
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel synthetic route for (parent) phosphetanes, phospholanes, phosphinanes and phosphepanes†
Stephan
Reichl
,
Gábor
Balázs
and
Manfred
Scheer
 *
*
Institute of Inorganic Chemistry, University Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: manfred.scheer@chemie.uni-regensburg.de
First published on 13th March 2023
Abstract
A novel synthetic route for (parent) phosphorus-containing cycloalkanes such as phosphetanes, phospholanes, phosphinanes and phosphepanes is reported. By using [K(dme)2]2[Cp*Fe(η4-P5)] (I) in combination with α,ω-dibromoalkanes CnH2nBr2 [n = 3–6], unique phosphetane, phospholane, phosphinane and phosphepane precursor complexes [Cp*Fe{η4-P5(CH2)n}] [n = 3–6] (2–5) are synthesised. They act as P-atom carriers and the corresponding phosphetane, phospholane, phosphinane and phosphepanes (6–9) can be released by nucleophiles i.e., potassium benzyl (KBn) or LiAlH4. The latter enables the selective synthesis of parent cyclic secondary phosphines (10) in an easy and straightforward way, including the first parent phospholane (10b).
Introduction
Heterocyclic chemistry is one of the prime topics in organic and inorganic chemistry, with billions of heterocyclic compounds synthesised and some of them widely used, e.g. in the pharmaceutical industry.1 On the other hand, phosphines are widely employed in (asymmetric) organocatalysis,2,3 which is still a major field, rewarded with two Nobel Prizes within the last two decades.4 To customise and tune (transition)metal complexes used in catalysis, a variety of phosphines have been and are still being synthesised.5–7While phosphorus heterocycles are of academic interest, such as e.g. the Wittig Reaction,8 bidentate6 and caged9 phosphines are on the rise as tuneable ligands in catalysis and Wittig-type reactions, which are widely used in industry.10 The importance of specific phosphorus heterocycles in transition-metal-assisted (asymmetric) catalysis is remarkable, as for instance the usage of phospholanes in DuPhos or BPE, representing chiral ligands for asymmetric catalysis (Scheme 1).11
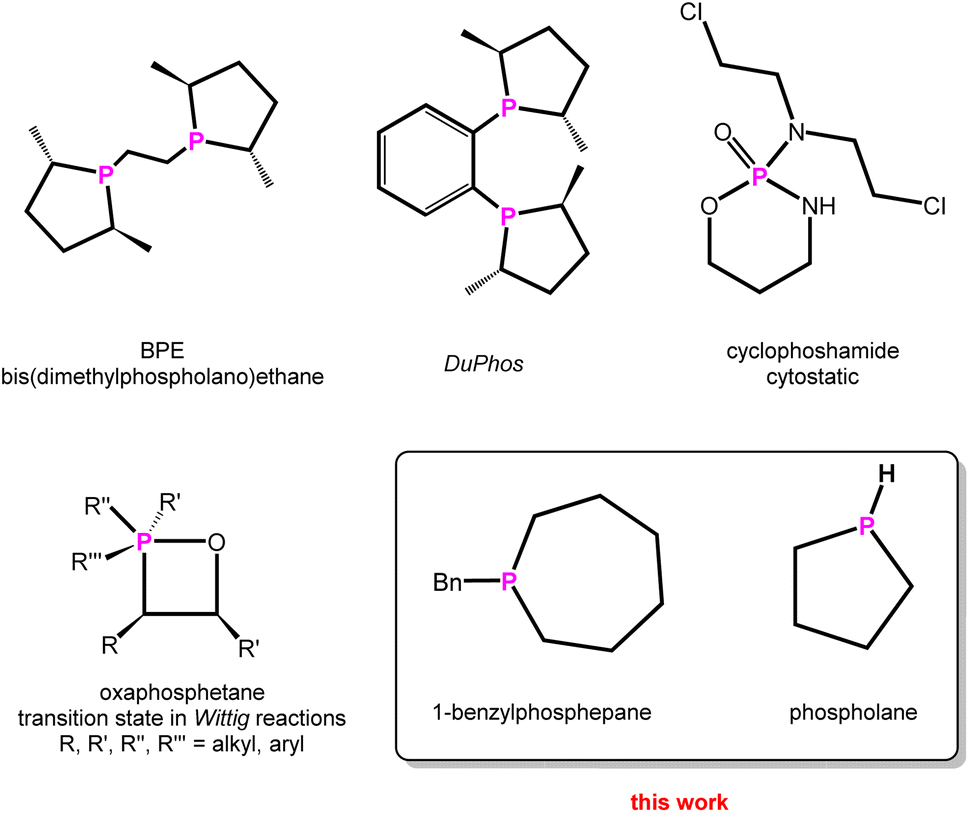 | ||
| Scheme 1 Selected phosphorus heterocycles used in organocatalysis,12 pharmacy,22 industry,10 and the parent phospholane. | ||
The synthesis of cyclic phosphines is anything but trivial. Generally speaking, special reaction conditions such as liquid ammonia as solvent,12 dilution conditions, slow fractional distillation over long columns for work-up or tedious multi-step syntheses are necessary.5,11,13 This is accompanied by a lack of selectivity and low yields. In addition, sterically demanding substituents such as tert-butyl groups in the carbon backbone or phenyl groups attached to the phosphorus atom in the starting materials are necessary to stabilise the phosphorus-containing heterocycles by these approaches, which makes access to the corresponding parent (P–H) compounds even more difficult.14,15 Very recently, Cummins reported the synthesis of the parent phosphirane (C2H5P), not coordinated to W(CO)5,16 based on a multistep Nickel-catalysed transfer reaction of tri-tert-butylphosphatetrahedrane.17 Other parent phosphorus-containing heterocycles are usually accessible by low yields, time-consuming synthesis or less selective methods.18,19 Specifically, secondary cyclic phosphines are prepared by hydrolysis of silylated species or protonation of suitable precursors.17,20,21 However, the latter usually also have to be synthesised in a complex manner, using rather unselective conventional routes with low overall yields.20,21 Therefore, there is an increasing need for a rational and direct synthesis of cyclic phosphines, especially of the parent derivatives (Scheme 1).
Herein we report the synthesis of the precursor compounds [Cp*Fe{η4-P5(CH2)n}] (Cp* = η5-C5Me5; 2: n = 3; 3: n = 4; 4: n = 5; 5: n = 6) and the straightforward, selective and easy synthesis of cyclic phosphines of different ring sizes – phosphetane (6), phospholane (7), phosphinane (8) and phosphepane (9), via nucleophilic phosphine abstraction by a pentaphosphaferrocene-mediated route.23 The reaction of the spiro compounds 2–5 with nucleophiles leads to the formation of unprecedented heterocyclic parent phosphines 10a–d including the first parent phospholane 10b.
Results and discussion
When reacting [K(dme)2]2[Cp*Fe(η5-P5)] (I)24 (dme = dimethoxyethane) with two equivalents of 1,3-dibromopropane, the 31P{1H} NMR spectrum of the reaction solution exhibits sets of signals corresponding to two different AMM'XX' spin systems in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (products 1 and 2). Chromatographic workup under inert conditions leads to the isolation of two complexes, [Cp*Fe{η4-P5(C3H6Br)2}] (1) and [Cp*Fe{η4-P5(C3H6)}] (2) (Scheme 2), but lowers the yield. Complex 1 represents the expected reaction product and the molecular structure in the solid state exhibits an η4-P5 moiety, bearing two 4-bromo-n-propyl substituents (Fig. 1 and S42†). In contrast, the XRD analysis of 2 reveals the formation of a spiro-cyclic ligand with a phosphetane-like moiety (Fig. 1). Its formation can be explained by the initial formation of the ionic compound [Cp*Fe{η-P5(C3H6Br)}]−, followed by an intramolecular salt metathesis reaction. This side reaction competes with the formation of 1. However, the formation of 1 cannot be supressed by using a 1:1 stochiometric ratio between I and 1,3-dibromopropane. By using a 1:2 ratio, the yields of both products 1 and 2 are similar (cf. ESI).†
:1 (products 1 and 2). Chromatographic workup under inert conditions leads to the isolation of two complexes, [Cp*Fe{η4-P5(C3H6Br)2}] (1) and [Cp*Fe{η4-P5(C3H6)}] (2) (Scheme 2), but lowers the yield. Complex 1 represents the expected reaction product and the molecular structure in the solid state exhibits an η4-P5 moiety, bearing two 4-bromo-n-propyl substituents (Fig. 1 and S42†). In contrast, the XRD analysis of 2 reveals the formation of a spiro-cyclic ligand with a phosphetane-like moiety (Fig. 1). Its formation can be explained by the initial formation of the ionic compound [Cp*Fe{η-P5(C3H6Br)}]−, followed by an intramolecular salt metathesis reaction. This side reaction competes with the formation of 1. However, the formation of 1 cannot be supressed by using a 1:1 stochiometric ratio between I and 1,3-dibromopropane. By using a 1:2 ratio, the yields of both products 1 and 2 are similar (cf. ESI).†
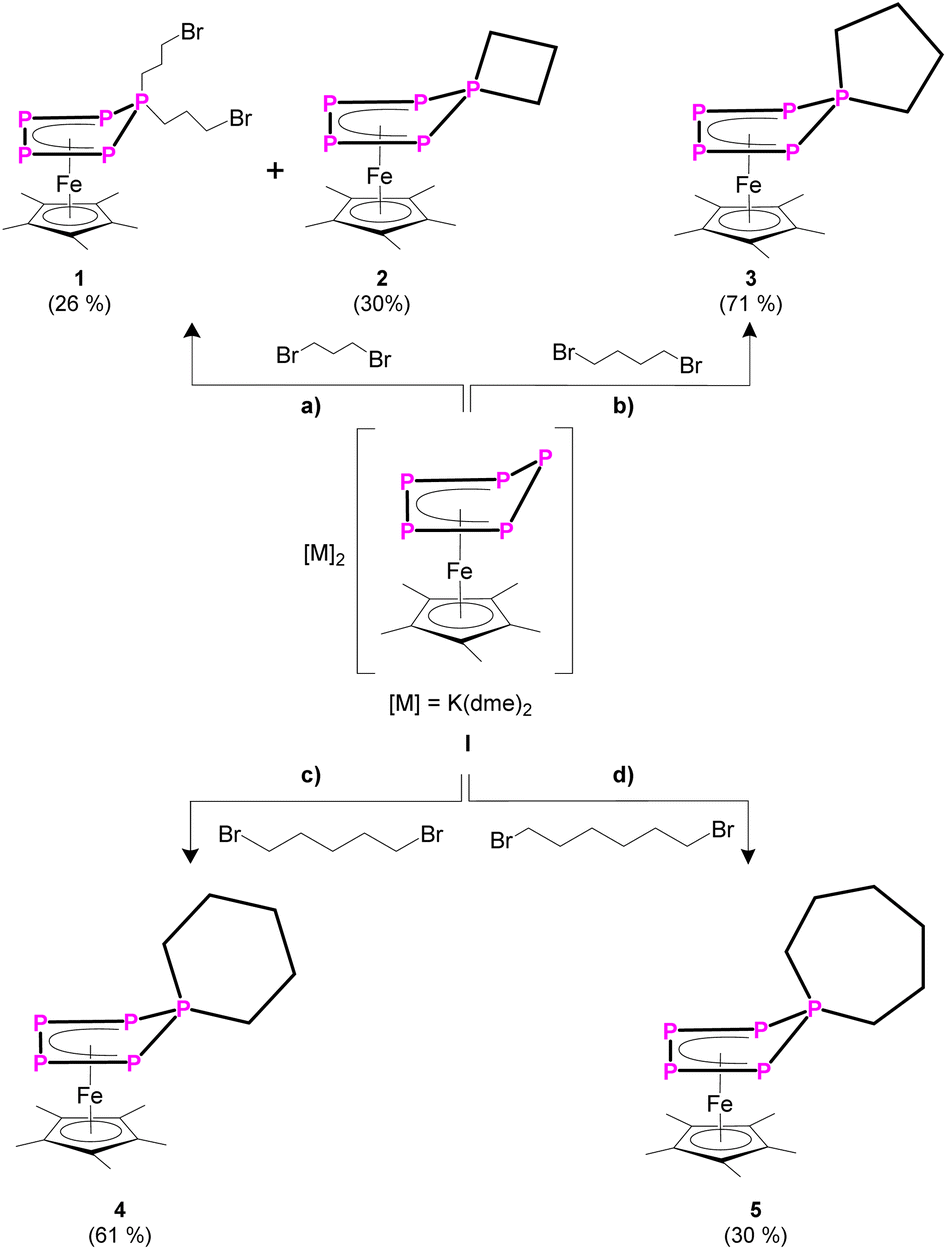 | ||
| Scheme 2 Reactivity of I towards: (a) 1,3-dibromopropane; (b) 1,4-dibromobutane; (c) 1,5-dibromopentane; (d) 1,6-dibromohexane. Yields are given in parentheses. | ||
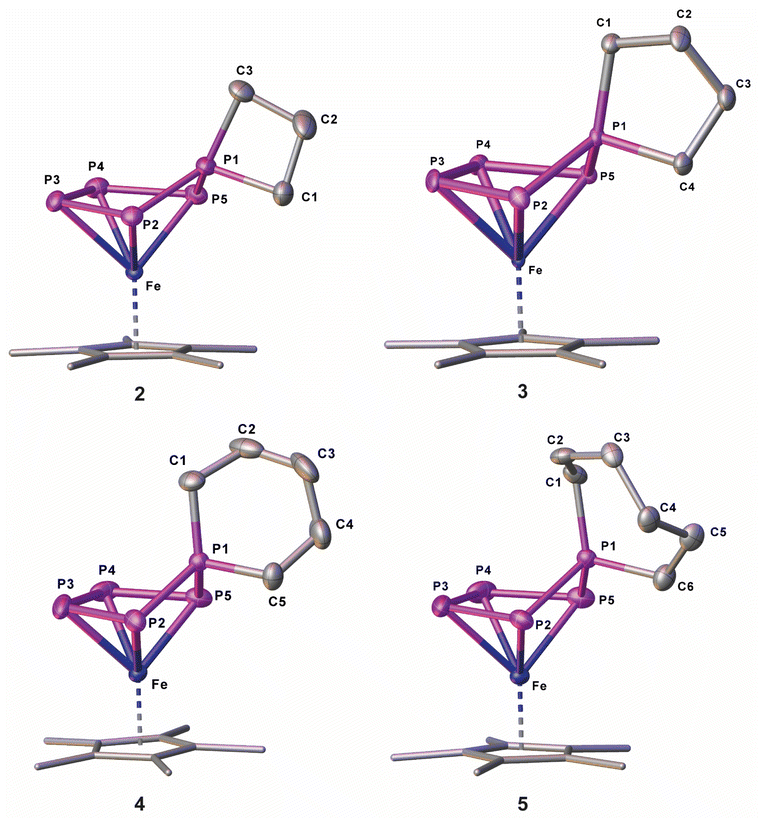 | ||
| Fig. 1 Molecular structure of 2–5 in the solid state; hydrogen atoms are omitted for clarity; thermal ellipsoids are drawn at 50% probability; Cp ligands are drawn in the wire frame model. | ||
Notably, the use of 1,2-dibromoethane does lead to a redox process in which [Cp*Fe(η5-P5)] (I′), KCl and ethene are formed.
When I is reacted with 1,4-dibromobutane without using dilution conditions, [Cp*Fe{η4-P5(C4H8)}] (3) can be isolated in 71% yield (Scheme 2). This shows that there is an intrinsic driving force for the formation of homocyclic products over disubstituted bromo-functionalised alkyl derivatives. The solid-state structure was proven by XRD (Fig. S44†) and reveals a spirocyclic phospholane-type ligand.
When reacting I with one equivalent of 1,5-dibromopentane (Scheme 2), the 31P{1H} NMR spectrum of the reaction solution exhibits two different (AMM′XX′) spin systems in a ratio of 10:1, in which the major compound can be attributed to complex 4 (Fig. S52†). After chromatographic workup, solely complex [Cp*Fe{η4-P5(C5H10)}] (4) can be isolated in 61% yield. Unfortunately, the minor product cannot be isolated and decomposes during the chromatographic workup.
Similarly, I reacts with one equivalent of 1,6-dibromohexane (Scheme 2), leading to [Cp*Fe{η4-P5(C6H12)}] (5), which was isolated in 30% yield. The 31P{1H} NMR spectrum of the reaction solution shows the formation of two other [Cp*Fe(η4-Pn)]-containing species with spin systems of higher order (Fig. S53†) in an overall ratio of 1:1 to complex 5. Mass spectrometric analysis of the reaction solution in combination with the 31P{1H} NMR data (Fig. S53†) of the reaction mixture strongly suggests the additional formation of [{Cp*Fe(η4-P5)}2(C6H12)2] in which the two {Cp*Fe(η4-P5)} moieties are bridged by two n-hexyl units. Unfortunately, the second compound decomposes during chromatographic workup and could not be isolated and further characterised.
Compounds 2–5 were characterised comprehensively by XRD, mass spectrometry, NMR spectroscopy and elemental analysis. They represent complexes of a rare class of phosphetane-, phospholane-, phosphinane- and phosphepane-like spirocyclic ligands containing complexes which can be easily synthesised.
Knowing that a doubly substituted phosphorus atom can be removed from the P5 unit of the Cp*Fe fragment,23 this strategy was also applied for compounds 2–5. It paved the way for a novel synthetic route for substituted phosphetane, phospholane, phosphepane and phosphinane derivatives, starting from white phosphorus, where the side product [K][Cp*Fe(η4-P4)] can be recycled in a “semi-catalytic-cyclic-process”.23 When reacting compound 2–5 with one equivalent of potassium benzyl (KBn) at −80 °C in THF, the corresponding phosphetane BnP(CH2)3 (6), phospholane BnP(CH2)4 (7), phosphinane BnP(CH2)5 (8) and phosphepane BnP(CH2)6 (9) (Scheme 3) can be isolated as colourless viscous liquids after extraction with n-pentane and slow removal of the solvent under reduced pressure in yields of 60–80% (Scheme 3). In addition, [K][Cp*Fe(η4-P4)] is formed. The identity of the phosphines was proven by NMR spectroscopy and, after oxidation with sulphur by the corresponding phosphine sulphides (cf. compounds 6′–9′), also by single crystal X-ray diffraction analysis (Fig. 2 and S47–S50†). Via this procedure, compounds 6–9 can be easily and selectively synthesised without the need of bulky substituents on the phosphorus atom or special starting materials, and at that in much better overall yields.
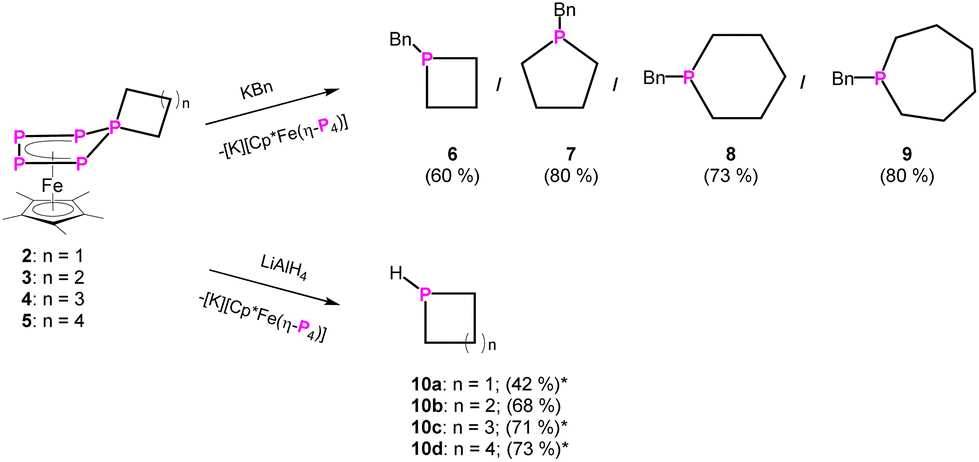 | ||
| Scheme 3 Reactivity of 2–5 towards KBn. Yields are given in parentheses (*NMR yield with PPh3 capillary as internal standard; cf. ESI;† note that the yields given in parentheses are not optimised). | ||
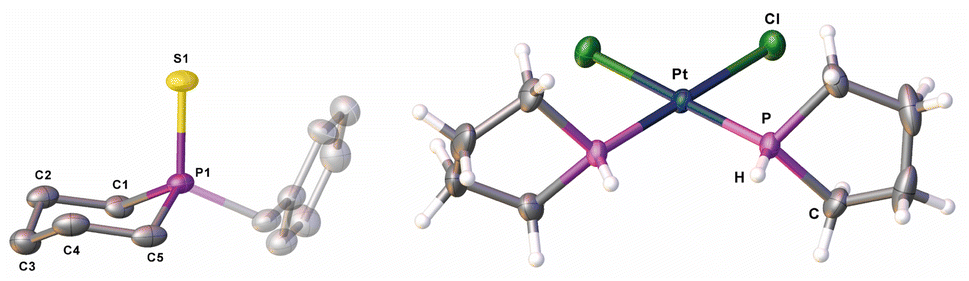 | ||
| Fig. 2 Molecular structure of 8′ (left) and 11 (right) in the solid state; hydrogen atoms of 8′ are omitted for clarity; thermal ellipsoids are drawn at 50% probability. | ||
Furthermore, we were interested in whether it is possible to use nucleophiles other than KBn to cleave off the phosphine. It has to be noted that MeLi and PhLi do not lead to the formation of the corresponding phosphines. However, using KPh shows the formation of phenylphospholane15 in the reaction of 3 with KPh.
An interesting class of substances are secondary phosphines, representing functionalisable compounds which can be converted to many different products e.g. phosphides or act as ligands themselves. As stated before, in order to synthesise cyclic secondary phosphines, harsh or cumbersome reactions conditions are needed. And even then, such reactions are not very selective, limited in their scope, and long-lasting workup by fractional distillation is necessary. Therefore, LiAlH4 was used as a hydride source to cleave off the P(CH2)n unit from 2–5. The reaction of 4 and 5 with LiAlH4 leads to the formation of the desired parent-phosphinine (HP(C5H10)) in 71% and -phosphepane (HP(C6H12)) in 73% yield, respectively, according to NMR spectroscopy (Scheme 3, Fig. S56 and S57†). The 31P/31P{1H} NMR data are in agreement with those products reported in the literature.20,21 To our surprise, (parent-)phospholane (HP(C4H8)) has not been reported so far. To validate the versatility of this synthetic procedure, we reacted 3 with LiAlH4 in THF-d8 (Scheme 4), leading to the parent phospholane (HP(C4H8)) (10b), which can be distilled off from the reaction mixture (1 × 10−3 mbar, 60 °C, 30 minutes) and isolated as a THF-d8 solution in 68% yield.
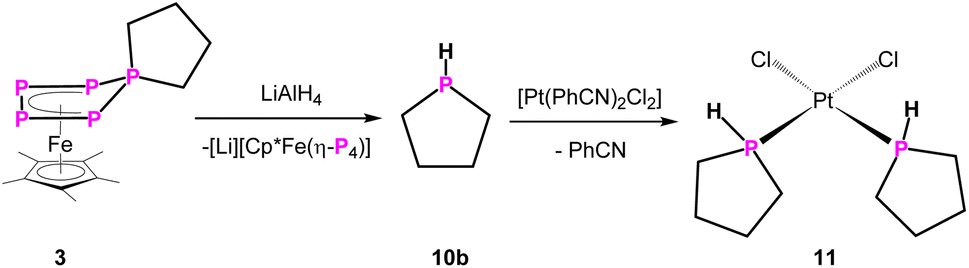 | ||
| Scheme 4 Reactivity of 3 towards LiAlH4. | ||
The 31P NMR spectra (THF-d8) of 10b show a doublet of triplets at δ = −70.8 ppm (1JP–H = 187 Hz, 2JP–H = 21 Hz) (Fig. S25 and S26†). To determine its molecular structure, compound 10b was reacted with [Pt(PhCN)2Cl2], leading to complex 11 (Scheme 4). The molecular structure of 11 in the solid state (Fig. 2) reveals the expected formation of the parent phospholane 10b, coordinating to a [PtCl2] unit in a κ1-fashion, forming the square planar cis-complex [({C4H8}PH)2PtCl2] (11) (Fig. 2).
The 31P{1H} NMR spectrum (CD2Cl2) of 11 shows a singlet at δ = −15.4 ppm with 195Pt satellites (1JP–Pt = 3384 Hz). The corresponding 31P NMR spectrum (CD2Cl2) of 11 shows a doublet (at δ = −15.4 ppm) with a 1JP–H coupling constant of 377 Hz. The NMR data are in agreement with those of similar secondary phosphines coordinating to platinum25 and prove, in combination with the corresponding FD-MS data of 11, unarguably the identity of the formerly unknown parent-phospholane HP(C4H8) (10b). It has to be noted that the reaction of 2 with LiAlH4 leads presumably to the formation of the desired (parent)phosphetane (HP(C3H6)). According to 31P NMR spectroscopy, this cyclic phosphine (10a) is formed (δ = −29.1 ppm (1JP–H = 164 Hz, 2JP–H = 18 Hz)) (Fig. S54†) in 42% yield alongside with unidentified volatile side products, which can unfortunately not be separated from 10a. 31P NMR spectra of the distillate suggest the additional formation of a di-phosphine, bearing one P–H bond, alongside of 10a (Fig. S58†).
Conclusions
In summary, the reaction of the dianionic polyphosphorus complex I with 1,3-dibromopropane yields complex 1, bearing two terminal bromine groups, as well as a spirocyclic ligand complex 2 containing a phosphetane-like moiety. When increasing the chain length of the di-bromoalkanes, the formation of the analogous two-fold substituted complex 1 is not observed. Instead, the intramolecular salt metathesis reaction is favoured and the corresponding free phospholane- and phosphinane-type spirocyclic ligands are obtained in high to moderate yields. When reacting I with 1,6-dibromohexane, a rare seven-membered phosphepane-like ligand complex 5 was isolated. Given the different alkyl chain lengths, the trend is towards yielding the functionalisation and ring formation for 1,3-dibromopropane, towards cyclisation exclusively when using 1,4-dibromobutane and towards ring formation and presumably linkage of two Cp*FeP5 moieties with longer alkyl chains. Moreover, complexes 2–5 represent very versatile precursors for the synthesis of the corresponding free phosphines by a nucleophilic phosphine abstraction reaction. The presented synthetic route offers great variability towards different reagents and can easily be extended, without the usage of large steric substituents. This procedure can be easily extended to other organo-substituted ring compounds. That way, the substituted phosphetane, phospholane, phosphinane and phosphepane (6–9) ring compounds could be synthesised in good yields, which are otherwise not accessible. In addition to this, we demonstrated that it is also possible to synthesise parent secondary phosphines derivatives 10a–dvia this route, as represented by the parent-phosphetane HP(C3H6) (10a) as well as the parent-phospholane HP(C4H8) (10b), which have been synthesised for the very first time. This synthetic strategy paves the way for having access to secondary phosphines, as a whole class of substances, in a straightforward way.Data availability
All experimental procedures, spectroscopic data, information on the theoretical calculations and crystallographic data can be found in the ESI.†Author contributions
The conceptualization (together with GB and MS), experimental work and writing of the manuscript of this work were achieved by SR. The entire work was supervised, guided, and revised by MS. The final manuscript was reviewed and edited by SR, GB and MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/38-3. Stephan Reichl is grateful to the Studienstiftung des Deutschen Volkes for his PhD fellowship.Notes and references
- L. D. Quin and J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, Wiley & Sons Ltd, 1st edn, 2010 Search PubMed.
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- L. Pignolet, Homogeneous Catalysis with Metal Phosphine Complexes, Springer US, Boston, MA, 1983 Search PubMed.
- (a) The Nobel Prize in Chemistry, 2001, https://www.nobelprize.org/prizes/chemistry/2001/summary/ Search PubMed; (b) The Nobel Prize in Chemistry, 2010, https://www.nobelprize.org/prizes/chemistry/2010/summary/ Search PubMed.
- P. C. J. Kamer and P. W. N. van Leeuwen, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- J. F. Teichert, Homogeneous Hydrogenation with Non-Precious Catalysts, Wiley, 2019 Search PubMed.
- K. Dimroth, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. B. T.-C. H. C. Rees, Elsevier, Oxford, 1984, pp. 493–538 Search PubMed.
- H. Shet, U. Parmar, S. Bhilare and A. R. Kapdi, Org. Chem. Front., 2021, 8, 1599–1656 RSC.
- G. Wittig and H. Pommer, German Pat., DBP954247, 1956 Search PubMed.
- A. Marinetti and D. Carmichael, Chem. Rev., 2002, 102, 201–230 CrossRef CAS PubMed.
- R. I. Wagner, L. D. Freeman, H. Goldwhite and D. G. Rowsell, J. Am. Chem. Soc., 1967, 89, 1102–1104 CrossRef CAS.
- R. Emrich and P. W. Jolly, Synthesis, 1993, 39–40 CrossRef CAS.
- R. A. Baber, M. F. Haddow, A. J. Middleton, A. G. Orpen, P. G. Pringle, A. Haynes, G. L. Williams and R. Papp, Organometallics, 2007, 26, 713–725 CrossRef CAS.
- F. Mathey and M. Regitz, in Comprehensive Heterocyclic Chemistry II, Elsevier, 1996, pp. 277–304 Search PubMed.
- A. A. Khan, P. Junker, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Chem. Commun., 2019, 55, 9987–9990 RSC.
- M.-L. Y. Riu, A. K. Eckhardt and C. C. Cummins, J. Am. Chem. Soc., 2022, 144, 7578–7582 CrossRef CAS PubMed.
- K. Issleib and S. Häusler, Chem. Ber., 1961, 94, 113–117 CrossRef CAS.
- F. Mathey, Compounds with One Saturated Carbon Heteroatom Bond, in Science of Synthesis, ed. F. Mathey and B. M. Trost, Georg Thieme Verlag, Stuttgart, 2009, vol. 42(5) Search PubMed.
- D. M. Schubert and A. D. Norman, Inorg. Chem., 1984, 23, 4130–4131 CrossRef CAS.
- D. M. Schubert, M. L. J. Hackney, P. F. Brandt and A. D. Norman, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 123, 141–160 CrossRef CAS.
- A. Emadi, R. J. Jones and R. A. Brodsky, Nat. Rev. Clin. Oncol., 2009, 6, 638–647 CrossRef CAS PubMed.
- S. Reichl, E. Mädl, F. Riedlberger, M. Piesch, G. Balázs, M. Seidl and M. Scheer, Nat. Commun., 2021, 12, 5774 CrossRef CAS PubMed.
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972–2976 CrossRef CAS PubMed.
- M. R. Eberhard, E. Carrington-Smith, E. E. Drent, P. S. Marsh, A. G. Orpen, H. Phetmung and P. G. Pringle, Adv. Synth. Catal., 2005, 347, 1345–1348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2232605–2232614. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00580a |
| This journal is © The Royal Society of Chemistry 2023 |