, Chem. Sci., 2013, 4, 677 RSC.
- A. Mardyukov, H. Quanz and P. R. Schreiner, Nat. Chem., 2017, 9, 71 CrossRef CAS PubMed.
- A. K. Eckhardt, F. R. Erb and P. R. Schreiner, Chem. Sci., 2019, 10, 802 RSC.
- D. Gerbig, H. P. Reisenauer, C.-H. Wu, D. Ley, W. D. Allen and P. R. Schreiner, J. Am. Chem. Soc., 2010, 132, 7273 CrossRef CAS PubMed.
- A. K. Eckhardt, M. M. Linden, R. C. Wende, B. Bernhardt and P. R. Schreiner, Nat. Chem., 2018, 10, 1141 CrossRef PubMed.
- B. Bernhardt, F. Dressler, A. K. Eckhardt, J. Becker and P. R. Schreiner, Chem.–Eur. J., 2021, 27, 6732 CrossRef CAS PubMed.
- M. Pettersson, E. M. S. Maçôas, L. Khriachtchev, J. Lundell, R. Fausto and M. Räsänen, J. Chem. Phys. A, 2002, 117, 9095 CrossRef CAS.
- G. Maier, J. Endres and H. P. Reisenauer, J. Mol. Struct., 2012, 1025, 2 CrossRef CAS.
- D. Gerbig and P. R. Schreiner, J. Chem. Phys. B, 2015, 119, 693 CrossRef CAS.
- M. Tsuge and L. Khriachtchev, J. Phys. Chem. A, 2015, 119, 2628 CrossRef CAS.
- P. R. Schreiner, J. P. Wagner, H. P. Reisenauer, D. Gerbig, D. Ley, J. Sarka, A. G. Császár, A. Vaughn and W. D. Allen, J. Am. Chem. Soc., 2015, 137, 7828 CrossRef CAS PubMed.
- H. Rostkowska, L. Lapinski and M. J. Nowak, Phys. Chem. Chem. Phys., 2018, 20, 13994 RSC.
- H. Rostkowska, L. Lapinski and M. J. Nowak, J. Phys. Chem. A, 2017, 121, 6932 CrossRef CAS PubMed.
- S. Góbi, I. Reva, I. P. Csonka, M. C. Nunes, G. Tarczay and R. Fausto, Phys. Chem. Chem. Phys., 2019, 21, 24935 RSC.
- S. Góbi, C. M. Nunes, I. Reva, G. Tarczay and R. Fausto, Phys. Chem. Chem. Phys., 2019, 21, 17063 RSC.
- E. M. Brás and R. Fausto, J. Photochem. Photobiol., A, 2018, 357, 185 CrossRef.
- E. M. Brás and R. Fausto, J. Mol. Struct., 2018, 1172, 42 CrossRef.
- D. Prusinowska, L. Lapinski, M. J. Nowak and L. Adamowicz, Spectrochim. Acta A, 1995, 51, 1809 CrossRef.
- M. J. Nowak, L. Lapinski, H. Rostkowska, A. Les and L. Adamowicz, J. Phys. Chem., 1990, 94, 7406 CrossRef CAS.
- M. J. Nowak, L. Lapinski, J. Fulara, A. Les and L. Adamowicz, J. Phys. Chem., 1991, 95, 2404 CrossRef CAS.
- L. Lapinski, H. Rostkowska, A. Khvorostov, M. Yaman, R. Fausto and M. J. Nowak, J. Chem. Phys. A, 2004, 108, 5551 CrossRef CAS.
- L. Lapinski, H. Rostkowska, A. Khvorostov and M. J. Nowak, Phys. Chem. Chem. Phys., 2003, 5, 1524 RSC.
- H. Rostkowska, L. Lapinski, A. Khvorostov and M. J. Novak, J. Phys. Chem. A, 2003, 107, 6373 CrossRef CAS.
- T. Schleif, J. Tatchen, J. F. Rowen, F. Beyer, E. Sanchez-Garcia and W. Sander, Chem.–Eur. J., 2020, 26, 10452 CrossRef CAS PubMed.
- A. K. Eckhardt and P. R. Schreiner, Angew. Chem., Int. Ed., 2018, 57, 5248 CrossRef CAS PubMed.
- B. Bernhardt, M. Ruth, H. P. Reisenauer and P. R. Schreiner, J. Phys. Chem. A, 2021, 125, 7023 CrossRef CAS.
- X. Song, Y. Meng and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 16744 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05388h |
| ‡ These authors contributed equally. |
| § Present address: Lehrstuhl für Organische Chemie II, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |