
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifaceted behavior of a doubly reduced arylborane in B–H-bond activation and hydroboration catalysis†
Sven E.
Prey‡
a,
Christoph
Herok‡
 b,
Felipe
Fantuzzi
bc,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Bernd
Engels
*b and
Matthias
Wagner
*a
b,
Felipe
Fantuzzi
bc,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Bernd
Engels
*b and
Matthias
Wagner
*a
aInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Frankfurt am Main D-60438, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bInstitut für Physikalische und Theoretische Chemie, Julius-Maximilians-Universität Würzburg, Würzburg D-97074, Germany. E-mail: bernd.engels@uni-wuerzburg.de
cSchool of Chemistry and Forensic Science, University of Kent, Canterbury CT2 7NH, UK
First published on 5th December 2022
Abstract
Alkali-metal salts of 9,10-dimethyl-9,10-dihydro-9,10-diboraanthrancene (M2[DBA-Me2]; M+ = Li+, Na+, K+) activate the H–B bond of pinacolborane (HBpin) in THF already at room temperature. For M+ = Na+, K+, the addition products M2[4] are formed, which contain one new H–B and one new B–Bpin bond; for M+ = Li+, the H− ion is instantaneously transferred from the DBA-Me2 unit to another equivalent of HBpin to afford Li[5]. Although Li[5] might commonly be considered a [Bpin]− adduct of neutral DBA-Me2, it donates a [Bpin]+ cation to Li[SiPh3], generating the silyl borane Ph3Si–Bpin; Li2[DBA-Me2] with an aromatic central B2C4 ring acts as the leaving group. Furthermore, Li2[DBA-Me2] catalyzes the hydroboration of various unsaturated substrates with HBpin in THF. Quantum-chemical calculations complemented by in situ NMR spectroscopy revealed two different mechanistic scenarios that are governed by the steric demand of the substrate used: in the case of the bulky Ph(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NtBu, the reaction requires elevated temperatures of 100 °C, starts with H–Bpin activation which subsequently generates Li[BH4], so that the mechanism eventually turns into “hidden borohydride catalysis”. Ph(H)CNPh, Ph2CO, Ph2CCH2, and iPrNCNiPr undergo hydroboration already at room temperature. Here, the active hydroboration catalyst is the [4 + 2] cycloadduct between the respective substrate and Li2[DBA-Me2]: in the key step, attack of HBpin on the bridging unit opens the bicyclo[2.2.2]octadiene scaffold and gives the activated HBpin adduct of the Lewis-basic moiety that was previously coordinated to the DBA-B atom.
NtBu, the reaction requires elevated temperatures of 100 °C, starts with H–Bpin activation which subsequently generates Li[BH4], so that the mechanism eventually turns into “hidden borohydride catalysis”. Ph(H)CNPh, Ph2CO, Ph2CCH2, and iPrNCNiPr undergo hydroboration already at room temperature. Here, the active hydroboration catalyst is the [4 + 2] cycloadduct between the respective substrate and Li2[DBA-Me2]: in the key step, attack of HBpin on the bridging unit opens the bicyclo[2.2.2]octadiene scaffold and gives the activated HBpin adduct of the Lewis-basic moiety that was previously coordinated to the DBA-B atom.
Introduction
The activation of chemical bonds by main-group compounds is not only conceptually appealing, but also holds great application potential. A prominent class of p-block catalysts is that of Frustrated Lewis Pairs (FLPs), which contain suitable combinations of sterically encumbered Lewis acids (LA) and bases (LB). Together, these functional units contribute the vacant orbital and electron lone pair which, in classical catalysis, are provided by one single transition metal center with partially filled d orbitals.1,2 More recently, alternative systems based on doubly boron-doped (hetero)arenes have been reported.3,4 Common to all of them is a central six-membered ring featuring an aromatic π-electron system and two mutually cooperating B atoms integrated therein at opposite positions. As an example, Kinjo et al. disclosed that the 1,3,2,5-diazadiborinine A (Fig. 1) adds the single bond of H3C–OSO2CF3, the double bonds of alkenes and carbonyls, as well as the triple bonds of alkynes across its B sites. As mode of action, Kinjo postulated that A has a B(I)/B(III) mixed-valence character,5–7 which would render it formally analogous to an LB/LA FLP. A related 1,4,2,5-diazadiborinine activates H–H, B–H, Si–H, and P–H bonds. Here, it was proposed that the two chemically equivalent B atoms “act as both nucleophilic and electrophilic centers, demonstrating ambiphilic nature”.8,9 Catalytic cycles based on these diazadiborinines have not yet been described, with one notable exception: in situ-generated [4 + 2] cycloadducts of A and Ph(Me)CO or H2CCH2 act as “electrostatic catalysts” to promote hydroboration of carbonyl compounds with pinacolborane (HBpin; see Fig. 3 below).7
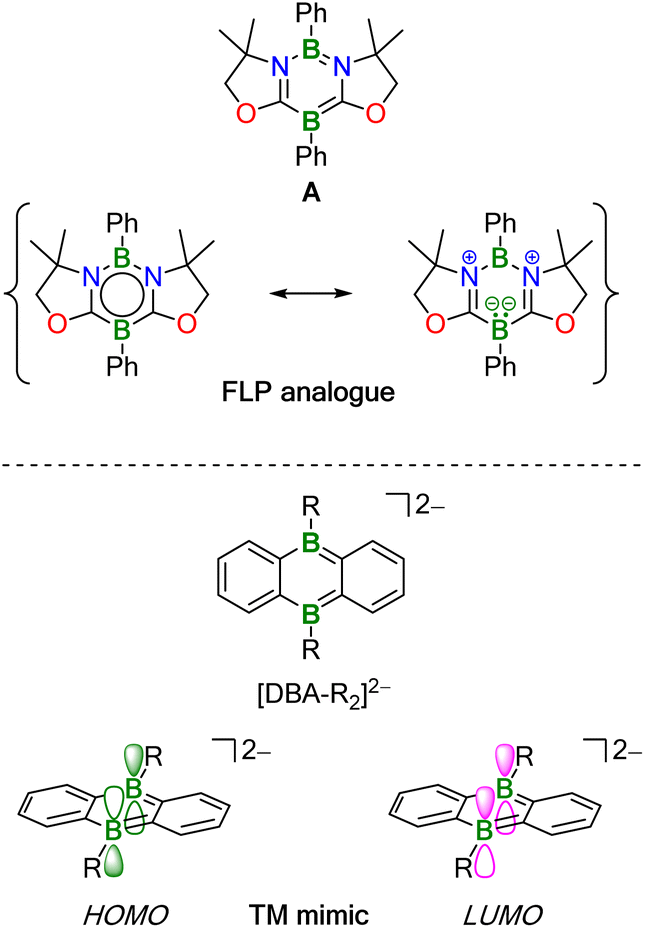 | ||
| Fig. 1 (Top) 1,3,2,5-Diazadiborinine A can be regarded as B(I)/B(III) mixed-valence system, which activates substrate molecules in a similar manner to LB/LA-FLPs. (Bottom) The frontier orbital symmetries of the 9,10-dihydro-9,10-diboraanthracene (DBA) dianion [DBA-R2]2− allow concerted transition metal-like (TM-like) reactions. | ||
Wagner and coworkers have introduced 9,10-dihydro-9,10-diboraanthracene (DBA) dianions [DBA-R2]2– into catalysis (Fig. 1; R = H, Me). These species have indistinguishable, sterically accessible B atoms along with high-lying HOMO-energy levels and are therefore particularly reactive. Apart from reacting with H–H and C–H bonds, [DBA-R2]2− dianions undergo facile [4 + 2] cycloaddition with various unsaturated organic molecules.10–14
The HOMO and LUMO of [DBA-R2]2− have the same local symmetries about the B atoms as, respectively, the LUMO and HOMO of H2, and quantum-chemical calculations suggest a concerted, transition metal-like (TM-like) bond-cleavage pathway (Fig. 1).11 H2 activation by [DBA-R2]2− is at the core of two recently developed catalytic hydrogenation- and H−-transfer cycles.13 In addition, it has been shown that [DBA-R2]2− can catalyze the disproportionation of CO2 to CO and [CO3]2−.12,15
In a joint experimental and theoretical effort, we are herein unveiling the capacity of the [DBA-R2]2− platform to activate B–H bonds and catalyze hydroborations. Besides our system, there are also numerous FLPs that catalyze hydroborations. Various modes of action have been discussed.16 The hydroboration reaction is therefore an ideal tool to put into context the behavior of [DBA-R2]2− dianions with those of other main group catalysts and to learn more about the subtleties of subvalent boron species.
Three key results are disclosed: (i) HBpin activation by [DBA-R2]2− is a means of forming new B–B bonds. (ii) Depending on the steric bulk of the substrate, the hydroboration mechanism differs between “hidden borohydride catalysis”17 and actual DBA-driven catalysis. (iii) To gain accurate theoretical insights into the mechanisms, one must explicitly include the counter cations and their coordinating solvent molecules in the calculations, since simple continuum approaches are too imprecise.
Results and discussion
All reactions were performed in (deuterated) tetrahydrofuran (THF, THF-d8).Activation of HBpin by M2[DBA-R2]
Pinacolborane (HBpin) was selected as the hydroboration reagent for the following reasons: (i) in contrast to, e.g., B2H6 or (9-BBN)2 (BBN = 9-borabicyclo[3.3.1]nonane), HBpin is strictly monomeric18 and does not form solvent adducts in THF, which facilitates the theoretical assessment of its reactivity. (ii) The alkyl boronic ester products (R′–Bpin) are comparatively stable to air and moisture, easy to purify, and have a wide range of applications.19 The aforementioned assets are the result of pronounced OB π donation, decreasing the electrophilicity of the B atom. On the other hand, less Lewis-acidic boranes are less prone to spontaneous addition to unsaturated substrates, so that a catalyst is often required. Thus, hydroboration reactions with HBpin provide the ideal setting to further explore the scope of M2[DBA-R2]-mediated reactions.
Our investigations into the activation of HBpin by M2[DBA-R2] were carried out with M+ = Li+–K+ as counter cations and R = H, Me as B-bonded substituents (Li2[1]–K2[1], Li2[2]–K2[2]; Scheme 1). In all cases, the reaction with HBpin was already instantaneous at room temperature, as judged by the rapid fading of the intensely colored M2[DBA-R2] solutions after addition of the borane (M+ = Li+: red; M+ = Na+, K+: green). According to in situ NMR spectroscopy, 1 equiv. of HBpin was sufficient to quantitatively convert Na2[1], K2[1], and K2[2] to Na2[3], K2[3], and K2[4], respectively, featuring newly formed B–H and B–Bpin bonds together with two tetracoordinate B centers on their DBA cores (type-I structures; Scheme 1). In contrast, equimolar mixtures of Li2[1] or Na2[2] with HBpin contained Li2[3] or Na2[4] and significant amounts of still unconsumed starting materials. Full conversion of the DBA-dianion salts to the corresponding type-I addition products required the use of excess HBpin. These [DBA-R2]2−-reactivity trends are the same as those previously reported for H2 activation:13 (i) M+ ions with larger charge-radius ratio have a higher tendency to form contact–ion pairs with [DBA-R2]2− in solution, which leads to the reactivity order Li2[DBA-R2] < Na2[DBA-R2] < K2[DBA-R2].11 (ii) Smaller B-bonded substituents R impede substrate access to [DBA-R2]2− less than bulkier groups, resulting in the reactivity order [2]2− < [1]2−.
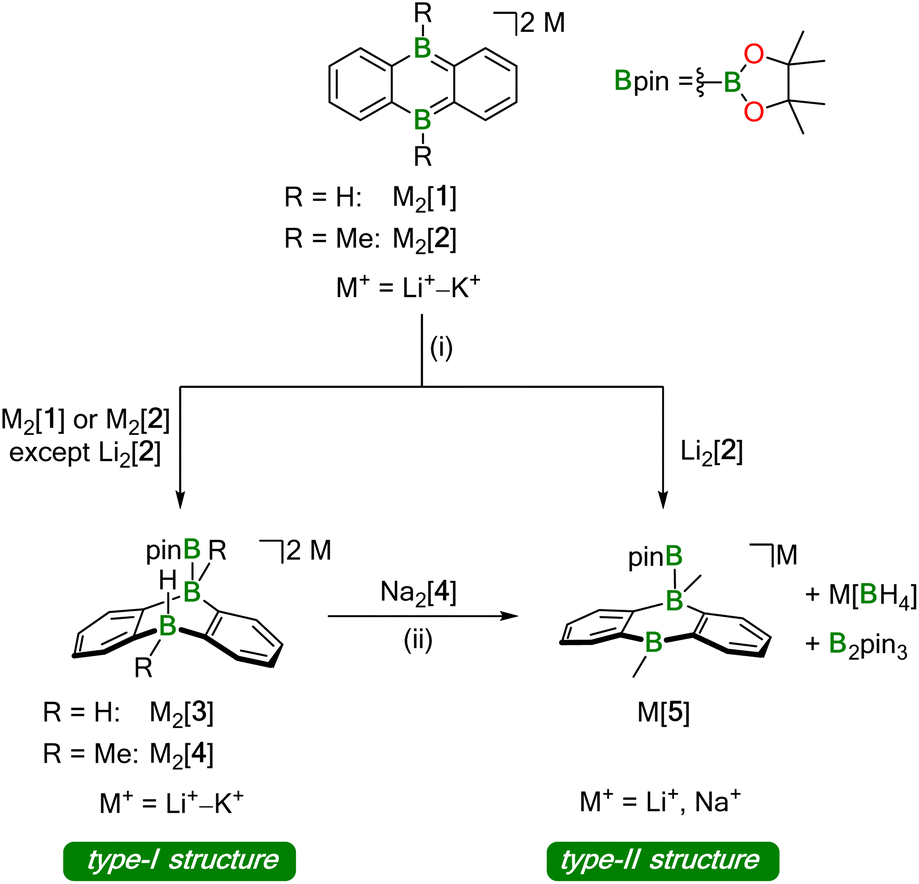 | ||
| Scheme 1 Addition of pinacolborane (HBpin) across the two B atoms of 9,10-dihydro-9,10-diboraanthracene (DBA) dianions to form B–B bonds. Depending on the nature of the counter cations, the dianionic type-I structures compete with monoanionic type-II structures, which are formed by transfer of the H− ligand onto a second equivalent of HBpin. (i) 1–5 equiv. HBpin (see ESI†), THF-d8, room temperature; (ii) prolonged storage in 1,2-dimethoxyethane (DME) during crystallization experiments. | ||
The accordingly least reactive Li2[2] is indeed a peculiarity, since a type-I product (putative Li2[4]) could not even be detected as an intermediate. Rather, the monoanion salt Li[5] was formed without H− ligand at the DBA moiety (type-II structure; Scheme 1). Again, an excess of HBpin was necessary to enforce quantitative transformation of Li2[2] to Li[5], which is accompanied by a formal release of LiH. According to quantum-chemical calculations, this would be an energetically unfavorable process20,21 and it is, therefore, reasonable to assume that excess HBpin acts as H− scavenger. The NMR-spectroscopically observed formation of [BH4]− and B2pin3 (ref. 22) supports this assumption, since H− is known to induce a corresponding decay of HBpin.17 In summary, we propose that Li2[2] and HBpin are in a dynamic addition–elimination equilibrium, which is shifted towards Li[5] formation by H− transfer to HBpin and the subsequent irreversible decomposition of the resulting [H2Bpin]− adduct. Consistent with this view, addition of LiH to a THF-d8 solution of Li[5] leads back to Li2[2] and HBpin (NMR-spectroscopic control).
The characteristic NMR data of type-I/II structures are exemplarily discussed with reference to the Na2[4]/Li[5] couple. The 11B NMR spectrum of Na2[4] contains three signals. Two of them appear in the chemical shift range of tetracoordinate B nuclei [−22.0 ppm (BBpin), −17.4 ppm (d, 1JBH = 68 Hz, BH)] and the third is characteristic of a tricoordinate B center [42.6 ppm (broad, Bpin)].23 The δ(11B) values of Li[5] prove the presence of one tetra- and two tricoordinate B atoms [−19.7 ppm (BBpin), 41.6 ppm (Bpin), 61.8 ppm (BC3)]. Accordingly, only the 1H{11B} NMR spectrum of Na2[4] shows the signal of a B-bonded H atom (2.14 ppm). The 1H integral values in the spectra of both Na2[4] and Li[5] are consistent with the presence of one Bpin substituent in each of these molecules. In line with the proposed average Cs symmetry of Na2[4] and Li[5] in solution, their 13C{1H} NMR spectra exhibit only six resonances in the aromatic region. Single crystals of the type-II compound [Li(thf)2][5] were grown by gas-phase diffusion of n-hexane into a C6H6 solution of thf-solvated Li[5] (Fig. 2). Attempts at the crystallization also of type-I compounds gave specimens suitable for X-ray diffraction only from 1,2-dimethoxyethane (DME) solutions of thf-solvated Na2[4] (room temperature, 1–2 d). Since X-ray analysis revealed that the crystals consisted of [Na(thf)2][5] as opposed to Na2[4] (Fig. 2), it appears that Na2[4] is also susceptible to formal NaH elimination (triggered by residual HBpin under the crystallization conditions). The DBA moiety of [Na(thf)2][5] has one sp3-hybridized [B(1)] and one sp2-hybridized B atom [B(2); 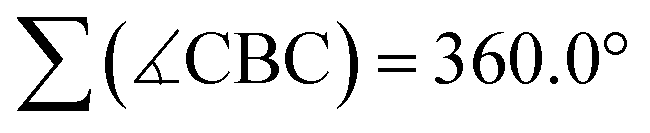 ]. At B(1), the Bpin substituent is attached in an axial position and with a bond length of B(1)–B(3) = 1.731(9) Å.24 The [Na(thf)2]+ cation is coordinated by an O atom belonging to Bpin and by the centroid of a phenylene ring, creating a contact–ion pair. The solid-state structure of [Li(thf)2][5] does not merit further discussion, given that it differs from that of [Na(thf)2][5] mainly in details of cation–anion association.20
]. At B(1), the Bpin substituent is attached in an axial position and with a bond length of B(1)–B(3) = 1.731(9) Å.24 The [Na(thf)2]+ cation is coordinated by an O atom belonging to Bpin and by the centroid of a phenylene ring, creating a contact–ion pair. The solid-state structure of [Li(thf)2][5] does not merit further discussion, given that it differs from that of [Na(thf)2][5] mainly in details of cation–anion association.20
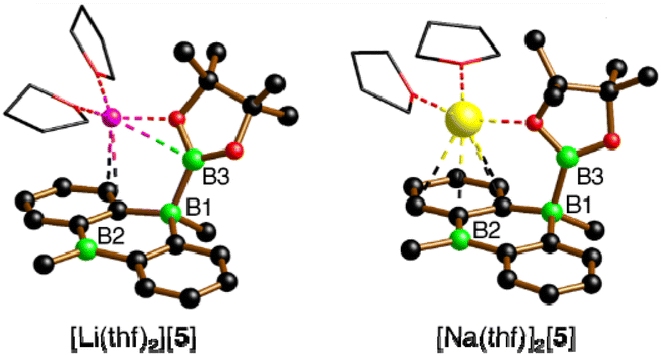 | ||
| Fig. 2 X-ray-crystallographically derived solid-state structures of the solvates [Li(thf)2][5] and [Na(thf)2][5], featuring one B–Bpin bond. C-bonded H atoms are omitted for clarity; the thf ligands are simplified as wireframes; Li: pink, B: green, C: black, O: red, Na: yellow spheres. | ||
Reactivities of M2[DBA-R2]/HBpin addition products
Hydridoborate ions [R3B–H]− and negatively charged adducts [R3B–Bpin]− are widely used sources of H− and [Bpin]− nucleophiles, respectively.25,26 Type-I compounds such as Na2[4] provide both functionalities in the same molecule, which raises the question of whether H− or [Bpin]− are preferentially transferred to, e.g., chlorosilanes as archetypal electrophiles. Treatment of freshly prepared Na2[4] with 1 equiv. of Et3SiCl resulted in selective H− abstraction to form Na[5] and Et3SiH, showing that H− wins the competition [cf. abstraction of MH by HBpin during the formation of M[5] (M+ = Li+, Na+)]. What about the possibility of [Bpin]− transfer in the absence of a competing H− ion? We have found that Li[5] is inert to Et3SiCl, Et3SiBr, and MeI at room temperature for several days and therefore does not appear to be a [Bpin]− donor. To probe if a polarity-inverted reactivity is present, Li[5] was next combined with the silanide salt Li[SiPh3], which indeed gave pinB–SiPh3 (ref. 27) in a clean reaction.28 Here, the highly delocalized byproduct [2]2− turns out to be a good enough leaving group to promote the transfer of a [Bpin]+ electrophile (Scheme 2). To confirm this remarkable result further, we also prepared the formal hydridoborate anion Li[6] through H−-adduct formation between the Lewis acid 2 and the H− source LiH and then removed H+ by deprotonation of Li[6] with Li[C(SiMe3)3]29 (Scheme 2).20 Again, [2]2− is liberated, accompanied by the formation of HC(SiMe3)3, which renders Li[6] a model system of Li[5] in which all reactive parts are stripped down to their absolute essence.30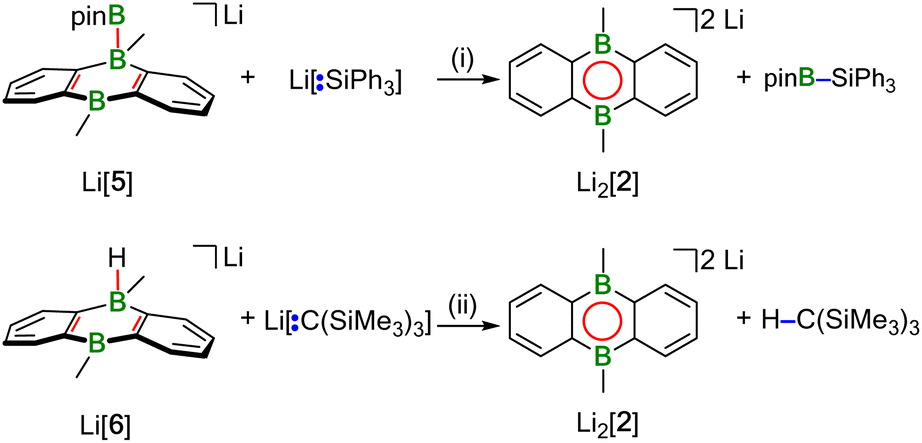 | ||
| Scheme 2 Reactions of Li[5] and Li[6] with an Si-centered nucleophile and a C-centered Brønsted base to demonstrate transfer of [Bpin]+ and H+ along with the suitability of [2]2− as leaving group. (i) THF-d8, room temperature, overnight; (ii) THF-d8, 115 °C, 35 h. | ||
The reactions between M2[1]/M2[2] and HBpin described up to this point are not only relevant for H–B-bond activation, but also represent B–B-bond formation reactions off the beaten track. The latter aspect gains additional weight because Na2[3] (one B–B bond) is prone to H−/[Bpin]− scrambling, thereby generating a DBA with two B–B bonds: When a DME solution of Na2[3] was stored at room temperature, [Na(dme)]2[7] precipitated in single-crystalline form (Scheme 3).
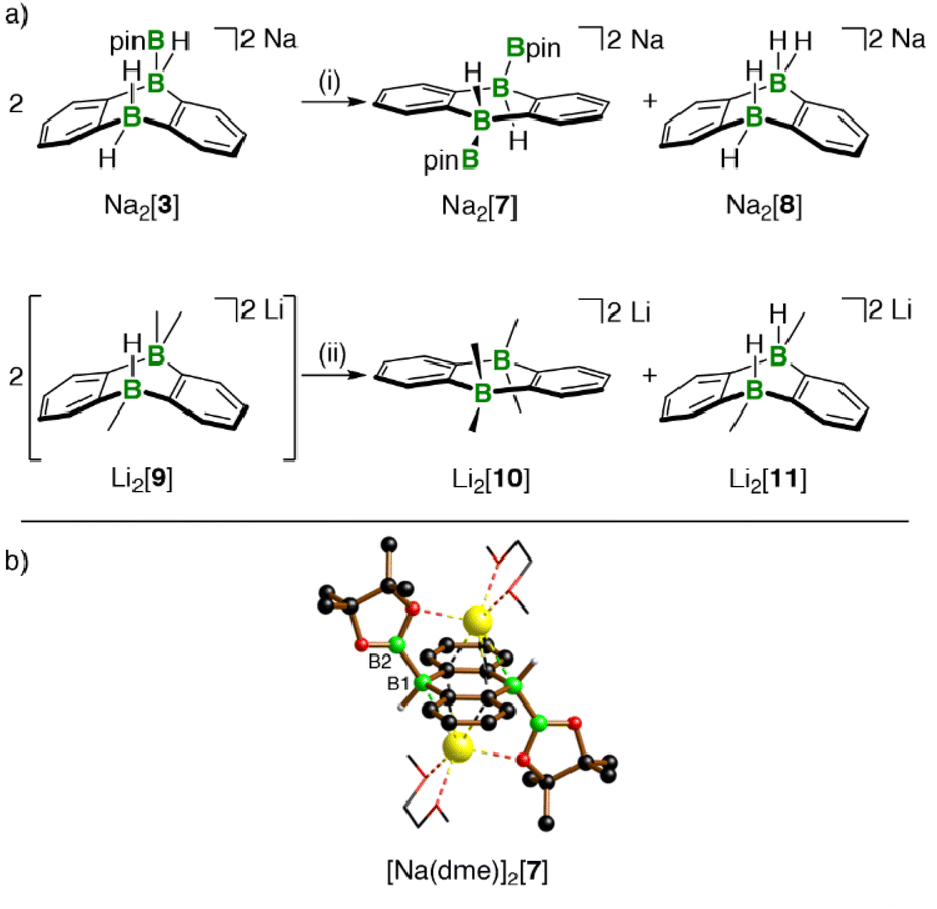 | ||
| Scheme 3 (a) Substituent-redistribution reaction at Na2[3] producing Na2[7] with two B–Bpin bonds. Substituent-redistribution reaction at Li2[9] leading to the pair of more symmetric compounds Li2[10]/Li2[11]. (b) X-ray-crystallographically derived solid-state structure of the solvate [Na(dme)]2[7], featuring two B–Bpin bonds. C-bonded H atoms are omitted for clarity; the dme ligands are shown as wireframes; B: green, C: black, O: red, Na: yellow spheres. (i) DME, room temperature, prolonged storage during crystallization experiments; (ii) THF-d8, room temperature. | ||
The dianion [7]2− can be regarded as Lewis pair {1·2[Bpin]−}; the necessary byproduct Na2[8]11,13 remained in the mother liquor and was detected by NMR spectroscopy after workup.
The 1H, 11B, and 13C{1H} NMR spectroscopic characterization of re-dissolved [Na(dme)]2[7] was in line with an average C2v or C2h symmetry but allowed no conclusion regarding a mutual cis or trans orientation of the two Bpin ligands. The assignment of [Na(dme)]2[7] as a Ci-symmetric trans complex (≙ C2h in solution) was finally achieved by X-ray crystallography (B(1)–B(2) = 1.711(3), Scheme 3). Note that the attempted synthesis of cis-Na2[7] from Na2[1] and B2pin2 failed, because the two compounds do not react with each other. Also the investigation of the transformation 2 Na2[3] → Na2[7] + Na2[8] was complemented by a model reaction, in which a Me group mimicked the Bpin group of Na2[3] (Scheme 3): Addition of MeLi (1 equiv.) to Li[6] did not lead to an NMR-spectroscopically detectable diadduct Li2[9], but rather to an equimolar mixture of the more symmetric Li2[10] and Li2[11]13 scrambling products,20 the former corresponding to Na2[7].
Taken together, a clear picture of the reactivity trends of the DBA substituents emerges from the model studies, which is of immediate importance with respect to the transfer of H or Bpin fragments onto substrates in the course of hydroboration reactions: a substituent R residing on the tetracoordinate B atom of a B(sp2),B(sp3)-DBA can be removed as an R+ fragment because the formerly B–R-bonding electron pair subsequently becomes part of a Clar's sextet31 within the central B2C4 ring. However, in a B(sp3),B(sp3)-DBA, this energetically favorable electron delocalization after R+ transfer is blocked by the second tetracoordinate B atom, which is why the same R now possesses mainly R− character.
Hydroboration reactions
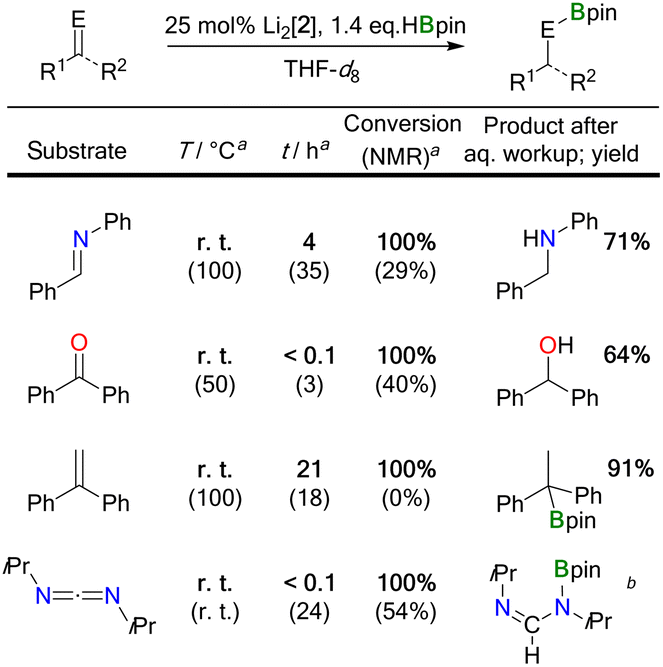
The substrates under investigation contained less polar (CC) as well as more polar (CN, CO) double bonds and possessed different steric demands (e.g., Ph(H)CNPh < Ph(H)CNtBu). Hydroborations were regularly performed with a loading of 25 mol% Li2[2] to facilitate the NMR-spectroscopic detection of reaction intermediates, side products, and byproducts; in the selected case of Ph2CO, it was confirmed that a Li2[2] loading of 5 mol% is sufficient for full conversion of the starting materials and thus the reactions are truly catalytic in the DBA. To ensure full comparability, we always added the HBpin to a mixture of freshly prepared Li2[2] and the unsaturated substrate.32
Two fundamentally different reaction mechanisms were found to be operative under these conditions.33 Which of the two comes into play depends decisively on the steric requirements of the substrate. Only in the case of the bulkiest substrate, the imine Ph(H)CNtBu, does HBpin activation by Li2[2] initiate the transformation; the actual catalytic cycle is a textbook example of “hidden borohydride catalysis”.17 In all other cases, [4 + 2] cycloaddition of the substrates' double bonds occurs prior to HBpin addition; subsequent hydroboration is then catalyzed by the cycloadducts.
NtBu, and HBpin showed the characteristic resonances of the above-mentioned HBpin-activation product Li[5], together with the signals of Li[BH4] and B2pin3. The resonances of the imine were still prominently visible, and the spectrum did not indicate transformation of this starting material. The situation remained unchanged for several hours at room temperature. Yet, heating the sample to 100 °C for 40 h led to 85% conversion of the imine, but it took another 50 h at 100 °C to drive the reaction to completion. The NMR data of the borylamine primary product agreed well with literature data;34 after in situ hydrolysis, we observed the resonances of the free amine Ph(H)2C–N(H)tBu.13 We took the initial appearance of the [BH4]− ion as a warning signal that the present case might be an example of hidden catalysis by the system BH3·thf/[BH4]−.17 Indeed, preliminary quantum-chemical calculations indicated that such a process might be at play (see below for an in-depth theoretical treatment of the reaction mechanism). To substantiate the assumption of hidden borohydride catalysis further, we adapted a test reaction recommended by Thomas and coworkers17 and repeated the experiment in the presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA), which is supposed to act as a BH3 scavenger. Subsequently, we detected the diagnostic signal of (BH3)2·tmeda35 and noted a significantly slower reaction. All in all, this confirmed our working hypothesis that the hydroboration of Ph(H)CNtBu by HBpin is only initiated by Li2[2], but catalyzed by BH3·thf/[BH4]−.
NPh, Ph2CO, and Ph2CCH2 with HBpin in the presence of Li2[2] proceeded quantitatively already at room temperature (Table 1). We found that the key elements of each reaction scenario are the same for all three substrates. It is therefore sufficient to discuss the experimental facts using Ph(H)CNPh as an example. Imine Ph(H)CNPh undergoes an instantaneous and quantitative [4 + 2] cycloaddition reaction with Li2[2] to afford the dianionic bicyclo[2.2.2]octadiene derivative Li2[12] (Scheme 4). As a distinct difference, the corresponding cycloaddition with the bulkier Ph(H)CNtBu is very slow at room temperature (and underlies a dynamic addition–elimination equilibrium at 100 °C).13 Consequently, under the prevailing reaction conditions, only in the case of Ph(H)CNtBu is free Li2[2] still available for initial HBpin activation, while in the other cases the actual active species must be a different one. Thus, the different steric demands of Ph(H)CNtBu and, e.g., Ph(H)CNPh lead to a bifurcation of the reaction mechanism into Mechanism I and a new Mechanism II.
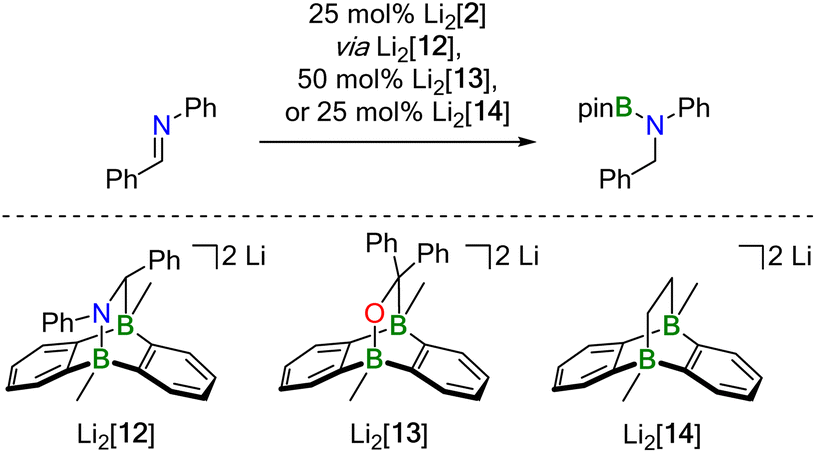 | ||
| Scheme 4 Crossover experiment to demonstrate that hydroboration of Ph(H)CNPh with HBpin can be mediated not only by Li2[2] (actual catalyst: Li2[12]), but also by pre-formed Li2[13] or Li2[14] without generating Ph2(H)C–OBpin or H3C–CH2Bpin as crossover side product, respectively (THF-d8, room temperature). Note that the high catalyst loadings were used for the sole purpose of being able to detect any catalyst degradation or trace formation of crossover products by NMR spectroscopy. | ||
Probable candidate catalysts under Mechanism II would be the respective [4 + 2] cycloadducts. This assumption is supported by crossover experiments in which the hydroboration of Ph(H)CNPh was efficiently mediated by pre-formed Li2[13] or Li2[14], obtained from Li2[2] and Ph2CO or H2CCH2, respectively. According to in situ NMR spectroscopy, the reactions furnished exclusively Ph(H)2C–N(Bpin)Ph (and no Ph2(H)C–OBpin or H3C–CH2Bpin), leaving Li2[13] or Li2[14] intact (Scheme 4).
At first glance, the molecular scaffolds of [12]2−, [13]2−, and [14]2− may appear similar to Kinjo's bicyclic compound B (Fig. 3a), which he employed as “electrostatic catalyst” for the hydroboration of various aldehydes and ketones. Here, the key to activation of the H–Bpin bond is believed to lie in electrostatic interactions between the negative partial charge on the borane's H atom and positively polarized regions in the binding pocket of B.7
 | ||
| Fig. 3 Comparison of Lewis structures and computed charge densities of (a) Kinjo's bicyclic catalyst B and (b) compounds [14]2− and Li2[14]. | ||
However, decisive differences between Kinjo's and our catalysts should arise from (i) the presence of electronegative N and O atoms in B, which are (largely) absent in [12]2−, [13]2−, and [14]2−, and (ii) the fact that B is a neutral compound whereas our catalysts are dianion salts. A comparison of the computed charge densities of B, [14]2−, and Li2[14] confirms this view (Fig. 3b): while positively polarized regions are indeed found in the binding pocket of B, they are completely missing in [14]2−. If the Li+ counter cations are included in the charge-density calculations, positively polarized areas also emerge for Li2[14] but remain largely associated with Li+. Although the charge distribution in Li2[14] does not directly correspond to the charge distribution in the binding pocket of B, it would nevertheless be conceivable that an interaction between the negatively polarized borane-H atom and one of the Li+ cations could still activate the H–Bpin bond.36 However, our calculations show that Li+ and HBpin preferentially interact via an O atom (and not the H atom) of the borane (Fig. S93†).37,38 Kinjo's mechanism is therefore not applicable to our case.
What is a plausible alternative? Although a dynamic equilibrium Li2[C] ⇄ Li2[2] + substrate does not exist at room temperature for the substrates Ph(H)CNPh, Ph2CO, and Ph2CCH2, it is conceivable that HBpin attack induces the reversible cleavage of one E–B(bridgehead) bond of Li2[C] (Scheme 5; E = CH2, NPh, O). In other words, there may be a competition between the B atoms of DBA and HBpin for the same E− donor. Precedence exists in the form of the acetone/Na2[2] cycloadduct Na2[15], which straightforwardly inserts CO2 into its O–B bond to afford Na2[16] (Scheme 5).12 In the putative negatively charged adduct [D]2−, the electronic situation at the H–B bond should be comparable to the case of, e.g., [BH4]−. Thus, hydride transfer from [D]2− to the unsaturated substrate, followed by “[Bpin]+” transfer, would yield the respective hydroboration product and regenerate the catalyst Li2[C]. Like Mechanism I, the essence of Mechanism II has also been confirmed by quantum-chemical calculations (vide infra).
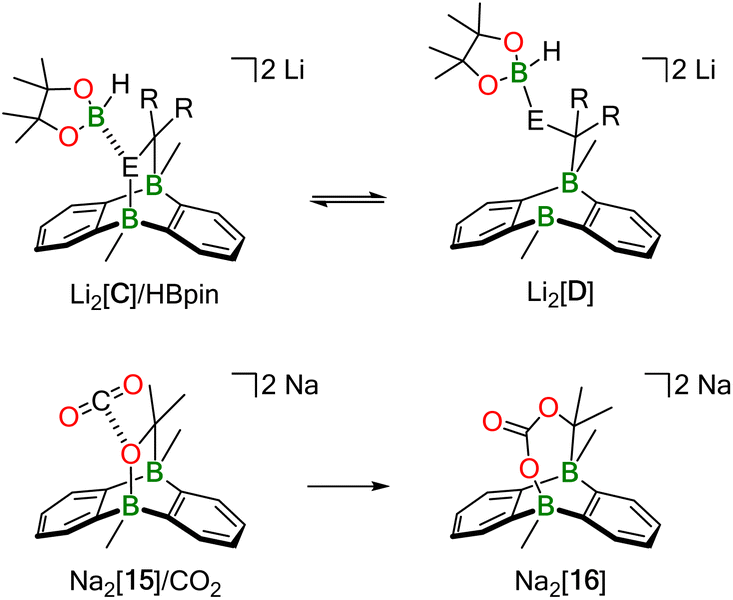 | ||
| Scheme 5 Conceptual relationship between H–Bpin activation by Li2[C] and CO2 activation by Na2[15] (R2C–E = H2C–CH2, Ph2C–CH2, Ph(H)C–NPh, Ph2C–O). | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CNiPr.
The carbodiimide iPrNCNiPr has two CN bonds and can therefore be singly or doubly hydroborated. So far, no catalyst has been described to promote both reaction scenarios.39–43 We now report that Li2[2] can catalyze the monohydroboration of iPrNCNiPr to give iPrNC(H)–N(Bpin)iPr (room temperature, instantaneous). The reaction is selective even in the presence of 2.4 equiv. of HBpin and proceeds via Mechanism II (the moderate steric bulk of the heterocumulene allows formation of its [4 + 2] cycloadduct, which has been fully characterized).20 Upon heating the sample to 100 °C (23 h), a second hydroboration furnishes iPr(pinB)N–CH2–N(Bpin)iPr. Due to the larger steric bulk of iPrNC(H)–N(Bpin)iPr compared to iPrNCNiPr, Mechanism I is operative in the second step (the [BH4]− ion was detected by in situ NMR spectroscopy).
CNiPr.
The carbodiimide iPrNCNiPr has two CN bonds and can therefore be singly or doubly hydroborated. So far, no catalyst has been described to promote both reaction scenarios.39–43 We now report that Li2[2] can catalyze the monohydroboration of iPrNCNiPr to give iPrNC(H)–N(Bpin)iPr (room temperature, instantaneous). The reaction is selective even in the presence of 2.4 equiv. of HBpin and proceeds via Mechanism II (the moderate steric bulk of the heterocumulene allows formation of its [4 + 2] cycloadduct, which has been fully characterized).20 Upon heating the sample to 100 °C (23 h), a second hydroboration furnishes iPr(pinB)N–CH2–N(Bpin)iPr. Due to the larger steric bulk of iPrNC(H)–N(Bpin)iPr compared to iPrNCNiPr, Mechanism I is operative in the second step (the [BH4]− ion was detected by in situ NMR spectroscopy).
Quantum chemical calculations
Since our above-mentioned experimental results exclude direct H− transfer from Li2[4] to Ph(H)CNtBu, a corresponding reaction mechanism was not considered theoretically. Rather, inspired by experiments of Clark and coworkers,51 we presumed that HBpin would be able to take up the H− ion provided by Li2[4], forming Li[H2Bpin], analogous to Clark's reactive species [tBuO(H)Bpin]−. HBpin could thereby act as H− shuttle to Ph(H)CNtBu. Indeed, this approach led to an overall exergonic reaction (−24 kcal mol−1) with the free energy of the highest-lying intermediate Li[H2Bpin] being +13 kcal mol−1 (Fig. S96†).
Despite its thermodynamic feasibility, this mechanism fails to explain the experimentally observed formation of [BH4]− and B2pin3. To take these two species into account, we further investigated their formation from decomposition of [H2Bpin]−.
We first calculated the complete reduction of 1 equiv. HBpin by 3 equiv. Li2[4]. The energetically costly formation of 1 equiv. Li2[pin] per equivalent of Li[BH4] generated renders this reaction exceedingly endergonic and thermodynamically out of scope (+36 kcal mol−1, Fig. S95†; H2pin = pinacol).
As an alternative approach, we propose a stepwise decomposition of Li[H2Bpin] via B–H/B–O σ-bond metathesis with 2 equiv. HBpin (Fig. S97†). Formation of the final products Li[BH4] and B2pin3 is exergonic by −21 kcal mol−1, likely due to the isodesmic nature of this reaction and to the formation of two stable BO3 motifs. Once formed, Li[BH4] takes on the role as the actual imine-reducing agent. On this basis, we can now establish a catalytic cycle for hydroboration of Ph(H)CNtBu by HBpin that is fully consistent with all our experimental results (Fig. 4): in a first step, transition metal-like addition of HBpin to precatalyst Li2[2] affords Li2[4], the starting point of catalytic Cycle I (Fig. 4a, right). While the newly formed B–B bond persists throughout all subsequent reaction steps, the H− ligand on Li2[4] is transferred to a second HBpin molecule from solution. The free energy of the intermediates Li[5] and Li[H2Bpin] amounts to +19 kcal mol−1 when referenced to the starting point of the cycle, Li2[4] (Fig. 4b, right). This energy penalty, which is equivalent to the most endergonic step of the reaction sequence, results from the loss of π conjugation in the H− carrier Li[H2Bpin]. As outlined above, decomposition of Li[H2Bpin] generates Li[BH4]. In a slightly exergonic reaction, the double bond of Ph(H)CNtBu inserts into a B–H bond of [BH4]−, affording the amide-borane adduct Int1. The next step involves a ligand exchange at the negatively charged N atom of Int1 through nucleophilic attack of Int1 on HBpin from solution, which releases BH3·thf and forms Int2.52Int2 possesses an activated H–Bpin bond (compare again Clark's intermediate) and thus represents the second highest thermodynamic barrier (+12 kcal mol−1) of Cycle I (Fig. 4b, right). For the last reaction step, there are two possible options, which leads to a bifurcation of the catalytic cycle into Cycles I and II (Fig. 4a). Following Cycle I, Li[5] abstracts the H− ligand from Int2 in a notably exergonic reaction (−26 kcal mol−1). As a result, the hydroboration product P is formed and Cycle I can start anew. The second possibility to produce P is H− transfer from Int2 to BH3·thf (generated in the preceding step), thereby entering Cycle II (left panels of Fig. 4a and b). The driving force to P is significantly higher along Cycle II (−38 kcal mol−1) than along Cycle I (−26 kcal mol−1). Moreover, since Cycle II starts from Li[BH4] rather than Li2[4], it bypasses the formation of Li[5] and Li[H2Bpin], the highest-lying intermediate of Cycle I. The steps between the formation of Li[BH4] and the formation of Int2 are equivalent in both cycles. Given that BH3·thf has the highest H− affinity of all species along Cycles I and II (Table S8†), it will take over the role as H− shuttle as soon as it is available, effectively making the DBAs Li2[4] and Li[5] obsolete. We, therefore, predict that only in the beginning the reaction proceeds with activation of HBpin by Li2[2]. After a few cycles, the mechanism switches to hidden catalysis, where BH3·thf is the catalytically active species. From this point on, Cycle II outcompetes Cycle I.
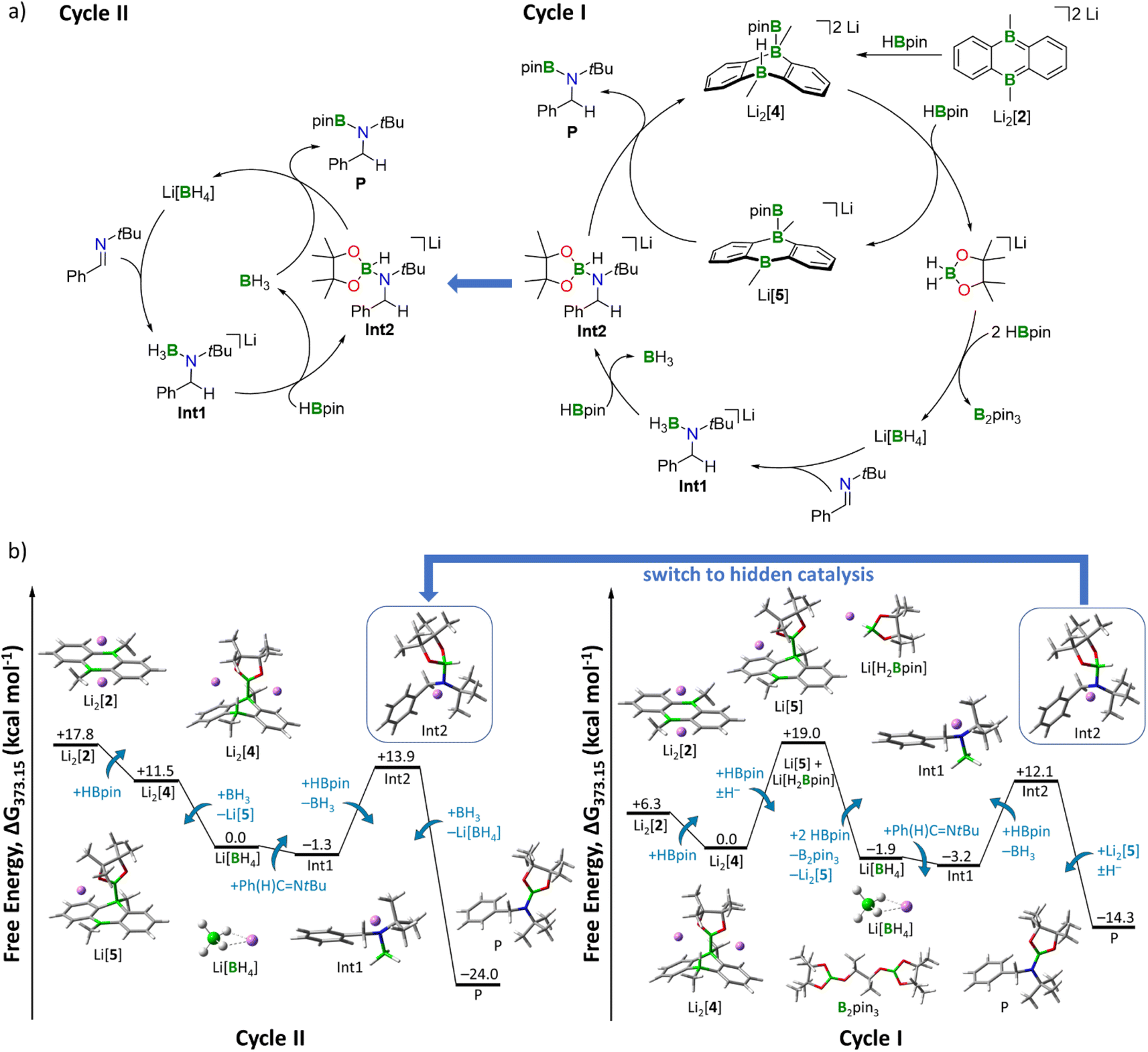 | ||
| Fig. 4 (a) (Right) Catalytic Cycle I (hydride shuttle: Li[H2Bpin]). (Left) Catalytic Cycle II, hidden catalysis (hydride shuttle: Li[BH4]). (b) (Right) Free energy diagram for catalytic Cycle I. (Left) Free energy diagram for hidden catalysis Cycle II including BH3·thf/Li[BH4]. Energies are referenced to the starting point of the respective cycle. Level of theory: SMD(solvent = THF)/ωB97XD/6-311++G(d,p) from optimized structures at SMD(solvent = THF)/ωB97XD/6-31+G(d,p). Explicit thf molecules are omitted for clarity. | ||
NtBu readily form bicyclo[2.2.2]octadiene derivatives by [4 + 2] cycloaddition to Li2[2] at room temperature. For the following reasons, the combination of Ph(H)CNPh and Li2[14] was chosen as representative model system for our theoretical study: (i) Li2[14] should be the most challenging candidate to evaluate the feasibility of the key ring-opening step in our proposed catalytic mechanism, because, unlike bridging O or NR units, CH2 fragments do not carry electron lone pairs as obvious sites of attack for incoming HBpin molecules (see Scheme 5). (ii) Li2[14] possesses a symmetric scaffold so that only one kind of reactive center has to be considered.
Our proposed catalytic cycle for the Li2[14]-mediated hydroboration of Ph(H)CNPh is shown in Fig. 5; the corresponding free energies of the intermediates/transition states along the reaction path are depicted in Fig. 6. Similar to Mechanism I, the first step of Mechanism II is again activation of HBpin, but the actual mode is different: instead of the previously observed transition metal-like cleavage of the H–Bpin bond, the borane is now nucleophilically attacked by a bridging CH2 group of Li2[14].53
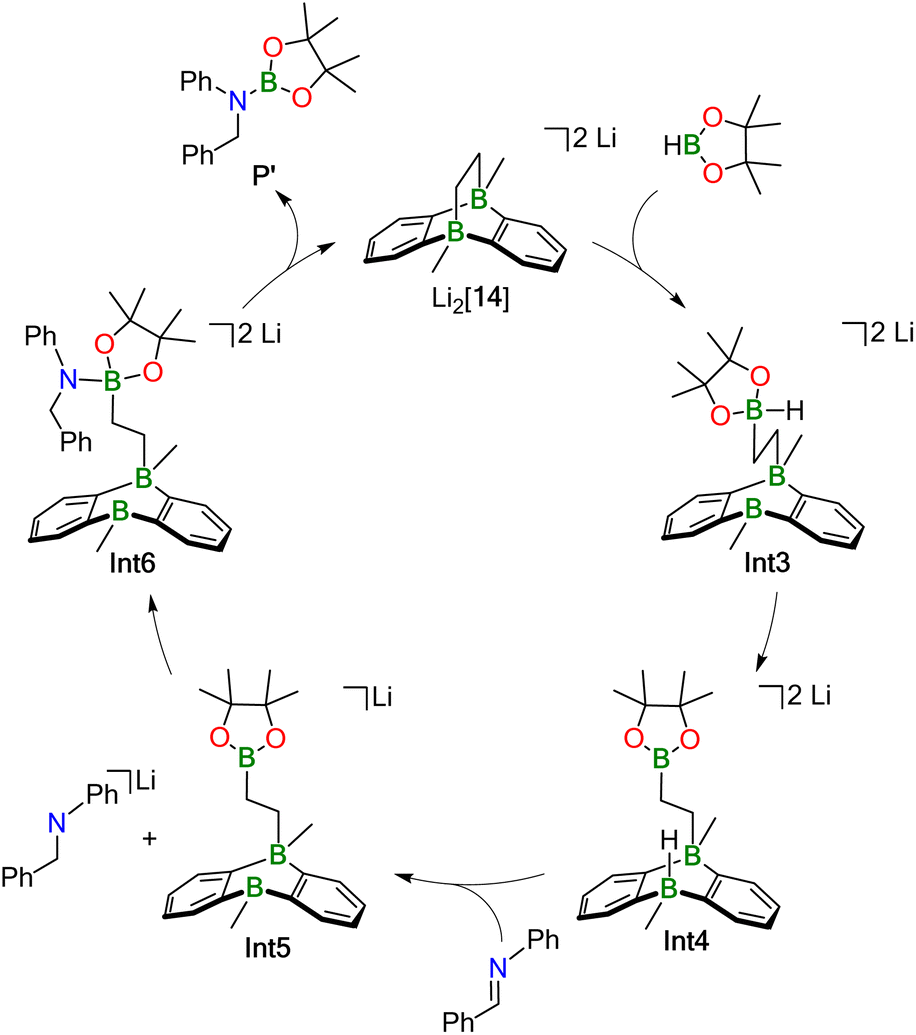 | ||
| Fig. 5 Hydroboration Mechanism II for less sterically demanding substrates. The corresponding energy diagram is given in Fig. 6. | ||
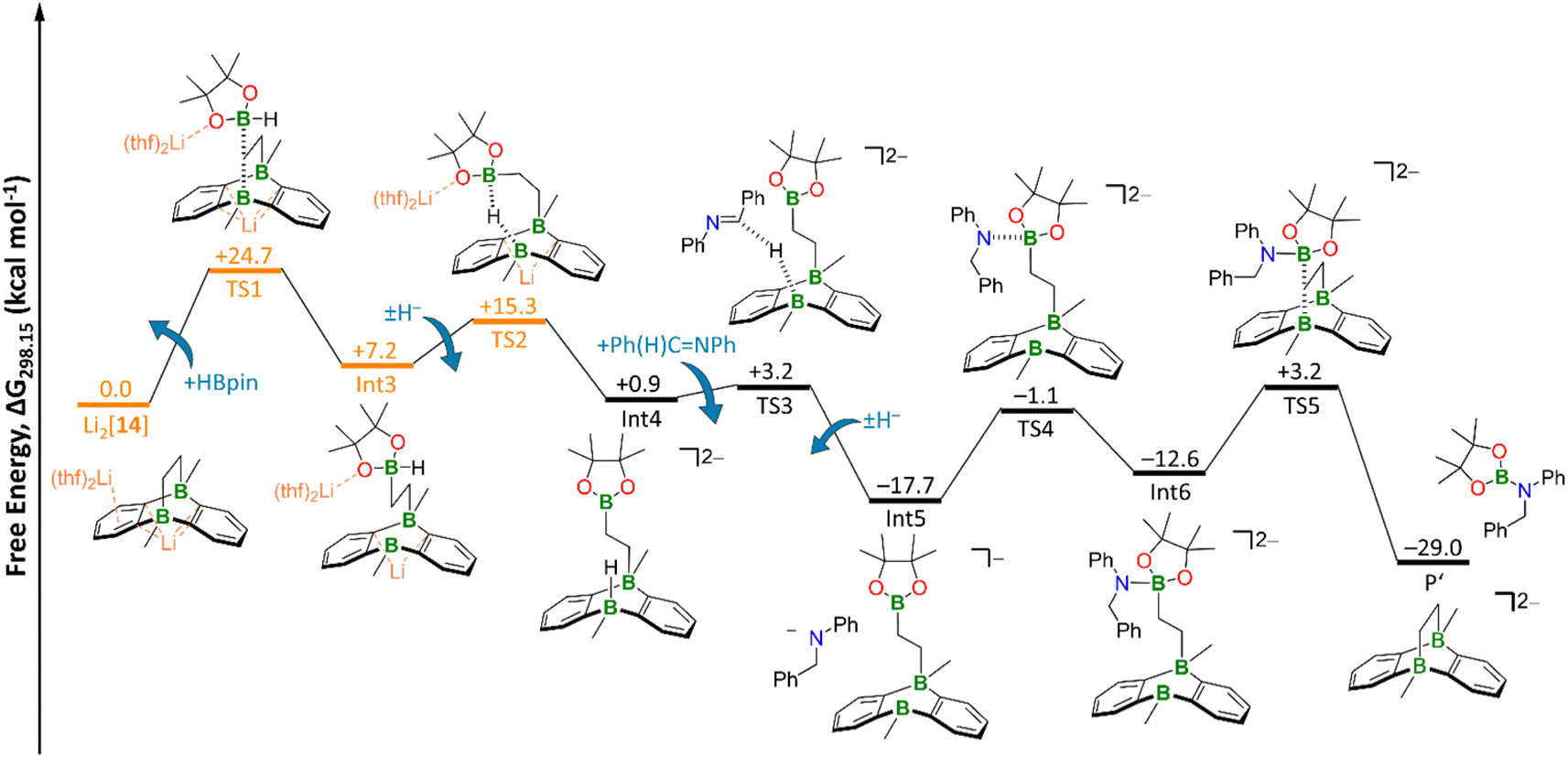 | ||
| Fig. 6 Free energy diagram of hydroboration Mechanism II. Orange: transition states and intermediate including counterions and explicit solvent molecules. The TS2 geometry was obtained via a relaxed scan (Fig. S100†) and subsequent restricted optimization of the counterions and explicit THF molecules. TS3 and TS4 geometries were obtained via relaxed scans (Fig. S101 and S102,† respectively). Level of theory: SMD(solvent = THF)/ωB97XD/6-311++G(d,p) from optimized structures at SMD(solvent = THF)/ωB97XD/6-31+G(d,p). | ||
As a consequence, the tricyclic scaffold is opened, leading to formation of a tetracoordinate C–(H)Bpin unit and a tricoordinate DBA-B center (Int3; Fig. 6). Int3 lies about +7 kcal mol−1 above the reactants. The corresponding barrier (TS1) amounts to about +25 kcal mol−1. The activated H− ligand of the C–(H)Bpin moiety in Int3 is subject to intramolecular H− transfer to the tricoordinate DBA-B center. This H− shift restores the π-conjugated BO2 motif in the Bpin residue and an electron octet on the DBA-B atom to afford the stabilized intermediate Int4, which lies only about +1 kcal mol−1 above the reactants. The corresponding barrier (TS2) is about +15 kcal mol−1 higher in energy than the reactants, i.e. considerably lower than the barrier of the first step. Int4 then acts as an H− donor to the imine, generating the amide [Ph(H2)C–NPh]−. The barrier of this step is about +3 kcal mol−1 and the resulting Int5 is −18 kcal mol−1 lower in energy than the reactants.54 [Ph(H2)C–NPh]− forms an adduct with the Bpin residue of Int5, affording Int6 (−13 kcal mol−1). The corresponding barrier lies −1 kcal mol−1 below the reactants and +17 kcal mol−1 above Int5. The latter barrier height would have to be overcome if the reaction energy of the Int4 → Int5 step is instantaneously dissipated into the solvent. If this is not the case, the barrier associated with TS4 would in fact be lower. In any case, the barrier of the Int5 → Int6 step is at least 8 kcal mol−1 lower than the rate-determining TS1 (+25 kcal mol−1). The catalytic cycle is completed by the restitution of the tricyclic framework of [14]2− and the simultaneous release of the hydroboration product Ph(H)2C–N(Bpin)Ph (P′). The barrier of this last step is about +3 kcal mol−1 above the reactants (+16 kcal mol−1 above the previous intermediate) while the exothermicity of the overall reaction is high (−29 kcal mol−1).
As a final remark on key technical details of the calculations, we emphasize that the first step of the mechanism, i.e., nucleophilic attack of the C2 bridge on HBpin, possesses by far the highest barrier (TS1; Fig. 6). It is about +10 kcal mol−1 higher than the second highest barrier (TS2) with respect to the reactants. Concerning the respective preceding intermediate, the difference is even larger (+25 kcal mol−1vs. +8 kcal mol−1) since Int3 lies about +7 kcal mol−1 above the reactants.
Hence, we investigated the influence of the counterions and their first solvation shell on the performance of the catalyst for both steps in more detail. Rather than modeling the effects of the Li+ counter cations and explicit THF solvent molecules, we tested the consequences of a mere inclusion of continuum effects by the SMD model. We found the resulting barrier for the trigonal-bipyramidal transition state (TS1) to increase by +7 kcal mol−1 to +32 kcal mol−1, which is too high considering the experimental conditions. Furthermore, TS2 is also destabilized by +4 kcal mol−1 (Fig. S98†). This effect can be rationalized by considering coordination of the Li+ cation via the O atom of the HBpin molecule. Electron density is thereby withdrawn from the adjacent B center, rendering it more electrophilic and, in turn, facilitating its attack on the electron-rich C2 bridge of Li2[14]. Furthermore, the tetracoordinate B center in the resulting Int3 is also stabilized by a Li+⋯O interaction (Fig. S98†). Since all other barriers are significantly lower than TS1 and TS2, we calculated them – as well as the corresponding intermediates – without explicit counterions and THF molecules to reduce calculation cost.
Conclusions
We have successfully expanded the range of [DBA-Me2]2−-catalyzed reactions to include hydroborations with pinacolborane (HBpin; DBA-Me2 = 9,10-dimethyl-9,10-dihydro-9,10-diboraanthracene). In comparison to analogous hydrogenation reactions, distinct differences become apparent: the general entry step for [DBA-Me2]2−-catalyzed hydrogenations is H–H-bond addition across the two B atoms of the doubly reduced arylborane. Thus, any competing [4 + 2] cycloaddition reaction between the unsaturated substrate (to be hydrogenated) and the [DBA-Me2]2− dianion leads to catalyst poisoning. Although H–Bpin-bond activation with [DBA-Me2]2− is also possible, it only plays a role in the hydroboration of sterically loaded substrates such as Ph(H)CNtBu and proceeds via HBpin degradation to [BH4]− and B2pin3, providing an example of “hidden borohydride catalysis”. In all other cases where [4 + 2] cycloaddition to [DBA-Me2]2− is not hampered by the steric demands of the substrates (e.g., Ph(H)CNPh), the cycloadducts are not dead ends, but rather constitute the active hydroboration catalysts. Using the hydroboration of Ph(H)CNPh with HBpin, mediated by the cycloadduct between [DBA-Me2]2− and H2CCH2, as representative model reaction, we have unveiled the underlying reaction mechanism in a joint experimental and theoretical effort: the 1,2-ethanediyl-bridged tricyclic catalyst can be viewed as intramolecular B–C adduct, in which a Lewis-acidic tricoordinate DBA-B center protects a Lewis-basic [:CH2–CH2–B(Me)Ar2]2− fragment to create a stable resting state. In the presence of HBpin, the tricycle can reversibly open to form an HBpin-alkyl adduct in which the H–Bpin bond is activated for H− transfer to the tricoordinate DBA-B center, from which H− migrates further to the polar Ph(H)CNPh bond. The resulting amide [Ph(H)2C–NPh]− takes up [Bpin]+ from [pinB–CH2–CH2–B(Me)Ar2]− to form Ph(H)2C–N(Bpin)Ph, thereby regenerating the catalyst and completing the catalytic cycle. According to thorough DFT calculations, our mechanistic proposal is energetically feasible under the experimentally applied reaction conditions (room temperature, THF-d8, Li+ counter cations). Quantum-chemical calculations by Kinjo and coworkers have led them to explicitly rule out such a scenario for their related neutral bicyclo[2.2.2]octadienes, assembled from 1,3,2,5-diazadiborinines rather than [DBA-Me2]2− dianions.7 Instead, they postulated H–Bpin-bond activation through electrostatic interactions with positively polarized regions of the still intact tricyclic catalyst. This comparison clearly demonstrates the multifaceted character of doubly B-doped bicyclo[2.2.2]octadienes and shows how seemingly subtle modifications of their molecular scaffolds can significantly alter their catalytic properties. In the future, it will be interesting to explore what else this class of compounds can contribute to the field of main group catalysis.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. E. P. performed all preparative experiments. C. H. performed all calculations. M. B. performed the X-ray crystal structure analyses. F. F., H.-W. L, B. E., and M. W. supervised the project. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Erker and D. W. Stephan, Frustrated Lewis Pairs I & II, Springer, Heidelberg, 2013 Search PubMed.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS.
- S. E. Prey and M. Wagner, Adv. Synth. Catal., 2021, 363, 2290–2309 CrossRef CAS.
- Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
- D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS PubMed.
- D. Wu, R. Ganguly, Y. Li, S. N. Hoo, H. Hirao and R. Kinjo, Chem. Sci., 2015, 6, 7150–7155 RSC.
- D. Wu, R. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Chem, 2017, 3, 134–151 CAS.
- B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS PubMed.
- B. Wang and R. Kinjo, Tetrahedron, 2018, 74, 7273–7276 CrossRef CAS.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 5762–5765 CrossRef CAS.
- E. von Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef CAS PubMed.
- E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2018, 57, 16491–16495 CrossRef CAS PubMed.
- E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2019, 141, 6082–6091 CrossRef CAS PubMed.
- E. von Grotthuss, F. Nawa, M. Bolte, H.-W. Lerner and M. Wagner, Tetrahedron, 2019, 75, 26–30 CrossRef CAS.
- Harman et al. have reported a number of remarkable (catalytic) transformations mediated by Au complexes of bidentate diphosphine ligands with DBA backbones: (a) J. W. Taylor, A. McSkimming, M.-E. Moret and W. H. Harman, Angew. Chem., Int. Ed., 2017, 56, 10413–10417 CrossRef CAS PubMed; (b) J. W. Taylor, A. McSkimming, L. A. Essex and W. H. Harman, Chem. Sci., 2019, 10, 9084–9090 RSC; (c) J. W. Taylor and W. H. Harman, Chem. Commun., 2020, 56, 4480–4483 RSC; (d) J. W. Taylor and W. H. Harman, Chem. Commun., 2020, 56, 13804–13807 RSC.
- (a) M. A. Dureen, A. Lough, T. M. Gilbert and D. W. Stephan, Chem. Commun., 2008, 4303–4305 RSC; (b) P. Eisenberger, A. M. Bailey and C. M. Crudden, J. Am. Chem. Soc., 2012, 134, 17384–17387 CrossRef CAS PubMed; (c) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed; (d) F.-G. Fontaine, M.-A. Courtemanche, M.-A. Légaré and É. Rochette, Coord. Chem. Rev., 2017, 334, 124–135 CrossRef CAS; (e) P. Vasko, I. A. Zulkifly, M. Á. Fuentes, Z. Mo, J. Hicks, P. C. J. Kamer and S. Aldridge, Chem.–Eur. J., 2018, 24, 10531–10540 CrossRef CAS PubMed; (f) A. Falconnet, M. Magre, B. Maity, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 17567–17571 CrossRef CAS PubMed; (g) Y. Lebedev, I. Polishchuk, B. Maity, M. Dinis Veloso Guerreiro, L. Cavallo and M. Rueping, J. Am. Chem. Soc., 2019, 141, 19415–19423 CrossRef CAS PubMed; (h) D. W. Stephan, Chem, 2020, 6, 1520–1526 CrossRef CAS.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS.
- K. Wade, Electron Deficient Compounds, Springer, New York, 1971 Search PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- See the ESI† for further details.
- The free Gibbs energy for the reaction [Li(thf)2]2[4] + THF → [Li(thf)2][5] + [Li(thf)3]H amounts to +27.4 kcal mol−1 at room temperature.
- W. Clegg, A. J. Scott, C. Dai, G. Lesley, T. B. Marder, N. C. Norman and L. J. Farrugia, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 2545–2547 CrossRef.
- The 11B NMR signal of the Bpin group in Na2[4] (42.6 ppm; THF-d8) may seem unusually downfield shifted, as the resonance of B2pin2 appears at 32.6 ppm (own measurement in THF-d8). However, the signals of Bpin groups attached to anionic, tetracoordinate B centers appear in the same range as that of Na2[4] (cf. K[B2pin2·OtBu] δ = 41.1; THF-d8): S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7099 CrossRef CAS PubMed . The calculated 11B NMR shifts of [Na(thf)2]2[4] are: δ = 45.8 (Bpin), −18.9 (BH), −23.3 (BBpin) (GIAO method at the SMD(solvent = THF)/ωB97XD/6-311++G(d,p) level)..
- Further B–Bpin bond lengths (Å) useful for comparison are (a) B2pin2 = 1.7041(15), (b) [K(18-c-6)][B2pin2OMe] = 1.753(2); [Me4N][B2pin2F]·THF = 1.736(8), and (c) [Mg(HC-{(Me)CN(2,6-iPr2C6H3)}2)][pinB–BPh3] = 1.718(7): (a) C. Kleeberg, A. G. Crawford, A. S. Batsanov, P. Hodgkinson, D. C. Apperley, M. S. Cheung, Z. Lin and T. B. Marder, J. Org. Chem., 2012, 77, 785–789 CrossRef CAS PubMed; (b) ref. 23; (c) A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef PubMed.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- C. Kleeberg and C. Borner, Eur. J. Inorg. Chem., 2013, 2013, 2799–2806 CrossRef CAS.
- Li[SiPh3] was chosen as the nucleophile, because its high steric demand disfavors the formation of a stable adduct with the tricoordinate DBA-B atom in Li[5]. Moreover, silylboranes such as Ph3Si–Bpin have considerable application potential: (a) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29–49 CrossRef CAS; (b) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS PubMed; (c) S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16–24 CrossRef; (d) C. Moberg, Synthesis, 2020, 52, 3129–3139 CrossRef CAS; (e) K. Hirano and M. Uchiyama, Adv. Synth. Catal., 2021, 363, 2340–2353 CrossRef CAS; (f) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- G. Becker, H.-M. Hartmann, A. Münch and H. Riffel, Z. Anorg. Allg. Chemie, 1985, 530, 29–42 CrossRef CAS.
- Quantitative deprotonation of Li[6] is also possible using Ph(H)2C–N(Li)tBu as the base. Given that the amide can be formed through H− transfer to Ph(H)CNtBu, this deprotonation experiment also marks the hitherto missing keystone of the reaction mechanism previously proposed for the catalytic hydrogenation of the imine by Li2[2]/H2: In contrast to conventional FLPs, the H2-activation product Li2[11] (Scheme 3) delivers first an H− and second an H+ ion (cf. ref. 13).
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, J. Am. Chem. Soc., 2010, 132, 3440–3451 CrossRef CAS PubMed.
- For the following reasons, Li2[2] was chosen despite its somewhat different behavior toward HBpin compared to the other M2[1]/M2[2] compounds: (i) the neutral Me-substituted DBA 2 is more convenient to handle than its H-substituted congener 1. (ii) We have already published the Li+ salts of some cycloadducts between [DBA-Me2]2− and unsaturated substrates, which are the actual catalytically active species and thus the focus of our mechanistic investigations (cf. ref. 12 and 13). (iii) Since our DFT calculations include the counter cations, the computational cost for the treatment of Li+ is lower than for the treatment of Na+/K+.
- We initially relied on this protocol because it is most similar to the one applied for our earlier hydrogenation reactions (cf. ref. 13), which we want to use for comparison. Later, it was found that adding the unsaturated substrate first is essential to exclude contributions of the hidden borohydride catalysis as an undesired background reaction.
- X. Wang and X. Xu, RSC Adv., 2021, 11, 1128–1133 RSC.
- A. R. Gatti and T. Wartik, Inorg. Chem., 1966, 5, 329–330 CrossRef CAS.
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed.
- Sen et al. identified a comparable Li+⋯O coordination as the key interaction in lithium phenolate-catalyzed HBpin additions to carbonyl compounds. The resulting decrease in O=B π donation leads to an increase in the Lewis acidity of the HBpin molecule and in turn facilitates its interaction with the carbonyl-O atom. We adopted such a scenario to Li2[14] and computed a corresponding reaction barrier of +31 kcal mol−1. Although this value is lower than that of the uncatalyzed reaction by –14 kcal mol−1, it is still higher than TS1 by +6 kcal mol−1. Since TS1 represents the highest barrier in our hydroboration Mechanism II (see the ESI† for details), also Sen’s mechanism can be ruled out for our case: M. K. Bisai, T. Das, K. Vanka and S. S. Sen, Chem. Commun., 2018, 54, 6843–6846 RSC.
- Hydroboration of Ph(H)CNPh with HBpin in THF-d8 required about the same time, regardless of whether Li2[14] or Na2[14] was used as the catalyst (NMR-spectroscopic control). We take this as justification that Li2[14] is a representative example catalyst to study Mechanism II. It should also be noted that the necessary explicit treatment of the counter cation’s coordination sphere is much more complex in the case of the larger Na+. Its coordination sphere includes six neighbors while the one of Li+ only contains four. Consequently, not only does the size of the system increase, but also a greater variety of coordination polyhedra can be expected.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem.–Eur. J., 2016, 22, 7158–7162 CrossRef CAS PubMed.
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155–13158 RSC.
- M. D. Anker, M. Arrowsmith, R. L. Arrowsmith, M. S. Hill and M. F. Mahon, Inorg. Chem., 2017, 56, 5976–5983 CrossRef CAS PubMed.
- A. Ramos, A. Antiñolo, F. Carrillo-Hermosilla, R. Fernández-Galán and D. García-Vivó, Chem. Commun., 2019, 55, 3073–3076 RSC.
- N. Sarkar, S. Bera and S. Nembenna, J. Org. Chem., 2020, 85, 4999–5009 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. von Ragué Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. Ben-Naim, J. Phys. Chem., 1978, 82, 792–803 CrossRef CAS.
- J. R. Pliego Jr and J. M. Riveros, Phys. Chem. Chem. Phys., 2002, 4, 1622–1627 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Peterson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Wallingfort, CT, USA, 2016 Search PubMed.
- I. P. Query, P. A. Squier, E. M. Larson, N. A. Isley and T. B. Clark, J. Org. Chem., 2011, 76, 6452–6456 CrossRef CAS PubMed.
- Another possible source of Bpin residues is Li[5]. Attack of Int1 at the Bpin substituent of Li[5] would lead to the immediate formation of P and Li2[2] via [Bpin]+ transfer and with release of BH3·thf (cf. the analogous reaction Li[SiPh3] + Li[5] → pinB–SiPh3 + Li2[2]; Scheme 2). According to our calculations this alternative scenario would lead to a decrease in the driving force of the overall reaction from −14 kcal mol−1 to −8 kcal mol−1. On the other hand, formation of Int2 (+15 kcal mol−1) would be bypassed. However, an interaction between the amide anion of Int1 and negatively charged [5]− is electrostatically unfavorable and the reactive B(pin) center of [5]− is sterically less accessible than that of HBpin. We therefore assume that the thermodynamically less favorable formation of Int2 (+15 kcal mol−1) is preferred by kinetics. Since the formation of Li[5] and Li[H2Bpin], which is the energetically most costly step of Cycle I, is not circumvented, a switch to Cycle II will take place in any case.
- An analogous attack of a B-bonded Me residue on HBpin or a B–H/B–C σ-bond metathesis leading directly to Int4 (Fig. 5) can be ruled out due to significantly higher reaction barriers (Fig. S98,†TSa and TSb).
- H− transfer to the alternative electrophile HBpin would be thermodynamically less favorable by +13 kcal mol−1. In general, HBpin appears to be the weakest H− acceptor of the investigated species (see Table S8†).
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, X-ray crystallographic data and computational details. CCDC 2207512–2207520. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05518j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |