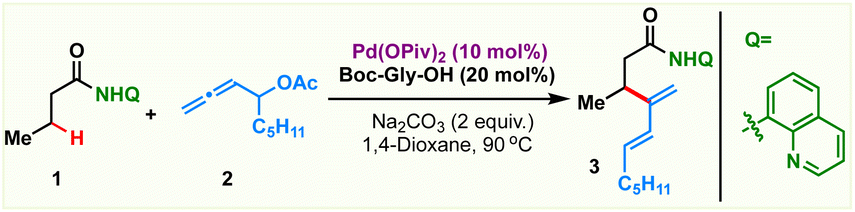DOI:
10.1039/D2SC05188E
(Edge Article)
Chem. Sci., 2023,
14, 955-962
Pd(II)-catalyzed β- and γ-C-(sp3)–H dienylation with allenyl acetates†
Received
18th September 2022
, Accepted 19th December 2022
First published on 20th December 2022
Abstract
Recent years have seen the emergence of transition metal catalyzed C–H activation as a powerful synthetic tool in organic chemistry. Allenes have fascinated synthetic chemists due to their unique reactivity. While directing group assisted functionalization of C(sp2)–H bonds with allenes is well documented in the literature, their coupling with more challenging aliphatic C(sp3)–H bonds remains elusive. In this regard, we hereby report a Pd(II) catalyzed 8-aminoquinoline directed aliphatic C(sp3)–H dienylation protocol using allenyl acetates. A variety of carboxylic acids including fatty acids and amino acids were efficiently functionalized at β and γ-positions to afford diversely functionalized 1,3-dienes. Preliminary mechanistic studies revealed the crucial role of the base in the success of the transformation. The reaction proceeds via regioselective 2,3-migratory insertion of the allene with the alkylpalladium(II) species followed by β-acetoxy elimination.
Introduction
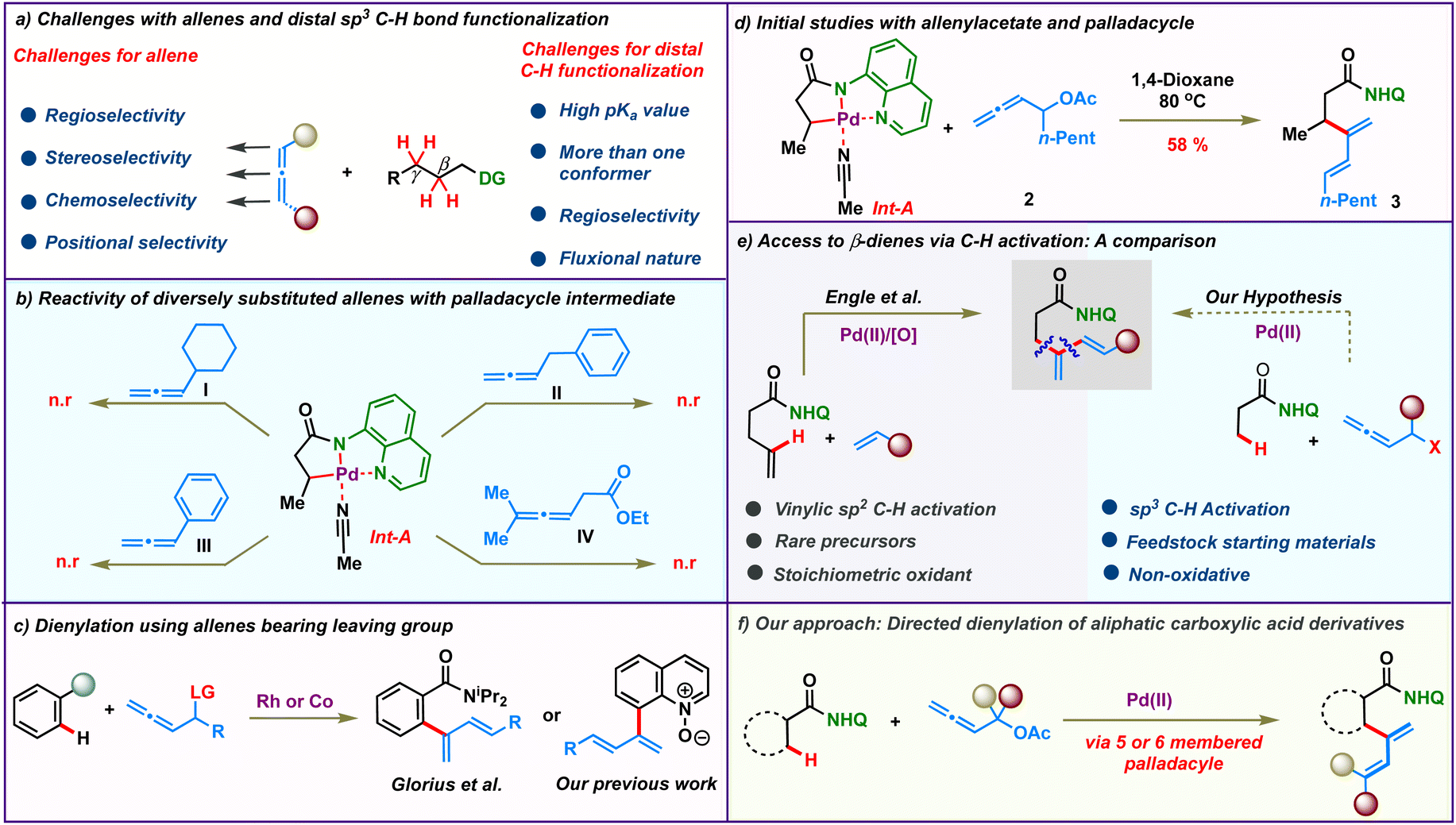
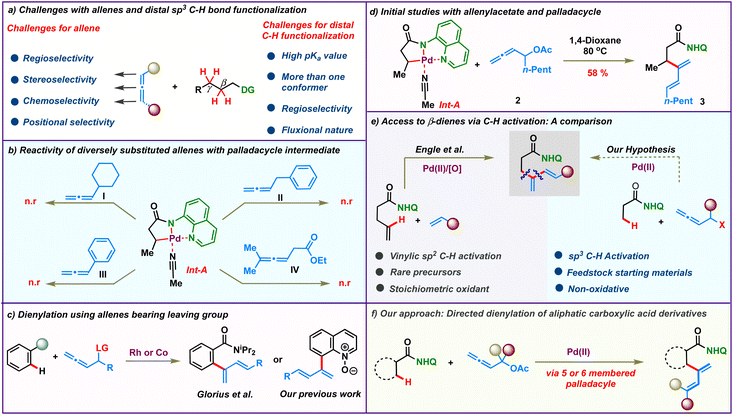
For all the myriad advancements in modern synthetic chemistry, the development of selective C–H bond functionalization remains an attractive challenge.1–3 Tremendous progress has been achieved in this area employing π-systems like alkenes and alkynes as coupling partners. In comparison, the study of allenes in transition metal catalyzed C–H activation reactions is mired with several obscurities and is less explored. The presence of two orthogonal double bonds in allenes leads to a variety of selectivity issues like chemo-, stereo-, regio- and positional selectivities (Scheme 1a, left).4 At the same time, addressing these issues would enable rapid access to molecular complexity from fairly simple precursors.5–9 As a result, although challenging, the study of allenes is quite rewarding and has long intrigued synthetic chemists. Efforts in this direction have led to elegant reports wherein allenes have been used as multifaceted coupling partners in transition metal catalyzed C(sp2)–H activations via the judicious choice of the metal catalysts and substituents.10,11 The selective functionalization of one of the double bonds of allenes based on the reaction conditions resulted in a variety of reaction outcomes like annulation, alkenylation and allylation.12–29
 |
| | Scheme 1 Challenges, initial studies, earlier work and overview of the work. | |
On the other hand, the low reactivity and non-polar character of aliphatic C(sp3)–H bonds along with the formation of conformationally feeble complexes post C–H activation make C(sp3)–H functionalization quite challenging (Scheme 1a, right).30–48 Consequently it is not surprising that the coupling of distal aliphatic C(sp3)–H bonds with allenes remains unreported and hence is an unanswered problem. To study this, we envisioned to probe the reactivity of allenes having different substituents in Pd-catalyzed C(sp3)–H bond activation. Initially, a palladacycle Int-A was isolated by treating 8-aminoquinoline butanamide 1 with stoichiometric Pd(OAc)2. Subsequently, we investigated the stoichiometric reactivity of the palladacycle Int-A with a variety of allenes to identify the best substrate combination (Scheme 1b). Allenes like cyclohexylallene, phenylallene, benzylallene, and allenyl ester failed to react with Int-A under various reaction conditions. We postulated that the complexes resulting from the carbopalladation of Int-A onto these allenes would have lacked sufficient conformational stability to afford further reactions. We then envisioned that allenes bearing a heteroatom like oxygen at the α-position could facilitate the migratory insertion by the coordinative interaction with the metal centre. These allenes were illustrated by Glorius and co-workers for Rh(iii)-catalyzed ortho-dienylation of benzamides.20 We have also engaged these allenes to access C-8 dienylated quinoline-N-oxides under Co(iii)-catalysis (Scheme 1c).21 So, we proceeded to study the reactivity of allenes bearing heteroatoms with Int-A hoping that the coordination of the heteroatom with the palladium post migratory insertion would provide some stability to the resulting palladacycle. Indeed, to our delight, β-dienylated derivative 3 was isolated in 58% when Int-A was reacted with allenyl acetate 2 in 1,4-dioxane at 80 °C (Scheme 1d). These initial results motivated us to further probe the catalytic version of the β-C(sp3)–H dienylation protocol.
Meanwhile, very few functionalities can rival the practicality offered by 1,3-dienes as building blocks in synthetic organic chemistry.49–53 Historically, they have been a linchpin in many of the most celebrated synthetic transformations like [4 + 2]-cycloaddition and other pericyclic reactions. Additionally, they are also found in the core of a plethora of commercially important molecules like natural products, drugs and advanced materials. Traditionally, they are synthesized using protocols like Wittig olefinations or cross coupling reactions.54–57 Recent years have witnessed the emergence of elegant alternative methodologies to access 1,3-dienes.58–61 However, these protocols suffer from the need for pre-functionalized substrates like boronic acids, halides or pseudohalides. Direct C–H activation to access dienes would bypass the need for such functionalized molecules and such an elegant protocol was realized by Engle's group in 2018 (Scheme 1e, left).62 They described an 8-aminoquinoline directed, oxidative Pd(II)-catalyzed C(alkenyl)–H olefination of alkenyl amides to access β-dienylated carboxylic acid derivatives. Though elegant, this protocol relied on the use of rare alkenyl carboxylic amides which require up to 5 steps for their synthesis. Furthermore, their protocol was limited for activated alkenes and require 3 equiv. of oxidants. We anticipated that similar β-dienylated acid derivatives with simple aryl or alkyl substituents could be directly obtained by C(sp3)–H dienylation of aliphatic carboxylic amides employing allenes. The feedstock availability of aliphatic carboxylic acids as petrochemical by-products, fatty acids and amino acids would make such a protocol highly pertinent and attractive (Scheme 1e, right). In this regard, we hereby report a Pd(II) catalyzed distal β- and γ-C-(sp3)–H dienylation of aliphatic carboxylic acid derivatives employing allenyl acetates (Scheme 1f). Bypassing the need for stoichiometric oxidants makes the process more alluring. The reaction proceeds via 2,3-migratory insertion of allenes with the C–H activated palladacycle, which then undergoes facile β-acetoxy elimination to afford the corresponding dienes.
Results and discussion
Optimization of the reaction
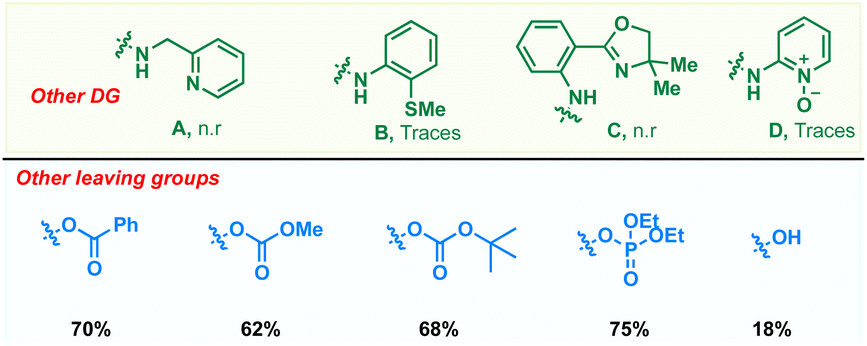
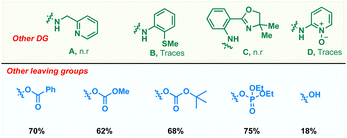
To realize an efficient catalytic C(sp3)–H dienylation protocol, 8-aminoquinoline butanamide 1 and allenyl acetate 2 were chosen as the model substrates (Table 1). Captivatingly, β-monodienylated compound 3 was observed in 30% yield when a mixture of 1 (1 equiv.) and 2 (1.5 equiv.) was treated with 10 mol% Pd(OAc)2 and 2 equiv. of Ag2CO3 in 1,4-dioxane at 90 °C. Encouraged by this, extensive optimization of various reaction parameters like the solvent, base, ligand and catalyst was carried out (see the ESI† for the full optimization details of each parameter). We found that the reaction of 1 with 2 in the presence of Pd(OPiv)2 (10 mol%), Boc–Gly–OH (20 mol%) and Na2CO3 (2 equiv.) in 1,4-dioxane at 90 °C is optimal, furnishing the product 3 in 78% isolated yield (entry 1). Other ligands like 2,2-bpy, 1,10-phen, and collidine led to inferior yields (entry 2). Substituting Pd(OPiv)2 with other Pd(II) salts was found to be deleterious for the transformation (entry 3). 1,4-Dioxane was found to be the most optimal solvent as reduced yields were observed in the case of other solvents like MeCN, HFIP, DCM and xylene (entry 4). Increasing or decreasing the reaction temperature from 90 °C also led to inferior yields (entries 6 and 7). The reaction failed to proceed in the absence of the palladium catalyst and only 40% of the product was isolated in the absence of the base (entries 8 and 9). Other bidentate directing groups failed to deliver the corresponding products under the standard conditions indicating that 8-aminoquinoline is the ideal directing group for the transformation. Finally other leaving groups on the allene such as hydroxy, benzoate, methyl carbonate, –OBoc and phosphonate were found to be inferior as compared to acetate, furnishing reduced yields of the diene 3.
Table 1 Optimization of the reaction parameters
|
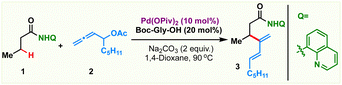
|
| Entry |
Variation from standard conditions |
Yielda (%) |
|
Yield is based on 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard.
Yield in parenthesis refers to isolated yield.
|
|
1
|
None
|
80 (78)
|
| 2 |
2,2-Bpy, collidine, and Fmoc–Gly–OH as ligands |
<70 |
| 3 |
Pd(OTf)2 and Pd(acac)2 as catalysts |
<45 |
| 4 |
Acetonitrile, HFIP, xylene, and DCM as solvents |
<49 |
| 5 |
NaOPiv, NaOAc, and KOAc as bases |
<35 |
| 6 |
80 °C |
71 |
| 7 |
100 °C |
75 |
| 8 |
Without Pd(II) |
Trace |
| 9 |
Without a base |
40 |
|
Substrate scope of β-sp3-C–H dienylation
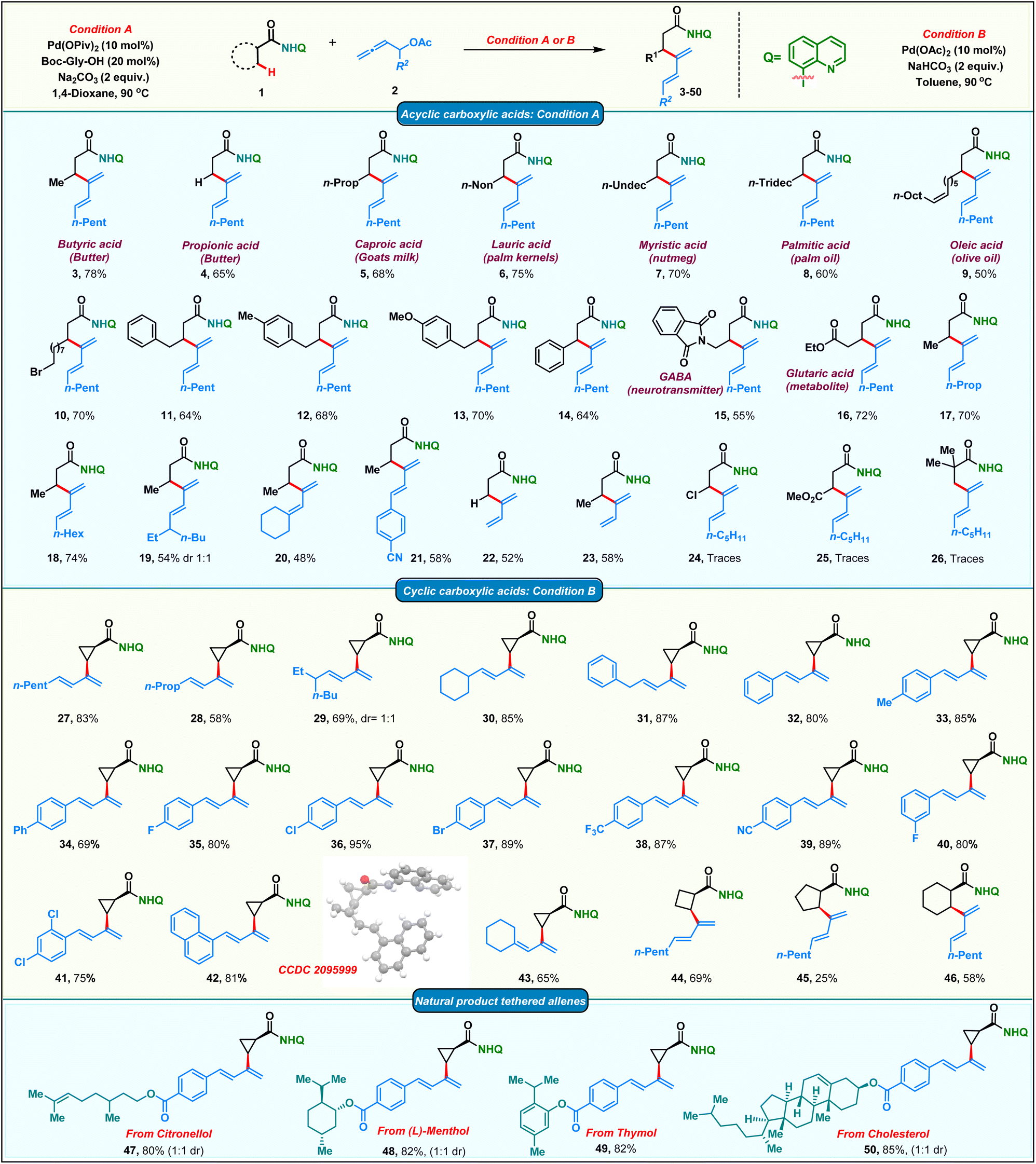
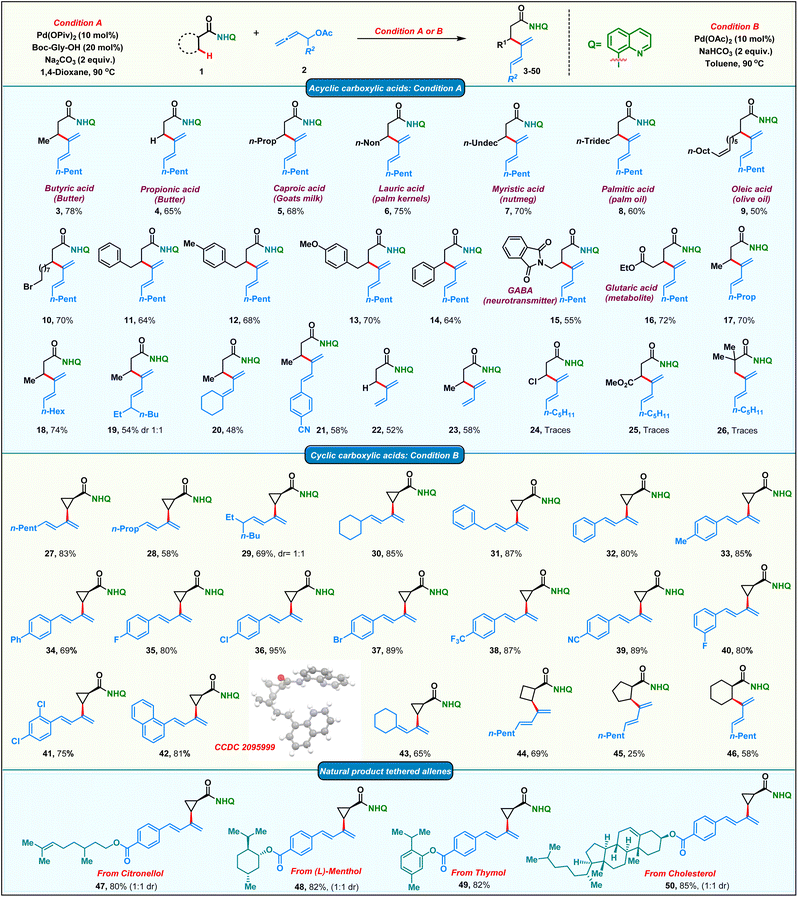
After having the optimized conditions in hand, we proceeded to probe the substrate scope of the protocol (Scheme 2). Initially, we studied the reactivity of various aliphatic acid derivatives 1 with allenyl acetate 2. Gratifyingly, we found that a variety of biologically relevant fatty acids like butyric acid, propionic acid, caproic acid, lauric acid, myristic acid, palmitic acid and oleic acid could be effortlessly converted to their corresponding β-dienylated derivatives 3–9 in moderate to good yields 50–78%. 11-Bromoundecanoic acid delivered the corresponding product 10 in 70%. Subsequently, the reactivity of various substituted 4-phenyl butanoic amides was tested and the corresponding products 11–13 were obtained in moderate to good yields (64–70%). Additionally, 3-phenyl propionic amide delivered the corresponding diene 14 in 64%. Functionalized acids like GABA (neurotransmitter) and glutaric acid (metabolite) upon appropriate protection also yielded the corresponding β-dienes 15 and 16 in good yields. To our delight, allenes bearing diverse alkyl chains such as linear, branched and cyclic were found to be amenable substrates in our protocol furnishing the products 17–20 in 48–70% yields. Phenyl substituted diene 21 was obtained in 58% from the corresponding allene. Delectably, mono-substituted butadienes 22 & 23 were obtained in moderate yields from unsubstituted allenyl acetate. Amide bearing electron withdrawing groups at the β-carbon failed to furnish the corresponding products 24 & 25. Furthermore, no product 26 formation was observed in the case of the pivaloylamide derivative, due to high steric bulk on the α-carbon, which would impede the C–H activation step.
 |
| | Scheme 2 Scope of β-sp3-C–H dienylation. | |
Substrate scope of cyclic carboxamides
On the other hand, the smallest carbocycle: cyclopropane, is found in the core of a variety of commercially important molecules like pharmaceuticals, natural products, agrochemicals and functional materials. Consequently, there is a growing demand for the synthesis of multisubstituted cyclopropanes. The synthesis of trans-disubstituted cyclopropanes is well documented in the literature. However, the corresponding cis-isomers require more exotic methods. Directing group assisted C–H functionalization has emerged as a versatile technology in recent years to access cis-disubstituted cyclopropanes.63 In this regard, we proceeded to carry out palladium catalyzed dienylation of an amide derived from cyclopropane carboxylic acid. To our delight, cis-dienylated product 27 was isolated in excellent yield under slightly modified conditions (see the ESI† for the optimization details). Encouraged by this, we screened the reactivity of a variety of substituted allenes with cyclopropane amide. Allenes bearing linear, branched and cyclic alkyl chains furnished the corresponding products 28–30 in appreciable yields. Diene 31 was obtained in 87% yield when a benzyl-substituted allene was employed. The dienylation of the methylene C–H bond of a cyclopropane with allenes bearing electronically varied phenyl rings was also found to be amenable affording the corresponding 2-cyclopropyl, arylated 1,3-dienes 32–42 in good to excellent 69–95% yields. Consequently, the protocol was extended to functionalize β-methylene C(sp3)–H bonds of four to six-membered cyclic carboxamides to isolate mono-dienylated products 44–46 in 25–69% yield with exclusively cis-stereochemistry. Furthermore, we found that the protocol was amenable for the late-stage modifications of complex natural products. Allenes tethered to citronellol, menthol, thymol and cholesterol afforded the corresponding dienes 47–50 in good yields adding significant value to the protocol.
Substrate scope of γ-sp3-C–H dienylation
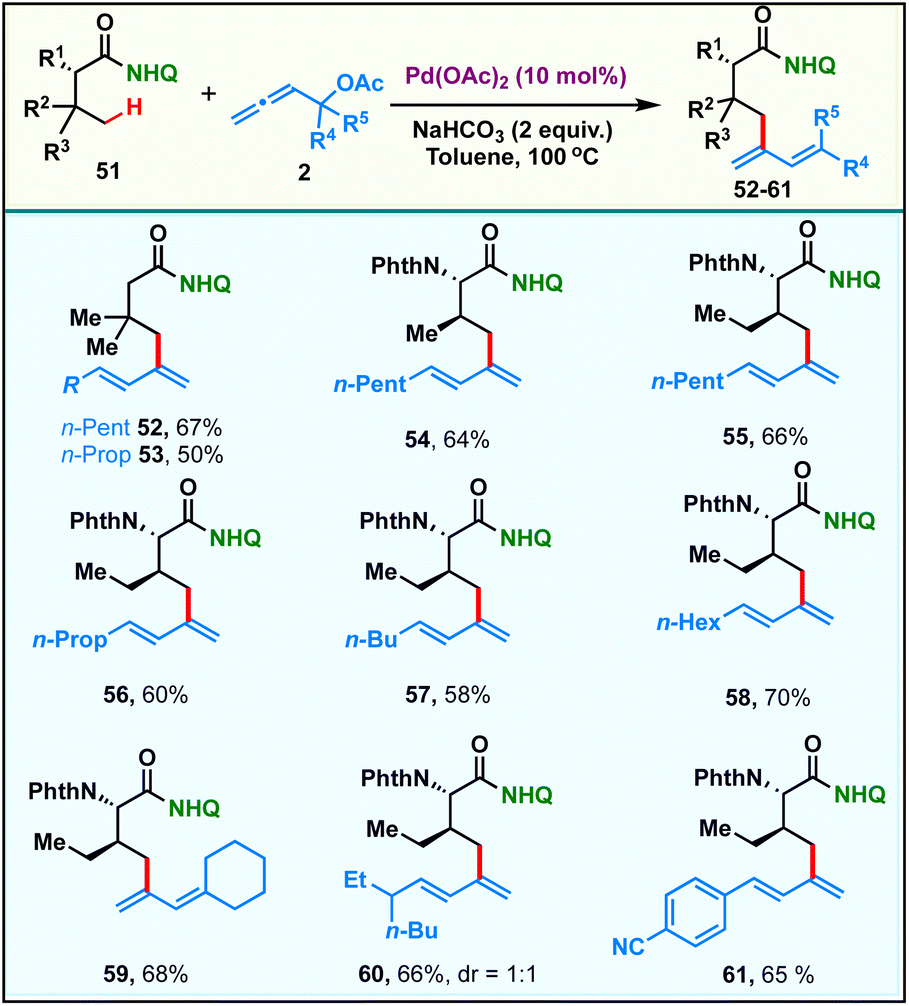
Recently, selective γ-sp3-C–H activation reactions have emerged as powerful strategies to carry out distal functionalizations. The formation of six membered metallacycle intermediates is typically less favored in comparison to five membered metallacycles, which adds significant challenges. Inspired by this, we proceeded to probe the scope of our current γ-sp3-C–H dienylation with carboxamides, which are sterically hindered at β-carbon. Thus, exploration began with the 8-aminoquinoline amide derived from 3,3-dimethylbutanoic 51, which under our standard reaction conditions provided mono-γ-dienylation products 52 & 53 in 67% and 50% yields respectively (Scheme 3). Since α-amino acids form the basis of life and biological research, the incorporation of a new functionality onto them which can alter their properties would be of tremendous value. Under the present reaction conditions, amides derived from α-amino acids possessing primary γ-C–H bonds, like L-valine and L-isoleucine, afforded the respective γ-selective dienylation products 54–61 in good to moderate yields.
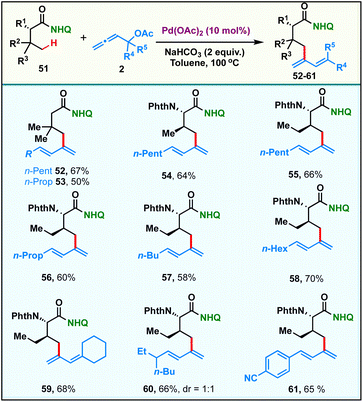 |
| | Scheme 3 Scope of γ-sp3-C–H dienylation. | |
Synthetic utility of the protocol
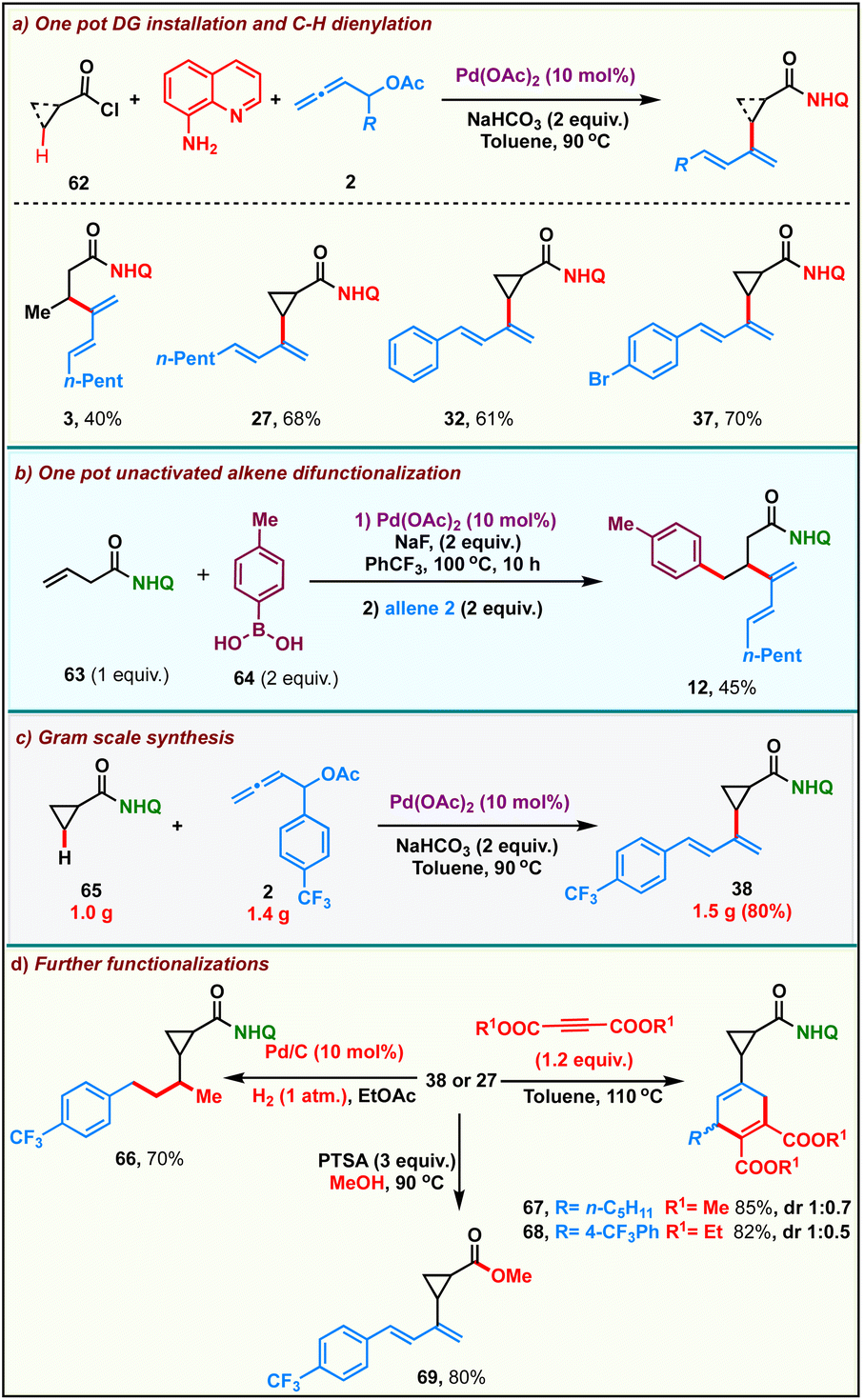
After successfully establishing the scope of the developed protocol, we proceeded to probe its practicality. We carried out a step-economical one-pot multicomponent reaction comprising the installation of the bidentate directing group followed by Pd(II)-catalyzed sp3-C–H activation and β-dienylation. Accordingly, the reaction of a mixture of aliphatic/cyclic acid chloride 62, 8-aminoquinoline with allenyl acetate 2 in the presence of 10 mol% Pd(OAc)2 and 2 equiv. of NaHCO3 in toluene at 90 °C, directly afforded the corresponding β-C–H dienylated N-(quinolin-8-yl)carboxamide derivatives in moderate yields (Scheme 4a). We were also able to develop a one-pot palladium catalyzed hydroarylation/dienylation sequence onto the alkenyl carboxamide derivative 63 to furnish the product 12 in 45% over two steps (Scheme 4b). The reaction could be scaled up without appreciable loss in the yields of the product: 1.0 g of cyclopropyl benzamide 65 upon reaction with 1.4 g of allene 2 delivered 1.5 g of diene 38 in 80% (Scheme 4c). Furthermore, we found that the dienes could participate in a variety of transformations (Scheme 4d). Catalytic hydrogenation of diene 38 afforded 1-phenyl 3-cyclopropyl butane derivative 66 in 70%. We found that the diene could effortlessly participate in the Diels–Alder reaction with alkynes affording the adducts 67 and 68 in 85% and 82% respectively. The amide could be alcoholyzed under acidic conditions to deliver the ester 69 in 80%.
 |
| | Scheme 4 One-pot synthesis, scale-up and further functionalizations. | |
Preliminary mechanistic studies
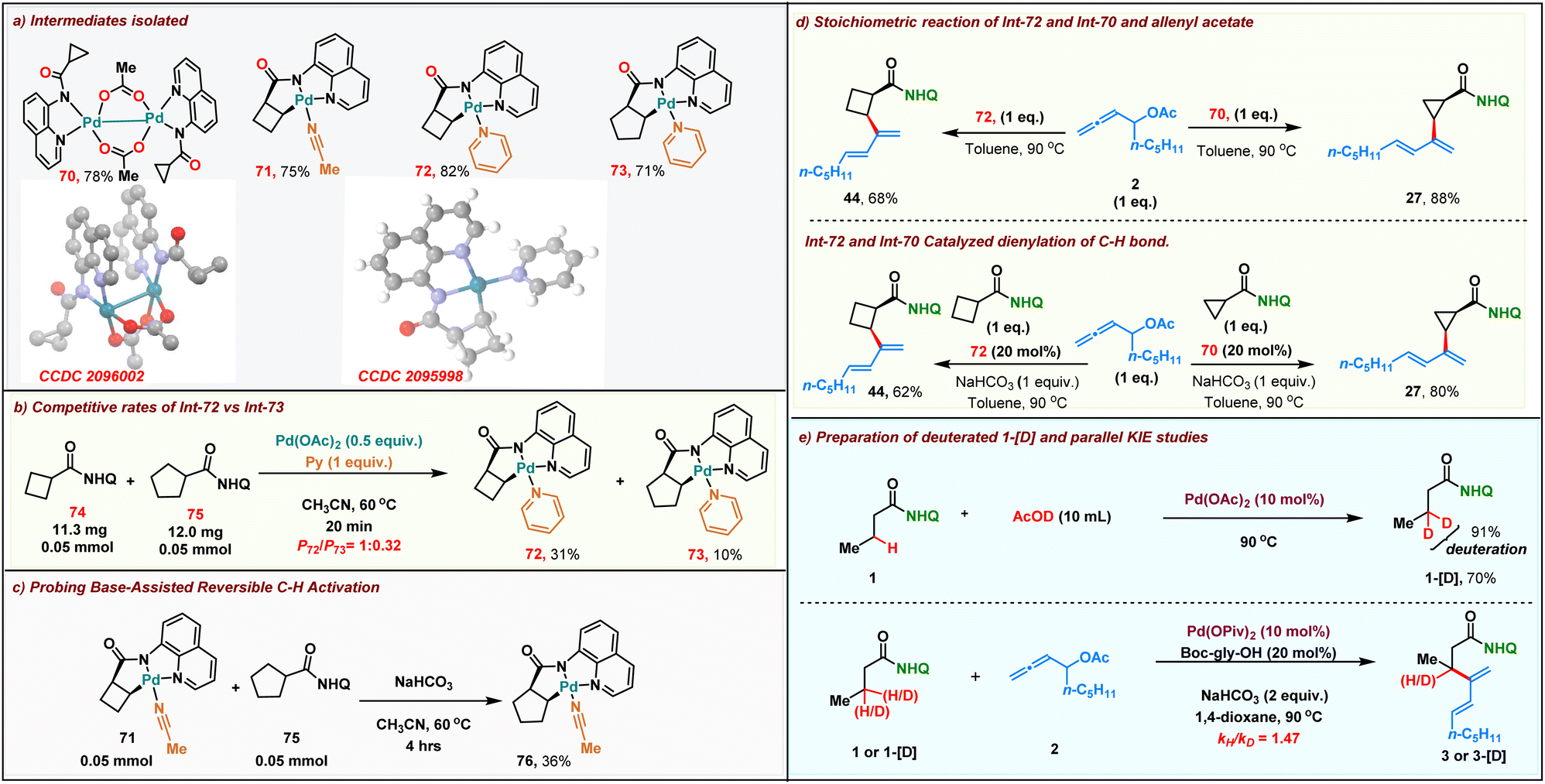
We then proceeded to carry out preliminary studies to shed light onto the reaction mechanism. Initially we proceeded to synthesize the alkyl palladium reactive intermediates involved in our reaction. A stoichiometric reaction of cyclopropyl amide and Pd(OAc)2 in acetonitrile resulted in the formation of dimeric palladium complex 70 within 20 min at room temperature. The structure of the dimeric palladium complex was unambiguously confirmed by single-crystal X-ray crystallography. Furthermore, we performed a stoichiometric reaction of cyclobutyl and cyclopentyl amides with Pd(OAc)2, resulting in the formation of the corresponding five-membered palladacycle intermediate 71–73 (Scheme 5a). We then compared the rate of formation of the intermediates 72 & 73 with stoichiometric Pd(OAc)2. As expected, the more reactive cyclo-butane ring C–H bond led to the formation of 72 faster than 73 (31% vs. 10% in crude NMR) (Scheme 5b). To study the role of the base in the reaction, alkyl palladium complex 71 derived from cyclobutane amide was reacted with cyclopentyl carboxamide 75 in the absence and presence of a base. Remarkably, the formation of the aryl palladium complex 76 (36% yield in crude NMR) was observed when NaHCO3 was employed as the base (Scheme 5c). Meanwhile in the absence of NaHCO3, we failed to detect the formation of complex 76. These results confirm that palladacycle formation is readily reversible under the reaction conditions. Both the dimeric intermediate 70 and the five-membered palladacycle 72 upon reacting with the allenyl acetate 2 in toluene at 90 °C furnished the corresponding dienes 27 and 44 in 88% and 68% yields, respectively (Scheme 5d, top). Furthermore, both the Pd(II)-intermediates were also found to be catalytically competent for the dienylation protocol in the presence of 1 equiv. of NaHCO3 (Scheme 5d, bottom). This indicated that both the dimeric and palladacycle intermediates 70 and 72 were chemically and kinetically compatible with the reaction. To further probe the mechanism, we proceeded to carry out parallel kinetic isotopic studies, for which β-deuterated 1-[D] was synthesized in 70% yield with 91% deuterium incorporation by treating 1 with AcOD under Pd catalysis. Parallel kinetic experiments were then performed with 1 and 1-[D] which revealed a kH/kD value of 1.47, indicating that the C–H activation might not be involved in the rate determining step in the reaction.
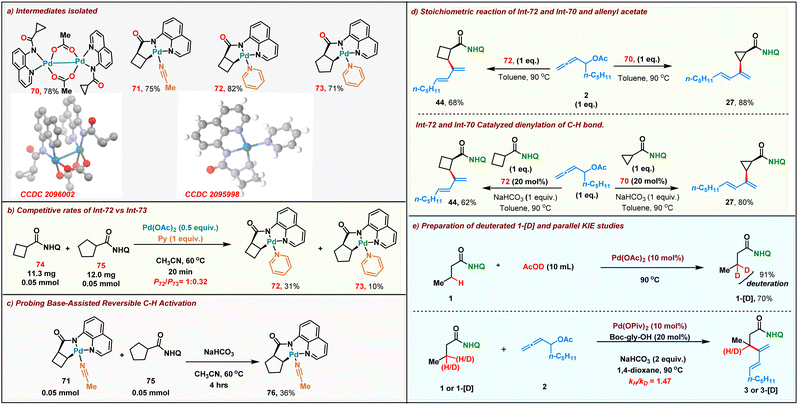 |
| | Scheme 5 Isolation of intermediates and mechanistic studies. | |
Proposed reaction mechanism
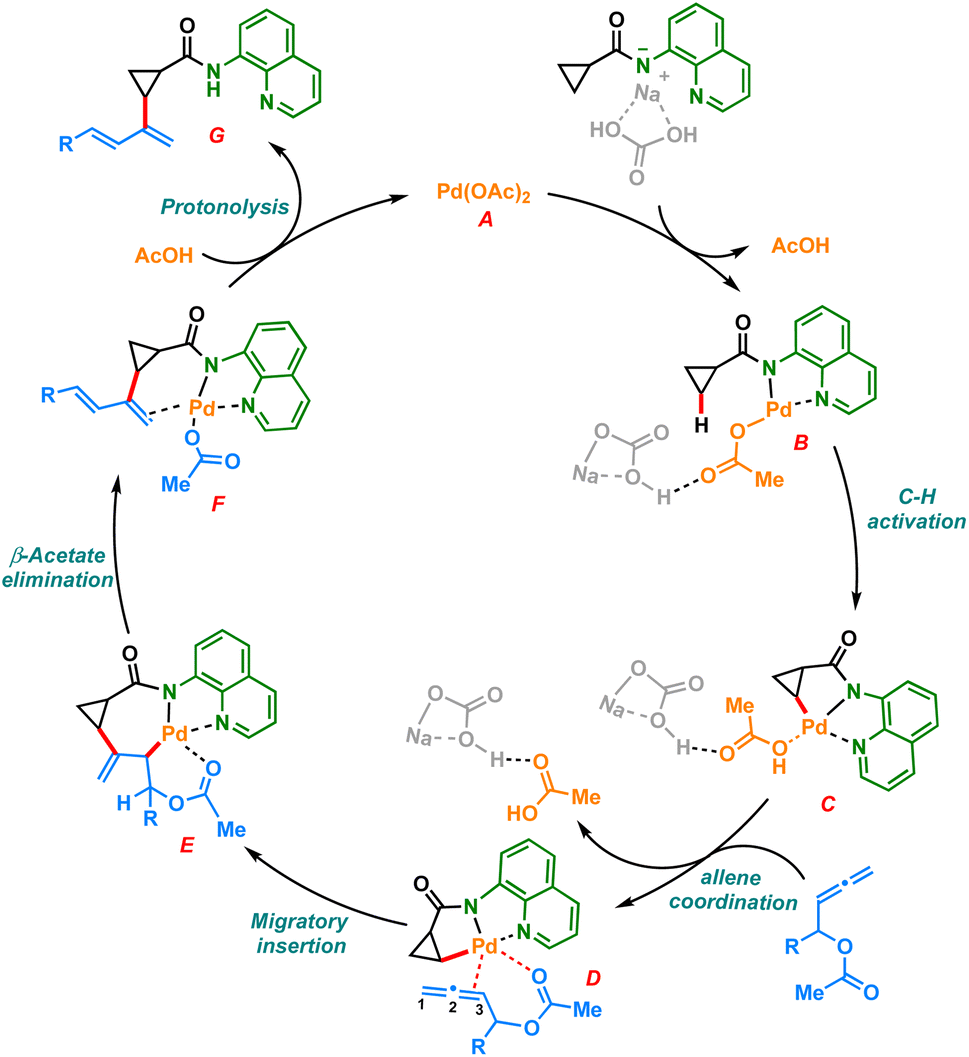
Based on previous literature and our mechanistic studies, the sp3 C–H dienylation reaction of carboxamide is likely to start with substrate catalyst binding, forming the intermediate B (Scheme 6). The amino quinoline directed C–H activation assisted by the base would form a five membered palladacycle intermediate C. The palladacycle C coordinates with allenyl acetate to form the intermediate D, which undergoes selective 2,3 migratory insertion and generates the σ-allyl palladium species E. The intermediate E upon β-acetoxy elimination could furnish the desired dienylated product G after the protonation of F.
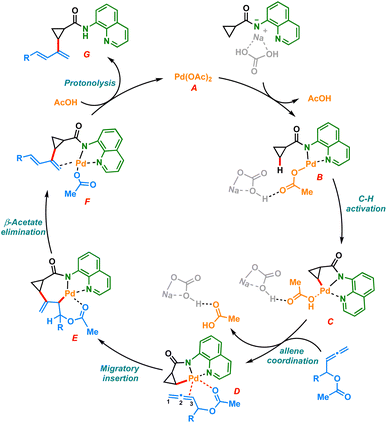 |
| | Scheme 6 Plausible mechanism. | |
Conclusion
In conclusion, we have developed an efficient Pd(II)-catalyzed dienylation of aliphatic C(sp3)–H bonds with allenyl acetates using 8-aminoquinoline as the directing group. A variety of aliphatic carboxylic acid derivatives were efficiently functionalized at b- and g-positions to afford diversely functionalized 1,3-dienes. The feedstock availability of carboxylic acids coupled with the fact that the current reaction was amenable to a variety of biologically and commercially important carboxylic acids adds to the value of this protocol. Delectably, the reaction was found to be scalable and the products could be further diversified efficiently. Detailed mechanistic studies signified the crucial roles of the base. The reaction proceeds via a sequence of regioselective 3,2-migratory insertion of alkyl palladium species onto the allene followed by β-acetoxy elimination to furnish the corresponding products.
Data availability
All data supporting the findings of this study are available within the article and the ESI.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (CCDC) under access no. CCDC: 2095999 (42), CCDC: 2096002 (70) and CCDC: 2095998 (72). Data relating to the materials and methods, experimental procedures, and NMR spectra are available in the ESI.† All other data are available from the authors upon reasonable request.
Author contributions
R. K. S. and A. M. N. conducted all the experiments and characterized the new compounds. R. K. S., A. M. N. and C. M. R. V. designed the experiments and wrote the manuscript. C. M. R. V. directed the research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This activity is supported by SERB, India (CRG/2019/005059). R. K. S. and A. M. N. are grateful to IIT Bombay for their fellowships.
References
-
P. H. Dixneuf and H. Doucet, C–H Bond Activation and Catalytic Functionalization I, Springer, 2016, p. 1 Search PubMed.
- T. Brückl, R. D. Baxter, Y. Ishihara and P. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed.
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682 CrossRef CAS PubMed.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS PubMed.
- R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef CAS PubMed.
- J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS PubMed.
- C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed.
- S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed.
- J. M. Alonso and P. Almendros, Chem. Rev., 2021, 121, 4193 CrossRef CAS PubMed.
- S. R. Kumar and C.-H. Cheng, Asian J. Org. Chem., 2018, 7, 1151 CrossRef.
- R. K. Shukla, A. M. Nair and C. M. R. Volla, Synlett, 2021, 32, 1169 CrossRef CAS.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181 CrossRef CAS PubMed.
- R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597 CrossRef CAS PubMed.
- H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed.
- N. Casanova, K. P. Del-Rio, R. García-Fandiño, J. L. Mascareñas and M. Gulías, ACS Catal., 2016, 6, 3349 CrossRef CAS PubMed.
- N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361 CrossRef CAS PubMed.
- C. Wang, A. Wang and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 9935 CrossRef CAS PubMed.
- S. Nakanowatari, T. Müller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15891 CrossRef CAS PubMed.
- N. Thrimurtulu, R. Nallagonda and C. M. R. Volla, Chem. Commun., 2017, 53, 1872 RSC.
- H. Wang, B. Beiring, D.-G. Yu, K. D. Collins and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12430 CrossRef CAS PubMed.
- R. K. Shukla, A. M. Nair, S. Khan and C. M. R. Volla, Angew. Chem., Int. Ed., 2020, 59, 17042 CrossRef CAS PubMed.
- J. Mo, T. Muller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 7719 CrossRef CAS PubMed.
- A. M. Messinis, L. H. Finger, L. Hu and L. Ackermann, J. Am. Chem. Soc., 2020, 142, 13102 CrossRef CAS PubMed.
- D. A. Petrone, M. Isomura, I. Franzoni, S. L. Rössler and E. M. Carreira, J. Am. Chem. Soc., 2018, 140, 4697 CrossRef CAS PubMed.
- X. Vidal, J. L. Mascareñas and M. Gulías, Org. Lett., 2021, 23, 5323 CrossRef CAS PubMed.
- S.-Y. Chen, X.-L. Han, J.-Q. Wu, Q. Li, Y. Chen and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 9939 CrossRef CAS PubMed.
- S.-Y. Chen, Q. Li and H. Wang, J. Org. Chem., 2017, 82, 11173–11181 CrossRef CAS PubMed.
- A. Singh, R. K. Shukla and C. M. R. Volla, Chem. Sci., 2022, 13, 2043 RSC.
- A. Singh, R. K. Shukla and C. M. R. Volla, Org. Lett., 2022, 24, 8936 CrossRef CAS PubMed.
- R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS PubMed.
- H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031 CAS.
- J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887 RSC.
- Q. Zhang and B.-F. Shi, Chem. Sci., 2021, 12, 841 RSC.
- O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- S. Li, R.-Y. Zhu, K.-J. Xiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 4317 CrossRef CAS PubMed.
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed.
- S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef CAS PubMed.
- J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750 CrossRef CAS PubMed.
- N. Thrimurtulu, S. Khan, S. Maity, C. M. R. Volla and D. Maiti, Chem. Commun., 2017, 53, 12457 RSC.
- Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS PubMed.
- Y.-J. Liu, Y.-H. Liu, Z.-Z. Zhang, S.-Y. Yan, K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2016, 55, 13859 CrossRef CAS PubMed.
- H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755 CrossRef CAS PubMed.
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS PubMed.
- J. He, T. Shigenari and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 6545 CrossRef CAS PubMed.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS PubMed.
- E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650 CrossRef CAS PubMed.
- X. H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774 CrossRef CAS PubMed.
- S. R. Sardini and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 9823 CrossRef CAS PubMed.
- A. Tortajada, R. Ninokata and R. Martin, J. Am. Chem. Soc., 2018, 140, 2050 CrossRef CAS PubMed.
- V. A. Schmidt, C. R. Kennedy, M. J. Bezdek and P. J. Chirik, J. Am. Chem. Soc., 2018, 140, 3443 CrossRef CAS PubMed.
- Y. Wang and F. G. West, Synthesis, 2002, 1, 99 Search PubMed.
- E. I. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef CAS PubMed.
- S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 466 CrossRef CAS PubMed.
- X. H. Hu, J. Zhang, X. F. Yang, Y. H. Xu and T. P. Loh, J. Am. Chem. Soc., 2015, 137, 3169 CrossRef CAS PubMed.
- A. M. Olivares and D. J. Weix, J. Am. Chem. Soc., 2018, 140, 2446 CrossRef CAS PubMed.
- C. Zheng, D. Wang and S. Stahl, J. Am. Chem. Soc., 2012, 134, 16496 CrossRef CAS PubMed.
- N. J. McAlpine, L. Wang and B. P. Carrow, J. Am. Chem. Soc., 2018, 140, 13634 CrossRef CAS PubMed.
- V. T. Nguyen, H. T. Dang, H. H. Pham, V. D. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2018, 140, 8434 CrossRef CAS PubMed.
- M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805 CrossRef CAS PubMed.
- R. Parella, B. Gopalakrishnan and S. A. Babu, Org. Lett., 2013, 15, 3238 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Chandra M. R.
Volla
and
Chandra M. R.
Volla