
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the thermodynamic–kinetic synergistic separation of propyne/propylene in anion pillared cage MOFs with entropy–enthalpy balanced adsorption sites†
Yunjia
Jiang‡
a,
Lingyao
Wang‡
a,
Tongan
Yan‡
 b,
Jianbo
Hu‡
c,
Wanqi
Sun
a,
Rajamani
Krishna
d,
Dongmei
Wang
a,
Zonglin
Gu
e,
Dahuan
Liu
b,
Xili
Cui
c,
Huabin
Xing
c and
Yuanbin
Zhang
*a
b,
Jianbo
Hu‡
c,
Wanqi
Sun
a,
Rajamani
Krishna
d,
Dongmei
Wang
a,
Zonglin
Gu
e,
Dahuan
Liu
b,
Xili
Cui
c,
Huabin
Xing
c and
Yuanbin
Zhang
*a
aKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, College of Chemistry and Life Sciences, Zhejiang Normal University, Jinhua 321004, China. E-mail: ybzhang@zjnu.edu.cn
bState Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China
cDepartment of Chemistry, Zhejiang University, 38 Zheda Road, 310027 Hangzhou, P. R. China
dVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, Netherlands
eCollege of Physical Science and Technology, Yangzhou University, Jiangsu 225009, China
First published on 28th November 2022
Abstract
Propyne/propylene (C3H4/C3H6) separation is an important industrial process yet challenged by the trade-off of selectivity and capacity due to the molecular similarity. Herein, record C3H4/C3H6 separation performance is achieved by fine tuning the pore structure in anion pillared MOFs. SIFSIX-Cu-TPA (ZNU-2-Si) displays a benchmark C3H4 capacity (106/188 cm3 g−1 at 0.01/1 bar and 298 K), excellent C3H4/C3H6 IAST selectivity (14.6–19.3) and kinetic selectivity, and record high C3H4/C3H6 (10/90) separation potential (36.2 mol kg−1). The practical C3H4/C3H6 separation performance is fully demonstrated by breakthroughs under various conditions. 37.8 and 52.9 mol kg−1 of polymer grade C3H6 can be produced from 10/90 and 1/99 C3H4/C3H6 mixtures. 4.7 mol kg−1 of >99% purity C3H4 can be recovered by a stepped desorption process. Based on the in situ single crystal analysis and DFT calculation, an unprecedented entropy–enthalpy balanced adsorption pathway is discovered. MD simulation further confirmed the thermodynamic–kinetic synergistic separation of C3H4/C3H6 in ZNU-2-Si.
Introduction
Propylene (C3H6) is the world's second largest volume hydrocarbon with the global production capacity exceeding 140 million tons in 2020. It is a basic olefin feedstock for the manufacture of various polymers and chemicals such as polypropylene and propylene oxide.1 Originating from the cracking of crude oil, C3H6 is inevitably mixed with a small amount of propyne (C3H4), which must be reduced to a ppm level before further processing as it can severely poison the C3H6 polymerization catalysts.2 The state-of-the-art industrial technologies for the removal of C3H4 from C3H6 rely on noble-metal catalyst based selective hydrogenation, which suffers from several drawbacks such as high cost, low efficiency and potential secondary pollution. On the other hand, C3H4 that can be manufactured from the catalytic or thermal pyrolysis of C3H6 is also a fundamental material for speciality fuel and chemical intermediates.3 To recover C3H4, solvent extraction is the current dominant technology, which is not only energy intensive but also associated with pollution. Thus, it is of urgent importance to develop new technologies for efficient C3H4/C3H6 separation.Adsorptive separation based on porous solid adsorbents has been recognized as a promising alternative technology for gas/vapor separation because of its eco-friendly nature and energy efficiency.4–10 However, due to the great similarity in the molecular size (C3H4: 4.16 × 4.01 × 6.51 Å3, C3H6: 4.65 × 4.16 × 6.44 Å3) and polarizability (C3H4: 55.5 × 10−25 cm3, C3H6: 62.6 × 10−25 cm3), the adsorptive separation of C3H4/C3H6 by molecular recognition is still very challenging.11 Only two examples of zeolites are reported and their C3H4 capacity is relatively low.10 Recently, metal–organic frameworks (MOFs) with tuneable pore size/shape and chemistry have emerged as a new class of porous materials for the separation of C3H4/C3H6.12–27 Among them, pcu type anion pillared MOFs (APMOFs) with strong Lewis basic functional sites display the benchmark separation performance.12–18 Nonetheless, the trade-off between the capacity and selectivity is still a critical problem to overcome. For example, SIFSIX-3-Ni (pore size = 4.2 Å, Scheme 1a) as a single-molecule trap for C3H4 can afford extremely high C3H4/C3H6 selectivity (>200), but the capacity of C3H4 is only 67 cm3 g−1; SIFSIX-1-Cu (pore size = 8.0 Å, Scheme 1b) can accommodate a large amount of C3H4 (201 cm3 g−1) by cooperative host–guest interactions, but the separation selectivity is <10. Besides, these pillared layered SIFSIX MOFs are not chemically stable and some are even sensitive to humid air, which hinders the practical applications.8d,28 On the other hand, the kinetic separation of C3H4/C3H6 has never been reported.
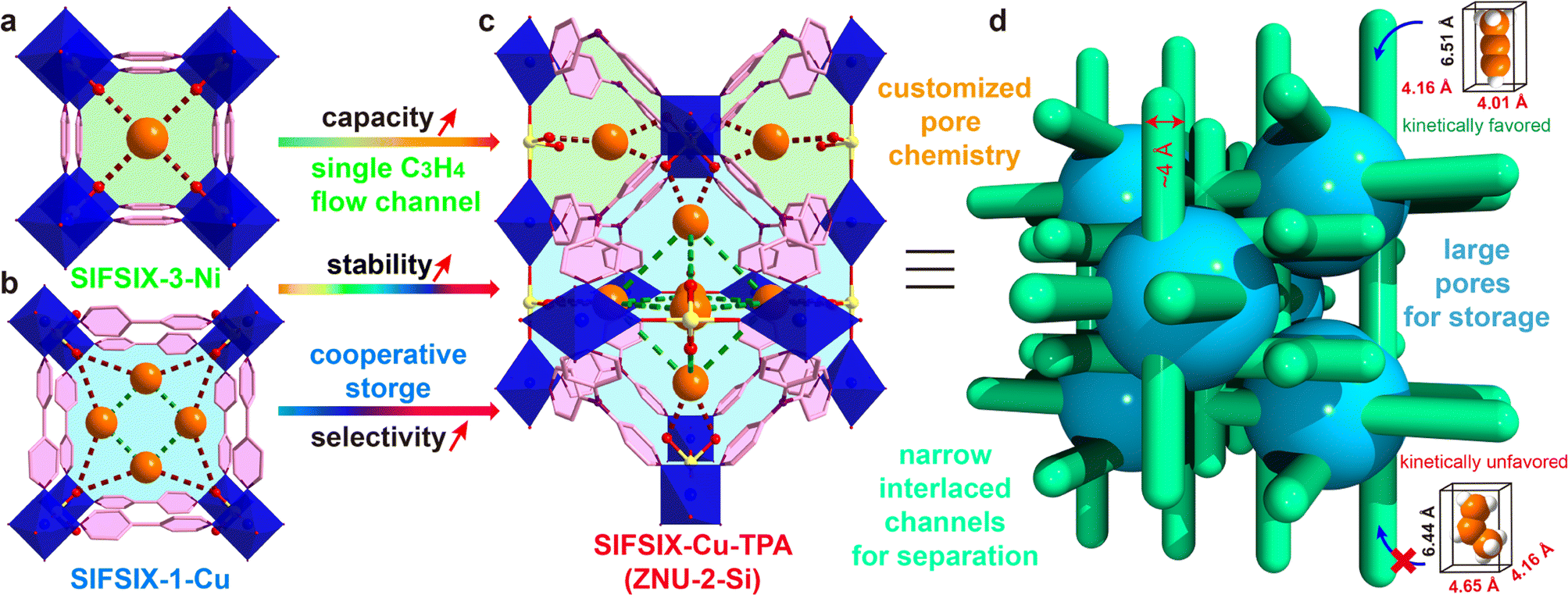 | ||
| Scheme 1 Strategies to overcome the trade-off of capacity and selectivity in C3H4/C3H6 separation in a stable cage-like APMOF by the thermodynamic–kinetic synergism mechanism. (a) Structure of SIFSIX-3-Ni. (b) Structure of SIFSIX-1-Cu. (c) Structure of SIFSIX-Cu-TPA. (d) Illustration of the thermodynamic–kinetic cooperation for C3H4/C3H6 separation. | ||
Anion pillared cage-like MOFs with ith-d topology are a new class of APMOFs.27,29 The rational combination of anion pillars and tridentate organic linkers provides anion sustained cage-like APMOFs with ultrahigh chemical stability. In 2021, Wu et al. discovered the first pillar-cage ith-d MOF SIFSIX-Cu-TPA (Scheme 1c) with a complete SiF62− cross-link for CO2/C2H2 separation.29 Soon after, our group reported independently an isomorphic MOF termed ZNU-2 (TIFSIX-Cu-TPA).27 Considering the slight change of the organic linker, the metal ion or anion pillar in pcu-type APMOFs can lead to a dramatic separation difference, we envision that tuning the pore aperture and pore window in cage-like APMOFs can also be applied to tune the gas adsorption properties. Nonetheless, the length and angle matching between the tridentate ligand and anion pillar is very significant to construct the pillar embedding structures. Pillar-cage Tripp-Cu-SIFSIX with the overlong organic linker 2,4,6-tris(4-pyridyl)pyridine is not stable upon guest removal since the mononuclear Cu ion center is only half sustained by SiF62− and coordination unsaturated.30 [Cu3(SiF6)3(TMTPB)4] (FJI-W1) with triangular ligand 1,3,5-trimethyl-2,4,6-tris(4-pyridyl)benzene belongs to pillar-layer APMOFs that feature 1D hexagonal channels.26 Thus, only the modification of the anion pillar or metal ion (size difference < 0.1 Å) is a good alternative to fine-tune the pore structure and chemistry while retaining the topology. Furthermore, the integration of large cages and narrow interlaced channels has the potential to show a kinetic difference for C3H4 and C3H6 molecules with a slight diameter difference (Scheme 1d), which has not been explored in theory yet.
With this in mind, herein we prepared three isomorphic APMOFs using SiF62−, TiF62−, and NbOF52− as the pillars, and investigated the C3H4/C3H6 adsorption and separation performance. To our delight, these three reticular MOFs exhibit quite distinctive but ordered C3H4 adsorption capacity as well as C3H4/C3H6 selectivity. The pore size follows the sequence of SIFSIX-Cu-TPA < TIFSIX-Cu-TPA < NbOFFIVE-Cu-TPA while the C3H4 adsorption capacity and the separation selectivity are both SIFSIX-Cu-TPA > TIFSIX-Cu-TPA > NbOFFIVE-Cu-TPA. Benchmark high uptakes of C3H4 are observed both at low pressure (106 STP cm3 g−1 at 0.01 bar and 298 K) and normal pressure (188 STP cm3 g−1 at 1 bar and 298 K) on SIFSIX-Cu-TPA. The C3H4 storage density reached 0.60 and 0.65 g cm−3 at 298 and 278 K, 89% and 97% of the liquid C3H4 density. The calculated IAST selectivity is 14.6–19.3 depending on the ratio of C3H4/C3H6 (1/99–50/50). Record high C3H4/C3H6 (10/90) separation potential (36.2 mol kg−1) is obtained, which is 65% higher than the previous benchmark of NKMOF-11 without anion functionalities. The modestly high C3H4 adsorption heat of 43.2 kJ mol−1 is advantageous for both C3H4 adsorption and facile regeneration. Simulated breakthroughs indicated SIFSIX-Cu-TPA displays the best separation performance for C3H4/C3H6 (10/90) mixtures. Practical separations of C3H4/C3H6 (50/50, 10/90, 1/99) mixtures were also confirmed by breakthrough experiments. Notably, the practical separation performance is even superior to that of simulation due to the kinetic enhancement, which has never been reported in C3H4/C3H6 separation. 37.8 and 52.9 mol kg−1 of C3H6 is produced from the 10/90 and 1/99 C3H4/C3H6 mixtures, respectively. The productivity is increased to 79.2 mol kg−1 when the process temperature decreased to 278 K. Such high productivity has never been achieved by chemically stable porous materials. 4.7 mol kg−1 of >99% purity C3H4 can be recovered. Repeated breakthrough experiments under dry or humid conditions showed the complete retention of separation performance, confirming the high stability of SIFSIX-Cu-TPA for practical separations. The in situ single crystal structure of C3H4-loaded SIFSIX-Cu-TPA directly demonstrates the C3H4 binding configuration under near-saturation conditions, which is distinct from the previous study.27 Comprehensive modelling studies including Grand Canonical Monte Carlo (GCMC) simulations, Molecular Dynamics (MD) simulations and Density Functional Theory (DFT) calculations were completely applied to investigate the adsorption/separation process, which indicated that the contracted channel serves as a single molecule flow channel that differentiates C3H4/C3H6 kinetically while the large cage provides high affinity for C3H4 adsorption by cooperative host–guest and guest–guest interactions. To the best of our knowledge, the kinetic separation of C3H4/C3H6 has for the first time been revealed by MD simulations. The obvious thermodynamic–kinetic synergism in breakthroughs has never been reported in porous materials for C3H4/C3H6 separation. Moreover, our study unprecedentedly disclosed the important role of entropy effects on C3H4 adsorption and gas cluster assembly in the pores while the GCMC and DFT based gas binding configuration may not reflect the practical gas binding sites due to the neglect of the entropy effect.
Results and discussion
The single crystals of isostructural SIFSIX-Cu-TPA (ZNU-2-Si), TIFSIX-Cu-TPA (ZNU-2-Ti) and NbOFFIVE-Cu-TPA (ZNU-2-Nb) are all produced by layering a MeOH solution of TPA onto an aqueous solution of CuX (X = SiF62−, TiF62−, NbOF52−). ZNU-2-Nb has been reported for the first time (Fig. 1a and b). All of these three coordination complexes crystallize in three-dimensional (3D) frameworks with the cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n space group (Table S1†). The frameworks consist of large icosahedral cages (∼8.5 Å, Fig. 1c) with 12 outlets and narrow interlaced channels (∼4 Å, Fig. 1d) that connect four independent cages (Fig. S6 and S7†). The large cages have abundant Lewis basic F binding sites in the surface for C3H4 adsorption and storage. Such interconnected 3D channel pores (Fig. 1e) are distinct from those of pillar-layer MOFs (e.g. SIFSIX-3-Ni) with straight 1D channels. Due to the tiny size difference of anion pillars, the pore aperture and channel diameter also show a very slight difference (<0.05 Å), which is reflected in the N⋯N and Cu⋯Cu distances (Fig. 1f). As the channel diameter is very close to the cross-sections (4.01 × 4.16 Å2 for C3H4 and 4.65 × 4.16 Å2 for C3H6), a slight shrinking of the channel may provide a much enhanced kinetic difference in C3H4/C3H6 adsorption. Thus, ZNU-2-Si with a reduced channel diameter has the potential to show a remarkable kinetic difference in C3H4 and C3H6 adsorption. In brief, ZNU-2-Si features the most promising structure to offer benchmark C3H4/C3H6 separation performance by thermodynamic–kinetic synergism.
n space group (Table S1†). The frameworks consist of large icosahedral cages (∼8.5 Å, Fig. 1c) with 12 outlets and narrow interlaced channels (∼4 Å, Fig. 1d) that connect four independent cages (Fig. S6 and S7†). The large cages have abundant Lewis basic F binding sites in the surface for C3H4 adsorption and storage. Such interconnected 3D channel pores (Fig. 1e) are distinct from those of pillar-layer MOFs (e.g. SIFSIX-3-Ni) with straight 1D channels. Due to the tiny size difference of anion pillars, the pore aperture and channel diameter also show a very slight difference (<0.05 Å), which is reflected in the N⋯N and Cu⋯Cu distances (Fig. 1f). As the channel diameter is very close to the cross-sections (4.01 × 4.16 Å2 for C3H4 and 4.65 × 4.16 Å2 for C3H6), a slight shrinking of the channel may provide a much enhanced kinetic difference in C3H4/C3H6 adsorption. Thus, ZNU-2-Si with a reduced channel diameter has the potential to show a remarkable kinetic difference in C3H4 and C3H6 adsorption. In brief, ZNU-2-Si features the most promising structure to offer benchmark C3H4/C3H6 separation performance by thermodynamic–kinetic synergism.
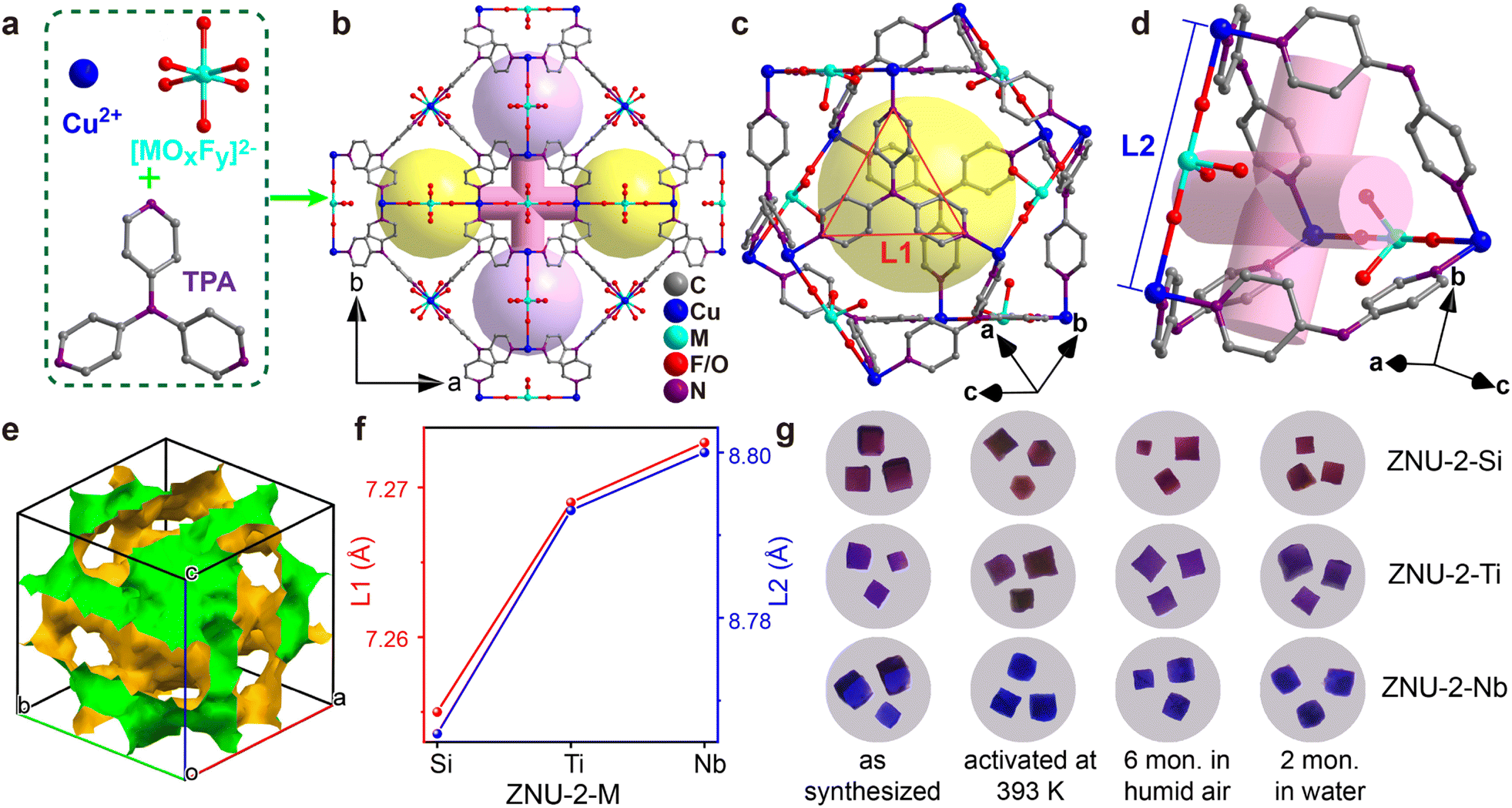 | ||
| Fig. 1 Porous structure and stability test of ZNU-2-M (M = Si, Ti, Nb). (a) Basic units to construct ZNU-2-M. (b) Structure of ZNU-2-M with cage-like pores and interlaced channels. (c) Structure of the icosahedral CuII cage with the N⋯N distance (L1) of TPA highlighted. (d) Structure of the interlaced channel between four cages with the Cu⋯Cu distance (L2) highlighted. (e) The voids of ZNU-2-Si illustrating the interlinked 3D channels. (f) Comparison of the L1 and L2 among ZNU-2-M. (g) Optical microscopy of single crystals of ZNU-2-M (M = Si, Ti, Nb) after different treatments. | ||
Before gas adsorption experiments, the chemical and thermal stability of ZNU-2-M is fully studied since stability is a prerequisite for practical use in real-world systems. To our delight, all three materials are highly stable in humid air and water as indicated by the PXRD patterns (Fig. S12, S14 and S15†). To provide straightforward evidence, we take photographs of the single crystals of ZNU-2-M after different treatments (Fig. 1g, S86, S88 and S90†). As shown, the single crystals are still of high quality after being left in humid air for 6 months, soaking in water for 2 months, soaking in acidic and basic aqueous solutions or exposure to humid HCl vapor at 323 K for 3 h. Heating at 393 K under vacuum provides desolvated ZNU-2-M with the same crystal structure as indicated by single crystal and powder XRD analysis (Fig. 1g, S11, S14, S15 and S86–S90†). TGA curves showed that the framework of ZNU-2-M is stable at 523, 581, and 573 K (250, 308 and 300 °C), respectively (Fig. S13–S15†). The weight reduction between room temperature and 423 K is ascribed to the loss of solvents (MeOH/H2O) in the pores of ZNU-2-M.
Encouraged by the ultrahigh stability of ZNU-2-M, we are interested in investigating their permanent porosity as well as unary gas adsorption difference between C3H4 and C3H6. At first, N2 gas adsorption experiments at 77 K were conducted (Fig. S16–S19†), which indicated their microporous character with pore size distribution in the range of 6.27–9.84 Å, 6.56–9.40 Å, and 7.85–9.40 Å, respectively, very close to the pore aperture of ∼8.5 Å calculated from the single crystal structure. The BET surface areas and pore volumes are 1339/1380/1281 m2 g−1 and 0.565/0.575/0.521 cm3 g−1, for ZNU-2-Si, ZNU-2-Ti and ZNU-2-Nb respectively. These BET surface areas are all superior to the benchmark of SIFSIX-1-Cu (1128 m2 g−1) in pillar-layer APMOFs.14
Single-component C3H4 adsorption isotherms were then collected at 298 K (Fig. 2a). At 1 bar, the C3H4 uptakes were 188, 171 and 162 cm3 g−1 for ZNU-2-Si, ZNU-2-Ti and ZNU-2-Nb, corresponding to 4.52, 4.25, and 4.34 C3H4 molecules adsorbed per anion (Fig. 2b). Such a high C3H4/anion ratio means every free F site can bind 1.13, 1.06, and 1.09 C3H4 molecules, much higher than those of SIFSIX-2-Cu-i (2.57), TIFSIX-14-Cu-i (2.31), ZU-62 (2.30) and SIFSIX-3-Ni (1.09) (Fig. 2b). The adsorption capacities under 0.01 and 0.1 bar were further compared with those of other MOFs (Fig. 2c). The C3H4 uptake of ZNU-2-Si at 0.01 bar is record high at 106 cm3 g−1. This uptake is even much higher than the saturated capacities (1 bar) of most MOFs, such as ELM-12 (61.4 cm3 g−1),19 SIFSIX-3-Ni (66.8 cm3 g−1),14 NKMOF-11 (69.4 cm3 g−1),20 GeFSIX-14-Cu-i (75.3 cm3 g−1),17 Ca-based MOF (67.4 cm3 g−1),25 UTSA-200 (81.1 cm3 g−1),15 ZU-62 (82.0 cm3 g−1)18 and TIFSIX-14-Cu-i (86.5 cm3 g−1).17 Interestingly, a good negative linear relationship between the C3H4 uptakes under low pressure (0.1 bar) and the N⋯N/Cu⋯Cu distances is observed (Fig. 2d). Such a structure–capacity relationship has never been reported before. Then C3H4 and C3H6 adsorption isotherms on ZNU-2-M at 278, 298 and 308 K were all collected (Fig. 2e). The C3H6 adsorption capacities are much lower than those of C3H4, especially in low pressure regions. The C3H4/C3H6 selectivity on ZNU-2 at 298 K was calculated by using Ideal Adsorbed Solution Theory (IAST). As shown in Fig. 2f, the selectivity of ZNU-2-Si for 1/99 C3H4/C3H6 is 14.64, which is higher than that of ZNU-2-Ti (12.53), ZNU-2-Nb (9.84), ZIF-8 (1.9),15 FJI-W1 (2.2),26 Cu-BTC (3.2),15 MIL-100(Cr) (4.5),15 and SIFSIX-1-Cu (9.0)14 (Fig. 2f). The increased ratio of C3H4 in the gas mixture results in increased C3H4/C3H6 selectivity, which is 16.05 and 19.29 for 10/90 and 50/50C3H4/C3H6 mixtures, respectively. The simultaneous increase of the C3H4/C3H6 selectivity along the uptakes or C3H4 ratios suggests the existence of cooperative interactions inside ZNU-2-Si.
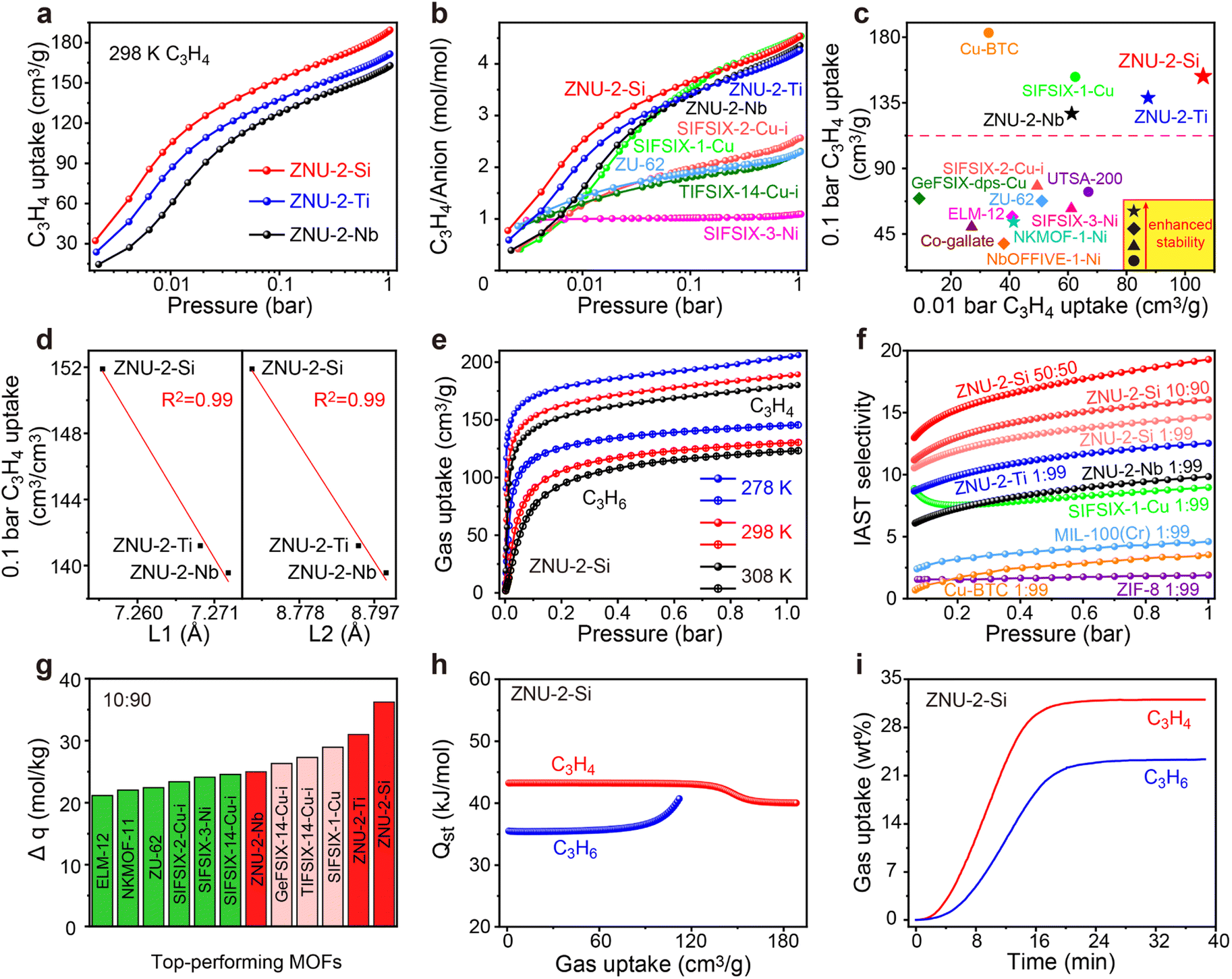 | ||
| Fig. 2 (a) C3H4 adsorption isotherms in the ZNU-2 family at 298 K. (b) Comparison of the C3H4 adsorption isotherms of the ZNU-2 family with fluorinated anion hybrid microporous materials. (c) Comparison of the low pressure C3H4 uptake and stability among top-performing MOFs. (d) Plot of C3H4 uptake at 0.1 bar vs. N⋯N distance (L1)/Cu⋯Cu distance (L2). (e) C3H4 and C3H6 adsorption isotherms for ZNU-2-Si at 278, 298 and 308 K. (f) Comparison of IAST selectivity of the ZNU-2 family with other MOFs showing high C3H4 capacity (>100 cm3 g−1). (g) Comparison of ZNU-2's IAST based separation potential (Δq = C3H4 uptake × 9 − C3H6 uptake) for C3H4/C3H6 (10/90) mixtures with reported top performing MOFs. (h) The isosteric heat of adsorption, Qst, for C3H4 and C3H6 in ZNU-2-Si. (i) Adsorption kinetic curves of C3H4 and C3H6 in ZNU-2-Si. | ||
The static C3H4 and C3H6 uptakes from the 10/90 mixture of C3H4/C3H6 were calculated for the ZNU-2 family and other leading materials (Fig. S30, S35, S40 and S42–S48†). The separation potential (Δq = q1y2/y1 − q2)31 as a combined metric of both selectivity and capacity was utilized here for further comparison, which showed a trend of ZNU-2-Si (36.2 mol kg−1) > ZNU-2-Ti (31.0 mol kg−1) > SIFSIX-1-Cu (29.0 mol kg−1)14 > TIFSIX-14-Cu-i (27.3 mol kg−1)17 > GeFSIX-14-Cu-i (26.3 mol kg−1)17 > ZNU-2-Nb (25.0 mol kg−1) > SIFSIX-14-Cu-i (24.6 mol kg−1) > SIFSIX-3-Ni (24.1 mol kg−1)14 > SIFSIX-2-Cu-i (23.4 mol kg−1)14 > ZU-62 (22.4 mol kg−1)18 > NKMOF-11 (22.0 mol kg−1)20 > ELM-12 (21.2 mol kg−1)19 at 1 bar and 298 K (Fig. 2g). The isosteric enthalpies of adsorption (Qst) for ZNU-2-M were then calculated with the Clausius–Clapeyron equation. Qst values at near-zero loading for C3H4 and C3H6 were 43.2/43.0/41.6 and 35.5/34.5/32.4 kJ mol−1, respectively (Fig. 2h, S28, S33 and S38†). The Qst values for C3H4 in the ZNU-2 family are lower than those of most MOFs for C3H4/C3H6 separation such as ZU-62 (71.0 kJ mol −1),25 SIFSIX-3-Ni (68.0 kJ mol−1),14 NKMOF-1-Ni (65.1 kJ mol −1),25 Ca-based MOF (55.4 kJ mol −1),25 UTSA-200 (55.3 kJ mol−1),15 ELM-12 (60.6 kJ mol −1)19 and SIFSIX-2-Cu-i (46.0 kJ mol −1),14 but slightly higher than that of SIFSIX-1-Cu (37.2 kJ mol−1)14 (Table S18†). Such modestly high Qst not only facilitates preferential C3H4 adsorption, but also allows the facile recovery of C3H4 by desorption under mild conditions. To further compare the adsorption difference of C3H4 and C3H6 on ZNU-2-Si, we studied the kinetic adsorption behavior. The adsorption rate of C3H4 in ZNU-2-Si is faster than that of C3H6. This means that the intra-crystalline diffusion of C3H4 is faster than that of C3H6 (Fig. 2i). To the best of our knowledge, such kinetic difference has not been reported in pillar-layered APMOFs for C3H4/C3H6 separations. Besides, adsorption thermodynamics and diffusion are usually anti-synergistic as stronger adsorption often implies reduced mobility.32 Therefore, ZNU-2-Si with thermodynamic–kinetic synergism is highly promising to provide benchmark practical C3H4/C3H6 separation performance.
To obtain direct host–guest interaction information between ZNU-2-Si and adsorbed gases, we introduced C3H4 and C3H6 into the desolvated ZNU-2-Si and measured it in the single crystal X-ray diffractometer. Due to the high stability of ZNU-2-Si, the C3H4 and C3H6 loaded structures are ambiguously resolved (Fig. 3 and S10†). 24 C3H4 molecules and 18 C3H6 molecules are adsorbed per cell, equal to 4 C3H4 and 3 C3H6 molecules for every SIFSIX anion, consistent with the experimental results. After adsorption, the framework remained in the same cubic Pmn space group with negligible bond or angle changes (Table S2†). From the in situ crystals, C3H4 showed disorder into two overlapping configurations. The configuration with the alkynyl C–H end closer to SiF62− is chosen to be discussed in the following text. The hydrogen bond distances between the terminal C3H4 hydrogen and F atom of SiF62− are all 2.576 Å. Interestingly, no single C3H4 molecule is completely loaded in the narrow interlaced single molecule channel, which was previously considered as the best energy favored single molecule binding site.27 Instead, four C3H4 molecules are equally close to the interlaced channel while their alkynyl C–H ends are inside (Fig. 3b). On the other hand, all C3H4 molecules can be considered to locate in the large cage with their C–H end reaching outside (Fig. 3c). Therefore, every large cage can accommodate 12 C3H4 molecules. The C3H6 adsorption sites are very close to that for C3H4. Due to the high symmetry, every free F atom is able to bind 0.75 C3H6 molecules (Fig. S10†). Strangely, the hydrogen bond distance (2.108 Å) between terminal ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and the F atom is even shorter than the
CH2 and the F atom is even shorter than the ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H⋯F distance. Thus, DFT calculations are applied to directly compare their binding energies, which will be discussed in the next part.
C–H⋯F distance. Thus, DFT calculations are applied to directly compare their binding energies, which will be discussed in the next part.
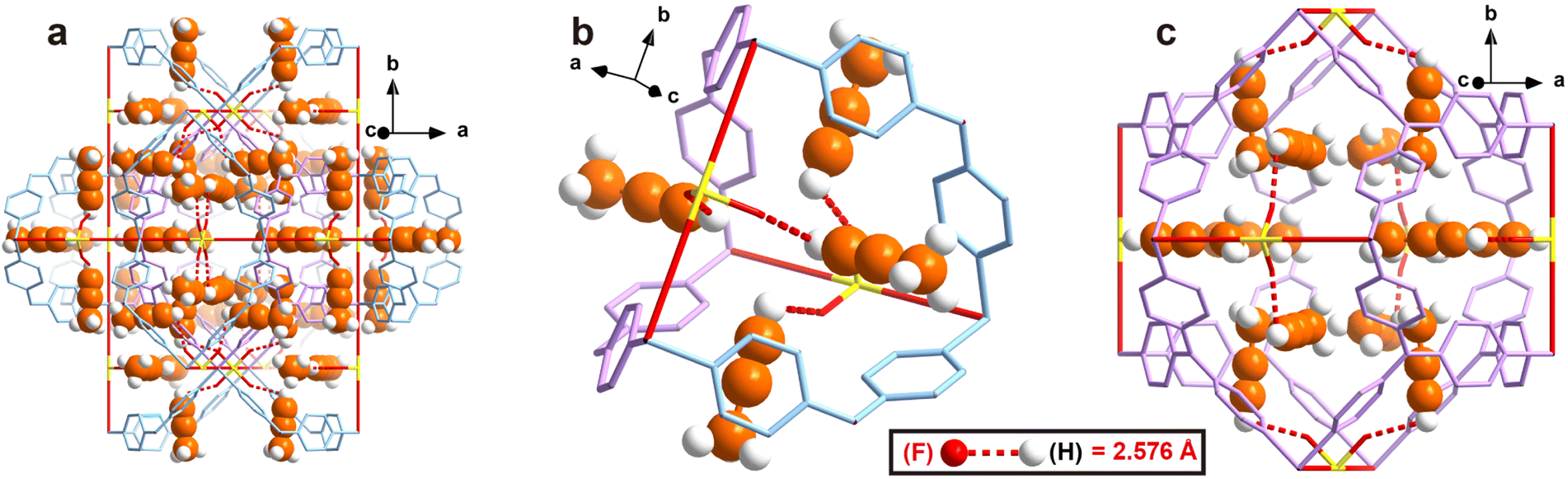 | ||
| Fig. 3 Single crystal structure of C3H4 loaded ZNU-2-Si. (a) A holistic view. (b) View around the interlaced channel. (c) View around the cage. | ||
The structure of ZNU-2-Si with less C3H4 loading is also obtained by controlling the adsorption pressure at 0.01 bar. However, due to the ultrahigh adsorption uptake of C3H4 under low pressure, there is still a large amount of C3H4 observed in the cage of ZNU-2-Si and the binding sites are the same. The only difference is the occupancy of every C3H4 molecule is only ca. 50%, equal to 6 C3H4 molecules in every cage. Such uptake is close to the experimental adsorption capacity (106 cm3 g−1) under 0.01 bar.
In most of the literature, bond length is used to compare the interaction strength. However, in our case, the C–H⋯F distance (2.576 Å) is longer than the CH⋯F distance (2.108 Å), making it difficult to judge which interaction is stronger as the acidity of the C–H hydrogen is stronger than that of CH2. Thus, crystallography based DFT calculation is applied to calculate the bonding energy. First of all, we calculate the bonding energy between the framework and single gas molecule. To our delight, the results indicated the binding energy between a single C3H4 molecule and ZNU-2-Si is −39.35 kJ mol−1 (Fig. 4a) while that for C3H6 is only −34.26 kJ mol−1 (Fig. 4b), indicating the interaction between C3H4 and ZNU-2-Si is stronger. The binding energy difference (5.09 kJ mol−1) is also close to the experimental Qst difference (7.7 kJ mol−1).
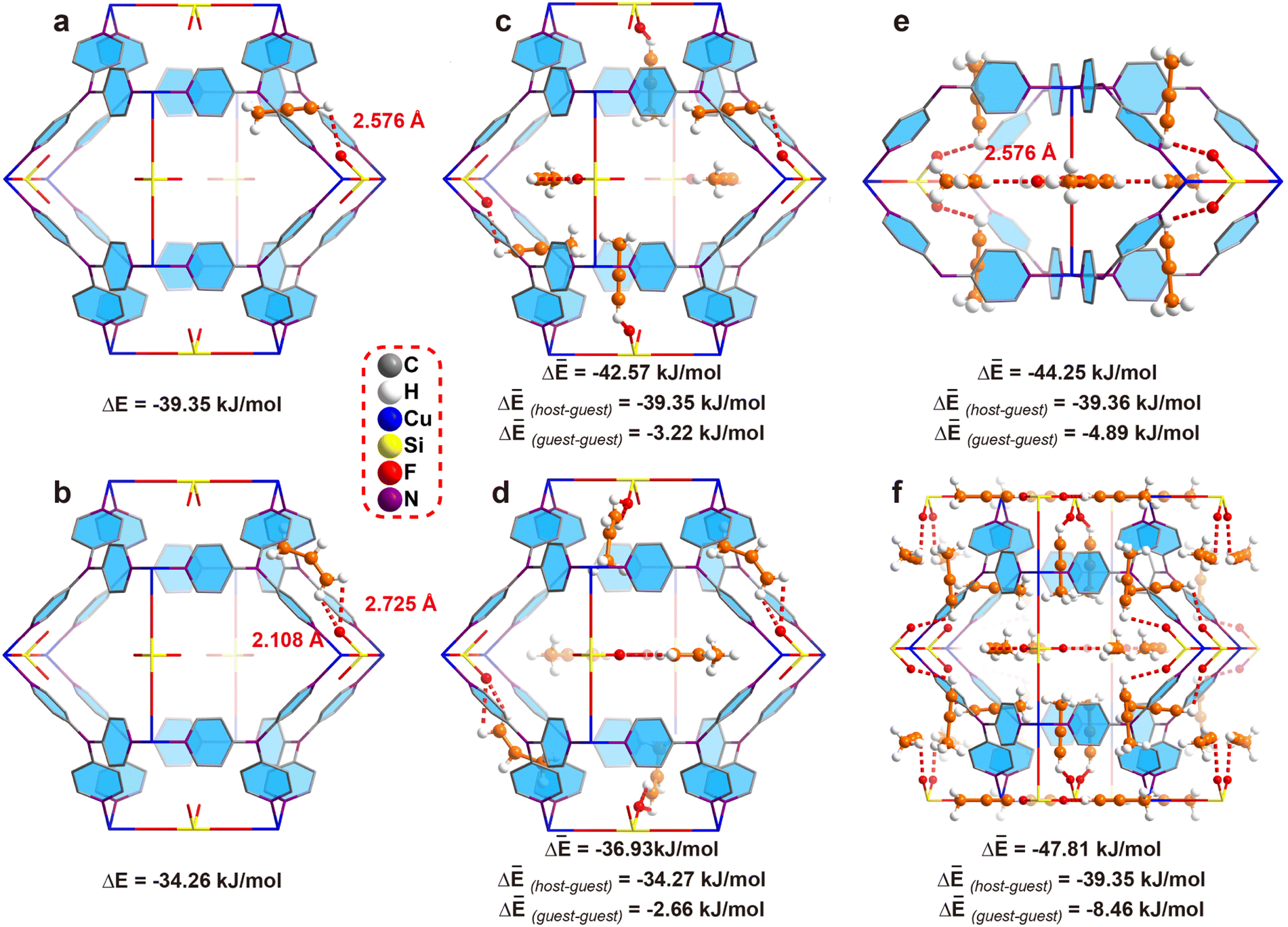 | ||
| Fig. 4 The DFT-D calculated interaction energy of ZNU-2-Si and gas molecules under different loadings based on the single crystal structure. (a and b) A C3H4 or C3H6 molecule located in a cage. (c and d) Six C3H4 or C3H6 molecules located in a cage. (e) 8 C3H4 molecules located near two neighbouring interlaced channels. (f) 24 C3H4 molecules in a unit cell. | ||
The binding energies of ZNU-2-Si and six gas molecules were also calculated, which are −42.57 and −36.93 kJ mol−1 for C3H4 and C3H6 (Fig. 4c and d), respectively. These binding energies can be separated into two parts: ZNU-2-Si⋯gas (host–guest) interaction energy and gas⋯gas (guest–guest) interaction energy. For C3H4, the ZNU-2-Si⋯C3H4 binding energy is still −39.35 kJ mol−1 and the interaction energy of six C3H4⋯C3H4 molecules is −3.22 kJ mol−1 (Fig. 4c). For C3H6, the ZNU-2-Si⋯C3H6 binding energy is −34.27 kJ mol−1 and the interaction energy of six C3H6⋯C3H6 molecules is −2.66 kJ mol−1 (Fig. 4d). These results indicated that the C3H4⋯C3H4 interactions are stronger than C3H6⋯C3H6 interactions in the confined cavity while the ZNU-2-Si⋯gas molecules remained nearly unchanged with the loading increase.
To gain more insight into the C3H4⋯C3H4 cluster, we choose another two models with different C3H4 molecules for comparison. Fig. 4e displays the structure of 8 C3H4 molecules in two neighboring interlaced channels, where the C3H4⋯C3H4 interaction energies increased to −4.89 kJ mol−1. Fig. 4f displays the complete loading of C3H4 molecules in the cages (i.e. 24 C3H4 molecules in a unit cell), where the C3H4⋯C3H4 interaction energies are further increased to −8.46 kJ mol−1. These results unambiguously revealed the boosted C3H4 adsorption behavior in ZNU-2-Si through cooperative guest⋯guest interactions.
To gain more insight into the gas adsorption behavior, GCMC simulations were performed, which indicated two distinct binding sites: one located completely in the interlaced channel and the other completely inside the cage. Moreover, the results indicated that 30 C3H4 molecules can be adsorbed in a single unit cell at 298 K and 1 bar (Fig. S68†), equal to 209 cm3 g−1 for ZNU-2-Si, similar to the experimental value of 188 cm3 g−1. DFT calculations were then applied to identify the adsorption configuration and binding energies of C3H4 in ZNU-2-Si. Fig. 5a shows that the C3H4 molecule in the first binding site is completely in the interlaced channel. The three hydrogen atoms from the methyl group in C3H4 strongly interact with three F atoms at the sharing edges of four different cages. The hydrogen bond distances are 2.24, 2.73, 2.86 and 2.89 Å. Besides, multiple additional weak van der Waals interactions exist with the C⋯H distances of 2.68, 2.77 and 2.85 Å (Fig. S56†). All of these interactions contribute to a high binding energy of −55.31 kJ mol−1. The second binding site located inside the cage adsorbs C3H4 by two strong hydrogen bonds between the terminal hydrogen of C3H4 and two adjacent F atoms with distances of 2.29 and 2.31 Å (Fig. 5b). This binding energy is −42.87 kJ mol−1. The binding energy for the second C3H4 molecule inside the cage increases to −46.45 kJ mol−1. Thus, the average binding energy of two C3H4 molecules inside the cavity is −44.66 kJ mol−1. In addition, the binding energies increase to −48.98, −49.72, and −50.55 kJ mol−1 for accommodation of 6, 10 and 13 C3H4 molecules in a cage, respectively (Fig. 5c–f). To provide direct comparison, the GCMC simulation result with 24 C3H4 molecules located both in the narrow channel and the large cage was chosen as a model for optimization. DFT calculation indicated the average bonding energy is −50.85 kJ mol−1 (Fig. S69a†), which is still higher than that (−47.81 kJ mol−1) based on the single crystal structure. Moreover, the GCMC optimized C3H4 configurations (i.e. 6 C3H4 molecules completely in the 6 narrow channels and 18 C3H4 molecules in two large cages) do not display distinct changes under DFT optimization.
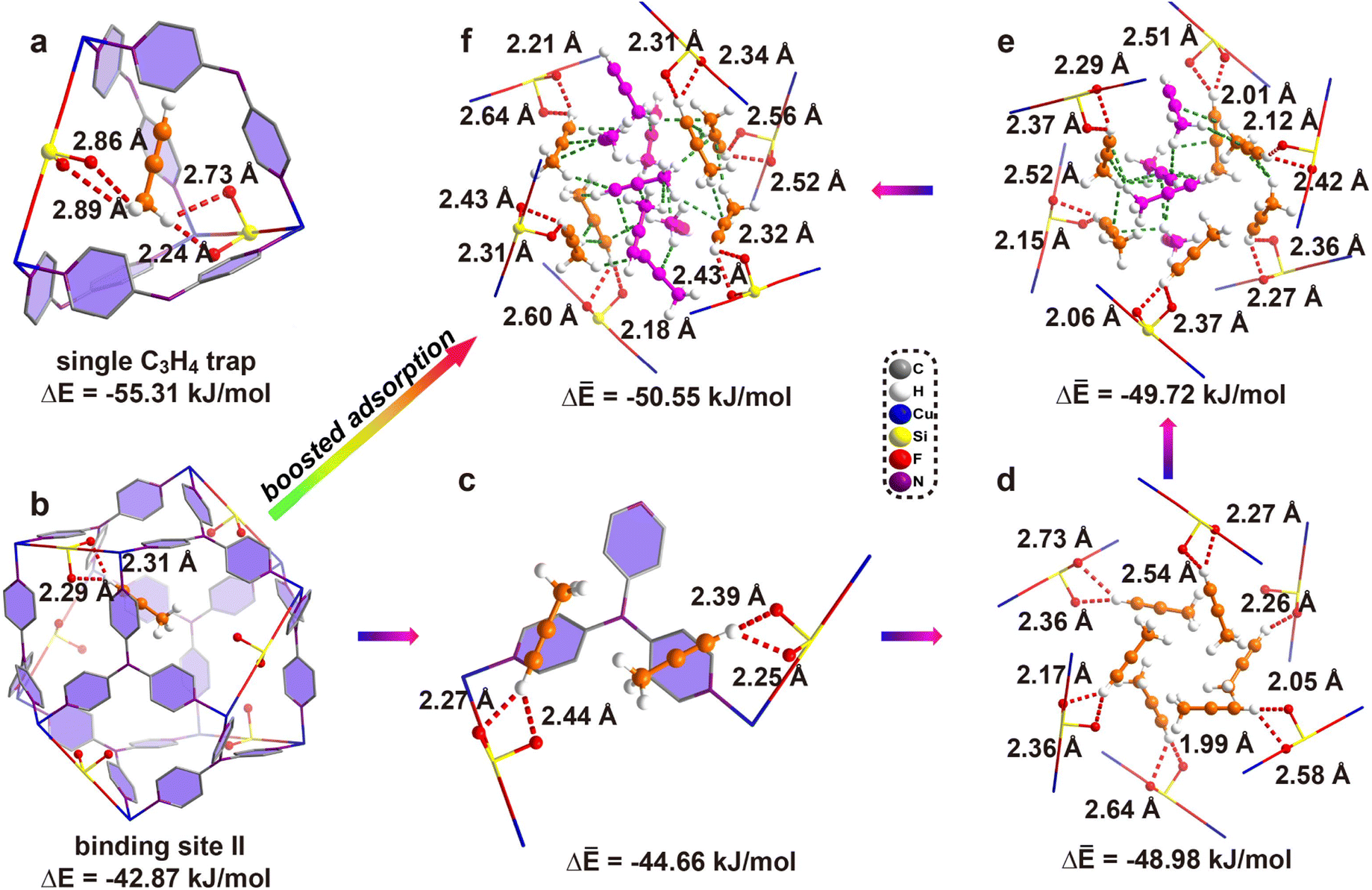 | ||
| Fig. 5 The DFT-D optimized C3H4 adsorption configuration based on GCMC simulation and bonding energy of C3H4 in ZNU-2-Si. (a) Binding site I inside the interlaced channel. (b) Binding site II inside the cage. (c–f) 2, 6, 10, and 13 C3H4 molecules adsorbed inside the cage. | ||
As described above, the GCMC based DFT calculation obviously provided more energy favorable binding sites for C3H4 molecules compared to those based on the single crystal structure. Then why do C3H4 molecules not follow this pathway for accommodation? Analysis of the cage-channel structure indicates that the narrow interlaced channel is the only passage that connects cages. Gas molecules in cage I must pass through the intersection to reach cage II. Thus, once the intersection is occupied, the diffusion is limited. Moreover, the entropy of C3H4 in the interlaced channel is the lowest because the rotation is highly restricted in the narrow channel. The diffusion of C3H4 from the intersection to the large cages is entropy favorable. Therefore, the experimentally observed C3H4 binding configuration is an entropy–enthalpy balanced result. As GCMC simulations neglect the influence of diffusion or entropy effect, they may not reflect the real binding sites in biporous materials.33 Binding site I (Fig. 5a) can be the exact location for the adsorption of the first C3H4 molecule under extremely low pressure. Once the pressure or number of C3H4 molecules increases, the diffusion or entropy effect becomes obvious, and the symmetrical binding sites in Fig. 4 to provide higher entropy are favored. On the other hand, the final C3H4 adsorption configuration can be considered as the result of competitive adsorption of C3H4 from different cages. Due to the high symmetry of the framework, four C3H4 molecules in the neighbouring large cages show the same potential to enter the interlaced channel to be strongly trapped but this narrow channel can only accommodate a single C3H4 completely. Thus, four C3H4 molecules squeeze their smaller C–H ends into the interlaced channel but leave their larger C–CH3 ends outside of the channel. We further calculate the bonding energy between 24 C3H4 molecules and ZNU-2-Si based on single crystal structures with all molecules relaxed (Fig. S69b†). In this case, the binding energy of −50.42 kJ mol−1 is only slightly inferior to the GCMC result (−50.85 kJ mol−1), which is easy to be covered by the entropy penalty. In brief, GCMC based DFT calculations can provide some information on the initial adsorption while in situ single crystal structures give the direct adsorption behavior under the measured conditions.
To gain some insight into the distinct adsorption kinetic difference of C3H4 and C3H6 in ZNU-2-Si as well as to provide more evidence for the entropy effect, MD simulations were carried out. The configurations of C3H4 and C3H6 molecules are based on the GCMC simulations and the whole framework is considered flexible except the Cu atoms. Fig. 6a–c illustrate the MSD in the x, y and z directions for 1, 4, and 7 C3H4 or C3H6 molecules per cage of ZNU-2-Si respectively. These graphs show that within the period of 5000 ps, the C3H4 molecules can migrate to other cages through the interlaced channels freely independent of the pressure, namely the number of C3H4 molecules located in a cage (Fig. 6d, S70, S71 and S73†). However, the C3H6 molecules can only move inside the original cage and are not able to spread to other cages until the number of molecules accommodated in a single cage reaches 5 (Fig. 6e, S70, S72 and S74†). MD-derived C3H4 and C3H6 diffusion coefficients in ZNU-2-Si were further calculated. The values are 4.72 × 10−11/6.79 × 10−14, 4.89 × 10−11/4.64 × 10−13, and 7.55 × 10−11/2.50 × 10−11 m2 s−1 for 1, 4 and 7 C3H4 or C3H6 molecules located inside a cage. Therefore, the diffusion coefficient of C3H4 is much higher than that of C3H6, especially under low pressure with the number of the gas molecules in a cage less than 5 (Table S20†). In other words, the diffusion rate of C3H4 in ZNU-2-Si is much faster than that of C3H6. The C3H4/C3H6 kinetic selectivity is as high as ∼695 under low pressures and ∼3.0 under high pressures. Such high kinetic selectivity has never been found in porous materials for C3H4/C3H6 separation, which is highly related to its unique framework structure with large cavities and narrow channels. Since the cages are connected by narrow interlaced channels, the gas molecules must pass through the channels when they need to diffuse from one cage to another. As the cross-section of C3H6 (4.65 × 4.16 Å2) is larger than that of C3H4 (4.01 × 4.16 Å2), larger pressure is needed to expand the channel sizes (original size ∼ 4 Å) by the rotation of the pyridine ring. Therefore, the narrow interlaced channels can be regarded as molecular sieves to allow the C3H4 molecules to pass through while prohibiting the migration of the C3H6 molecules under certain pressures. Only when the pressure increased to a higher degree did the gate opening (i.e. ligand rotation) allow C3H6 to diffuse fast within different cages.
 | ||
| Fig. 6 MD simulations. (a–c) MSD plot of C3H4 and C3H6 molecules in ZNU-2-Si with 1, 4 and 7 molecules in a single cage. (d and e) Snapshots of MD simulation of C3H4 (d) and C3H6 (e) molecules at 0, 2500, and 5000 ps. | ||
We also tried MD simulation considering the framework is completely rigid. In this case, it is very difficult for both C3H4 and C3H6 molecules to diffuse from one cage to another due to the limitation of the over-contracted intersection (∼4 Å). Therefore, the free energies of C3H4 and C3H6 moving from the narrow channels to the large cages were compared by calculating the potential of mean force (PMF). The results are presented in Fig. S75† which showed that C3H4 has a lower free energy barrier than C3H6, suggesting the diffusion of C3H4 is much easier than C3H6.
To evaluate the practical separation performance of ZNU-2-Si for selective C3H4/C3H6 separation, transient breakthrough simulations were conducted for the 10/90 C3H4/C3H6 mixture. The results showed that highly efficient separations could be accomplished by ZNU-2-Si (Fig. 7a). The productivity of C3H6 (>99.996% purity) in a single adsorption process is also calculated for ZNU-2-Si and other benchmark materials, which showed ZNU-2-Si has the record C3H6 productivity of 30.8 mol kg−1 (Fig. 7b), consistent with the separation potential ΔqIAST based on the static gas adsorption isotherms. Experimental breakthrough studies with the C3H4/C3H6 (10/90) mixture flowed over a ZNU-2-Si packed column with a flow rate of 4 mL min−1 at 298 K were then carried out. The experimental results were superior to the simulated one and 37.8 mol kg−1 of high purity C3H6 can be produced (Fig. 7c). Such enhancement can be attributed to the existence of the kinetic effect, which has never been reported in C3H4/C3H6 separation. For isomorphic ZNU-2-Ti, the kinetic enhancement is not obvious. The experimental C3H4 productivity (25.50 mol kg−1) is even slightly lower than that of the simulation (25.93 mol kg−1). The difference between ZNU-2-Si and ZNU-2-Ti can be accounted for by the reduced channel aperture in ZNU-2-Si that increases the diffusion difference in C3H4/C3H6 adsorption.
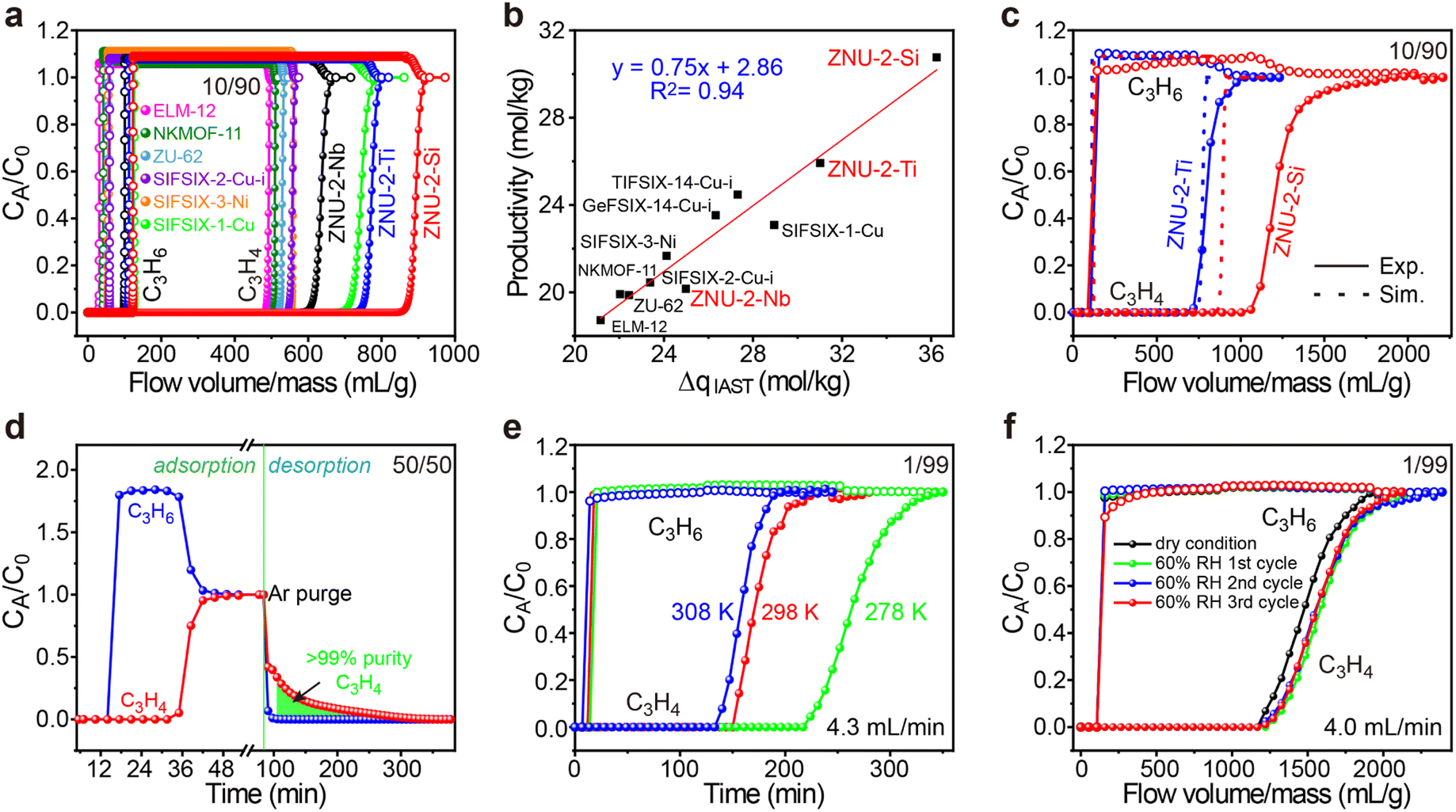 | ||
| Fig. 7 (a) Simulated breakthrough curves of ZNU-2-Si and other top-performing materials for C3H4/C3H6 (10/90) at 298 K. (b) Plots of the calculated productivity of C3H6 in >99.996% purity and separation potential ΔqIAST. (c) Comparison of the experimental and simulated breakthrough curves of ZNU-2-Si and ZNU-2-Ti for C3H4/C3H6 (10/90). (d) Experimental breakthrough curves and desorption curves of ZNU-2-Si for C3H4/C3H6 (50/50) at 298 K. (e) Experimental breakthrough curves of ZNU-2-Si for C3H4/C3H6 (1/99) at 278, 298, and 308 K. (f) Experimental breakthrough curves of ZNU-2-Si for C3H4/C3H6 (1/99) at 298 K under dry and humid conditions (activation conditions of ZNU-2-Si between cycles: Ar flow rate 20 mL min−1 at 393 K). | ||
To thoroughly identify the separation performance of ZNU-2-Si, we conducted more breakthrough experiments under various conditions. C3H4/C3H6 mixtures containing a higher ratio (50%) or lower ratio (1%) of C3H4 were tested. In both cases, clean C3H4/C3H6 separations were achieved. For the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 C3H4/C3H6 mixture, the retention time of C3H4 is over twice that of C3H6. 7.06 mol kg−1 of C3H4 was captured in the column with a purity of ∼86% (Fig. 7d). Controlling the desorption conditions, 4.7 mol kg−1 of >99% purity C3H4 can be recovered from the column by evacuation after blowing C3H6 out firstly (Fig. 7d and S81†). This record high dynamic productivity of C3H4 is impossible to obtain by other APMOFs due to their low C3H4 capacity. For the 1:99 C3H4/C3H6 mixture, C3H6 broke out at ∼18 min and became saturated immediately while C3H4 was not detected until ∼156 min and reached saturation slowly (Fig. 7e). The calculated experimental productivity of C3H6 from the 1:99 C3H4/C3H6 mixture at 298 K is 52.9 mol kg−1, much higher than those of SIFSIX-1-Cu (5.0 mol kg−1), ELM-12 (15.0 mol kg−1), SIFSIX-3-Ni (20.0 mol kg−1), SIFSIX-2-Cu-i (25.5 mol kg−1) and ZNU-2-Ti (42.0 mol kg−1). Upon lowering the experimental temperature to 278 K, the productivity of C3H6 increased to 79.20 mol kg−1, exceeding that of UTSA-200 (62.9 mol kg−1, 298 K)15 and NKMOF-11 (74.4 mol kg−1, 298 K)20 (Fig. 7e and S79†). Due to its extremely high water stability, we further carried out the breakthrough experiments under humid conditions. The C3H4/C3H6 (1:99) mixture was firstly bubbled into a bottle full of water and then introduced into the column packed with ZNU-2-Si. The humidity was measured constantly, which was stable at ∼60% after reaching equilibrium. From the repetitive humidity tests, it can be concluded that the influence of moisture is negligible for C3H4/C3H6 separation in ZNU-2-Si (Fig. 7f). Finally, the breakthrough experiments were conducted for six cycles, and the excellent separation capacity of ZNU-2-Si was retained, indicating that ZNU-2-Si possesses a high cycling stability (Fig. S84 and S85†). In summary, ZNU-2-Si sets a new record for practical simultaneous C3H6 purification and C3H4 recovery/storage by the combination of high productivity of polymer grade C3H6, large amount recovery of C3H4, retention of separation performance under humid conditions, outstanding recycling capacity and facile regeneration conditions.
:50 C3H4/C3H6 mixture, the retention time of C3H4 is over twice that of C3H6. 7.06 mol kg−1 of C3H4 was captured in the column with a purity of ∼86% (Fig. 7d). Controlling the desorption conditions, 4.7 mol kg−1 of >99% purity C3H4 can be recovered from the column by evacuation after blowing C3H6 out firstly (Fig. 7d and S81†). This record high dynamic productivity of C3H4 is impossible to obtain by other APMOFs due to their low C3H4 capacity. For the 1:99 C3H4/C3H6 mixture, C3H6 broke out at ∼18 min and became saturated immediately while C3H4 was not detected until ∼156 min and reached saturation slowly (Fig. 7e). The calculated experimental productivity of C3H6 from the 1:99 C3H4/C3H6 mixture at 298 K is 52.9 mol kg−1, much higher than those of SIFSIX-1-Cu (5.0 mol kg−1), ELM-12 (15.0 mol kg−1), SIFSIX-3-Ni (20.0 mol kg−1), SIFSIX-2-Cu-i (25.5 mol kg−1) and ZNU-2-Ti (42.0 mol kg−1). Upon lowering the experimental temperature to 278 K, the productivity of C3H6 increased to 79.20 mol kg−1, exceeding that of UTSA-200 (62.9 mol kg−1, 298 K)15 and NKMOF-11 (74.4 mol kg−1, 298 K)20 (Fig. 7e and S79†). Due to its extremely high water stability, we further carried out the breakthrough experiments under humid conditions. The C3H4/C3H6 (1:99) mixture was firstly bubbled into a bottle full of water and then introduced into the column packed with ZNU-2-Si. The humidity was measured constantly, which was stable at ∼60% after reaching equilibrium. From the repetitive humidity tests, it can be concluded that the influence of moisture is negligible for C3H4/C3H6 separation in ZNU-2-Si (Fig. 7f). Finally, the breakthrough experiments were conducted for six cycles, and the excellent separation capacity of ZNU-2-Si was retained, indicating that ZNU-2-Si possesses a high cycling stability (Fig. S84 and S85†). In summary, ZNU-2-Si sets a new record for practical simultaneous C3H6 purification and C3H4 recovery/storage by the combination of high productivity of polymer grade C3H6, large amount recovery of C3H4, retention of separation performance under humid conditions, outstanding recycling capacity and facile regeneration conditions.
Conclusions
In conclusion, we reported a chemically stable MOF, ZNU-2-Si, with large three-dimensional pores and narrow interlaced channels for record propyne storage and propyne/propylene separation. Notable features of this work include: (1) benchmark C3H4 capacity of 106 cm3 g−1 under a low pressure of 0.01 bar; (2) extremely high C3H4 storage capacity (188 cm3 g−1, 298 K) and storage density (0.60/0.65 g cm−3 at 298/278 K) at 1.0 bar; (3) record high C3H4/C3H6 (10/90) separation potential (36.2 mol kg−1); (4) record high experimental C3H6 productivity (37.81 mol kg−1) from 10/90 C3H4/C3H6 mixtures; (5) record high >99% purity C3H4 recovery (4.7 mol kg−1) from a 50/50 C3H4/C3H6 mixture by a stepped desorption process; (6) benchmark experimental C3H6 productivity (52.9/79.2 mol kg−1 at 298/278 K) from 1/99 C3H4/C3H6 mixtures; (7) excellent breakthrough recyclability and performance retention under humid conditions; (8) unprecedented revelation of the adsorption and separation mechanism by in situ single crystal analysis and GCMC/MD simulations and DFT calculations. In general, our work not only proposes a strategy of using MOFs with large cages and narrow channels for thermodynamic–kinetic synergistic separation, but also highlights the importance of combining the in situ single crystal structure analysis and theoretical studies to investigate the adsorption/separation mechanism. These cage-like APMOFs with optimal pore chemistry and pore structures are supposed to be promising for many other challenging gas separations.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
Y. J.: synthesis, characterization, adsorption experiments, draft preparation; L. W.: single crystal structure measurement and analysis, funding; T. Y.: GCMC simulation, DFT calculation, MD simulation; J. H.: GCMC simulation, DFT calculation; W. S.: breakthrough experiments; R. K.: IAST calculation, breakthrough simulation; D. W.: discussion, advice; Z. G.: PMF calculation; D. L.: supervision of the theoretical study; X. C.: supervision of the theoretical study; H. X.: supervision of the theoretical study; Y. Z.: concept, supervision, draft preparation, funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21908193 and 22205207) and Jinhua Industrial Key Project (2021A22648).References
- (a) I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS; (b) M.-Y. Gao, B.-Q. Song, D. Sensharma and M. J. Zaworotko, SmartMat, 2021, 2, 38–55 CrossRef CAS.
- A. Chavel and G. Lefebvre, Petrochemical Process, Institut Francais du Petrole Publications, Gulf Publishing, 1989, pp. 199–208 Search PubMed.
- (a) Z. Miao, A. M. Esper, S. S. Nadif, S. A. Gonsales, B. S. Sumerlin and A. S. Veige, React. Funct. Polym., 2021, 169, 105088 CrossRef CAS; (b) J. L. Alterman and G. A. Kraus, Synthesis, 2022, 54, 655–657 CrossRef CAS.
- (a) H. Wang and J. Li, Acc. Chem. Res., 2019, 52, 1968–1978 CrossRef CAS; (b) B. R. Barnett, M. I. Gonzalez and J. R. Long, Trends Chem., 2019, 1, 159–171 CrossRef CAS; (c) X. Zhao, Y. Wang, D. S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef; (d) R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CrossRef CAS; (e) Z. Zhang, S. B. Peh, C. Kang, K. Chai and D. Zhao, EnergyChem, 2021, 3, 100057 CrossRef CAS; (f) S.-Q. Yang and T.-L. Hu, Coord. Chem. Rev., 2022, 468, 214628 CrossRef CAS.
- (a) Z. Shi, Y. Tao, J. Wu, C. Zhang, H. He, L. Long, Y. Lee, T. Li and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 2750–2754 CrossRef CAS; (b) W. Sun, J. Hu, S. Duttwyler, L. Wang, R. Krishna and Y. Zhang, Sep. Purif. Technol., 2022, 283, 120220 CrossRef CAS; (c) Y. Zhang, X. Cui and H. Xing, Mater. Chem. Front., 2021, 5, 5970–6013 RSC; (d) O. T. Qazvini and S. G. Telfer, ACS Appl. Mater. Interfaces, 2021, 13, 12141–12148 CrossRef CAS PubMed; (e) X.-W. Zhang, D.-D. Zhou and J.-P. Zhang, Chem, 2021, 7, 1006–1019 CrossRef CAS.
- (a) K. Chen, D. G. Madden, T. Pham, K. A. Forrest, A. Kumar, Q. Yang, W. Xue, B. Space, J. J. Perry IV, J. Zhang, X. Chen and M. J. Zaworotko, Angew. Chem., Int. Ed., 2016, 55, 10268–10272 CrossRef CAS; (b) S. Tu, L. Yu, D. Lin, Y. Chen, Y. Wu, X. Zhou, Z. Li and Q. Xia, ACS Appl. Mater. Interfaces, 2022, 14, 4242–4250 CrossRef CAS; (c) R. L. Siegelman, P. J. Milner, A. C. Forse, J. Lee, K. A. Colwell, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, J. Am. Chem. Soc., 2019, 141, 13171–13186 CrossRef CAS PubMed; (d) P. Hu, H. Wang, C. Xiong, H. Liu, J. Han, J. Zhou, Z. Zhao, Y. Wang and H. Ji, ACS Sustainable Chem. Eng., 2021, 9, 15897–15907 CrossRef CAS; (e) J. Li, G. L. Smith, Y. Chen, Y. Ma, M. Kippax-Jones, M. Fan, W. Lu, M. D. Frogley, G. Cinque, S. J. Day, S. P. Thompson, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, M. Schröder and S. Yang, Angew. Chem., Int. Ed., 2022, 61, 202207259 Search PubMed.
- (a) L. Wang, W. Sun, Y. Zhang, N. Xu, R. Krishna, J. Hu, Y. Jiang, Y. He and H. Xing, Angew. Chem., Int. Ed., 2021, 60, 22865–22870 CrossRef CAS; (b) H. Zeng, M. Xie, Y.-L. Huang, Y. Zhao, X.-J. Xie, J. Bai, M.-Y. Wan, R. Krishna, W. Lu and D. Li, Angew. Chem., Int. Ed., 2019, 58, 8515–8519 CrossRef CAS PubMed; (c) Z. Niu, X. Cui, T. Pham, G. Verma, P. C. Lan, C. Shan, H. Xing, K. A. Forrest, S. Suepaul, B. Space, A. Nafady, A. M. Al-Enizi and S. Ma, Angew. Chem., Int. Ed., 2021, 60, 5283–5288 CrossRef CAS; (d) J. Wang, Y. Zhang, Y. Su, X. Liu, P. Zhang, R.-B. Lin, S. Chen, Q. Deng, Z. Zeng, S. Deng and B. Chen, Nat. Commun., 2022, 13, 200 CrossRef CAS PubMed; (e) W. Sun, J. Hu, Y. Jiang, N. Xu, L. Wang, J. Li, Y. Hu, S. Duttwyler and Y. Zhang, Chem. Eng. J., 2022, 439, 135745 CrossRef CAS; (f) W. Lou, J. Li, W. Sun, Y. Hu, L. Wang, R. F. Neumann, M. Steiner, Z. Gu, B. Luan and Y. Zhang, Chem. Eng. J., 2023, 452, 139296 CrossRef CAS.
- (a) Y. Zhang, J. Hu, R. Krishna, L. Wang, L. Yang, X. Cui, S. Duttwyler and H. Xing, Angew. Chem., Int. Ed., 2020, 59, 17664–17669 CrossRef CAS PubMed; (b) R.-B. Lin, L. Li, H. Wu, H. Arman, B. Li, R.-G. Lin, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 8022–8028 CrossRef CAS PubMed; (c) W. Fan, S. Yuan, W. Wang, L. Feng, X. Liu, X. Zhang, X. Wang, Z. Kang, F. Dai, D. Yuan, D. Sun and H. Zhou, J. Am. Chem. Soc., 2020, 142, 8728–8737 CrossRef; (d) Q. Dong, Y. Huang, K. Hyeon-deuk, I.-Y. Chang, J. Wan, C. Chen, J. Duan, W. Jin and S. Kitagawa, Adv. Funct. Mater., 2022, 32, 2203745 CrossRef CAS.
- (a) G.-D. Wang, Y.-Z. Li, W.-J. Shi, L. Hou, Y.-Y. Wang and Z. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202205427 CAS; (b) X.-W. Gu, J.-X. Wang, E. Wu, H. Wu, W. Zhou, G. Qian, B. Chen and B. Li, J. Am. Chem. Soc., 2022, 144, 2614–2623 CrossRef CAS; (c) P. Zhang, Y. Zhong, Y. Zhang, Z. Zhu, Y. Liu, Y. Su, J. Chen, S. Chen, Z. Zeng, H. X., S. Deng and J. Wang, Sci. Adv., 2022, 8, eabn9231 CrossRef CAS; (d) L. Yang, L. Yan, W. Niu, Y. Feng, Q. Fu, S. Zhang, Y. Zhang, L. Li, X. Gu, P. Dai, D. Liu, Q. Zheng and X. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202204046 CAS; (e) P.-Q. Liao, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8697 CrossRef.
- (a) C. He, R. Krishna, Y. Chen, J. Yang, J. Li and L. Li, Chin. J. Chem. Eng., 2021, 37, 217–221 CrossRef; (b) Y. Chai, X. Han, W. Li, S. Liu, S. Yao, C. Wang, W. Shi, I. Da-Silva, P. Manuel, Y. Cheng, L. D. Daemen, A. J. Ramirez-Cuesta, C. C. Tang, L. Jiang, S. Yang, N. Guan and L. Li, Science, 2020, 368, 1002–1006 CrossRef CAS.
- (a) H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef; (b) K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC; (c) L. Yang, S. Qian, X. Wang, X. Cui, B. Chen and H. Xing, Chem. Soc. Rev., 2020, 49, 5359–5406 RSC.
- X. Li, H. Bian, W. Huang, B. Yan, X. Wang and B. Zhu, Coord. Chem. Rev., 2022, 470, 214714 CrossRef CAS.
- H.-M. Wen, L. Li, R.-B. Lin, B. Li, B. Hu, W. Zhou, J. Hu and B. Chen, J. Mater. Chem. A, 2018, 6, 6931–6937 RSC.
- L. Yang, X. Cui, Q. Yang, S. Qian, H. Wu, Z. Bao, Z. Zhang, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2018, 30, 1705374 CrossRef.
- L. Li, H. M. Wen, C. He, R. B. Lin, R. Krishna, H. Wu, W. Zhou, J. Li, B. Li and B. Chen, Angew. Chem., Int. Ed., 2018, 57, 15183–15188 CrossRef CAS PubMed.
- Z. Zhang, Q. Ding, J. Cui, X. Cui and H. Xing, Small, 2020, 16, 2005360 CrossRef CAS.
- L. Yang, X. Cui, Y. Zhang, Q. Yang and H. Xing, J. Mater. Chem. A, 2018, 6, 24452–24458 RSC.
- L. Yang, X. Cui, Z. Zhang, Q. Yang, Z. Bao, Q. Ren and H. Xing, Angew. Chem., Int. Ed., 2018, 57, 13145–13149 CrossRef CAS PubMed.
- L. Li, R. B. Lin, R. Krishna, X. Wang, B. Li, H. Wu, J. Li, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 7733–7736 CrossRef CAS.
- Y.-L. Peng, T. Wang, C. Jin, P. Li, S. Suepaul, G. Beemer, Y. Chen, R. Krishna, P. Cheng, T. Pham, B. Space, M. J. Zaworotko and Z. Zhang, J. Mater. Chem. A, 2021, 9, 2850–2856 RSC.
- Z. T. Lin, Q. Y. Liu, L. Yang, C. T. He, L. Li and Y. L. Wang, Inorg. Chem., 2020, 59, 4030–4036 CrossRef CAS PubMed.
- Y. L. Peng, C. He, T. Pham, T. Wang, P. Li, R. Krishna, K. A. Forrest, A. Hogan, S. Suepaul, B. Space, M. Fang, Y. Chen, M. J. Zaworotko, J. Li, L. Li, Z. Zhang, P. Cheng and B. Chen, Angew. Chem., Int. Ed., 2019, 58, 10209–10214 CrossRef CAS PubMed.
- T. Ke, Q. Wang, J. Shen, J. Zhou, Z. Bao, Q. Yang and Q. Ren, Angew. Chem., Int. Ed., 2020, 59, 12725–12730 CrossRef CAS PubMed.
- Z. Li, L. Li, L. Guo, J. Wang, Q. Yang, Z. Zhang, Y. Yang, Z. Bao and Q. Ren, Ind. Eng. Chem. Res., 2020, 59, 13716–13723 CrossRef CAS.
- L. Li, L. Guo, F. Zheng, Z. Zhang, Q. Yang, Y. Yang, Q. Ren and Z. Bao, ACS Appl. Mater. Interfaces, 2020, 12, 17147–17154 CrossRef CAS.
- S. Zou, Z. Di, H. Li, Y. Liu, Z. Ji, H. Li, C. Chen, M. Wu and M. Hong, Inorg. Chem., 2022, 61, 7530–7536 CrossRef CAS PubMed.
- Y. Jiang, J. Hu, L. Wang, W. Sun, N. Xu, R. Krishna, S. Duttwyler, X. Cui, H. Xing and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200947 CAS.
- (a) D. O'Nolan, A. Kumar and M. J. Zaworotko, J. Am. Chem. Soc., 2017, 139, 8508–8513 CrossRef; (b) S. Noro, R. Kitaura, M. Kondo, S. Kitagawa, T. Ishii, H. Matsuzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568–2583 CrossRef CAS PubMed.
- H. Li, C. Liu, C. Chen, Z. Di, D. Yuan, J. Pang, W. Wei, M. Wu and M. Hong, Angew. Chem., Int. Ed., 2021, 60, 7547–7552 CrossRef CAS PubMed.
- M. Lusi, P. B. A. Fechine, K.-J. Chen, J. J. Perry IV and M. J. Zaworotko, Chem. Commun., 2016, 52, 4160–4162 RSC.
- (a) R. Krishna, RSC Adv., 2017, 7, 35724–35737 RSC; (b) R. Krishna, ACS Omega, 2020, 5, 16987–17004 CrossRef CAS PubMed.
- R. Krishna and J. M. van Baten, ACS Omega, 2022, 7, 13050–13056 CrossRef CAS PubMed.
- M. Xie, Z. Lu, W. Lu and D. Li, Inorg. Chem. Front., 2022, 9, 2697–2705 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2190367–2190372. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05742e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |