
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNonspecific interactions between Cas12a and dsDNA located downstream of the PAM mediate target search and assist AsCas12a for DNA cleavage†
Ruirui
Sun
ab,
Yuqian
Zhao
ac,
Wenjuan
Wang
d,
Jun-Jie Gogo
Liu
ac and
Chunlai
Chen
 *ab
*ab
aBeijing Advanced Innovation Center for Structural Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China. E-mail: chunlai@mail.tsinghua.edu.cn
bBeijing Frontier Research Center for Biological Structure, Tsinghua University, Beijing 100084, China
cTsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China
dTechnology Center for Protein Sciences, School of Life Sciences, Tsinghua University, Beijing 100084, China
First published on 10th March 2023
Abstract
Cas12a is one of the most commonly used Cas proteins for genome editing and gene regulation. The first key step for Cas12a to fulfill its function is to search for its target among numerous nonspecific and off-target sites. Cas12a utilizes one-dimensional diffusion along the contour of dsDNA to efficiently search for its target. However, due to a lack of structural information of the transient diffusing complex, the residues mediating the one-dimensional diffusion of Cas12a are unknown. Here, combining single-molecule and ensemble assays, we found that nonspecific interactions between Cas12a and dsDNA at the PAM downstream cause asymmetric target search regions of Cas12a flanking the PAM site, which guided us to identify a positive-charge-enriched alpha helix in the REC2 domain serving as a conserved element to facilitate one-dimensional diffusion-driven target search of AsCas12a, LbCas12a and FnCas12a. In addition, this alpha helix assists the target cleavage process of AsCas12a via stabilizing the cleavage states. Thus, neutralizing positive charges within this helix not only significantly slows target search but also enhances the specificity of AsCas12a both in vitro and in living cells. Similar behaviors are detected when residues mediating diffusion of SpCas9 are mutated. Thus, engineering residues mediating diffusion on dsDNA is a new avenue to optimize and enrich the versatile CRISPR-Cas toolbox.
Introduction
The clustered regularly interspaced short palindromic repeat (CRISPR) systems and CRISPR-associated (CRISPR-Cas) proteins are identified in archaea and bacteria as defense systems to target foreign nucleic acids.1–5 The widely used Cas9 and Cas12a proteins both belong to Class 2 CRISPR-Cas systems, whose effector modules only consist of a single protein.6,7 Cas9 and Cas12a have been widely used for genome editing, as they can be guided by single-stranded RNAs to recognize, to bind and to cleave their target adjacent to a PAM sequence in double-stranded DNA (dsDNA) with high specificity.8–13 In addition, dead Cas9 (dCas9), Cas9 nickase (nCas9) and dead Cas12a (dCas12a) have been engineered by mutating their nuclease domains while retaining their RNA-guided DNA-binding activities, which can be fused with other effector proteins and be repurposed for gene regulation, base editing, prime editing and so on.14–21 Using split-Cas9 proteins or guide RNAs with unique structural elements, the activity of Cas9 and Cas12a can be switched on by chemical or optical inputs.22–27For all Cas proteins and Cas protein-based effectors, the first key step is to locate their correct targets among numerous nonspecific and off-target sites in the complicated cellular environments. In general, DNA-binding proteins combine three-dimensional (3D) diffusion in the solution and one-dimensional (1D) diffusion along the contour of dsDNA to efficiently search for their cognate target sites on genomic DNA.28–31 Previous studies have confirmed that both Cas9 and Cas12a combine 3D and 1D diffusion to effectively facilitate their target search processes.32–36 1D diffusion of proteins on dsDNA is usually mediated by repetitive transient binding events driven by non-specific electrostatic interactions between them. Although numerous structures of Cas proteins in their apo states or in the complexes with DNA and RNA have been resolved,37–47 the transient Cas protein DNA binding domains mediating 1D diffusion-driven target search are less well characterized. Thus, engineering the residues mediating 1D diffusion of Cas proteins has never been used or even tested as a strategy to improve their specificity or to modify their functions.
In this study, we revealed that all three commonly used Cas12a orthologs, AsCas12a, LbCas12a and FnCas12a, display asymmetric 1D diffusion-driven target search regions flanking the PAM of their targets, suggesting that there are additional DNA-binding sites in Cas12a, besides the PI (PAM-interacting) domain, to mediate the 1D diffusion. A positive-charge-enriched alpha helix in the REC2 domain is identified as a conserved element to mediate 1D diffusion of all three Cas12a proteins. Neutralizing the positive charge in this helix not only significantly diminishes the contribution of 1D diffusion and reduces target search rates of all three Cas12a proteins, but also slows the target cleavage rate of AsCas12a by destabilizing its DNA-cleavage states. Lastly, by combining the non-specific binding sites between SpCas9 and dsDNA identified in our previous study,32 we demonstrated that modulating the residues mediating 1D diffusion of AsCas12a and SpCas9 improves their target specificity in vitro and in live cells. Thus, our work provides a new avenue to optimize and engineer versatile Cas protein-based tools.
Results and discussion
Asymmetric target search regions of Cas12a flanking PAM
Previous studies have shown that Cas12a displays 1D diffusion on dsDNA to search for its targets.34,35 Inspired by our previous report showing that the SpCas9 protein displays asymmetric target search regions flanking its PAM,32 we applied similar single-molecule fluorescence assays to examine whether Cas12a exhibits similar behaviors (Fig. 1). dsDNAs were immobilized on the slide surface via streptavidin and biotin linkage (Fig. 1a). Then, the Cy3-labeled Cas12a/crRNA was allowed to flow into the channel containing immobilized dsDNA, which contained 6 matched bases towards crRNA at its PAM proximal end. Such design permits transient partial R-loop formation at the target site to generate a fluorescence signal in single-molecule trajectories. The dwell times of the transiently formed Cas12a/crRNA/dsDNA complex were 40–200 ms (Fig. S1†), which are significantly longer than the transient binding between Cas12a and dsDNA without PAM and target sites (<5 ms, Fig. S2†). Thus, with 10 ms exposure time, only binding events of Cas12a on the correct target site were captured. The time from injection until the appearance of an individual Cy3 signal was recorded and assigned as the appearance time (Fig. 1b), representing the time spent by Cas12a/crRNA to search for its target via both 3D and 1D diffusion processes. The distributions of appearance time under various conditions were fitted by single-exponential decay to quantify apparent target search rates (Fig. 1c–h and S3†). The search rates of a series of dsDNA with different lengths enable us to quantify the 1D diffusion length.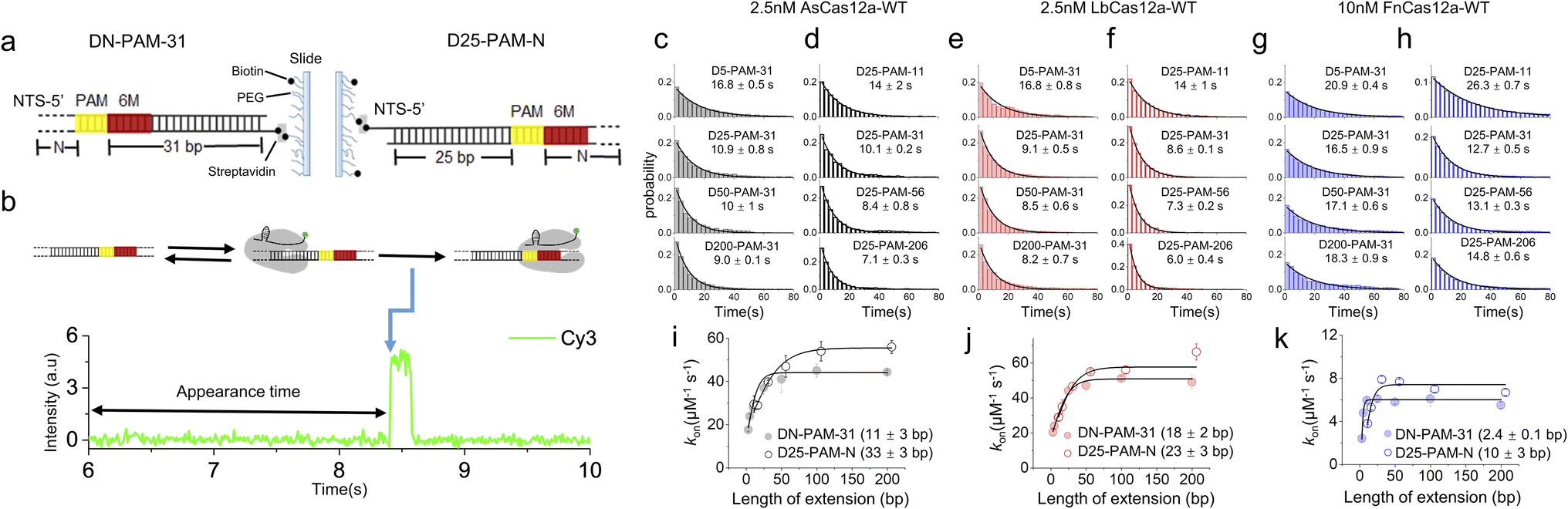 | ||
| Fig. 1 Single-molecule fluorescence assays quantifying 1D diffusion of AsCas12a, LbCas12a and FnCas12a. (a) dsDNAs with upstream and downstream extensions of PAM are named DN-PAM-31 and D25-PAM-N, respectively. 6-matched base pairs between crRNA and target DNA at the PAM-proximal end are shown in red. M is an abbreviation for matched base pairs at the PAM-proximal end. The PAM is shown in yellow. For DN-PAM-31 dsDNAs, the distance between PAM and the 3′ end of the NTS remains at 31 bp and the distance between PAM and the 5′ end of the NTS varies from 3 bp to 200 bp (left). For D25-PAM-N dsDNAs, the distance between PAM and the 5′ end of the NTS remains at 25 bp and the distance between PAM and the 3′ end of the NTS varies from 11 bp to 206 bp (right). (b) A representative single-molecule trace. The time from injecting Cy3-labeled crRNA/Cas12a onto immobilized dsDNA until the appearance of the binding event, indicated by the appearance of the Cy3 signal (blue arrow), was recorded. (c and d) Distributions of the appearance time of AsCas12a-WT on DN-PAM-31 dsDNAs (c) and D25-PAM-N dsDNAs (d), which were fitted by single exponential decay. Distributions of one repeat are shown. Values were mean ± SEM from three replicates. (e and f) Distributions of the appearance time of LbCas12a-WT on DN-PAM-31 dsDNAs (e) and D25-PAM-N dsDNAs (f). (g and h) Distributions of the appearance time of FnCas12a-WT on DN-PAM-31 dsDNAs (g) and D25-PAM-N dsDNAs (h). (i–k) The apparent target search rates (kon) of AsCas12a-WT (i), LbCas12a-WT (j) and FnCas12a-WT (k) were affected by the extension of dsDNA. Values were mean ± SEM from three replicates. | ||
For the target dsDNAs of the DN-PAM-31 series, the distance between PAM and the 3′ end of the non-target strand (NTS) remains constant at 31 bp and the distance between PAM and the 5′ end of the NTS varies from 3 bp to 200 bp (Fig. 1a left, Table S1†). The DN-PAM-31 series enable us to quantify the effect of PAM upstream extension on the target search process of Cas12a. Both AsCas12a and LbCas12a exhibit similar behaviors, whose apparent search rates increase by 2–3 fold with an extension of PAM upstream (Fig. 1i and j). The effective search lengths of AsCas12a and LbCas12a on PAM upstream are ∼11 bp and ∼18 bp, respectively, with a maximal search rate of 40–50 μM−1 s−1. However, for FnCas12a, its target search rate is significantly reduced (the maximal value ∼5.6 μM−1 s−1) and its effective search length is shrunk to a few bp (Fig. 1k). The salt concentration and crowding reagent only have a minor effect on the target search behaviors of FnCas12a (Fig. S4†). To verify that the Cy3 signal captured in our assay (Fig. 1b) is truly the result of Cas12a binding to the target site, we performed additional single-molecule fluorescence resonance energy transfer (FRET) measurements, in which FRET between Cy5-labeled dsDNA and Cy3-labeled crRNA was used to pinpoint the binding of Cas12a/crRNA on the target site (Fig. S5a and b†). Both the single-molecule FRET assay and the single-molecule fluorescence assay with only Cy3 signals had similar results, confirming the accuracy of our measurements (Fig. 1 and S5†).
Next, we used the D25-PAM-N series to quantify the effect of PAM downstream extension on the target search of Cas12a, in which the distance between PAM and the 5′ end of the NTS remains constant at 25 bp and the distance between PAM and the 3′ end of the NTS varies from 11 bp to 206 bp (Fig. 1a right, Table S1†). The downstream extension of PAM also accelerates target search rates of AsCas12a and LbCas12a by 2–3 fold to 55–60 μM−1 s−1 (Fig. 1i and j). The effective search lengths of AsCas12a and LbCas12a on PAM downstream are ∼33 bp and ∼23 bp, respectively, which are longer than their search lengths on PAM upstream (Fig. 1i and j). Although FnCas12a displays a slower target search rate than AsCas12a and LbCas12a, its search length on PAM downstream is still longer than the one on PAM upstream (Fig. 1k). Together, our single-molecule assays confirmed that Cas12a combines both 3D and 1D diffusion to accelerate the target search process on dsDNA. In addition, in line with our previous finding on SpCas9,32 AsCas12a, LbCas12a and FnCas12a all exhibit asymmetric target search regions flanking PAM, suggesting that, besides the PAM-interacting residues, there are additional nonspecific interactions between Cas12a and dsDNA at the PAM downstream to facilitate 1D diffusion.
Cas12a displays extremely weak binding towards isolated PAM sites randomly located on dsDNA (<30 ms),34 which is beyond the time resolution of our experimental design and will not contribute to our analysis. In our experiments, AsCas12a and FnCas12a shared the same crRNA sequence, whereas LbCas12a formed a binary complex with another different crRNA previously used48 (Table S2†). We confirmed that switching between these two crRNAs only has minor effects on the target search dynamics of Cas12a (Fig. S6†). Therefore, the distinctive target search dynamics among AsCas12a, LbCas12a and FnCas12a captured in Fig. 1 are mainly caused by the differences in protein sequences.
Critical residues mediating 1D diffusion of Cas12a
In our previous studies, SpCas9 exhibits similar asymmetric target search behaviours to those of Cas12a, guiding us to reveal a non-specific binding site between SpCas9 and intact dsDNA ∼8 bp downstream of the PAM to mediate 1D diffusion.32 Asymmetric target search of Cas12a shown in Fig. 1 indicated that the non-specific binding site mediating 1D diffusion of Cas12a on dsDNA is also located at the downstream of PAM. However, the target sequence of Cas9 is located at the PAM upstream, whereas the target sequence of Cas12a is located at the PAM downstream. Thus, in the published structures of the Cas12a/crRNA/dsDNA ternary complex, the PAM downstream dsDNA is already unwound to form an R-loop with crRNA.42,43 Furthermore, due to the instantaneous, fast and unstable search process of Cas12a, non-specific interaction between Cas12a and intact dsDNA has not been identified yet. Nevertheless, we hypothesized that the positively charged residues in the cavity formed by REC1, REC2, RuvC and Nuc domains to interact with the RNA/DNA heteroduplex and NTS might contribute to non-specific interactions with intact dsDNA to mediate 1D diffusion of Cas12a. Based on the published structure,42 we selected two positively charged residues and seven regions with enriched positively charged residues as candidates for further examination (Fig. 2a, Table S3†).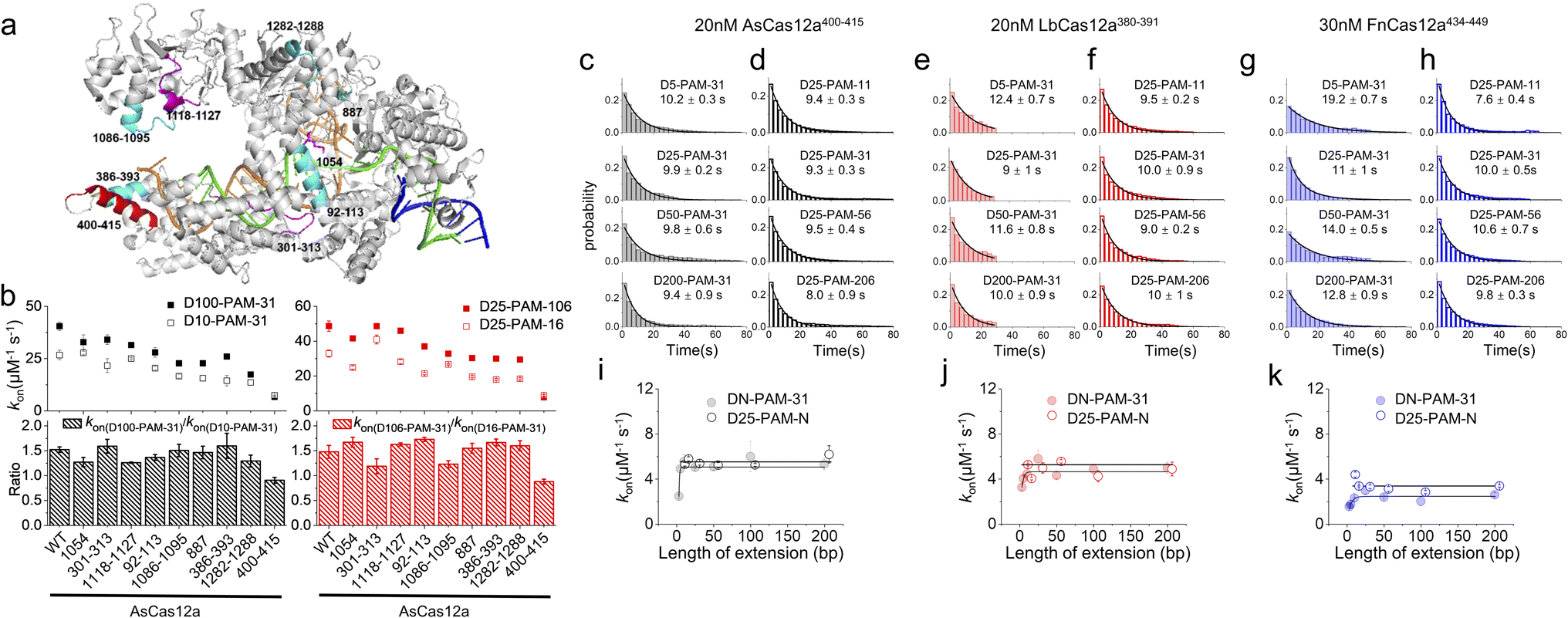 | ||
| Fig. 2 Residues mediating 1D diffusion and target search of Cas12a. (a) Structure of AsCas12a/crRNA/dsDNA (PDB: 5b43). The AsCas12a protein, crRNA, TS and NTS are shown in gray, orange, green and blue, respectively. Residues 1054, 301–313 and 1118–1127 are shown in magenta. Residues 92–113, 1086–1095, 887, 386–393, and 1282–1288 are shown in cyan. Residues 400–415 are shown in red. (b) The apparent target search rates (kon) of AsCas12a-WT and AsCas12a-mutants on dsDNAs with extensions of PAM upstream and downstream and their relative ratios. Values were mean ± SEM from three replicates. (c and d) Distributions of the appearance time of AsCas12a400–415 on DN-PAM-31 dsDNAs (c) and D25-PAM-N dsDNAs (d), which were fitted by single exponential decay. Distributions of one repeat are shown. Values were mean ± SEM from three replicates. (e and f) Distributions of the appearance time of LbCas12a380–391 on DN-PAM-31 dsDNAs (e) and D25-PAM-N dsDNAs (f). (g and h) Distributions of the appearance time of FnCas12a434–449 on DN-PAM-31 dsDNAs (g) and D25-PAM-N dsDNAs (h). (i–k) The apparent target search rates of AsCas12a400–415 (i), LbCas12a380–391 (j) and FnCas12a434–449 (k) were affected by the extension of dsDNA. Values were mean ± SEM from three replicates. | ||
Next, we generated nine AsCas12a mutants by mutating positively charged residues within the candidate regions to alanine (Table S3†). All of these mutants retained their abilities to recognize and to cleave the dsDNA target (Fig. S7†). Then, how extensions of PAM upstream and downstream affect apparent target search rates of these AsCas12a mutants were examined using the assays described above (Fig. 2b). AsCas12a1054, AsCas12a301–313 and AsCas12a1118–1127 had similar apparent target search rates as AsCas12a-WT. Although AsCas12a92–113, AsCas12a1086–1095, AsCas12a887, AsCas12a386–393 and AsCas12a1282–1288 displayed slower target search rates than AsCas12a-WT, the sensitivity of their search rates towards the extension of PAM upstream and downstream remained almost the same as that of AsCas12a-WT. On the other hand, AsCas12a400–415 not only exhibited the slowest target search rates among all mutants but also showed the least sensitivity towards the extension of the PAM upstream and downstream (Fig. 2b). With the DN-PAM-31 series and the D25-PAM-N series used in Fig. 1, we further confirmed that mutating positively charged residues lys400, lys403, lys406, lys408, arg411 and lys414 to alanine slowed the target search process by 7–8 fold and reduced the search length of 1D diffusion (Fig. 2c–d, i and S8a, b†). With improved time resolution (0.7 ms per frame), we revealed that the nonspecific binding rate of AsCas12a400–415 with dsDNA containing no PAM and no target is almost the same as that of AsCas12a-WT, indicating that mutations of the corresponding site do not affect the 3D collision between AsCas12a and dsDNA (Fig. S2a†). However, the transient dwell time of AsCas12a400–415 on nonspecific dsDNA is shorter (∼2 fold) than that of AsCas12a-WT, indicating that these positively charged residues contribute to nonspecific interactions between AsCas12a and dsDNA. Together, we identified that residues 400–415 are the key elements to non-specifically interact with dsDNA to mediate 1D diffusion of AsCas12a and to facilitate target search.
In the Cas12a/crRNA/DNA ternary complex, residues 400–415 of AsCas12a formed an alpha helix to interact with the RNA/DNA heteroduplex at the PAM distal end.42 Through sequence and structure alignment, we identified their corresponding sequences in LbCas12a and FnCas12a and generated LbCas12a380–391 and FnCas12a434–449, respectively, by mutating positively charged residues in the corresponding regions to alanine (Table S3†). AlphaFold predictions show that mutations at the REC2 domain do not disrupt the structure of Cas12a proteins (Fig. S9†). Similar to AsCas12a400–415, LbCas12a380–391 and FnCas12a434–449 both exhibited significantly reduced apparent search rates and shortened diffusional search lengths compared with their WT proteins (Fig. 2e–h, j and k and S8c–f†). Thus, this alpha helix serves as a conserved element among the three Cas12a orthologs to mediate 1D diffusion during the target search process.
AsCas12a, LbCas12a and FnCas12a all contain six positively charged residues in the alpha helix dominating their 1D diffusion on dsDNA (Table S3†). LbCas12a385–390, which was constructed by mutating only four arg and lys residues facing the RNA/DNA heteroduplex to alanine, displayed a ∼3.5 fold decrease in apparent target search rates compared to LbCas12a-WT and was still ∼2 fold faster than LbCas12a380–391 (Fig. S10†). LbCas12a372–391, having three additional nearby lys mutated to alanine, displayed a further reduced search rate compared to LbCas12a380–391 (Fig. S10†). With improved time resolution, we found that reducing positively charged residues within and near the alpha helix reduces the dwell time of LbCas12a on non-specific DNA and weakens the electrostatic interaction between LbCas12a and DNA (Fig. S2b†). These results confirmed that positively charged residues within and near this alpha helix all contribute to mediating 1D diffusion via non-specific electrostatic interactions with dsDNA.
FnCas12a shows significantly slower target search rates and shorter search lengths than AsCas12a and LbCas12a. We questioned whether their different target search behaviors are caused solely by this alpha helix and its orientation (Fig. S11a†). Thus, we created two chimera proteins, AsCas12a(400–415)Fn and FnCas12a(434–449)As, by switching their helix sequences (Table S3†). AsCas12a(400–415)Fn displayed slower search rates than AsCas12a-WT, but its rates were still ∼5 fold faster than that of FnCas12a-WT. FnCas12a(434–449)As exhibited even slower rates than FnCas12a-WT (Fig. S11†). Therefore, exchanging the sequences of this alpha helix cannot switch the behaviors of AsCas12a and FnCas12a. Consistent with our results, AlphaFold49 predicts that AsCas12a(400–415)Fn adopts a similar structure to that of AsCas12a-WT, whereas FnCas12a(434–449)As adopts a similar structure to that of FnCas12a-WT (Fig. S11f†). Thus, we speculate that these are additional factors that determine the orientation of this alpha helix causing differences among Cas12a orthologs.
Residues 400–415 of AsCas12a affect the dynamics of dsDNA cleavage
K414 in residues 400–415 of AsCas12a,42 R386 and K391 in residues 380–391 of LbCas12a50 and K447 in residues 434–449 of FnCas12a44 are shown to interact with the RNA/DNA heteroduplex in the Cas12a/crRNA/dsDNA ternary complex. Therefore, we questioned whether the helix mediating 1D diffusion of Cas12a also contributes to the target cleavage process of the ternary complex after target search and PAM recognition. We applied the single-molecule FRET assay developed in previous studies to examine the dynamic processes from Cas12a/crRNA/dsDNA ternary complex formation until the release of the dsDNA fragment after cleavage (Fig. 3a).34,51 In our assay, Cy3 was labeled at the 3′ end of crRNA and Cy5 was labeled at the target strand located 28 nt downstream of the PAM (Table S4†). After injecting the Cy3-labeled Cas12a/crRNA complex into the flow chamber containing immobilized Cy5-labeled dsDNA, the appearance of Cy3/Cy5 signals and change of Cy3/Cy5 FRET efficiencies were caused by the formation of the Cas12a/crRNA/dsDNA complex followed by a series of conformational changes. Based on single-molecule trajectories, five distinctive states were identified, including a non-fluorescence state (B) representing the binary complex before binding with dsDNA and four FRET states (T1, T2, T3 and T4) representing different conformational states of the ternary complex (Fig. 3a). Based on the previous assignment, T1 is the initiation state containing a partially formed R-loop, T2 is the NTS pre-cleavage state, and T3 and T4 are two target strand (TS) pre-cleavage states.34,51 Thus, the transition from B to T1 represents the overall target search process affected by 1D diffusion, whereas the transition from T1 to T3 and T4 states represent a series of conformational changes to complete dsDNA cleavage after Cas12a/crRNA/dsDNA ternary complex formation. From individual single-molecule trajectories, dwell times of these five states and transition rates among them were quantified (Fig. S12–S14†), from which the differences in free energies among these states and the energy barriers along the cleavage pathway were quantified and used to plot energy landscapes of different Cas12a with fully matched or partially matched dsDNA targets (Fig. 3).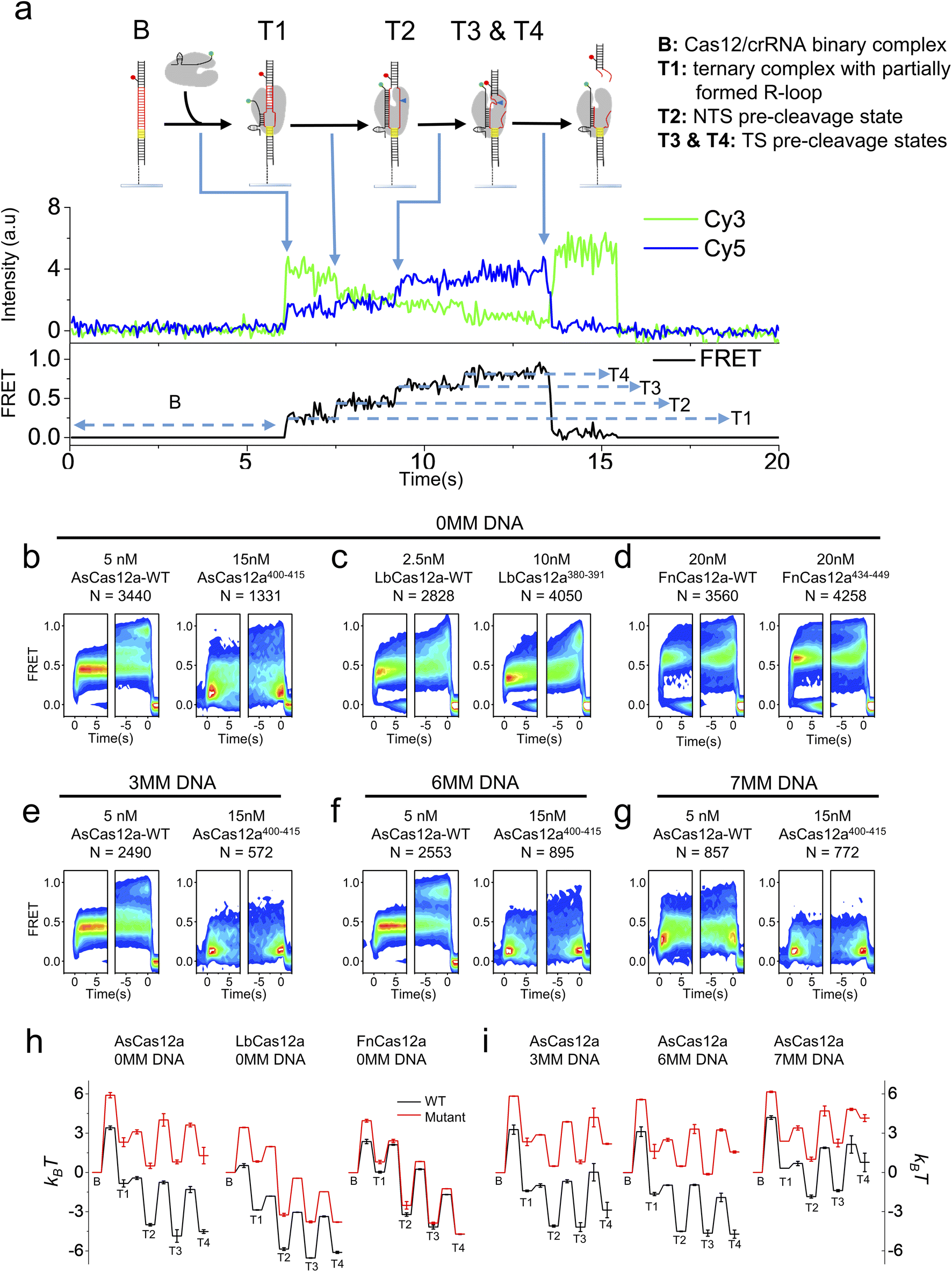 | ||
| Fig. 3 Conformational dynamics of Cas12a while mediating DNA cleavage. (a) Cartoons and a typical smFRET trajectory illustrating the corresponding states of Cas12a. Cy3 (green dots) was labeled at the 3′ end of crRNA, and Cy5 (red dots) was labeled at the target strand located 28 nt downstream of the PAM. Under 532 nm laser excitation, spontaneous appearance of Cy3 and FRET signals represents the binding of Cas12a on immobilized dsDNAs, leading to the transition from a Cas12a/crRNA binary complex (B state) to a ternary complex containing a partially formed R-loop (T1 state). The T1 state might further transit to the NTS pre-cleavage state (T2) and TS pre-cleavage states (T3 and T4). (b–g) Time-dependent FRET probability density plots of Cas12a-WT and Cas12a-mutants with fully matched dsDNA (b–d) and AsCas12a proteins with partially matched dsDNAs (e–g). MM is an abbreviation for mismatched base pairs at the PAM-distal end. Time-dependent FRET probability density plots were synchronized at the appearance of FRET (defined as t = 0 in the left panel of each sub-figure) or at the disappearance of FRET (defined as t = 0 in the right panel of each sub-figure), from which FRET changes of Cas12a after complex formation or before disassembly of the ternary complex can be carefully examined, respectively. (h) Energy landscapes of Cas12a-WTs and Cas12a-mutants with fully matched dsDNA. (i) Energy landscapes of AsCas12a-WT and AsCas12a400–415 with partially matched dsDNAs. | ||
In the presence of the fully- matched dsDNA targets, AsCas12a400–415 displayed significantly different FRET patterns from AsCas12a-WT showing that AsCas12a400–415 is much less likely to transit into high-FRET TS pre-cleavage states (T3 and T4) (Fig. 3b and S12†). Energy landscapes and transition rates showed that removing positively charged residues in the helix 400–415 of AsCas12a not only decreases the ternary complex formation rate by ∼9 fold but also destabilizes T3 and T4 states. After ternary complex formation, AsCas12a400–415 is less favorable to transit from T1 to the TS pre-cleavage states (T3 and T4) than AsCas12a-WT, indicated by its reduced transition rates from low FRET to high FRET states and elevated free energies of T2, T3 and T4 states (Fig. 3b, h and S14†). Although the ternary complex formation rate of LbCas12a380–391 was greatly reduced, unlike AsCas12a400–415, its transition rates between T1, T2, T3 and T4 states remained almost the same as that of LbCas12a-WT (Fig. S13 and S14†). Therefore, LbCas12a380–391 and LbCas12a-WT exhibited similar FRET patterns, illustrating similar conformational dynamics transiting from T1 to T4 (Fig. 3c and S12†). Consistent with the results shown above, disrupting positively charged residues in the helix 434–449 of FnCas12a had only minor effects on the ternary complex formation rate, transition rates among T1–T4 states and the overall energy landscapes (Fig. 3d, h and S13, S14†).
Among the three Cas12a orthologs, conformational dynamics of AsCas12a during target search and DNA cleavage is affected the most. When mismatches were introduced into the PAM-distal end between crRNA and dsDNA, the probabilities of AsCas12a400–415 to sample high-FRET TS pre-cleavage states were further reduced (Fig. 3e–g and S12–S14†). Quantification analysis showed that, in the presence of partially matched dsDNAs, the free energies of T3 and T4 states of AsCas12a400–415 are 4–6 kBT higher than those of AsCas12a-WT (Fig. 3i). Together, residues 400–415 of AsCas12a not only mediate 1D diffusion to facilitate target search but also play important roles in assisting the formation of DNA cleavage states. Disrupting positively charged residues in 400–415 decreases the cleavage activity of AsCas12a and increases its sensitivity towards mismatches between crRNA and dsDNA. Thus, AsCas12a400–415 might exhibit higher specificity than AsCas12a-WT, which will be examined in the following assays.
AsCas12a400–415 exhibits improved DNA cleavage and gene editing specificity
To examine the target specificity of AsCas12a-WT and AsCas12a400–415, we performed in vitro DNA cleavage assays (Fig. 4a, b, S15, S16a and Table S5†). Consistent with our speculation based on single-molecule FRET measurements, AsCas12a400–415 displayed lower cleavage activities, slower cleavage rates and higher specificity than AsCas12a-WT. Particularly, AsCas12a400–415 had no detectable cleavage targeting 7MM dsDNA containing 7 mismatches at the PAM-distal end in 16 min, whereas AsCas12a-WT displays moderate cleavage activity towards 7MM dsDNA. On the other hand, LbCas12a-WT and LbCas12a380–391 exhibited similar cleavage activities and specificity in in vitro assays (Fig. 4c, d and S16b, S17†). Our single-molecule FRET measurements showed that both AsCas12a400–415 and LbCas12a380–391 displayed a slower effective target search rate to form a ternary complex than their Cas12a-WT proteins, whereas only AsCas12a400–415 displayed a reduced probability to transit into DNA pre-cleavage states after ternary complex formation. Therefore, the specificity of AsCas12a is mainly modulated by its conformational dynamics after ternary complex formation and is not sensitive to the target search process.Next, we aimed to examine whether AsCas12a400–415 exhibited improved specificity in living cells. When targeting zrap (zinc resistance associated protein), an essential gene of Escherichia coli, AsCas12a-WT can tolerate up to 7 mismatched bases at the PAM-distal end without reduction of its editing efficiency. Although 6 mismatched bases located at the PAM-distal end have almost no influences on the editing efficiency of AsCas12a400–415, 7 mismatched bases significantly reduce its editing efficiency (Fig. 4e, f and S16c, Table S6†). In addition, we used GFP disruption assay with HEK293T cells harboring the GFP gene to examine the editing specificity of AsCas12a in mammalian cells (Fig. 4g). Two different sites were examined (Fig. 4h, i and S16d, e, Table S6†). For both sites, crRNA containing 3 PAM-distal mismatches towards the target exhibited similar editing efficiencies to that of the fully matched one, and the editing efficiencies of AsCas12a400–415 were always lower than those of AsCas12a-WT. 7 or 8 PAM-distal mismatches completely abolish editing of AsCas12a under our experimental conditions. When 6 PAM-distal mismatches were introduced, AsCas12a400–415 displayed much lower editing efficiency than that of AsCas12a-WT with site 1, whereas both AsCas12a400–415 and AsCas12a-WT had almost no editing with site 2. Together, AsCas12a400–415 exhibits improved editing specificity compared to AsCas12a-WT in both E. coli and HEK293T cells.
 | ||
| Fig. 4 Specificity of Cas12a and SpCas9 in vitro and in vivo. (a) DNA cleavage assays of AsCas12a-WT and AsCas12a400–415 at 25 °C for 16 min. Substrates that carry 0, 3, 6, 7 or 8 mismatched bases toward the crRNA at the PAM distal end are referred to as 3MM-, 6MM-, 7MM- and 8MM-dsDNA, respectively. (b) Cleavage percentage of AsCas12a-WT and AsCas12a400–415 towards fully matched and partially matched dsDNAs. The uncleaved (2281 bp) and cleaved (∼1140 bp) products were separated by 1% agarose gel. (c) DNA cleavage by LbCas12a-WT and LbCas12a380–391 at 25 °C for 16 min. (d) Cleavage percentage of LbCas12a-WT and LbCas12a380–391 towards fully matched and partially matched dsDNAs. (e) Schematic of bacterial interference assay targeting the zrap gene. Cleavage of the zrap gene results in cell death. (f) Cell death percentage using AsCas12a-WT or AsCas12a400–415 targeting the zrap gene. (g) Schematic of GFP disruption assay in HEK293T cells. Cleavage of the GFP gene causes loss of GFP fluorescence. (h and i) GFP disruption percentage using AsCas12a-WT or AsCas12a400–415 targeting site 1 (h) and site 2 (i) located within GFP. (j) DNA cleavage assays of SpCas9-WT and SpCas91151−1156 at 25 °C for 16 min. (k) Cleavage percentage of SpCas9-WT and SpCas91151−1156 targeting fully matched and partially matched substrates. The uncleaved (2188 bp) and cleaved (∼1090 bp) products were separated by 1% agarose gel. (l) Schematic of bacterial interference assay targeting the zrap gene using SpCas9. (m) Cell death percentage using SpCas9-WT and SpCas91151−1156 targeting the zrap gene. Significances were determined via a 2-tailed student's t-test between two groups. ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data were mean ± SEM from three replicates. | ||
Our previous studies showed that facilitated diffusion of SpCas9 is mediated by nonspecific interactions between residues 1151–1156 of SpCas9 and dsDNA.32 We questioned whether these nonspecific interactions also affect the specificity of SpCas9. Thus, we constructed SpCas91151−1156 by mutating all amino acids within this region to alanine (Tables S3†). Using the in vitro cleavage assay and the E. coli editing assay described above, we revealed that SpCas91151−1156 has better specificity than SpCas9-WT (Fig. 4j–m, S16f, g and S18, Tables S6–S7†). Therefore, our results showed that modulating residues mediating target search of Cas proteins on dsDNA is a potential alternative route to optimize their specificity.
The non-specific DNA binding site in the REC2 domain modulates target search and DNA cleavage
Class II type V CRISPR-Cas12a is a RNA-guided DNA endonuclease and provides an alternative to class II type II CRISPR-Cas9 for gene editing and engineering. Previous structural, biochemical and single-molecule studies have shed light on the working mechanisms of the Cas12a protein. It uses long-range hopping and 1D diffusion for a short distance on dsDNA to facilitate its target search process.34,35 After recognizing the PAM sequence, Cas12a undergoes a series of conformational changes to unwind dsDNA, to form an R-loop and to cleave the NTS and TS using its RuvC domain sequentially, leaving a 5′ overhang distal to the PAM site.7,34,42–45,48,52 In this study, we applied single-molecule assays with dsDNA strands to shed light on residues mediating 1D diffusion of Cas12a. We revealed that the three Cas12a orthologs display asymmetric target search regions flanking the PAM biased towards the downstream, suggesting that there are non-specific interactions between Cas12a and intact dsDNA at the PAM downstream during transient 1D diffusion.However, the target of Cas12a is also located the downstream of the PAM site, which is unwound to form an R-loop with crRNA in the Cas12a/crRNA/dsDNA ternary complex. Thus, we are unable to obtain a stable structure to illustrate non-specific interactions between Cas12a and the intact dsDNA at the PAM downstream as we did with SpCas9.32 After examining several potential sites, we identified that an alpha helix in the REC2 domain (residues 400–415 of AsCas12a, 380–391 of LbCas12a and 434–449 of FnCas12a) plays the key role in mediating 1D diffusion to facilitate target search (Fig. 5). Disrupting the non-specific interactions between this helix and dsDNA significantly diminishes the contribution of 1D diffusion, shortens the search length and reduces the apparent target search rate by up to 10 fold. The alpha helix (residues 400–415) of AsCas12a plays additional roles in stabilizing NTS and TS cleavage states. Neutralizing positively charged residues within this region destabilizes cleavage states by 4–6 kBT to slow cleavage. In summary, our study identified unknown non-specific interactions between the REC2 domain of Cas12a and dsDNA, which contributes to both target search and cleavage processes. Our findings provide further insights regarding the working mechanisms of Cas12a proteins and a new strategy to engineer them for future applications.
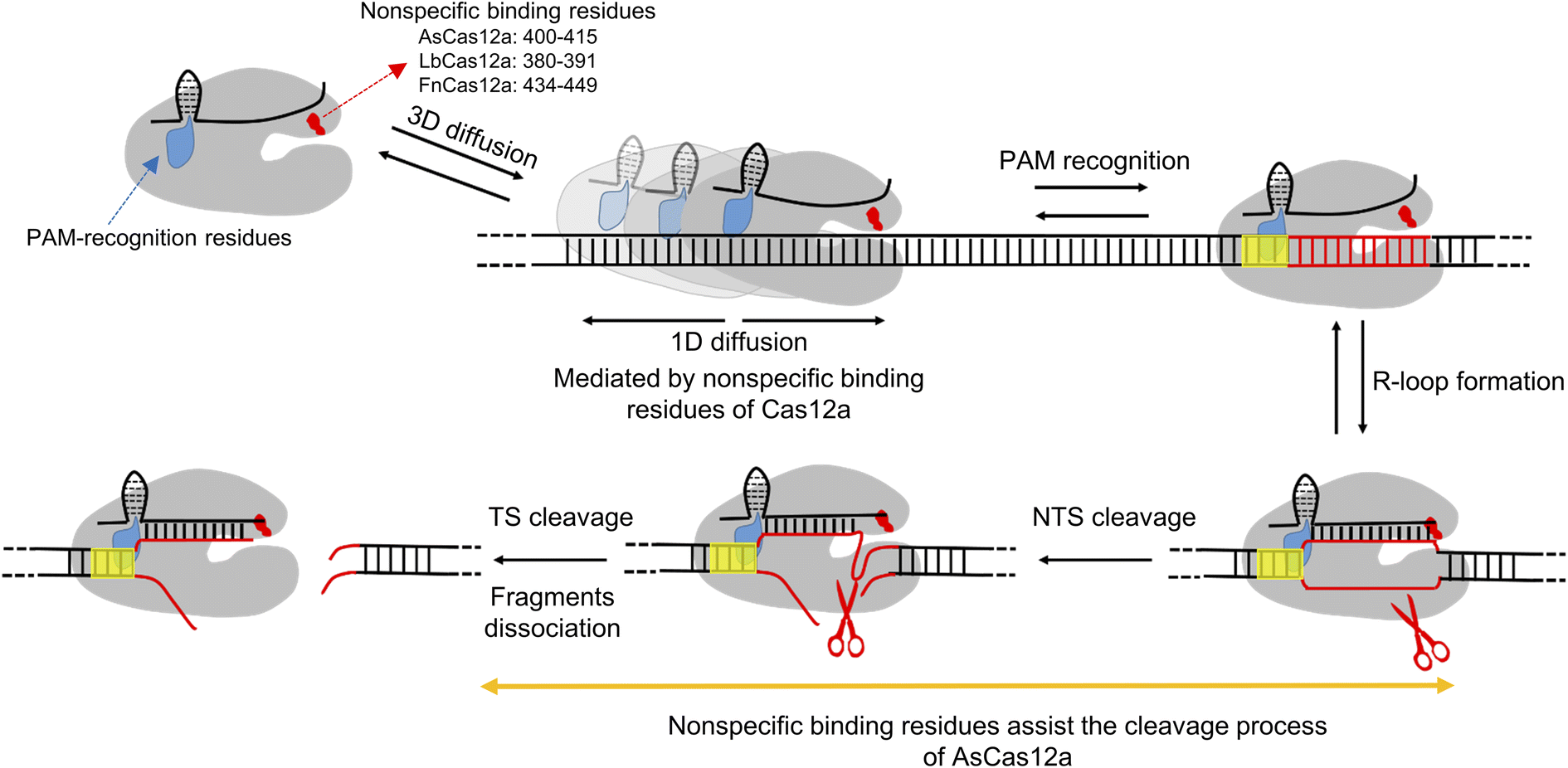 | ||
| Fig. 5 A scheme demonstrating the contribution of the DNA non-specific binding site in the REC2 domain to target search and DNA cleavage. A freely diffusing Cas12a/crRNA molecule binds to dsDNA through 3D random collision to form transient complexes mediated by the non-specific binding site and to initiate 1D diffusion in a relatively short range to search for PAM. PAM recognition causes R-loop formation and extension, which further triggers the sequential cleavage of the NTS and TS by the RuvC domain. For AsCas12a, the non-specific binding site plays an additional role besides mediating 1D diffusion, which is in stabilizing DNA cleavage states and facilitating DNA cleavage. Cas12a proteins are shown in gray. crRNAs and nonspecific dsDNAs are shown in black. The target DNA and PAM sequences are shown in red and yellow, respectively. | ||
AsCas12a, LbCas12a and FnCas12a share great similarities in their sequences and structures. However, previous studies have shown that FnCas12a is inefficient or inactive at multiple loci in human cells.7,53–55 Biochemical assays show that FnCas12a displays a slower target search rate than AsCas12a and LbCas12a, which is likely caused by its shortened 1D diffusion length (Fig. 1). Although all Cas12a proteins contain six positively charged residues in the non-specific binding alpha helix, contributions of the FnCas12a alpha helix to 1D diffusion are the smallest. Switching the sequences of the non-specific binding alpha helix between FnCas12a and AsCas12a cannot alter their differences in target search (Fig. S11†), suggesting that there are additional elements regulating the interactions between the alpha helix and intact dsDNA in the transient Cas12a/crRNA/dsDNA complex during 1D diffusion.
Weakening interactions between Cas proteins and DNA or RNA strands is a common strategy to enhance their specificity by sacrificing their nuclease activities.56–59 In agreement with this principle, both AsCas12a400–415 and SpCas91151−1156 exhibit weaker non-specific interactions with dsDNA, and higher specificity and slower cleavage rates than their wild-type proteins. On the other hand, although interactions with dsDNA are weakened in LbCas12a380–391, its apparent DNA cleavage rate still remain almost the same as that of LbCas12-WT and is much faster than that of AsCas12a400–415 and AsCas12a-WT (Fig. S15 and S17†), which explains why LbCas12a-WT and LbCas12a380–391 exhibit similar specificities.
To date, there are several routes to engineer Cas12a and Cas9 to enhance their functions or to achieve new functions. Disrupting electrostatic interactions between Cas proteins and the NTS/TS, truncating crRNA/sgRNA and partially substituting the RNA with DNA have been used to improve their specificity.56,57,60–62 Destabilizing the active conformation of Cas12a by mutating the bridge helix of Cas12a, changing the energy potential of crRNA base pairing to the target DNA by partial replacement of crRNA with DNA and disrupting potential interactions between Cas12a and the NTS by constructing an AsCas12a-K949A mutant can improve the specificity of Cas12a.60,61,63 Here, we demonstrated that modifying non-specific DNA binding sites of Cas proteins mediating 1D diffusion is a new additional strategy to engineer Cas12a and Cas9 and to modulate their specificities. In addition to cleavage specificity, evolving PAM-interacting domains can alter or expand their PAM compatibility.60,64 Mutations in catalytic sites can generate a nickase or catalytically dead Cas protein (dCas12a and dCas9), which can be fused with additional modulators to achieve site-specific transcription activation/repression, base editing and so on.14–16,18,20,21,65,66 Recently, the split-protein strategy and a chemically controlled autoinhibited RNA switch have been used to engineer temporally controlled Cas protein-based effectors responding to light or ligands.17,19,22–26 We speculated that the response speed of temporally controlled Cas protein-based effectors might be adjustable by modifying their non-specific DNA binding sites. The engineering strategy we described in this study is compatible with other engineering methods to expand the CRISPR-Cas toolbox.
Conclusions
In this work, we establish single-molecule fluorescence assays to the dissect dynamics of Cas12a complexes during target search and cleavage processes. We pinpoint a positive-charge-enriched alpha helix in the REC2 domain serving as a conserved element to interact with intact dsDNA during the target search process and to mediate one-dimensional diffusion of AsCas12a, LbCas12a and FnCas12a. In addition, this alpha helix assists the target cleavage process of AsCas12a via stabilizing the cleavage states. We further demonstrate that engineering residues mediating diffusion not only significantly affects the target search process but also modulates the target specificity of AsCas12 and SpCas9 both in vitro and in living cells. All in all, our work provides a novel strategy to engineer Cas proteins, which is compatible with current strategies, to enrich the versatile Cas protein-based tools.Experimental
Reagents
DNA oligonucleotides and crRNA with amine modification on the specific site were purchased from Sangon Biotech and Takara, respectively. Suflo-Cyanine3 (NHS ester) and Suflo-Cyanine5 (NHS ester) were purchased from Lumiprobe. UltraPure™ 1 M Tris–HCI pH 7.5, 1 M MgCl2 and 5 M NaCl were purchased from Invitrogen. Other common materials and reagents were purchased from Sigma or Amresco.Plasmid construction, and protein expression and purification
LbCas12a, AsCas12a and FnCas12a were expressed in Escherichia coli strain BL21 (DE3) using the expression plasmid pGEX-6P-1, a gift from Dr Zhiwei Huang,41 which contains an N-terminal GST tag and a precise protease cleavage site (HRV 3C site). Protein mutations were constructed by point mutations or homologous recombination confirmed by DNA sequencing. Cas12a wild-type or mutants were purified as previously described.41,51 Briefly, E. Coli BL21 (DE3) contained the Cas12a expression plasmid grown in an LB medium (100 μg mL−1 ampicillin) at 37 °C to an OD600 of 0.6–0.8. Protein expression was induced by 0.7 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and cultured for 12–16 h at 16 °C. The cells were harvested, and then sonicated in lysis buffer (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 1 mM MgCl2, and 1 mM TCEP) with 1 mM protease-inhibitor PMSF (Sigma). Cell debris was removed by centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The supernatant was incubated with glutathione sepharose 4B (GS4B) beads (GE Healthcare) in lysis buffer, and then eluted by using GSH elution buffer (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 1 mM MgCl2, 1 mM TCEP, and 20 mM GSH). The GST tag was removed by incubating the protein with HRV 3C protease (protein:protease = 25:1) overnight. The protein was further purified by ion-exchange chromatography with a Source S column (GE Healthcare), and eluted with a linear gradient of 0.15–1 M NaCl (buffer A: 50 mM MES pH 6.0, 150 mM NaCl, 1 mM TCEP; buffer B: 50 mM MES pH 6.0, 1 M NaCl, and 1 mM TCEP). The purity of the protein was verified by SDS-PAGE electrophoresis. The protein was concentrated in lysis buffer, flash-frozen in aliquots, and stored at −80 °C.
000 rpm. The supernatant was incubated with glutathione sepharose 4B (GS4B) beads (GE Healthcare) in lysis buffer, and then eluted by using GSH elution buffer (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 1 mM MgCl2, 1 mM TCEP, and 20 mM GSH). The GST tag was removed by incubating the protein with HRV 3C protease (protein:protease = 25:1) overnight. The protein was further purified by ion-exchange chromatography with a Source S column (GE Healthcare), and eluted with a linear gradient of 0.15–1 M NaCl (buffer A: 50 mM MES pH 6.0, 150 mM NaCl, 1 mM TCEP; buffer B: 50 mM MES pH 6.0, 1 M NaCl, and 1 mM TCEP). The purity of the protein was verified by SDS-PAGE electrophoresis. The protein was concentrated in lysis buffer, flash-frozen in aliquots, and stored at −80 °C.
SpCas9 wild-type and mutants were expressed in Escherichia coli strain BL21 (DE3) using the expression plasmid pMJ806 (Addgene plasmid # 39312) and purified as previously described.67 Briefly, cells were sonicated in lysis buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 1 mM TCEP, and 10% glycerol) with 1 mM PMSF and debris was removed by centrifugation. The supernatant was incubated with Ni-NTA agarose (Qiagen) in lysis buffer, and then eluted with elution buffer (20 mM HEPES pH 7.5, 250 mM KCl, 1 mM TCEP, and 150 mM imidazole). After removing the His6-MBP affinity tag, the protein was further purified by using a Source S column, and eluted with a linear gradient of 0–1 M KCl (buffer A: 20 mM HEPES pH 7.5, 0 M KCl, 1 mM TCEP; buffer B: 20 mM HEPES pH 7.5, 1 M KCl, and 1 mM TCEP). The protein was concentrated in storage buffer (50 mM Tris–HCl pH 7.5, 150 mM KCl, and 1 mM TCEP), flash-frozen in aliquots, and stored at −80 °C.
Labeling of nucleic acids
Nucleic acids were labeled with fluorophores by reacting with N-hydroxysuccinimido (NHS)-ester derived fluorophores. Briefly, DNA or crRNA strands were dissolved to 0.4–1 mM in 10 mM NaHCO3 (Sigma) buffer and mixed with NHS-fluorophores at a 1:10 molar ratio. Then the labeling mixture was kept at room temperature (∼23 °C) for two hours in the dark. The labeled products were purified by ethanol precipitation and resuspended in nuclease-free water (Life tech). The labeling efficiency was calculated by measuring the absorption of nucleic acids at 260 nm and the absorption of fluorophores at 549 nm or 649 nm. Overall, the labeling efficiencies of nucleic acids used in this study were 80–90%.
In vitro cleavage assay
dsDNA (2281bp) for Cas12a and dsDNA (2188 bp) for Cas9 with target sites were generated from the plasmid by PCR amplification and purified using 1% agarose gel. The protospacer and PAM sequences for Cas12a or Cas9 were located at the center of the dsDNA. crRNA or sgRNA was pre-annealed at 85 °C for 10 min and then slowly cooled down to room temperature by 0.2 °C s−1. To ensure that all proteins formed binary complexes with their corresponding RNAs, we mixed Cas proteins with a 2× molar excess of the annealed-RNAs in the reaction buffer (Cas12a: 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT; Cas9: 50 mM Tris–HCl pH 7.5, 100 mM KCl, 5 mM MgCl2, and 1 mM DTT) at 25 °C for 25 minutes. For cleavage activity measurement of Cas12a proteins, 100 nM Cas12a/crRNA was used to cleave the 7 nM dsDNA at 25 °C for 16 min and 30 min in the reaction buffer. Cleavage was terminated by adding 6× DNA loading buffer (15% Ficoll®-400, 66 mM EDTA, 20 mM Tris–HCl pH 8.0, 0.12% SDS, and 0.09% bromophenol blue). For cleavage assays of dsDNA with mismatches at the PAM distal end as shown in Fig. 4 and S16,† 100 nM Cas12a/crRNA or SpCas9/sgRNA was used to cleave the 7 nM dsDNAs with fully matched or mismatched target sites at 25 °C for 16 min. For time-dependent cleavage assays of dsDNA with mismatches at the PAM distal end as shown in Fig. S15, S17 and S18,† 50 nM Cas12a/crRNA or 100 nM SpCas9/sgRNA was used to cleave the 7 nM dsDNA at 25 °C for 16 s, 32 s, 1 min, 2 min, 4 min, 8 min, 16 min, 30 min and 60 min, respectively. Uncleaved and cleaved products were separated by using 1% agarose gel and cleavage percentages were determined by Gaussian fitting.Bacterial-based negative selection assay
For a negative selection assay based on the zrap gene in Escherichia coli for AsCas12a, a plasmid engineered from BPK764,64 with a chloramphenicol resistance gene capable of expressing crRNA as well as Cas12a, was transformed into BL21 which has a kanamycin resistance gene in its genome. Then, it was incubated on ice for 25 min, heat shocked at 42 °C for 1 min 30 s, followed by 60 min recovery in LB media. Transformants were then plated on LB agar containing 50 μg mL−1 kanamycin and 25 μg mL−1 chloramphenicol in the absence of IPTG. Cleavage of the zrap gene can result in cell death. The plasmid expressing crRNA without a spacer sequence was used as the control. The survival ratio was obtained by counting the number of surviving colonies and compared with that of the control.For a negative selection assay based on the zrap gene in Escherichia coli for SpCas9, BPK764 that can express SpCas9 was transformed into BL21. Then, the plasmid engineered from pBSTNAV68 that can transcribe sgRNA targeting the conserved zrap gene was transformed into BL21, which was plated on LB agar containing 100 μg mL−1 ampicillin and 25 μg mL−1 chloramphenicol. pBSTNAV without sgRNA sequences was used as a control. The cleavage of essential zrap genes will result in the death of the bacteria; otherwise, the bacteria will survive.
Genome editing in fluorescent reporter human cells
The GFP stable expressed HEK293T reporter cells were seeded into 96-well plates and transfected 12–18 h later at 60–70% confluency according to the manufacturer's protocol with lipofectamine 3000 (Life Technologies) and 200 ng of plasmid DNA encoding Cas12a and crRNA. 24 h after transfection, GFP HEK293T reporter cells that were successfully transfected were selected by adding 1.5 mg mL−1 puromycin to the cell culture media for 48 h. A fresh medium with puromycin was added to the transfected HEK293T reporter cells for an additional 24 h before replacing them with cell culture media without puromycin. The cells were passaged regularly to maintain sub-confluent conditions and then analyzed in 96-well round bottom plates on an Attune NxT Flow Cytometer with an autosampler. The cells were analyzed on the flow cytometer after 7 days to track the disruption of the GFP gene in cells.Acquisition of single-molecule fluorescence data
All our smFRET experiments were performed at 25 °C. Unless specifically mentioned, all complex formations and single-molecule imaging were carried out in the reaction buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, and 1 mM DTT) with an oxygen scavenging system (3 mg mL−1 glucose, 100 μg mL−1 glucose oxidase (Sigma-Aldrich), 40 μg mL−1 catalase (Roche), 1 mM cyclooctatetraene (COT, Sigma-Aldrich), 1 mM 4-nitrobenzylalcohol (NBA, Sigma-Aldrich), and 1.5 mM 6-hydroxy-2,5,7,8-tetramethyl-chromane-2-carboxylic acid (Trolox, Sigma-Aldrich)). Single-molecule FRET measurements were performed on an objective-type TIRF microscope, based on a Nikon Eclipse Ti-E with an EMCCD camera (Andor iXon Ultra 897), and solid state 532 and 640 nm excitation lasers (Coherent Inc. OBIS Smart Lasers).69 Fluorescence emission from the probes was collected by using the microscope and spectrally separated by using interference dichroic (T635lpxr, Chroma) and bandpass filters, ET585/65m (Chroma, Cy3) and ET700/75m (Chroma, Cy5). All smFRET movies were collected using Cell Vision software (Beijing Coolight Technology).Single-molecule fluorescence experiments quantifying the target search rate
DN-PAM-31 or D25-PAM-N dsDNAs were generated from the plasmid by PCR amplification, and then purified using agarose or native PAGE gels.For a single-molecule fluorescence assay, the biotinylated dsDNAs were immobilized on the slide via biotin-streptavidin interaction. The unbound DNAs were removed by washing with reaction buffer. To ensure all crRNA formed the binary complex, Cy3-labeled crRNA was pre-incubated with excess Cas12a (Cy3-RNA:Cas12a = 1:3 molar ratio) at 1 μM in the reaction buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, and 1 mM DTT) at 25 °C for 25 min. Then, the binary complex was diluted to the corresponding concentration. The target search processes were recorded in the reaction imaging buffer. For experiments with different salt concentrations, the buffer was 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, and 1 mM DTT supplied with 20 mM to 100 mM NaCl. For the experiment with a crowding agent, the buffer was 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 5% PEG-8000 (w/v). The target search rates of Cas12a/crRNA were determined by recording each appearance time when an individual binding event occurred. All movies were acquired at 10 ms per frame with shrunk exposure area to eliminate the dead time between frames.
For a single-molecule FRET assay, Cy5-labeled dsDNAs were immobilized on the surface. FRET signals between Cy3-labeled crRNA and Cy5-labeled dsDNA were used to probe the binding of Cas12a/crRNA on the target site. All movies were acquired at 10 ms per frame. The apparent target search rates kon were calculated as follows:
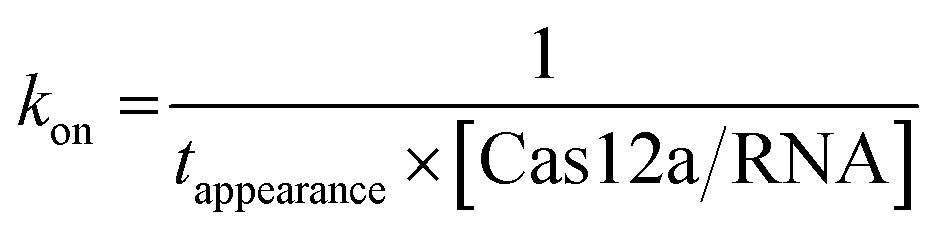
The apparent target search rates kon as a function of the extension length of dsDNA as shown in Fig. 1i–k and S10c† were fitted to the theoretical sliding length dependence curve:70
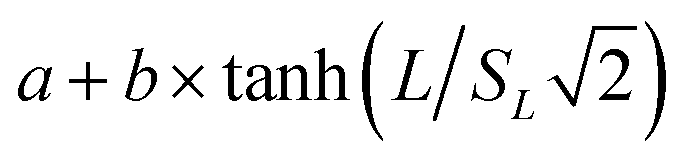
Single-molecule FRET experiments capturing the dynamics of the Cas12a ternary complex
For a single-molecule FRET assay to capture the conformational dynamics of the Cas12a ternary complex on fully matched or partially matched DNAs, dsDNA was immobilized on the cover slide. The binary complex was formed by incubating Cy3-labeled RNA and Cas12a with a molar ratio of 1:3 at 25 °C in the reaction buffer. The collection of movies started several seconds before the Cy3-labeled crRNA/Cas12a binary complex was allowed to flow and were recorded with 500 ms per frame without dead time between frames.
Single-molecule FRET data analysis
The ollected movies were analyzed by the custom-made software program developed as an ImageJ plugin (http://rsb.info.nih.gov/ij). Fluorescence spots were fitted by using a 2D Gaussian function within a 9-pixel by 9-pixel area, matching the donor and acceptor spots using a variant of the Hough transform.71 The background subtracted total volume of the 2D Gaussian peak was used as raw fluorescence intensity I.For a single-molecule FRET assay to detect the conformational dynamics of the Cas12a ternary complex, FRET traces which displayed anti-correlation behaviors between donor and acceptor fluorescent signals were picked and kinetics information was further analyzed by a Hidden Markov Model based software.72 Four FRET states from low to high FRET values were identified as T1–T4 states. The freely diffused binary complex state before Cas12a/crRNA binds to dsDNA was the B (binary) state. Transition rates (k) among FRET states were extracted from their dwell times. Relative free energies were calculated via
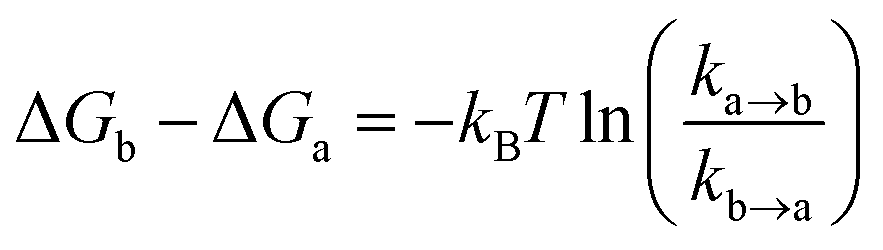 ln(ka→b) + 0.5 kBT.
ln(ka→b) + 0.5 kBT.
Data availability
The data that support the findings of this study are in the ESI.† All other data are available from the corresponding author upon reasonable request.Author contributions
R. S., W. W. and C. C. designed the experiments; R. S. prepared the materials and performed the biochemical and single-molecule experiments and editing experiments in Escherichia coli, Y. Z. performed the editing experiment in HEK293T cells, and R. S. and C. C. wrote the paper with input from Y. Z., W. W. and J. J. G. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [21922704, 21877069, 22277063 and 22061160466 to C. C., 22007054 to W. W. and 32150018 to J. J. G. L.], the Beijing Advanced Innovation Center for Structural Biology (to C. C. and J. J. G. L.), the Beijing Frontier Research Center for Biological Structure (to C. C.) and the Tsinghua-Peking Joint Center for Life Sciences (to J. J. G. L.).References
- H. Deveau, J. E. Garneau and S. Moineau, Annu. Rev. Microbiol., 2010, 64, 475–493 CrossRef CAS PubMed.
- P. Horvath and R. Barrangou, Science, 2010, 327, 167–170 CrossRef CAS PubMed.
- R. Sorek, C. M. Lawrence and B. Wiedenheft, Annu. Rev. Biochem., 2013, 82, 237–266 CrossRef CAS PubMed.
- B. Wiedenheft, S. H. Sternberg and J. A. Doudna, Nature, 2012, 482, 331–338 CrossRef CAS PubMed.
- P. Pausch, B. Al-Shayeb, E. Bisom-Rapp, C. A. Tsuchida, Z. Li, B. F. Cress, G. J. Knott, S. E. Jacobsen, J. F. Banfield and J. A. Doudna, Science, 2020, 369, 333–337 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- P. D. Hsu, E. S. Lander and F. Zhang, Cell, 2014, 157, 1262–1278 CrossRef CAS PubMed.
- R. Barrangou and J. A. Doudna, Nat. Biotechnol., 2016, 34, 933–941 CrossRef CAS PubMed.
- H. Kim, S. T. Kim, J. Ryu, B. C. Kang, J. S. Kim and S. G. Kim, Nat. Commun., 2017, 8, 14406 CrossRef CAS PubMed.
- M. A. Moreno-Mateos, J. P. Fernandez, R. Rouet, C. E. Vejnar, M. A. Lane, E. Mis, M. K. Khokha, J. A. Doudna and A. J. Giraldez, Nat. Commun., 2017, 8, 2024 CrossRef PubMed.
- J. Yin, R. Lu, C. Xin, Y. Wang, X. Ling, D. Li, W. Zhang, M. Liu, W. Xie, L. Kong, W. Si, P. Wei, B. Xiao, H. Y. Lee, T. Liu and J. Hu, Nat. Commun., 2022, 13, 1204 CrossRef CAS PubMed.
- A. C. Komor, Y. B. Kim, M. S. Packer, J. A. Zuris and D. R. Liu, Nature, 2016, 533, 420–424 CrossRef CAS PubMed.
- N. M. Gaudelli, A. C. Komor, H. A. Rees, M. S. Packer, A. H. Badran, D. I. Bryson and D. R. Liu, Nature, 2017, 551, 464–471 CrossRef CAS PubMed.
- J. W. Nelson, P. B. Randolph, S. P. Shen, K. A. Everette, P. J. Chen, A. V. Anzalone, M. An, G. A. Newby, J. C. Chen, A. Hsu and D. R. Liu, Nat. Biotechnol., 2022, 40, 402–410 CrossRef CAS PubMed.
- Y. Nihongaki, Y. Furuhata, T. Otabe, S. Hasegawa, K. Yoshimoto and M. Sato, Nat. Methods, 2017, 14, 963–966 CrossRef CAS PubMed.
- M. Kampmann, ACS Chem. Biol., 2018, 13, 406–416 CrossRef CAS PubMed.
- Y. E. Tak, B. P. Kleinstiver, J. K. Nunez, J. Y. Hsu, J. E. Horng, J. Gong, J. S. Weissman and J. K. Joung, Nat. Methods, 2017, 14, 1163–1166 CrossRef CAS PubMed.
- X. Wang, C. Ding, W. Yu, Y. Wang, S. He, B. Yang, Y. C. Xiong, J. Wei, J. Li, J. Liang, Z. Lu, W. Zhu, J. Wu, Z. Zhou, X. Huang, Z. Liu, L. Yang and J. Chen, Cell Rep., 2020, 31, 107723 CrossRef CAS PubMed.
- H. R. Kempton, K. S. Love, L. Y. Guo and L. S. Qi, Nat. Chem. Biol., 2022, 18, 742–750 CrossRef CAS PubMed.
- Y. Nihongaki, T. Otabe, Y. Ueda and M. Sato, Nat. Chem. Biol., 2019, 15, 882–888 CrossRef CAS PubMed.
- Y. Nihongaki, F. Kawano, T. Nakajima and M. Sato, Nat. Biotechnol., 2015, 33, 755–760 CrossRef CAS PubMed.
- N. Huynh, S. Wang and K. King-Jones, Insect Biochem. Mol. Biol., 2020, 120, 103336 CrossRef CAS PubMed.
- Y. Liu, J. Han, Z. Chen, H. Wu, H. Dong and G. Nie, Nat. Commun., 2017, 8, 2095 CrossRef PubMed.
- K. Kundert, J. E. Lucas, K. E. Watters, C. Fellmann, A. H. Ng, B. M. Heineike, C. M. Fitzsimmons, B. L. Oakes, J. Qu, N. Prasad, O. S. Rosenberg, D. F. Savage, H. El-Samad, J. A. Doudna and T. Kortemme, Nat. Commun., 2019, 10, 2127 CrossRef PubMed.
- W. Tang, J. H. Hu and D. R. Liu, Nat. Commun., 2017, 8, 15939 CrossRef CAS PubMed.
- A. D. Riggs, S. Bourgeois and M. Cohn, J. Mol. Biol., 1970, 53, 401–417 CrossRef CAS PubMed.
- O. G. Berg, R. B. Winter and P. H. von Hippel, Biochemistry, 1981, 20, 6929–6948 CrossRef CAS PubMed.
- R. B. Winter and P. H. von Hippel, Biochemistry, 1981, 20, 6948–6960 CrossRef CAS PubMed.
- E. Marklund, B. van Oosten, G. Mao, E. Amselem, K. Kipper, A. Sabantsev, A. Emmerich, D. Globisch, X. Zheng, L. C. Lehmann, O. G. Berg, M. Johansson, J. Elf and S. Deindl, Nature, 2020, 583, 858–861 CrossRef CAS PubMed.
- M. Yang, R. Sun, P. Deng, Y. Yang, W. Wang, J. G. Liu and C. Chen, Chem. Sci., 2021, 12, 12776–12784 RSC.
- V. Globyte, S. H. Lee, T. Bae, J. S. Kim and C. Joo, EMBO J., 2019, 38, e99466 CrossRef PubMed.
- Y. Jeon, Y. H. Choi, Y. Jang, J. Yu, J. Goo, G. Lee, Y. K. Jeong, S. H. Lee, I. S. Kim, J. S. Kim, C. Jeong, S. Lee and S. Bae, Nat. Commun., 2018, 9, 2777 CrossRef PubMed.
- M. Losito, Q. M. Smith, M. D. Newton, M. E. Cuomo and D. S. Rueda, Phys. Chem. Chem. Phys., 2021, 23, 26640–26644 RSC.
- D. L. Jones, P. Leroy, C. Unoson, D. Fange, V. Ćurić, M. J. Lawson and J. Elf, Science, 2017, 357, 1420–1424 CrossRef CAS PubMed.
- F. Jiang, K. Zhou, L. Ma, S. Gressel and J. A. Doudna, Science, 2015, 348, 1477–1481 CrossRef CAS PubMed.
- M. Jinek, F. Jiang, D. W. Taylor, S. H. Sternberg, E. Kaya, E. Ma, C. Anders, M. Hauer, K. Zhou, S. Lin, M. Kaplan, A. T. Iavarone, E. Charpentier, E. Nogales and J. A. Doudna, Science, 2014, 343, 1247997 CrossRef PubMed.
- C. Anders, O. Niewoehner, A. Duerst and M. Jinek, Nature, 2014, 513, 569–573 CrossRef CAS PubMed.
- F. Jiang, D. W. Taylor, J. S. Chen, J. E. Kornfeld, K. Zhou, A. J. Thompson, E. Nogales and J. A. Doudna, Science, 2016, 351, 867–871 CrossRef CAS PubMed.
- D. Dong, K. Ren, X. Qiu, J. Zheng, M. Guo, X. Guan, H. Liu, N. Li, B. Zhang, D. Yang, C. Ma, S. Wang, D. Wu, Y. Ma, S. Fan, J. Wang, N. Gao and Z. Huang, Nature, 2016, 532, 522–526 CrossRef CAS PubMed.
- T. Yamano, H. Nishimasu, B. Zetsche, H. Hirano, I. M. Slaymaker, Y. Li, I. Fedorova, T. Nakane, K. S. Makarova, E. V. Koonin, R. Ishitani, F. Zhang and O. Nureki, Cell, 2016, 165, 949–962 CrossRef CAS PubMed.
- S. Stella, P. Alcon and G. Montoya, Nature, 2017, 546, 559–563 CrossRef CAS.
- D. C. Swarts, J. van der Oost and M. Jinek, Mol. Cell, 2017, 66, 221–233 CrossRef CAS PubMed.
- S. Stella, P. Mesa, J. Thomsen, B. Paul, P. Alcon, S. B. Jensen, B. Saligram, M. E. Moses, N. S. Hatzakis and G. Montoya, Cell, 2018, 175, 1856–1871 CrossRef CAS.
- P. Gao, H. Yang, K. R. Rajashankar, Z. Huang and D. J. Patel, Cell Res., 2016, 26, 901–913 CrossRef CAS.
- W. Sun, J. Yang, Z. Cheng, N. Amrani, C. Liu, K. Wang, R. Ibraheim, A. Edraki, X. Huang, M. Wang, J. Wang, L. Liu, G. Sheng, Y. Yang, J. Lou, E. J. Sontheimer and Y. Wang, Mol. Cell, 2019, 76, 938–952 CrossRef CAS PubMed.
- D. Singh, J. Mallon, A. Poddar, Y. Wang, R. Tippana, O. Yang, S. Bailey and T. Ha, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5444–5449 CrossRef CAS PubMed.
- P. Cramer, Nat. Struct. Mol. Biol., 2021, 28, 704–705 CrossRef CAS PubMed.
- T. Yamano, B. Zetsche, R. Ishitani, F. Zhang, H. Nishimasu and O. Nureki, Mol. Cell, 2017, 67, 633–645 CrossRef CAS.
- L. Zhang, R. Sun, M. Yang, S. Peng, Y. Cheng and C. Chen, iScience, 2019, 19, 492–503 CrossRef CAS PubMed.
- I. Strohkendl, F. A. Saifuddin, J. R. Rybarski, I. J. Finkelstein and R. Russell, Mol. Cell, 2018, 71, 816–824 CrossRef CAS PubMed.
- D. Kim, J. Kim, J. K. Hur, K. W. Been, S. H. Yoon and J. S. Kim, Nat. Biotechnol., 2016, 34, 863–868 CrossRef CAS PubMed.
- M. Tu, L. Lin, Y. Cheng, X. He, H. Sun, H. Xie, J. Fu, C. Liu, J. Li, D. Chen, H. Xi, D. Xue, Q. Liu, J. Zhao, C. Gao, Z. Song, J. Qu and F. Gu, Nucleic Acids Res., 2017, 45, 11295–11304 CrossRef CAS PubMed.
- E. Toth, B. C. Czene, P. I. Kulcsar, S. L. Krausz, A. Talas, A. Nyeste, E. Varga, K. Huszar, N. Weinhardt, Z. Ligeti, A. E. Borsy, E. Fodor and E. Welker, Nucleic Acids Res., 2018, 46, 10272–10285 CAS.
- B. P. Kleinstiver, V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng and J. K. Joung, Nature, 2016, 529, 490–495 CrossRef CAS PubMed.
- I. M. Slaymaker, L. Gao, B. Zetsche, D. A. Scott, W. X. Yan and F. Zhang, Science, 2016, 351, 84–88 CrossRef CAS PubMed.
- J. S. Chen, Y. S. Dagdas, B. P. Kleinstiver, M. M. Welch, A. A. Sousa, L. B. Harrington, S. H. Sternberg, J. K. Joung, A. Yildiz and J. A. Doudna, Nature, 2017, 550, 407–410 CrossRef CAS PubMed.
- V. Pattanayak, S. Lin, J. P. Guilinger, E. Ma, J. A. Doudna and D. R. Liu, Nat. Biotechnol., 2013, 31, 839–843 CrossRef CAS PubMed.
- L. Gao, D. B. T. Cox, W. X. Yan, J. C. Manteiga, M. W. Schneider, T. Yamano, H. Nishimasu, O. Nureki, N. Crosetto and F. Zhang, Nat. Biotechnol., 2017, 35, 789–792 CrossRef CAS PubMed.
- H. Kim, W. J. Lee, Y. Oh, S. H. Kang, J. K. Hur, H. Lee, W. Song, K. S. Lim, Y. H. Park, B. S. Song, Y. B. Jin, B. H. Jun, C. Jung, D. S. Lee, S. U. Kim and S. H. Lee, Nucleic Acids Res., 2020, 48, 8601–8616 CrossRef CAS PubMed.
- Y. Fu, J. D. Sander, D. Reyon, V. M. Cascio and J. K. Joung, Nat. Biotechnol., 2014, 32, 279–284 CrossRef CAS.
- E. Worle, L. Jakob, A. Schmidbauer, G. Zinner and D. Grohmann, Nucleic Acids Res., 2021, 49, 5278–5293 CrossRef PubMed.
- B. P. Kleinstiver, M. S. Prew, S. Q. Tsai, V. V. Topkar, N. T. Nguyen, Z. Zheng, A. P. Gonzales, Z. Li, R. T. Peterson, J. R. Yeh, M. J. Aryee and J. K. Joung, Nature, 2015, 523, 481–485 CrossRef PubMed.
- X. Li, Y. Wang, Y. Liu, B. Yang, X. Wang, J. Wei, Z. Lu, Y. Zhang, J. Wu, X. Huang, L. Yang and J. Chen, Nat. Biotechnol., 2018, 36, 324–327 CrossRef CAS PubMed.
- B. P. Kleinstiver, A. A. Sousa, R. T. Walton, Y. E. Tak, J. Y. Hsu, K. Clement, M. M. Welch, J. E. Horng, J. Malagon-Lopez, I. Scarfo, M. V. Maus, L. Pinello, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2019, 37, 276–282 CrossRef CAS PubMed.
- M. Yang, S. Peng, R. Sun, J. Lin, N. Wang and C. Chen, Cell Rep., 2018, 22, 372–382 CrossRef CAS PubMed.
- L. Ponchon, M. Catala, B. Seijo, M. El Khouri, F. Dardel, S. Nonin-Lecomte and C. Tisne, Nucleic Acids Res., 2013, 41, e150 CrossRef CAS PubMed.
- S. Peng, R. Sun, W. Wang and C. Chen, Angew. Chem., 2017, 56, 6882–6885 CrossRef CAS PubMed.
- P. Hammar, P. Leroy, A. Mahmutovic, E. G. Marklund, O. G. Berg and J. Elf, Science, 2012, 336, 1595–1598 CrossRef CAS PubMed.
- J. Illingworth and J. Kittler, IEEE Trans. Pattern Anal. Mach. Intell., 1987, 9, 690–698 CAS.
- I. K. Darcy, J. Chang, N. Druivenga, C. McKinney, R. K. Medikonduri, S. Mills, J. Navarra-Madsen, A. Ponnusamy, J. Sweet and T. Thompson, BMC Bioinf., 2006, 7, 435 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05463a |
| This journal is © The Royal Society of Chemistry 2023 |