
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTurn-on fluorescent capsule for selective fluoride detection and water purification†
Yannan
Lin
,
Kang
Du
,
Michael R.
Gau
and
Ivan J.
Dmochowski
 *
*
Department of Chemistry, University of Pennsylvania, 231 S. 34th St., Philadelphia, Pennsylvania 19104−6323, USA. E-mail: ivandmo@sas.upenn.edu
First published on 30th November 2022
Abstract
It has been a long-standing challenge to develop organic molecular capsules for selective anion binding in water. Here, selective recognition of aqueous fluoride was achieved through triple protonation of a hemicryptophane (L), which is composed of a fluorescent cyclotriveratrylene (CTV) cap and tris(2-aminoethyl)amine (tren) as the anion binding site. Fluoride encapsulation by [3H-L]3+ was evidenced by 1H NMR, 19F NMR, LC-MS, and X-ray crystallography. In addition, [3H-L]3+ exhibited a ‘turn-on’ fluorescence signal (λem = 324 nm) upon fluoride addition. An apparent association constant KA = (7.5 ± 0.4) × 104 M−1 and a detection limit of 570 nM fluoride were extracted from the fluorescence titration experiments in citrate buffer at pH 4.1. To the best of our knowledge, [3H-L]3+ is the first example of a metal-free molecular capsule that reports on fluoride binding in purely aqueous solutions with a fluorescence response. Finally, the protonated capsule was supported on silica gel, which enabled adsorptive removal of stoichiometric fluoride from water and highlights real-world applications of this organic host–guest chemistry.
Introduction
Fluoride in saliva helps to remineralize the tooth enamel that is degraded by acid-releasing oral bacteria;1,2 however, fluoride accumulates in humans in trace quantities3 and is not essential for life. Fluoride binds to the sodium/iodide symporter and inhibits iodine absorption in humans.4 Prolonged exposure to high levels of fluoride produces brittle bones and joint stiffness, a condition known as skeletal fluorosis.5 Excess consumption can also damage the liver and kidneys.6,7 To ensure that fluoride levels in public water supplies remain within permissible limits (<1.5 mg L−1),8 strategies have been sought for sensitive fluoride detection and water remediation. Techniques such as the selective electrode method, 19F NMR analysis, and reverse-phase HPLC are available for fluoride determination.9–11 Compared to other analytical methods, fluoride detection using fluorescent sensors12–21 offers the advantages of simple operation, good sensitivity, and compatibility with biological systems. For example, chemosensors based on fluoride-triggered cleavage of Si–O or Si–C bonds have been extensively investigated,22–25 and a number of them have been reported to detect aqueous fluoride in water.26–35 These sensors can achieve high signal-to-noise ratios but require relatively long acquisition times and lack a dynamic, reversible response. Some of these probes also suffer from poor anion selectivity with severe interference by cyanide ions.36 As an alternative strategy, fluorescent probes that are designed based on molecular recognition and host–guest chemistry can operate in a reversible fashion. Indeed, most of these chemosensors are only operative in organic or organo-aqueous solvents due to the high hydration enthalpy of fluoride ion (ΔH° = −465 kJ mol−1)37 and/or the hydrophobicity of the probes. So far, only a few cases in which fluoride recognition was achieved in 100% aqueous medium have been reported in the literature, which includes using coordinatively unsaturated metal complexes that display different optical properties upon fluoride binding.38–45 In addition, fluoride recognition by synthetic organic receptors in water can be achieved by using polyammonium macrocycles and molecular capsules.46–51 On one hand, the macrocycles report fluoride binding with fluorescence response, but they cannot isolate a single bound fluoride anion from water and achieve selective and stoichiometric fluoride recognition. On the other hand, the molecular capsules exhibit strong affinity for fluoride but are not capable as fluorescent or colorimetric chemosensors for fluoride detection without relying on metal ion binding. For many applications in water purification and biological sensing, there are advantages to working with metal-free sensors. With these considerations in mind, we were motivated to develop a metal-free molecular capsule that can selectively detect stoichiometric fluoride in pure water with a reversible fluorescence response. To our knowledge, no such capsule has yet been reported.Meanwhile, fluoride removal from drinking water is an expensive and challenging process because the fluoride ions are highly soluble in water. At present, numerous methods have been developed including electrodialysis, reverse osmosis, ion exchange, precipitation–coagulation, and adsorption.52,53 Among these defluoridation strategies, the adsorption technique has been considered as the most promising treatment method for water remediation due to its low cost, ease of operation, and excellent efficiency.54,55 To date, a wide variety of metal-containing adsorbents such as activated alumina, zeolites, rare earth metal oxides/hydroxides, and metal–organic frameworks (MOFs) have been reported.56–60 While many of these materials exhibit fast adsorption rate and high adsorption capacity for fluoride, they could pose severe threats to human health because some metals may leach into water during the adsorption process.61,62 To avoid potential health risk of metal leaching, a number of metal-free fluoride sorbents such as activated charcoal, chitosan beads, and carbon nanotubes have also been reported. However, they generally suffer from poor adsorption capacity or high cost.63–66 Therefore, it is advantageous to develop a metal-free adsorbent that enables efficient water defluoridation and a process that includes efficient recycling of materials.
In this context, we sought to turn a known hemicryptophane L (ref. 67) into a fluorescent fluoride receptor by taking advantage of its cyclotriveratrylene (CTV) cap as the fluorophore and the tripodal tren unit as the fluoride binding site. By triply protonating the capsule to generate [3H-L]3+, we previously enhanced its affinity for a series of size-matched polar molecules and modulated guest binding with sulfonate counteranions in aprotic organic solvents.68 To expand its host–guest chemistry, [3H-L]3+ can be made water-soluble through the judicious choice of counteranions. Here, we report a protonated tren-based hemicryptophane [3H-L][OMs]3 (−OMs = methanesulfonate anion) that operates as a ‘turn-on’ fluorescent probe for selective detection of aqueous fluoride and also as a highly efficient fluoride sorbent when supported on silica gel. Multifunctional systems that can be used for both detection and removal of aqueous fluoride have been rarely explored.
Results and discussion
The ability of [3H-L][OMs]3 to bind anions in solution was first investigated in D2O by 1H NMR spectroscopy. As some of the anions may significantly change the acidity of the solution after titration, all spectra were recorded at the same pD to ensure that the hemicryptophane remained at a constant protonation state throughout the experiments. We chose to work at pD 4.5, the pD value of the aqueous solution of [3H-L][OMs]3 when dissolved in D2O. At this pD value, the receptors exist as a mixture of triply and doubly protonated capsules (Fig. S1–S3 and Table S1†). When [3H-L][OMs]3 was exposed to a variety of common anions including excess Cl−, Br−, NO3−, CH3CO2−, and H2PO4− (as the sodium salts), only minor 1H NMR spectroscopic changes were observed (Fig. 1a–f). This indicated a lack of anion encapsulation by [3H-L]3+, which is attributed to the rigidity conferred by the CTV and the small diameter of the portals that the anions must traverse to bind stably within the capsule (Scheme 1). | ||
| Fig. 1 1H NMR spectra (600 MHz, 298 K, D2O, pD 4.5 ± 0.1) of 0.5 mM [3H-L][OMs]3 before (a) and after the addition of 10 eq. (b) NaCl; (c) NaBr; (d) NaNO3; (e) NaOAc; (f) NaH2PO4; (g) NaF. | ||
 | ||
| Scheme 1 Structures of L and [3H-L][OMs]3. | ||
In contrast, introducing sulfate or fluoride to [3H-L][OMs]3 gave rise to significantly different observations. While the addition of sulfate led to immediate precipitation due to the formation of insoluble salts, the addition of fluoride resulted in dramatic spectroscopic changes that were consistent with anion binding (Fig. 1g). As the protonated capsule was titrated with NaF, only one set of resonances was seen for all proton signals during the whole experiment (Fig. S4†). This observation led us to conclude that the fluoride binding equilibrium is fast on the 1H NMR timescale. Over the course of the spectral titration, the peaks corresponding to the proton resonances of Ha, Hb, Hc and Hd diverged into three sets of well-separated sharp multiplets while undergoing a noticeable downfield shift, consistent with fluoride binding at the tren moiety. As the protons He and Hf on the p-phenylene linkers display well-defined signals during the entire titration experiment, their chemical shifts were plotted as a function of the host–guest ratio (Fig. S5†). However, both sets of chemical shifts changed linearly with the amount of NaF added, up to 1 equivalent of NaF. In order to determine an association constant (KA) for F− binding to [3H-L][OMs]3, the titration needs to be done at lower concentration with a more sensitive analytical technique.
The encapsulation of F− in the cavity of [3H-L][OMs]3 was also evidenced by 19F NMR spectroscopy. While the ‘free’ fluoride resonates at −124.4 ppm (Fig. 2a), the addition of 2 equivalents of [3H-L][OMs]3 to the NaF solution eliminated the ‘free’ fluoride signal and induced the appearance of a second peak that was 52 ppm downfield shifted (Fig. 2b). Subsequent addition of excess NaF to the mixture restored the ‘free’ fluoride signal but barely shifted this new peak (Fig. 2c). Given the large overall charge of the protonated tren unit (3+) and the substantial deshielding effect it confers on the encapsulated species, the peak at −72.4 ppm in the 19F NMR spectrum can be reasonably assigned as the ‘bound’ fluoride signal. The chemical shift of this ‘bound’ fluoride is comparable to the one reported by Bowman-James et al. in a p-xylyl-linked bis-tren construct (δ = −78 ppm).47 It should be noted that no additional fluoride species such as HF or HF2− were observed throughout the experiment (Fig. S6†).
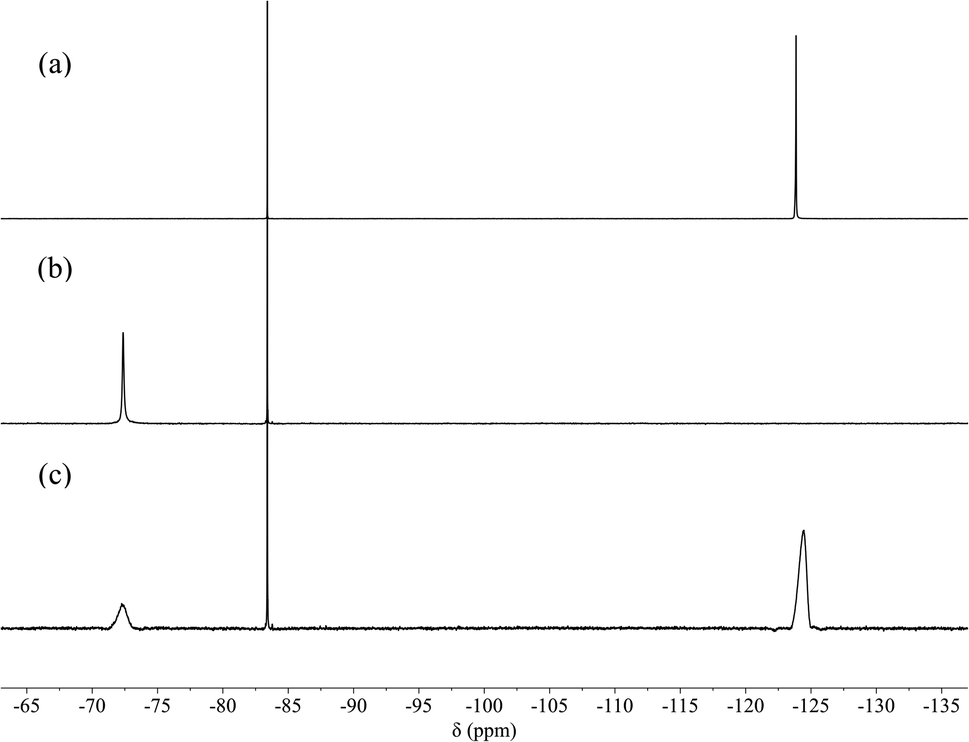 | ||
| Fig. 2 19F NMR spectra (565 MHz, 298 K, D2O, pD 4.5 ± 0.1, hexafluoroacetone as external reference) of (a) 0.75 mM NaF (1 eq.); (b) 1.5 mM [3H-L][OMs]3 (2 eq.) with 0.75 mM NaF (1 eq.); (c) 1.5 mM [3H-L][OMs]3 (2 eq.) with 4.5 mM NaF (6 eq.). | ||
To obtain more information about fluoride binding to the protonated hemicryptophane in the solution phase, an aqueous solution of [3H-L][OMs]3 was mixed with excess fluoride and subject to LC-MS. The positive mode of ESI-MS revealed that the capsule binds fluoride in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, with an m/z peak at 410.71 that was assigned to [L + 2H+]2+ and an m/z peak at 841.97 that was assigned to [L + 2H+ + F−]+ (Fig. 3). No other species were detected by mass spectrometry besides the fluoride-bound and unbound capsules, confirming the lack of reactivity between [3H-L][OMs]3 and F−.
:1 stoichiometry, with an m/z peak at 410.71 that was assigned to [L + 2H+]2+ and an m/z peak at 841.97 that was assigned to [L + 2H+ + F−]+ (Fig. 3). No other species were detected by mass spectrometry besides the fluoride-bound and unbound capsules, confirming the lack of reactivity between [3H-L][OMs]3 and F−.
 | ||
| Fig. 3 Positive ESI-MS spectrum of [3H-L][OMs]3 with 30 eq. NaF in H2O. | ||
Consistent with the observation in solution, fluoride binding to the protonated capsule was also confirmed in the solid state (Fig. 4). Single crystals suitable for structural analysis were grown by diffusing diethyl ether into a mixture of [3H-L][OMs]3 and 1.2 equiv. of NaF in methanol. The fluoride-encapsulated complex crystalized in the ![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) 3 space group with a F− ion residing on the same C3 axis with L. The occupancy of F− was found to be 1 with reasonable refinement parameters, consistent with the stoichiometric encapsulation of fluoride in solution. Importantly, the short F−⋯NH2+ distance, 2.629(4) Å, suggests the presence of strong H bonds between F− and the protonated tren moiety and provides structural evidence for the high F− affinity observed in solution. More details on structural refinements can be found in ESI (Table S2†).
3 space group with a F− ion residing on the same C3 axis with L. The occupancy of F− was found to be 1 with reasonable refinement parameters, consistent with the stoichiometric encapsulation of fluoride in solution. Importantly, the short F−⋯NH2+ distance, 2.629(4) Å, suggests the presence of strong H bonds between F− and the protonated tren moiety and provides structural evidence for the high F− affinity observed in solution. More details on structural refinements can be found in ESI (Table S2†).
 | ||
| Fig. 4 Crystal structure of the hemicryptophane with a single fluoride encapsulated within the protonated tren unit. Atoms are shown as ball and stick model for clarity. Gray, blue, red, green, and light-yellow represent the C, N, O, F, and H atoms, respectively. Solvent molecules, other counteranions and H atoms are omitted for clarity, except for those involved in the hydrogen bonds as light-yellow spheres. | ||
Although the encapsulation of F− by [3H-L][OMs]3 was unambiguously disclosed by NMR, LC-MS, and XRD, all the experiments were run at low millimolar concentrations. Given that the CTV has been used as a fluorophore to assess xenon complexation with numerous cryptophanes,69,70 we suspected that the selective fluoride binding revealed by 1H NMR may also induce a fluorescence response, which could potentially enable fluoride detection in the micromolar regime. In order to verify this hypothesis, an aqueous solution of 15 μM [3H-L][OMs]3 was excited at 280 nm and the emission spectra were collected in the presence of the same series of anions. To ensure that the capsules were characterized at the same protonation state as in the NMR experiments, the fluorescence spectra were recorded at pH 4.1, which is equivalent to pD 4.5.71 As expected, with the addition of Cl−, Br−, NO3−, CH3CO2−, H2PO4− and SO42−, only minor changes in the emission spectrum of [3H-L][OMs]3 were observed in the citrate buffer (Fig. 5a) or pure water (Fig. S7†). We noted that titrating excess SO42− to the capsule no longer led to sample precipitation due to the much lower concentration. In contrast, the introduction of excess F− led to a 5-fold fluorescence enhancement of the solution.
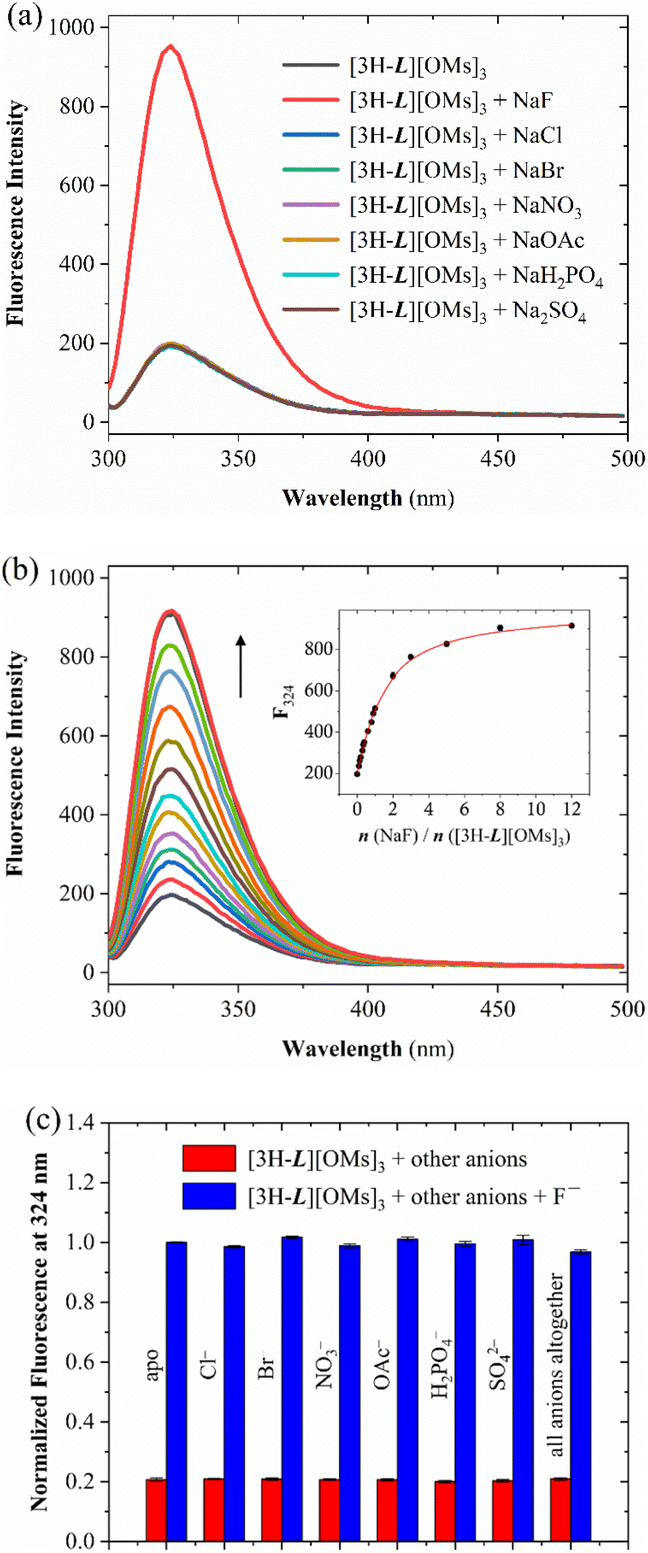 | ||
| Fig. 5 (a) Fluorescence spectra at 298 K of an aqueous solution of 15 μM [3H-L][OMs]3 with 10 eq. of different anions titrated in citrate buffer (0.1 M, H2O) at pH 4.1. (b) Fluorescence titration of 15 μM [3H-L][OMs]3 with NaF at 298 K in citrate buffer (0.1 M, H2O) at pH 4.1. Inset: the fluorescence intensity at 324 nm as a function of the equivalents of added NaF. The black spheres and red line represent experimental data and a global fit given by Bindfit program,78 respectively. (c) Fluorescence response of [3H-L][OMs]3 to F− in the presence of competing anions in citrate buffer (0.1 M, H2O) at pH 4.1. The red bars show the fluorescence intensity of 15 μM [3H-L][OMs]3 at 324 nm on the addition of respective anions (10 eq.). The blue bars represent the fluorescence intensity of 15 μM [3H-L][OMs]3 with 10 eq. F− at 324 nm on the addition of respective competing anions (10 eq.). The error bars represent standard deviations of averaging three measurements. | ||
Several experiments were undertaken to investigate this fluoride-specific fluorescence ‘turn-on’ effect. By comparing the emission spectrum of an aqueous solution of [3H-L][OMs]3 before and after degassing, we concluded that the fluorescence enhancement associated with F− binding is not associated with changes in dioxygen quenching (Fig. S8–S9†).72 We also considered whether this turn-on effect is related to the encapsulation of water, which acts as an ‘universal’ fluorescence quencher for organic fluorophores73 and has been reported to bind at the tren in a number of protonated azacryptands74,75 and calix[6]arenes.76 Thus, we hypothesized that fluoride binding to the capsule could displace the water and thereby enhance the fluorescence of [3H-L][OMs]3. To investigate this possible mechanism, fluorescence titration experiments were performed to determine the association constant (KA) between [3H-L][OMs]3 and F− in D2O and H2O under the same protonation states. Based on the 1:1 host–guest binding model that was suggested by LC-MS, crystal structure, and Job plot analysis (Fig. S10†), the apparent KA for fluoride was calculated to be (1.4 ± 0.1) × 105 M−1 in citrate buffer (0.1 M, D2O) at pD 4.5 (Table 1 and Fig. S11†), and (7.5 ± 0.4) × 104 M−1 in citrate buffer (0.1 M, H2O) at pH 4.1 (Fig. 5b) with a detection limit (LOD) of 0.6 μM (Fig. S12–S13†). On one hand, these values represent a lower estimate of KA because water molecules are believed to compete with fluoride for binding. In addition, we noticed that the KA values that were determined in citrate buffer were lower than those determined in pure H2O or D2O (Fig. S14–S17†). This could be attributed to the non-specific interactions of the cationic capsule with citrates at the periphery of the binding cavity, which could weaken the affinity of the protonated tren to fluoride. On the other hand, the fact that a stronger fluoride binding affinity was recorded in D2O than in H2O matches the expectation that the lighter H2O can make stronger hydrogen bonds to the tren unit and is more difficult than D2O to displace with the fluoride ion. We conclude that the lack of turn-on fluorescence signal with the larger anions provides further confirmation that they remain outside the protonated capsule, unable to displace water from the interior cavity.
| Aqueous conditions | K A (M−1) | LOD (μM) |
|---|---|---|
| Citrate buffer (0.1 M, H2O) at pH 4.1 | (7.5 ± 0.4) × 104 | 0.6 |
| Citrate buffer (0.1 M, D2O) at pD 4.5 | (1.4 ± 0.1) × 105 | N/A |
| H2O at pH 4.1 ± 0.1 | (4.0 ± 1.3) × 105 | 0.1 |
| D2O at pD 4.5 ± 0.1 | (6.0 ± 1.7) × 105 | N/A |
| Citrate buffer (0.1 M, H2O) at pH 7.0 | (3.2 ± 0.1) × 103 | 6.2 |
| PBS (10×, H2O) at pH 7.0 | (2.3 ± 0.2) × 103 | 11.2 |
To explore the practical application of [3H-L][OMs]3 as an anion-selective fluoride sensor, several cross-contamination experiments were carried out in citrate buffer (0.1 M, H2O) at pH 4.1. When [3H-L][OMs]3 was mixed with 10 equivalents of other anions individually or altogether, the fluorescence response of the capsule to fluoride was similar to that in pure buffer (Fig. 5c). This observation provided further evidence that the detection of fluoride was selective and not affected by many common anions. In addition, we investigated the potential application of [3H-L][OMs]3 in biologically relevant conditions at pH 7.0 under which the receptors exist as a mixture of monoprotonated and neutral capsules. Although the hemicryptophane exhibited significantly lower affinity and higher LOD for F− at neutral pH (Fig. S18–S23†), cross-contamination experiments reaffirmed that the detection of fluoride by [3H-L][OMs]3 was minimally influenced by other coexisting anions in citrate buffer or PBS at pH 7.0 (Fig. S24–S25†).
Encouraged by the high affinity and great selectivity exhibited by [3H-L][OMs]3 to fluoride, we proceeded to explore the potential of this compound to remove aqueous fluoride on solid support. First, we sought to immobilize the protonated capsules on weakly acidic silica gel, which has been successfully employed in a previous study to support metalated hemicryptophanes for catalytic purposes.77 By equilibrating a [3H-L][OMs]3 solution of known concentration with the silica for 1 h, the capsule-immobilized silica was separated from the supernatant by centrifugation. Given that the amount of capsule left in the supernatant solution can be determined by UV-vis spectroscopy (Fig. S26–S27†), the amount of hemicryptophane adsorbed to the silica gel via electrostatic interaction was determined by quantifying the solution depletion. While elevating the solution pH weakens the capability of the capsule to bind fluoride through lowering the protonation state of the tren, a higher pH results in a more negatively charged silica surface that was found to be more favorable for hemicryptophane adsorption (Fig. S28†). As such, pH 5 was chosen as the optimal condition to ensure sufficient capsule immobilization as well as effective fluoride binding capability. The adsorption efficiency of the protonated capsule to silica gel at pH 5 under ambient conditions was calculated to be 7.7 μmol g−1.
Once the protonated capsule was immobilized on the solid support, an aqueous solution of NaF (26.6 mg L−1, 3.5 μmol of F− in total) was incubated with the [3H-L][OMs]3-adsorbed silica. After 1 h incubation at pH 5.0 under ambient conditions, we discovered that 71% of the aqueous fluoride was removed based on a relative peak integration analysis in the 19F NMR spectra (Fig. 6a and b). In the meantime, a control experiment was performed under the same conditions, where the same fluoride solution was incubated with blank silica. This revealed that 8% of the fluoride removal occurred at the silica gel (Fig. 6c), which is a well-known adsorbent for fluoride.79 The remaining 63% of the fluoride removal was thereby attributed to the adsorption on the protonated capsule. The number of moles of fluoride (2.2 μmol) adsorbed by the hemicryptophane was essentially the same as the number of moles of [3H-L][OMs]3 (2.2 μmol) immobilized on the silica, which is consistent with the expected 1:1 binding stoichiometry. These experiments demonstrated that the protonated capsules retain their ability to encapsulate fluoride on the solid support.
 | ||
| Fig. 6 19F NMR spectra (565 MHz, 298 K, D2O, pD 5.4 ± 0.1) of (a) NaF solution before the incubation; (b) NaF solution after being incubated with the silica-supported [3H-L][OMs]3; (c) NaF solution after being incubated with blank silica. | ||
After the process of fluoride adsorption, a simple procedure was developed to recycle the capsule L. The fluoride-complexed hemicryptophane-supported silica was first suspended in DI water and mixed with the same volume of chloroform. Subsequent addition of NaOH raised the pH of the aqueous solution to 14 and completely deprotonated the capsule. As the neutral cage L is barely soluble in water, the hemicryptophane released the encapsulated fluoride and eventually went into the organic layer (Fig. S29†). This protocol provides an efficient (95% recovery) method for recycling the neutral tren-hemicryptophane after the process of water defluoridation.
Conclusions
In summary, we have developed a water-soluble tren-hemicryptophane capsule as a highly sensitive and selective fluoride sensor in a medium of 100% water. The hemicryptophane displays an exclusive ‘turn-on’ fluorescence response to the addition of NaF, which allows us to determine its apparent KA with fluoride by fluorescence titration and highlights the great selectivity of [3H-L][OMs]3 for F− recognition over a variety of common anions of larger sizes and different shapes in the range of pH 4.1–7.0. To the best of our knowledge, it is the first time that a metal-free molecular capsule was reported as a fluorescent probe that reports on fluoride binding in purely aqueous solutions. The capsule L provides an exemplar for organic host–guest chemistry in water, illustrating selective, stoichiometric anion binding.In addition, we have immobilized the capsule on silica gel and demonstrated the efficient adsorptive removal of fluoride from water. Via a simple procedure in which an acid–base reaction and liquid–liquid extraction were involved, the hemicryptophane can be recovered from the solid support in 95% yield after the defluoridation process. The compound reported in this work shows technological potential for real-world applications because of its rapid response to fluoride binding with great selectivity and high sensitivity, as well as its capability of efficient fluoride removal without risk of leaching metals into drinking water during the adsorption process.
In contrast with a few other examples of molecular capsules,80,81 our system is unique because the fluorophore is incorporated as part of the hosting scaffold and the anion binding site can also serve as the water-solubilizing group. A similar strategy has been shown to work in a few synthetic receptors that possess a more solvent-exposed binding site.50,51 The current study advances the design of synthetic cavitands as fluorescent probes for anion recognition in water.
Data availability
Data for this work, including general experimental procedures, characterization data (NMR, fluorescence, and UV-vis spectra), and XRD data are provided in the ESI.†Author contributions
Y. L. designed and performed the experiments. K. D. and M. R. G. collected and refined XRD data. I. J. D. supervised this project. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH grant R35-GM-131907 to I. J. D., and by UPenn Mitchell fellowship to Y. L. We thank the University of Pennsylvania NMR facility for spectrometer time.References
- D. P. Depaola, M. P. Faine and C. A. Palmer, Modern Nutrition in Health and Disease, ed. M. E. Shils, J. A. Olson, M. Shike and A. C. Ross, Williams & Wilkins, Baltimore, 9th edn, 1999 Search PubMed.
- J. D. B. Featherstone, Community Dent. Oral Epidemiol., 1999, 27, 31–40 CrossRef CAS PubMed.
- F. L. Cerklewski, Nutr. Res., 1997, 17, 907–929 CrossRef CAS.
- D. T. Waugh, Int. J. Environ. Res. Public Health, 2019, 16, 1086 CrossRef CAS.
- K. A. Krishnamachari, Prog. Food Nutr. Sci., 1986, 10, 279–314 CAS.
- S. Ayoob and A. K. Gupta, Crit. Rev. Environ. Sci. Technol., 2006, 36, 433–487 CrossRef CAS.
- X. Z. Xiong, J. L. Liu, W. H. He, T. Xia, P. He, X. M. Chen, K. Di Yang and A. G. Wang, Environ. Res., 2007, 103, 112–116 CrossRef CAS PubMed.
- A. K. Tolkou, N. Manousi, G. A. Zachariadis, I. A. Katsoyiannis and E. A. Deliyanni, Sustainability, 2021, 13, 7061 CrossRef CAS.
- H. Yahyavi, M. Kaykhaii and M. Mirmoghaddam, Crit. Rev. Anal. Chem., 2016, 46, 106–121 CrossRef CAS PubMed.
- Y. S. Solanki, M. Agarwal, A. B. Gupta, S. Gupta and P. Shukla, Sci. Total Environ., 2022, 807, 150601 CrossRef CAS.
- L. Li, H. Liu, J. Tang, P. Zhang and Y. Qian, Anal. Chem., 2022, 94, 13762–13769 CrossRef CAS.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS.
- E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS.
- Y. Jiao, B. Zhu, J. Chen and X. Duan, Theranostics, 2015, 5, 173–187 CrossRef CAS PubMed.
- P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2018, 354, 2–27 CrossRef CAS.
- J. Han, J. Zhang, M. Gao, H. Hao and X. Xu, Dyes Pigm., 2019, 162, 412–439 CrossRef CAS.
- S. H. Park, N. Kwon, J. H. Lee, J. Yoon and I. Shin, Chem. Soc. Rev., 2020, 49, 143–179 RSC.
- H. M. Tay and P. Beer, Org. Biomol. Chem., 2021, 19, 4652–4677 RSC.
- D. A. McNaughton, M. Fares, G. Picci, P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2021, 427, 213573 CrossRef CAS.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS.
- A. B. Descalzo, D. Jiménez, D. Beltràn, P. Amorós, M. D. Marcos, R. Martínez-Màñez and J. Soto, Chem. Commun., 2002, 2, 562–563 RSC.
- L. Gai, J. Mack, H. Lu, T. Nyokong, Z. Li, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2015, 285, 24–51 CrossRef CAS.
- P. Chen, W. Bai and Y. Bao, J. Mater. Chem. C, 2019, 7, 11731–11746 RSC.
- S. Dhiman, M. Ahmad, N. Singla, G. Kumar, P. Singh, V. Luxami, N. Kaur and S. Kumar, Coord. Chem. Rev., 2020, 405, 213138 CrossRef CAS.
- S. Y. Kim, J. Park, M. Koh, S. B. Park and J. I. Hong, Chem. Commun., 2009, 4735–4737 RSC.
- R. Hu, J. Feng, D. Hu, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2010, 49, 4915–4918 CrossRef CAS PubMed.
- F. Du, B. Liu, J. Tian, Q. Li and R. Bai, Chem. Commun., 2013, 49, 4631–4633 RSC.
- F. Zheng, F. Zeng, C. Yu, X. Hou and S. Wu, Chem.–Eur. J., 2013, 19, 936–942 CrossRef CAS PubMed.
- Y. C. Huang, C. P. Chen, P. J. Wu, S. Y. Kuo and Y. H. Chan, J. Mater. Chem. B, 2014, 2, 6188–6191 RSC.
- L. Li, Y. Ji and X. Tang, Anal. Chem., 2014, 86, 10006–10009 CrossRef CAS PubMed.
- S. Liu, J. Zhang, D. Shen, H. Liang, X. Liu, Q. Zhao and W. Huang, Chem. Commun., 2015, 51, 12839–12842 RSC.
- Q. Zhao, C. Zhang, S. Liu, Y. Liu, K. Y. Zhang, X. Zhou, J. Jiang, W. Xu, T. Yang and W. Huang, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- B. Qiu, Y. Zeng, L. Cao, R. Hu, X. Zhang, T. Yu, J. Chen, G. Yang and Y. Li, RSC Adv., 2016, 6, 49158–49163 RSC.
- X. Yu, L. Yang, T. Zhao, R. Zhang, L. Yang, C. Jiang, J. Zhao, B. Liu and Z. Zhang, RSC Adv., 2017, 7, 53379–53384 RSC.
- Y. Chen, L. Zhao, H. Fu, C. Rao, Z. Li and C. Liu, New J. Chem., 2017, 41, 8734–8738 RSC.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1993, 89, 713–718 RSC.
- M. Cametti, A. D. Cort and K. Bartik, ChemPhysChem, 2008, 9, 2168–2171 CrossRef CAS.
- A. Dalla Cort, G. Forte and L. Schiaffino, J. Org. Chem., 2011, 76, 7569–7572 CrossRef CAS PubMed.
- L. M. P. Lima, A. Lecointre, J. F. Morfin, A. De Blas, D. Visvikis, L. J. Charbonnière, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2011, 50, 12508–12521 CrossRef CAS.
- A. Brugnara, F. Topić, K. Rissanen, A. D. La Lande, B. Colasson and O. Reinaud, Chem. Sci., 2014, 5, 3897–3904 RSC.
- T. Liu, A. Nonat, M. Beyler, M. Regueiro-Figueroa, K. Nchiminono, O. Jeannin, F. Camerel, F. Debaene, S. Cianférani-Sanglier, R. Tripier, C. Platas-Iglesias and L. J. Charbonnière, Angew. Chem., Int. Ed., 2014, 53, 7259–7263 CrossRef CAS.
- O. A. Blackburn, A. M. Kenwright, A. R. Jupp, J. M. Goicoechea, P. D. Beer and S. Faulkner, Chem.–Eur. J., 2016, 22, 8929–8936 CrossRef CAS PubMed.
- H. Y. Zheng, X. Lian, S. J. Qin and B. Yan, ACS Omega, 2018, 3, 12513–12519 CrossRef CAS PubMed.
- X. Wang, C. Chu, Y. Wu, Y. Deng, J. Zhou, M. Yang, S. Zhang, D. Huo and C. Hou, Sens. Actuators, B, 2020, 321, 128455 CrossRef CAS.
- B. Dietrich, B. Dilworth, J. M. Lehn, J. P. Souchez, M. Cesario, J. Guilhem and C. Pascard, Helv. Chim. Acta, 1996, 79, 569–587 CrossRef CAS.
- M. A. Hossain, J. M. Llinares, S. Mason, P. Morehouse, D. Powell and K. Bowman-James, Angew. Chem., Int. Ed., 2002, 41, 2335–2338 CrossRef.
- C. A. Ilioudis, D. A. Tocher and J. W. Steed, J. Am. Chem. Soc., 2004, 126, 12395–12402 CrossRef CAS PubMed.
- B. G. Zhang, P. Cai, C. Y. Duan, R. Miao, L. G. Zhu, T. Niitsu and H. Inoue, Chem. Commun., 2004, 2206–2207 RSC.
- R. Chelli, G. Pietraperzia, A. Bencini, C. Giorgi, V. Lippolis, P. R. Salvi and C. Gellini, Phys. Chem. Chem. Phys., 2015, 17, 10813–10822 RSC.
- A. S. Oshchepkov, T. A. Shumilova, S. R. Namashivaya, O. A. Fedorova, P. V. Dorovatovskii, V. N. Khrustalev and E. A. Kataev, J. Org. Chem., 2018, 83, 2145–2153 CrossRef CAS.
- M. Mohapatra, S. Anand, B. K. Mishra, D. E. Giles and P. Singh, J. Environ. Manage., 2009, 91, 67–77 CrossRef CAS.
- S. Jagtap, M. K. Yenkie, N. Labhsetwar and S. Rayalu, Chem. Rev., 2012, 112, 2454–2466 CrossRef CAS.
- P. Loganathan, S. Vigneswaran, J. Kandasamy and R. Naidu, J. Hazard. Mater., 2013, 248–249, 1–19 CrossRef CAS.
- M. Habuda-Stanić, M. Ravančić and A. Flanagan, Materials, 2014, 7, 6317–6366 CrossRef.
- A. Bhatnagar, E. Kumar and M. Sillanpää, Chem. Eng. J., 2011, 171, 811–840 CrossRef CAS.
- E. W. Wambu, W. O. Ambusso, C. Onindo and G. K. Muthakia, J. Water Reuse Desalin., 2016, 6, 1–29 CrossRef CAS.
- Q. Gao, J. Xu and X. H. Bu, Coord. Chem. Rev., 2019, 378, 17–31 CrossRef CAS.
- K. Wan, L. Huang, J. Yan, B. Ma, X. Huang, Z. Luo, H. Zhang and T. Xiao, Sci. Total Environ., 2021, 773, 145535 CrossRef CAS.
- G. Nabar, K. Mahajan, M. Calhoun, A. Duong, M. Souva, J. Xu, C. Czeisler, V. Puduvalli, J. Otero, B. Wyslouzil and J. Winter, Int. J. Nanomed., 2018, 13, 351–366 CrossRef CAS.
- P. Mondal and S. George, Rev. Environ. Sci. Biotechnol., 2015, 14, 195–210 CrossRef CAS.
- W. Z. Gai and Z. Y. Deng, Environ. Sci.: Water Res. Technol., 2021, 7, 1362–1386 RSC.
- M. A. Menkouchi Sahli, S. Annouar, M. Tahaikt, M. Mountadar, A. Soufiane and A. Elmidaoui, Desalination, 2007, 212, 37–45 CrossRef CAS.
- M. H. Dehghani, G. A. Haghighat, K. Yetilmezsoy, G. McKay, B. Heibati, I. Tyagi, S. Agarwal and V. K. Gupta, J. Mol. Liq., 2016, 216, 401–410 CrossRef CAS.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. Del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- J. He, Y. Yang, Z. Wu, C. Xie, K. Zhang, L. Kong and J. Liu, J. Environ. Chem. Eng., 2020, 8, 104516 CrossRef CAS.
- Y. Makita, K. Sugimoto, K. Furuyoshi, K. Ikeda, S. Fujiwara, T. Shin-ike and A. Ogawa, Inorg. Chem., 2010, 49, 7220–7222 CrossRef CAS.
- Y. Lin, M. R. Gau, P. J. Carroll and I. J. Dmochowski, J. Org. Chem., 2022, 87, 5158–5165 CrossRef CAS.
- P. A. Hill, Q. Wei, R. G. Eckenhoff and I. J. Dmochowski, J. Am. Chem. Soc., 2007, 129, 9262–9263 CrossRef CAS.
- S. D. Zemerov, Y. Lin and I. J. Dmochowski, Anal. Chem., 2021, 93, 1507–1514 CrossRef CAS.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
- J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Chem. Sci., 2021, 12, 1352–1362 RSC.
- P. S. Lakshminarayanan, D. K. Kumar and P. Ghosh, Inorg. Chem., 2005, 44, 7540–7546 CrossRef CAS.
- M. A. Hossain, P. Morehouse, D. Powell and K. Bowman-James, Inorg. Chem., 2005, 44, 2143–2149 CrossRef CAS PubMed.
- U. Darbost, M. N. Rager, S. Petit, I. Jabin and O. Reinaud, J. Am. Chem. Soc., 2005, 127, 8517–8525 CrossRef CAS PubMed.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J.-P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- N. K. Mondal, R. Bhaumik, A. Banerjee, J. K. Datta and T. Baur, Int. J. Environ. Sci., 2012, 2, 1643–1661 CAS.
- J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC.
- Y. Liu, W. Zhao, C. H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2171670. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05352g |
| This journal is © The Royal Society of Chemistry 2023 |