
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 3-borylated cyclobutanols from epihalohydrins or epoxy alcohol derivatives†
Tyler R.
McDonald
 and
Sophie A. L.
Rousseaux
*
and
Sophie A. L.
Rousseaux
*
Department of Chemistry, University of Toronto. 80 St. George Street, Toronto, ON, Canada. E-mail: sophie.rousseaux@utoronto.ca
First published on 21st December 2022
Abstract
There is an increasing interest in cyclobutanes within the medicinal chemistry community. Therefore, methods to prepare cyclobutanes that contain synthetic handles for further elaboration are of interest. Herein, we report a new approach for the synthesis of 3-borylated cyclobutanols via a formal [3 + 1]-cycloaddition using readily accessible 1,1-diborylalkanes and epihalohydrins or epoxy alcohol derivatives. 1-Substituted epibromohydrin starting materials provide access to borylated cyclobutanols containing substituents at three of the four positions on the cyclobutane core, and enantioenriched epibromohydrins lead to enantioenriched cyclobutanols with high levels of enantiospecificity (>98%). Finally, derivatization studies demonstrate the synthetic utility of both the OH and Bpin handles.
Cyclobutanes are featured in a wide range of structurally interesting and biologically relevant natural products.1 Within the pharmaceutical industry, cyclobutanes have been gathering attention as they provide a rigid, sp3 rich, well-defined 3-dimensional backbone that enables them to act as bioisosteres for aromatic rings,2a,b while also allowing researchers to “escape flatland”.2
As interest in incorporating cyclobutanes into target molecules increases, so does the need to develop efficient methods for their synthesis. The preparation of borylated cyclobutanes is particularly desirable, as the boron moiety acts as a convenient synthetic handle for further elaboration via a range of C–C,3 C–N,4 C–O,5 or carbon–halogen6 bond forming processes. Current strategies to synthesize borylated cyclobutanes typically fall into four broad categories: (a) [2 + 2]-cycloadditions,7 (b) strain release/increase reactions,8 (c) C–H functionalizations,9 and (d) borylmetalation of alkenes10 (Scheme 1a).11 While significant work has gone towards the development of these methods, they often require starting materials that are not readily available and/or already contain the cyclobutane core. As such, finding new methods to rapidly access cyclobutanes is desirable.
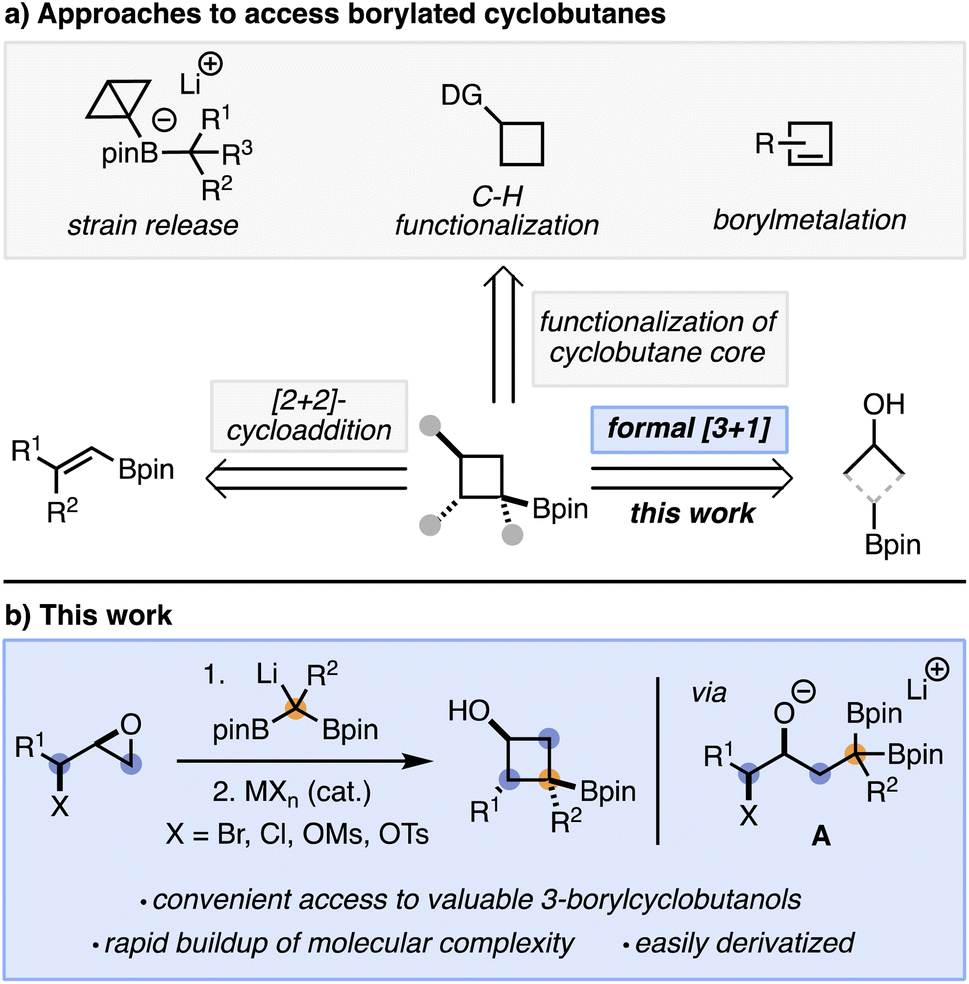 | ||
| Scheme 1 (a) Conventional methods to access borylated cyclobutanes. (b) This work. | ||
We hypothesized that a formal [3 + 1]-cycloaddition using 1,1-diborylalkanes and epoxy alcohol derivatives as stable and readily accessible starting materials could provide a convenient approach to borylated cyclobutanols while addressing some of the limitations with current methodologies (e.g. starting material synthesis). It has been known since the 1960s that, compared to boronic esters, 1,1-diborylalkanes are effective nucleophiles upon activation with a Lewis base.11a This behavior has been attributed to the second boronic ester being able to stabilize, through resonance, the negative charge resulting from Lewis base coordination and deborylation.11a Based on important contributions from the groups of Meek and Morken,11e,12 we anticipated that adding lithiated 1,1-diborylalkanes to epihalohydrins or epoxy alcohol derivatives would result in a ring opening reaction to generate an alkoxide (A, Scheme 1b), which could then act as a Lewis base and trigger the cyclization reaction to form the desired product (Scheme 1b).13–15
Herein, we report the realization of this strategy and demonstrate that the resulting cyclobutanes, which contain two complementary heteroatom handles (OH and Bpin), can be readily derivatized to form a range of cyclobutane products.3k The reaction works well with substituted 1,1-diborylalkanes and alkyl C3-biselectrophiles and is, to the best of our knowledge, the only method to directly access 3-borylated cyclobutanols.
Cyclobutanes are notoriously difficult to form via cyclization reactions: not only are they highly strained, with strain energies comparable to cyclopropanes (∼27 kcal mol−1),16 but substitutions to form cyclobutanes have high entropic and stereoelectronic barriers when compared to cyclopropane formation.17 With these challenges in mind, we began our studies by evaluating the reaction of 1a with epibromohydrin (2a) in the presence of various metal salts, solvents, and ligands (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | MXn | Solvent | Ligand | % 3a | d.r. |
| a Yields and diastereomeric ratios determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. b Toluene/HMPA ratio of 15/1 (v/v); HMPA (0.10 mL) was added to the reaction 10 minutes after adding the metal. For full details, see ESI (Table S1). | |||||
| 1 | — | THF | — | 58 | 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | ZnCl2 | THF | — | 67 | 4:1 |
| 3 | ZnCl2 | THF | bpy | 60 | 5:1 |
| 4 | ZnCl2 | THF | TMEDA | 78 | 5:1 |
| 5 | CuCl | THF | — | 75 | 2:1 |
| 6 | Zn(OTf)2 | THF | — | 77 | 4:1 |
| 7 | Zn(Cn)2 | THF | — | 96 | 6:1 |
| 8 | Zn(Cn)2 | Toluene | — | <1 | — |
| 9 | Zn(Cn)2 | Toluene/HMPAb | — | 55 | 4:1 |
|
|||||
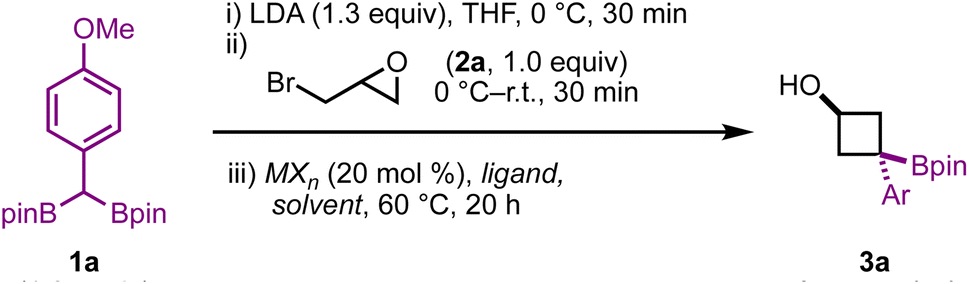
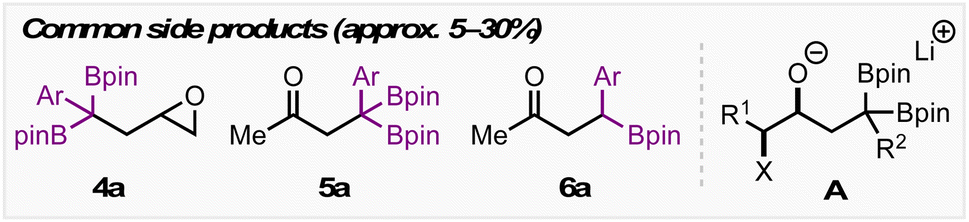
We initially screened a range of Zn salts with the idea that the boronate complex formed from A would undergo transmetalation prior to cyclization. Transmetalations of this type have been reported before,12 however initial control reactions revealed that the reaction proceeds in the absence of a metal additive. Empirically, we found that the addition of Zn(II) salts was beneficial for yield and reproducibility. These Zn(II) salts may play a role in sequestering reactive organolithium species (e.g. excess LDA or lithiated 1,1-diborylalkane) before heating – thereby limiting detrimental side reactions. The notable success of Zn(CN)2 leads us to suspect that cyanide may also play a role as a Lewis base to trigger deborylation and subsequent cyclization. Throughout optimization, side products resulting from direct substitution of the C–X bond (4a, Table 1), epoxide formation from intermediate A (4a), and semipinacol rearrangement from intermediate A (5a and 6a) were observed in yields ranging from approx. 5–30% depending on the reaction conditions employed. Using 20 mol% ZnCl2, 3a was obtained in 67% yield with a 4:1 diastereoselectivity. Various ligands were also investigated, with bpy giving similar results to the reaction free of exogenous ligand, and TMEDA giving a modest improvement in yield. Moving to Zn(OTf)2 resulted in an improved 77% yield, however we found Zn(CN)2 to be particularly effective for this transformation, with 3a being obtained in 96% yield and a 6:1 d.r. Switching to a non-polar, non-coordinating solvent such as toluene resulted in a complete shutdown of reactivity, however the addition of HMPA – which is known to facilitate substitution reactions in non-polar solvents18 – led to product formation, albeit in lower yield than when THF was used.
We next explored the scope of the reaction (Scheme 2). Overall, 1-aryl-1,1-diborylalkanes led to borylated cyclobutanols in good to excellent yields. Phenyl-substituted 1,1-diborylmethane give 3b in 85% yield and 5:1 d.r. 4-, 3-, and 2-anisyl derivatives were all competent in this reaction, leading to products 3a, 3c, and 3d respectively, in good to excellent yields. The reaction was tolerant of substitution at the ortho position(s) of the aromatic ring, as demonstrated with products 3d, 3e, and 3f. 1,1-Diborylalkanes bearing halogenated arenes are compatible with this chemistry, as seen with 3h and 3i. For 3h, ZnCl2 was found to be a more effective additive (cf. 44% yield with Zn(CN)2).19 Silyl and benzyl ether-protected phenol derivatives were compatible with the reaction conditions, leading to 3j and 3k in 74% and 51% yield, respectively. Additionally, amides (3l) and O-heterocycles (3m) are also tolerated. Notably, for 3l the reaction was performed with the magnesiated 1,1-diborylalkane, generated from magnesium diisopropylamide. Using 1,1-diborylmethane as a starting material resulted in the non-substituted borylated cyclobutanol 3n in 72% yield and 3:1 d.r.; it should be noted that CuCl was required for this substrate. Currently, 1,1-diborylalkane starting materials bearing simple alkyl substituents (e.g. R = benzyl) do not efficiently undergo the transformation.20 The inclusion of directing groups such as ethers or thioethers can partially remedy this challenge, resulting in the formation of 3o and 3p in 30% and 47% yield, respectively, when 1.0 equivalent of CuCl is used. 1,1-Diborylalkanes bearing heterocycles (e.g. pyrimidine) were unable to be tested in the reaction, as they underwent rapid protodeborylation during preparation. This decomposition is consistent with reports detailing similar systems.21
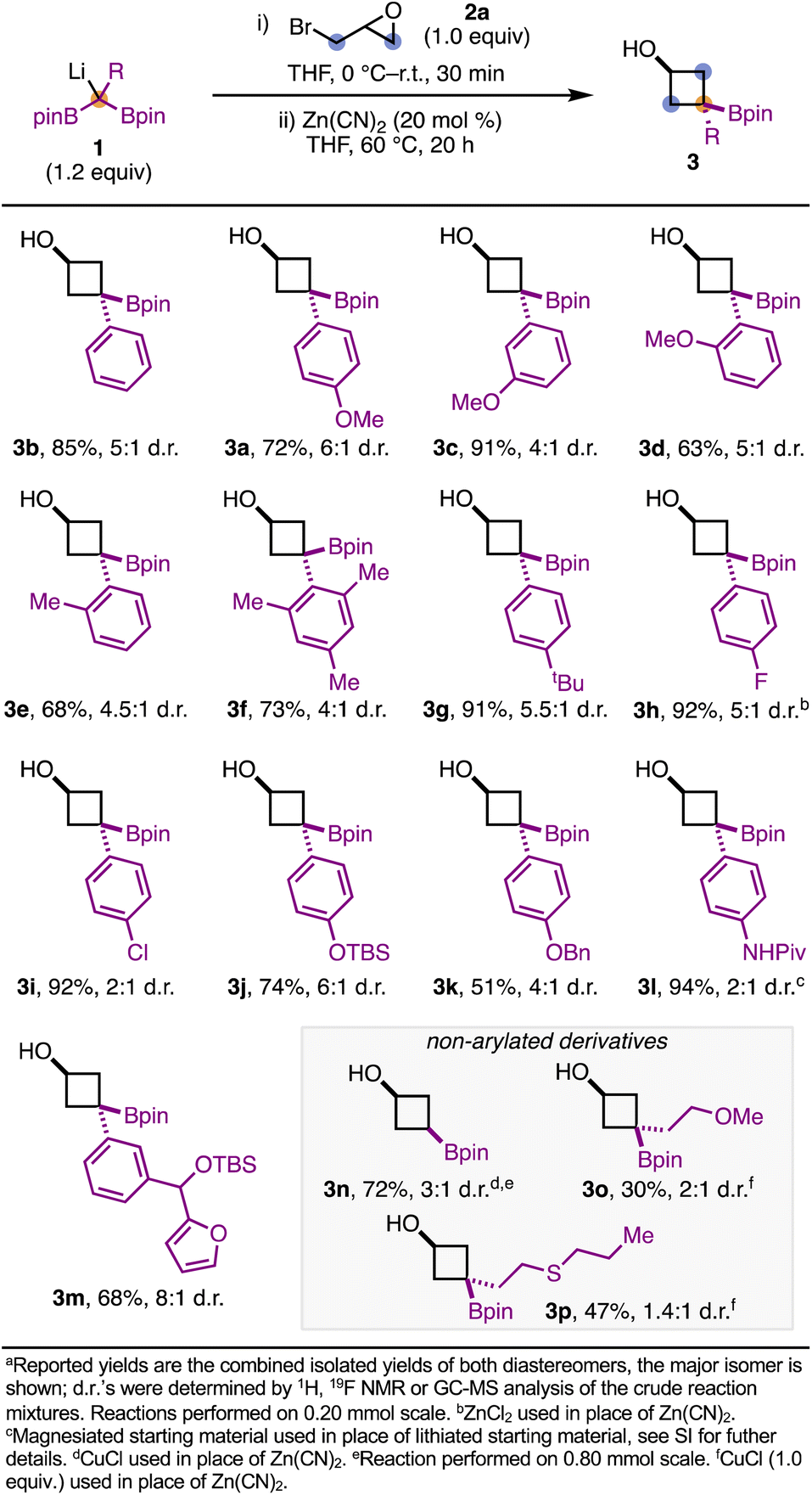 | ||
| Scheme 2 Scope of 1,1-diborylalkanes.a | ||
With respect to the 1,3-biselectrophile reaction partner, a range of different leaving groups were compatible with this chemistry (Scheme 3a). Along with epibromohydrins, epichlorohydrins were also efficient in this reaction, leading to product 3h in 87% yield and 5:1 d.r. Epoxy mesylates and tosylates were also competent, although the epoxy mesylate starting material benefited from the addition of one equivalent of LiBr. Substituted epibromohydrins were tolerated in this reaction, resulting in products (3q, 3r, 3s, and 3t) with at least one substituent at three of the four positions on the cyclobutane core (Scheme 3b and c). Notably, in all cases where substituted epibromohydrins were used, only a single diastereomer was isolated.
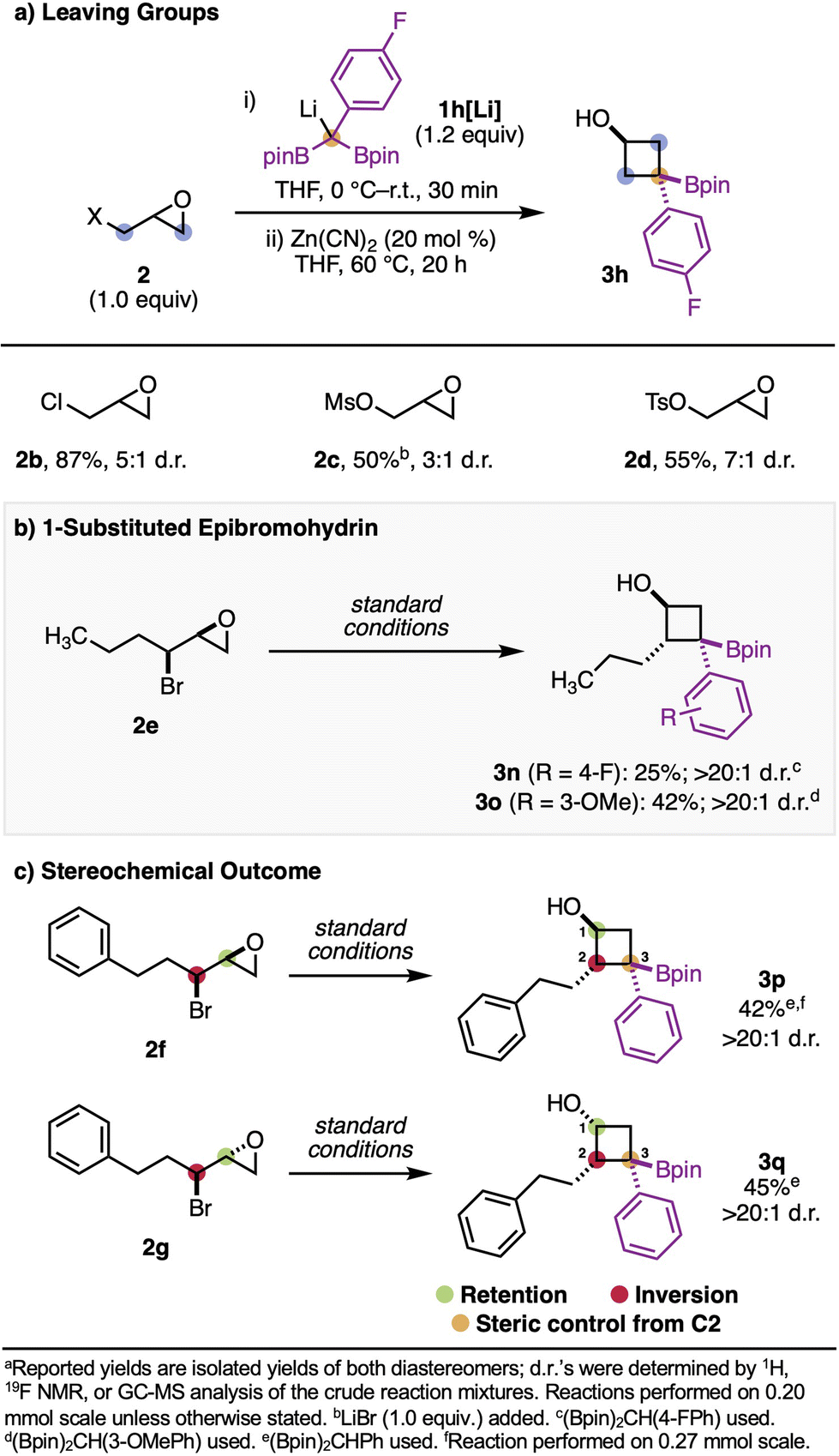 | ||
| Scheme 3 Scope of electrophiles.a | ||
The stereochemical relationship between the alcohol and the substituent at the 2-position of the cyclobutane core is determined by the stereochemical relationship of the starting epibromohydrin: syn-epibromohydrins result in trans products (3q, 3r, 3s), and anti-epibromohydrins result in cis products (3t) (Scheme 3c). Since α-boryl anions are able to planarize, we questioned whether the C3 stereocenter was controlled by steric effects from the substituent at the 2-position or by the alcohol at the 1-position (through coordination). When diastereomers 2f and 2g were tested, we found that the relative stereochemistry at C3 was primarily controlled by the substituent at C2, presumably through steric effects (Scheme 3c). This may explain why substituted epibromohydrins result in a single diastereomer of product and non-substituted epibromohydrins or epoxy alcohol derivatives do not – when there is a substituent, the steric interactions become greater and therefore selectivity is improved.
To further verify that the stereochemical information in the starting material was translated to the product, enantioenriched 1-substituted epibromohydrin 2h was used as a substrate. The reaction was found to proceed with high enantiospecificity (>98% es, Scheme 4). This is significant because enantioenriched epibromohydrins can be readily accessed via the corresponding allylic alcohols, thereby providing a synthetically convenient approach to enantioenriched, polysubstituted borylated cyclobutanols. These examples further highlight how this method enables rapid buildup of complexity from readily accessible, stereochemically defined starting materials.
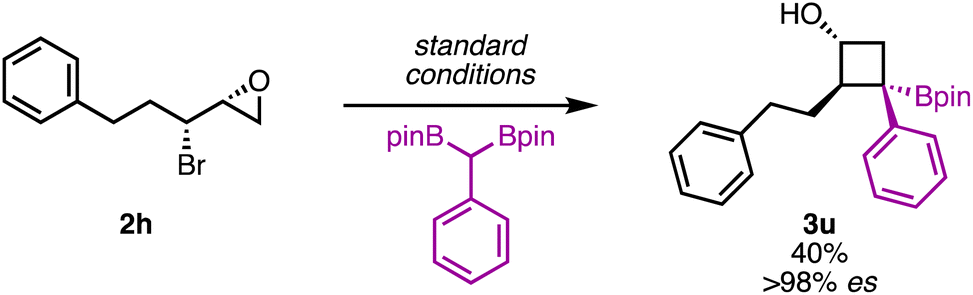 | ||
| Scheme 4 Stereospecificity using an enantioenriched epibromohydrin. | ||
The products of these reactions contain both an alcohol and a boronic ester as synthetic handles, priming them for derivatization. We focused our efforts on derivatizations that may be of particular interest to medicinal chemists. For the Bpin handle (Scheme 5a), initial TBS protection of the alcohol followed by Zweifel olefination efficiently yielded alkenylated products 3x and 3y, with 3y bearing a newly formed all-carbon quaternary stereocenter. The incorporation of heterocycles, such as furan, was possible using conditions reported by Aggarwal and coworkers, generating 3z in 80% yield.3e Oxidation of the boronic ester using sodium perborate tetrahydrate cleanly yielded cyclobutanol 3aa in 83% yield, and the presence of both a protected and an unprotected alcohol in this product should enable selective, orthogonal functionalization. Finally, a Matteson homologation resulted in the homologated product 3ab in 50% yield.
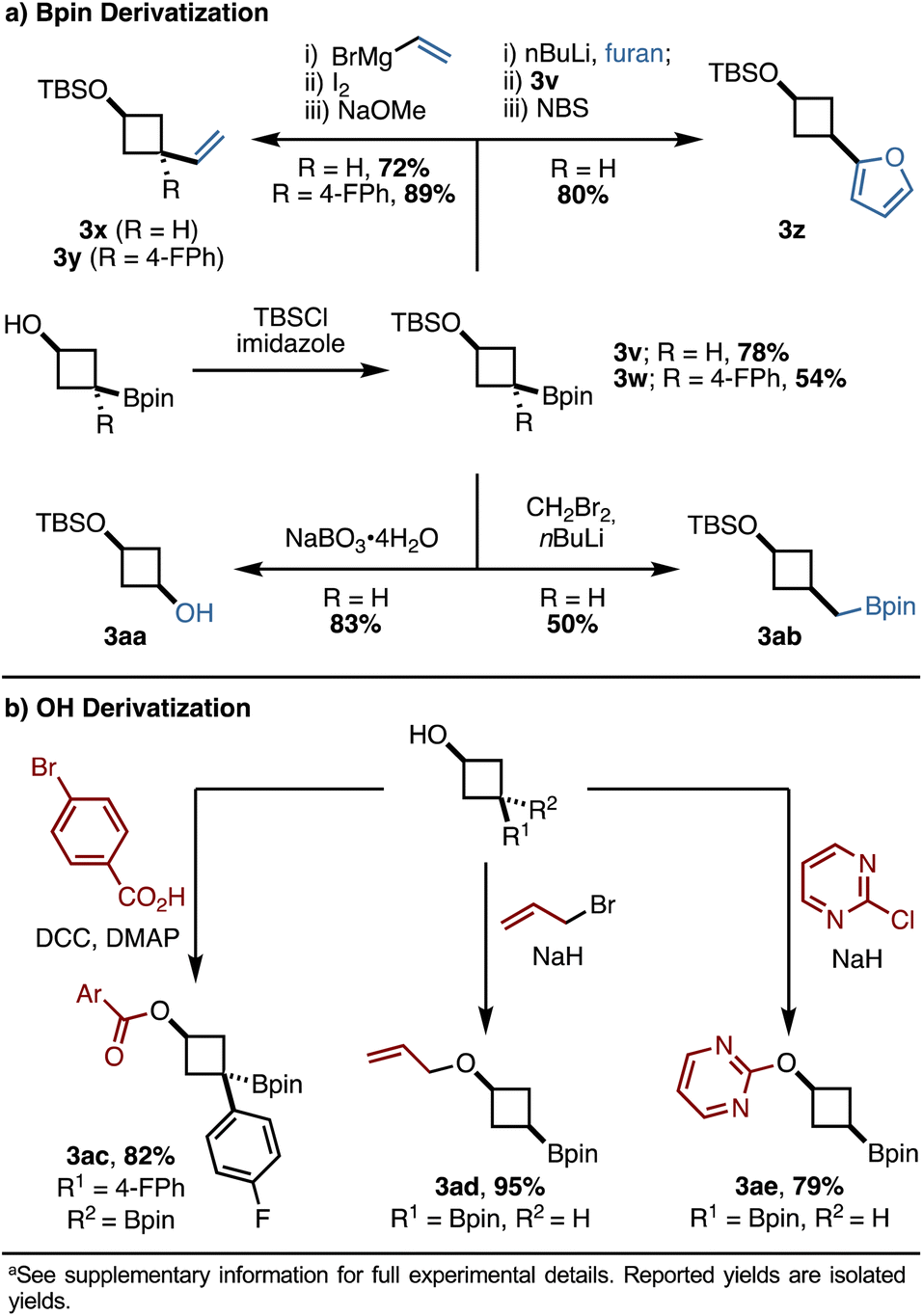 | ||
| Scheme 5 Product derivatization studies.a | ||
Functionalization of the alcohol handle was also explored (Scheme 5b). We found that esterification via DCC coupling efficiently produced 3ac in 82% yield. Etherification and SNAr reactions proceeded smoothly and led to products 3ad and 3ae, respectively, in good to excellent yields. These transformations suggest that the alkoxide can act as an effective nucleophile despite its potential sequestration as a boronate complex. Furthermore, the incorporation of heteroaromatics (3z, 3ae) via derivatization partially addresses the aforementioned difficulties in accessing heteroarene-substituted 1,1-diborylalkanes.
Throughout our studies, we noticed different reactivity patterns depending on the 1,1-diborylalkane that was being used. These differences manifested in three main observations: (i) both Zn and Cu salts effectively convert aryl substituted 1,1-diborylalkanes [(Bpin)2CHAr] to the desired cyclobutanols, whereas only Cu salts were effective with alkyl- and non-substituted starting materials; (ii) the use of Cu salts led to a diminished d.r., and (iii) aryl substituted 1,1-diborylalkanes lead to product in the absence of a metal additive, which was not the case for alkyl- and non-substituted 1,1-diborylalkanes. The observation that the reaction proceeds without the addition of a metal additive is noteworthy as it suggests that lithium-based Lewis bases can trigger boryl migration. In contrast, previous work has shown that in similar systems, Li alkoxides are rarely able to effectively promote C-to-O boryl-migrations in the absence of a transition metal.11e,15e Indeed, in their report detailing ring opening reactions with LiCH(Bpin)2 and subsequent substitutions with allylic electrophiles, Meek and coworkers found that CuCl was required, suggesting that Lewis base coordination alone was insufficient for the reaction to proceed.12 In our case, the additional stabilization provided by an aryl substituent may allow the transfer to occur, suggesting that this reaction may proceed through Lewis base activation and does not require transmetalation. With this in mind, the observation that CuCl is required for unsubstituted and alkyl-substituted 1,1-diborylalkane starting materials suggests that there may be a change in mechanism for these substrates, and that transmetalation is necessary for product formation (Fig. S16 and S17†). Further mechanistic studies are currently ongoing in our laboratory to better understand the unique interplay between the substrate structure and reaction mechanism.
In summary, we have developed a formal [3 + 1]-cycloaddition to generate 3-borylated cyclobutanols. This reaction takes advantage of epihalohydrins and epoxy alcohol derivatives as C3-biselectrophiles and lithiated 1,1-diborylalkanes as C1-bisnucleophiles. 1-Substituted epibromohydrins resulted in the formation of highly substituted borylated cyclobutanols, allowing for rapid buildup of molecular complexity within a single transformation. When 1-substituted epibromohydrins are used, a single diastereomer is obtained, with the stereochemistry at the C3 position being controlled by the substituent at C2. The reaction proceeded with high levels of stereospecificity, and when enantioenriched 1-substituted epibromohydrins were used, enantioenriched products were obtained. Finally, both the alcohol and the boronic ester were demonstrated to be orthogonal synthetic handles, allowing for convenient derivatization and elaboration of the products. We anticipate that these synthetic handles may alleviate some of the challenges regarding incorporation of cyclobutanes into more complex molecular scaffolds and are continuing to explore their potential in our laboratory.
Data availability
The synthetic procedures, characterization, and spectral data supporting this article have been uploaded as part of the ESI.†Author contributions
T. R. M. and S. A. L. R. contributed to project conception, experiment design and analysis, and the writing of this manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank NSERC (Discovery Grants and Canada Research Chair programs), the Canada Foundation for Innovation (Project No. 35261), the Ontario Research Fund, the Alfred P. Sloan Foundation, and the University of Toronto for generous financial support of this work. We also acknowledge the Canada Foundation for Innovation (Project No. 19119) and the Ontario Research Fund for funding the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers. T. R. M thanks NSERC for a graduate scholarship (CGS D). We would like to thank John J. Monteith for verifying reaction reproducibility.References
- (a) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC; (b) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254–259 RSC; (c) Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China: Chem., 2016, 59, 1126–1141 CrossRef CAS; (d) A. Sergeiko, V. V. Poroikov, L. O. Hanuš and V. M. Dembitsky, Open Med. Chem. J., 2008, 2, 26–37 CrossRef CAS.
- (a) K. C. Nicolaou, D. Vourloumis, S. Totokotsopoulos, A. Papakyriakou, H. Karsunky, H. Fernando, J. Gavrilyuk, D. Webb and A. F. Stepan, ChemMedChem, 2016, 11, 31–37 CrossRef CAS; (b) A. F. Stepan, C. Subramanyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414–3424 CrossRef CAS; (c) K. C. Nicolaou, Angew. Chem., Int. Ed., 2014, 53, 9128–9140 CrossRef CAS; (d) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS; (e) D. Blanco-Ania, S. A. Gawade, L. J. L. Zwinkels, L. Maartense, M. G. Bolster, J. C. J. Benningshof and F. P. J. T. Rutjes, Org. Process Res. Dev., 2016, 20, 409–413 CrossRef CAS; (f) P. Arya, R. Joseph, Z. Gan and B. Rakic, Chem. Biol., 2005, 12, 163–180 CrossRef CAS PubMed; (g) S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS; (h) R. Breinbauer, I. R. Vetter and H. Waldmann, Angew. Chem., Int. Ed., 2002, 41, 2878–2890 CrossRef CAS; (i) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1484 CrossRef CAS PubMed; (j) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef CAS.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS; (b) D. A. Evans, T. C. Crawford, R. C. Thomas and J. A. Walker, J. Org. Chem., 1976, 41, 3947–3953 CrossRef CAS; (c) R. J. Armstrong, C. García-Ruiz, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 786–790 CrossRef CAS PubMed; (d) Y. Wang, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 4270–4274 CrossRef CAS . For heteroaryl cross-couplings, see:; (e) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS; (f) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef CAS PubMed; (g) J. Llaveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958–10961 CrossRef CAS . For reviews on cross-couplings and functionalizations of boronic esters, see:; (h) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (i) A. Suzuki, Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (j) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS; (k) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC; (l) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CrossRef CAS.
- (a) S. N. Mlynarski, A. S. Karns and J. P. Morken, J. Am. Chem. Soc., 2012, 134, 16449–16451 CrossRef CAS; (b) A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054–16057 CrossRef CAS.
- (a) H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 2544 CrossRef CAS; (b) A. G. Davies and B. P. Roberts, J. Chem. Soc. B, 1967, 17 RSC; (c) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS; (d) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142 CrossRef CAS; (e) G. W. Kabalka, T. M. Shoup and N. M Goudgaon, Tetrahedron Lett., 1989, 30, 1483 CrossRef CAS; (f) S. Radomkit and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 53, 3387 CrossRef CAS PubMed.
- (a) H. C. Brown and C. F. Lane, J. Chem. Soc. D, 1971, 521, 2544–2551 Search PubMed; (b) H. C. Brown, N. R. De Lue, G. W. Kabalka and H. C. Hedgecock Jr, J. Am. Chem. Soc., 1976, 98, 1290–1291 CrossRef CAS; (c) H. C. Brown and C. F. Lane, Tetrahedron, 1988, 44, 2763–2772 CAS; (d) C. Sandford, R. Rasappan and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10100–10103 CrossRef CAS; (e) R. Larouche-Gautheir, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2011, 133, 16794–16797 CrossRef.
- (a) R. H. Fish, J. Org. Chem., 1969, 34, 1127–1128 CrossRef CAS; (b) W. G. Hollis Jr, W. C. Lappenbusch, K. A. Everberg and C. M. Woleben, Tetrahedron Lett., 1993, 34, 7517–7520 CrossRef; (c) S. C. Coote and T. Bach, J. Am. Chem. Soc., 2013, 135, 14948–14951 CrossRef CAS; (d) S. C. Coote, A. Pöthig and T. Bach, Chem.–Eur. J., 2015, 21, 6906–6912 CrossRef CAS PubMed; (e) R. A. Kleinnijenhuis, B. J. J. Timmer, G. Lutteke, J. M. M. Smits, R. de Gelder, J. H. van Maarseveen and H. Hiemstra, Chem.–Eur. J., 2016, 22, 1266–1269 CrossRef CAS; (f) M. M. Parsutkar, V. V. Pagar and T. V. RajanBabu, J. Am. Chem. Soc., 2019, 141, 15367–15377 CrossRef CAS PubMed; (g) O. P. Demchuck, O. V. Hryshchuk, B. V. Vashchenko, A. V. Kozytskiy, A. V. Tymtsunik, I. V. Komarov and O. O. Grygorenko, J. Org. Chem., 2020, 85, 5927–5940 CrossRef PubMed; (h) S. O. Scholz, J. B. Kidd, L. Capaldo, N. E. Flikweert, R. M. Littlefield and T. P. Yoon, Org. Lett., 2021, 23, 3496–3501 CrossRef CAS; (i) Y. Liu, D. Ni, B. G. Stevenson, V. Tripathy, S. E. Braley, K. Raghavachari, J. R. Swierk and M. K. Brown, Angew. Chem., Int. Ed., 2022, 61, e202200725 CAS; (j) Y. Liu, D. Ni and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 18790–18796 CrossRef CAS.
- (a) Z. X. Giustra, X. Yang, M. Chen, H. F. Bettinger and S.-Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 18918–18922 CrossRef CAS PubMed; (b) M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS; (c) A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117–122 CrossRef CAS; (d) R. Davenport, M. Silvi, A. Noble, Z. Hosni, N. Fey and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 6525–6528 CrossRef CAS PubMed; (e) S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 16766–16775 CrossRef CAS PubMed; (f) D. P. Hari, J. C. Abell, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 5515–5520 CrossRef CAS PubMed; (g) L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2021, 60, 212–216 CrossRef CAS.
- (a) R. Murakami, K. Tsunoda, T. Iwai and M. Sawamura, Chem.–Eur. J., 2014, 20, 13127–13131 CrossRef CAS PubMed; (b) J. He, Q. Shao, Q. Wu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 3344–3347 CrossRef CAS; (c) X. Chen, L. Chen, H. Zhao, Q. Gao, Z. Shen and S. Xu, Chin. J. Chem., 2020, 38, 1533–1537 CrossRef CAS; (d) R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. Hartwig, Science, 2020, 368, 736–741 CrossRef CAS.
- (a) H. Ito, T. Toyoda and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 5990–5992 CrossRef CAS PubMed; (b) M. Guisán-Ceinos, A. Parra, V. Martín-Heras and M. Tortosa, Angew. Chem., Int. Ed., 2016, 55, 6969–6972 CrossRef; (c) J. A. M. Mercer, C. M. Cohen, S. R. Shuken, A. M. Wagner, M. W. Smith, F. R. Moss III, M. D. Smith, R. Vahala, A. Gonzalez-Martinez, S. G. Boxer and N. Z. Burns, J. Am. Chem. Soc., 2016, 138, 15845–15848 CrossRef CAS PubMed; (d) K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 851–855 CrossRef CAS PubMed; (e) H. A. Clement, M. Boghi, R. M. McDonald, L. Bernier, J. W. Coe, W. Farrell, C. J. Helal, M. R. Reese, N. W. Sach, J. C. Lee and D. G. Hall, Angew. Chem., Int. Ed., 2019, 58, 18405–18409 CrossRef CAS; (f) E. N. Hancock, E. L. Kuker, D. J. Tantillo and M. K. Brown, Angew. Chem., Int. Ed., 2020, 59, 436–441 CrossRef CAS; (g) L. Nóvoa, L. Trulli, A. Parra and M. Tortosa, Angew. Chem., Int. Ed., 2021, 60, 11763–11768 CrossRef PubMed; (h) A. K. Simlandy, M.-Y. Lyu and M. K. Brown, ACS Catal., 2021, 11, 12815–12820 CrossRef CAS; (i) K. Nguyen, H. A. Clement, L. Bernier, J. W. Coe, W. Farrell, C. J. Helal, M. R. Reese, N. W. Sach, J. C. Lee and D. G. Hall, ACS Catal., 2021, 11, 404–413 CrossRef CAS; (j) L. Nóvoa, L. Trulli, I. Fernández, A. Parra and M. Tortosa, Org. Lett., 2021, 23, 7434–7438 CrossRef.
- For other miscellaneous methods, see: (a) S. P. Rhodes and H. C. Brown, J. Am. Chem. Soc., 1969, 91, 4306–4307 CrossRef; (b) I. D. Gridnev and A. Meller, J. Org. Chem., 1998, 63, 3599–3606 CrossRef CAS; (c) H.-W. Man, W. C. Hiscox and D. S. Matteson, Org. Lett., 1999, 1, 379–381 CrossRef CAS PubMed; (d) T. C. Atack, R. M. Lecker and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 9521–9523 CrossRef CAS PubMed; (e) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581–10584 CrossRef CAS PubMed; (f) X.-F. Zhou, Y.-D. Wu, J.-J. Dai, Y.-J. Li, Y. Huang and H.-J. Xu, RSC Adv., 2015, 5, 46672–46676 RSC; (g) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS; (h) C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870–3874 CrossRef CAS; (i) J. C. Beck, C. R. Lacker, L. M. Chapman and S. E. Reisman, Chem. Sci., 2019, 10, 2315–2319 RSC; (j) D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2019, 141, 14126–14130 CrossRef CAS; (k) J. Michalland, N. Casaretto and S. Z. Zard, Angew. Chem., Int. Ed., 2022, 61, e202113333 CrossRef CAS.
- S. A. Murray, M. Z. Liang and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 14061–14064 CrossRef CAS PubMed.
- (a) J. J. Eisch, S. K. Dua and M. Behrooz, J. Org. Chem., 1985, 50, 3674–3676 CrossRef CAS; (b) J. E. Jeffery, F. Kerrigan, T. K. Miller, G. J. Smith and G. B. Tometzki, J. Chem. Soc., Perkin Trans. 1, 1996, 2583–2589 RSC; (c) W.-C. Cheng, C. Halm, J. B. Evarts, M. M. Olmstead and M. J. Kurth, J. Org. Chem., 1999, 64, 8557–8562 CrossRef CAS; (d) J. Vogelgesang, A. Frick, G. Huttner and P. Kircher, Eur. J. Inorg. Chem., 2001, 949–971 CrossRef CAS; (e) P. Grongsaard, P. G. Bulger, D. J. Wallace, L. Tan, Q. Chen, S. J. Dolman, J. Nyrop, R. S. Hoerrner, M. Weisel, J. Arredondo, T. Itoh, C. Xie, X. Wen, D. Zhao, D. J. Muzzio, E. M. Bassan and C. S. Shultz, Org. Process Res. Dev., 2012, 16, 1069–1081 CrossRef CAS; (f) M. Jung and V. N. G. Lindsay, J. Am. Chem. Soc., 2022, 144, 4764–4769 CrossRef CAS.
- For reviews on the synthesis and reactivity of 1,1-diborylalkanes, see: (a) D. S. Matteson, Synthesis, 1975, 3, 147–157 CrossRef; (b) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306–1327 CrossRef CAS; (c) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC; (d) Y. Lee, S. Han and S. H. Cho, Acc. Chem. Res., 2021, 54, 3917–3929 CrossRef CAS; (e) C. Zhang, W. Hu and J. P. Morken, ACS Catal., 2021, 11, 10660–10680 CrossRef CAS; (f) A. B. Cuenca and E. Fernández, Chem. Soc. Rev., 2021, 50, 72–86 RSC.
- For miscellaneous examples of reactions using 1,1-diborylalkanes, see: (a) W. Jo, J. Kim, S. Choi and S. H. Cho, Angew. Chem., Int. Ed., 2016, 128, 9842–9846 CrossRef; (b) A. Ebrahim-Alkhalil, Z.-Q. Zhang, T.-J. Gong, W. Su, X.-Y. Lu, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 4891–4893 RSC; (c) R. Gava and E. Fernández, Chem.–Eur. J., 2019, 25, 8013–8017 CrossRef CAS PubMed; (d) O. Salvado, R. Gava and E. Fernández, Org. Lett., 2019, 21, 9247–9250 CrossRef CAS; (e) B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed.
- J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC.
- P. Deslongchamps, Stereoelectronic Effects in Organic Chemistry, Elsevier Science Limited, University of Michigan, 1983 Search PubMed.
- R. R. Dykstra, Hexamethylphosphoric Triamide in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, 2001 Search PubMed.
- When Zn(CN)2 is used for this substrate the major side product is protodemetalated starting material. We suspected that this may have been due to the Lewis basicity of the cyanide anion, which prompted us to attempt the reaction using less Lewis basic ZnCl2.
- This is in agreement with previous observations made by Meek and coworkers. See ref 12. Instead, the product resulting from semi-pinacol rearrangement of intermediate A is obtained as the major side-product.
- X. Ma and Y. Jiang, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-96vc8. This content is a preprint and has not been peer-reviewed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06088d |
| This journal is © The Royal Society of Chemistry 2023 |