
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Next-generation membrane-active glycopeptide antibiotics that also inhibit bacterial cell division†
Paramita
Sarkar
a,
Kathakali
De
a,
Malvika
Modi
b,
Geetika
Dhanda
a,
Richa
Priyadarshini
 b,
Julia E.
Bandow
c and
Jayanta
Haldar
*a
b,
Julia E.
Bandow
c and
Jayanta
Haldar
*a
aAntimicrobial Research Laboratory, New Chemistry Unit and School of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bengaluru, 560064, Karnataka, India. E-mail: jayanta@jncasr.ac.in; Tel: +91 802208 2565
bDepartment of Life Sciences, School of Natural Sciences, Shiv Nadar University, Dadri 201314, UP, India
cApplied Microbiology, Faculty of Biology and Biotechnology, Ruhr University Bochum, Universitätsstraße 150, 44780 Bochum, Germany
First published on 6th January 2023
Abstract
Resistance to vancomycin, a life-saving drug against Gram-positive bacterial infections necessitates developing alternative therapeutics. Herein, we report vancomycin derivatives that assimilate mechanisms beyond D-Ala–D-Ala binding. The role of hydrophobicity towards the structure and function of the membrane-active vancomycin showed that alkyl-cationic substitutions favored broad-spectrum activity. The lead molecule, VanQAmC10 delocalized the cell division protein MinD in Bacillus subtilis, implying an impact on bacterial cell division. Further examination of wild-type, GFP-FtsZ, or GFP-FtsI producing- and ΔamiAC mutants of Escherichia coli revealed filamentous phenotypes and delocalization of the FtsI protein. The findings indicate that VanQAmC10 also inhibits bacterial cell division, a property previously unknown for glycopeptide antibiotics. The conjunction of multiple mechanisms contributes to its superior efficacy against metabolically active and inactive bacteria, wherein vancomycin is ineffective. Additionally, VanQAmC10 exhibits high efficacy against methicillin-resistant Staphylococcus aureus (MRSA) and Acinetobacter baumannii in mouse models of infection.
Introduction
Vancomycin is a critically important antibiotic, that was the drug of last resort against infections caused by multidrug-resistant Gram-positive bacteria.1 It belongs to the glycopeptide class of antibiotics that consists of naturally occurring and semi-synthetic products that inhibit bacterial cell wall biosynthesis by binding to the D-Ala–D-Ala terminus of the cell wall precursor pentapeptide.1,2 However, widespread resistance to vancomycin that involves modification of the target peptide to D-Ala–D-Lac/D-Ser, concomitant with a reduction in binding affinity, and/or thickening of the cell wall has been reported.3 To incorporate additional mechanisms, second-generation glycopeptide antibiotics, telavancin, dalbavancin, and oritavancin, were equipped with lipophilic substitutions that abetted destabilization of the bacterial membranes.4 Despite reports of various modified glycopeptides that overcome inherited resistance,3,5–10 there are only a few reports of derivatives that are effective against Gram-negative bacteria and dormant bacteria.11–15 The presence of an outer membrane in Gram-negative bacteria (GNB) precludes entry of numerous antibiotics including glycopeptides.16 Although the conjugation of membrane-interacting moieties onto glycopeptide antibiotics has shown activity against vancomycin-resistant Gram-positive bacteria, they do not necessarily result in activity against Gram-negative bacteria.17We had developed C-terminally modified cationic lipophilic vancomycin derivatives that were effective against both vancomycin-resistant Gram-positive and the intrinsically resistant Gram-negative bacteria.18–20 Their broad-spectrum antibacterial activity was attributed to the incorporation of membrane–perturbation properties through the conjugation of lipophilic cationic moieties. In this report, we first examine the structure–activity relationship of alkyl and aryl moieties as hydrophobic substituents and identify the most selective compound. The effect of various hydrophobic group substitutions on membrane perturbing ability, the antibacterial activity of exponential growth, and quiescent bacteria was therefore examined to gain insights for better designs. Of most significance, here, a study of the effect of the lead compound on the membrane, cell division, and associated proteins was performed to better understand the modes of action of this new semisynthetic glycopeptide antibiotic. The protein localization and morphological changes were evaluated upon treatment of B. subtilis producing GFP-MinD protein, E. coli producing various GFP-tagged cell division proteins and an amidase lacking mutant. Further, the potential of the lead compound as a preclinical candidate has been demonstrated in the murine thigh infection model against MRSA and chronic burn-wound infection against A. baumannii.
Results
Structure–activity relationship study
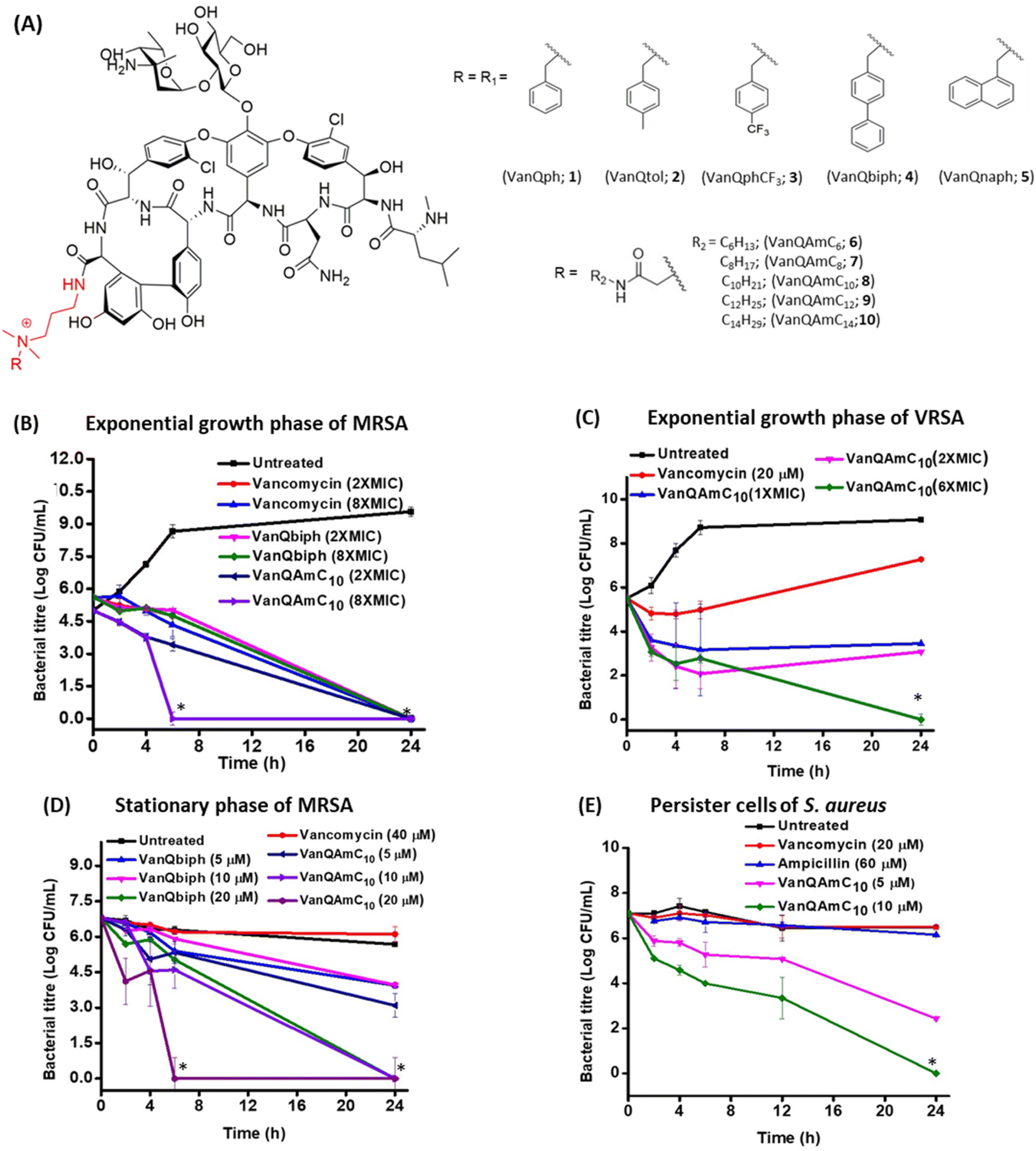 | ||
| Fig. 1 (A) Structure of cationic lipophilic vancomycin derivatives. Bactericidal kinetics of (B) VanQbiph and VanQAmC10 against exponential growth phase MRSA, (C) VanQAmC10 against VRSA, (D) VanQbiph and VanQAmC10 against stationary phase cells of MRSA, and (E) VanQAmC10 against persister cells of S. aureus. Against MRSA, MIC of VanQAmC10 = 0.4 μM, MIC of VanQbiph = 0.5 μM; MIC of VanQAmC10 against VRSA = 1.6 μM and ‘*’ indicates <50 CFU mL−1. | ||
| Compound | Minimum inhibitory concentration (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gram-positive bacteria | Gram-negative bacteria | |||||||||
| MRSA | VRSA 1 | VRSA 4 | VRSA 12 | VRE 51575 | VRE 51559 | AB 1425 | PA R590 | AB R674 | HC50 (μM) | |
| a MRSA, methicillin-resistant S. aureus (ATCC 33591); VRSA, vancomycin-resistant S. aureus; VRE vancomycin-resistant E. faecium (VanA phenotype, ATCC 51559); vancomycin-resistant E. faecalis (VanB phenotype, ATCC 51575); ND, not determined. MIC against VRE and VRSA, HC50 of VanQAmC10 was previously reported.18 | ||||||||||
| Vancomycin | 0.6 | 345 | 345 | 345 | 750 | 250 | 100 | 100 | 100 | N.D |
| VanQph (1) | 1 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | 25.5 | >500 |
| VanQtol (2) | 1 | >30 | 16 | >30 | >30 | >30 | >30 | >30 | 25.3 | >500 |
| VanQphCF3 (3) | 1 | >30 | 15.8 | >30 | >30 | >30 | >30 | 24.6 | 24.6 | >500 |
| VanQbiph (4) | 0.5 | 7.8 | 3.9 | 15.7 | 12.3 | 6.1 | 15.7 | >30 | 24.5 | >500 |
| VanQnaph (5) | 1 | >30 | 3.9 | >30 | 25 | 25 | >30 | >30 | 24.8 | >500 |
| VanQAmC6 (6) | 0.8 | >30 | 6.3 | >30 | >30 | 25 | 25 | 12.5 | 12.5 | >500 |
| VanQAmC8 (7) | 0.8 | 3.1 | 3.1 | >30 | 25 | 12.5 | 12.5 | 6.3 | 12.5 | >500 |
| VanQAmC10 (8) | 0.4 | 3.1 | 1.6–3.1 | 1.6 | 4 | 4 | 6.3 | 3.1 | 10 | >500 |
| VanQAmC12 (9) | 3.12 | 1.5 | 0.7 | 3 | 3.1 | 1.5 | 6.3 | 6.3 | 6.3 | 350 |
| VanQAmC14 (10) | 6.25 | 3 | 1.5 | 3 | 3.1 | 1.5 | 6.3 | 6.3 | 12.5 | 150 |
Among the amido-alkyl substituted compounds, 6–8 exhibited MICs similar to that of vancomycin against MRSA while the longer chain variants, 9 and 10 showed reduced activity. Against VRSA, the lower chain length variants, 6 and 7 were less effective; VanQAmC10 (8) showed a significant 115–490-fold improvement in activity as compared to vancomycin. 9 and 10 had similar MICs and were slightly more effective than 8. Against VRE, VanQAmC10 (8) showed activity similar to that against VRSA (∼160-fold improvement in activity as compared to vancomycin). In general, the series of amido-alkyl derivatives, 6–10 exhibited an increase in activity with longer chains, with no further enhancement in activity between longer chain substituted derivatives 9 and 10. The amido-alkyl chain containing compounds exhibited better activity than those with aromatic substitutions. This indicated that the more flexible hydrophobic alkyl chain moieties resulted in better activity. VanQbiph (4), VanQAmC10 (8), and VanQAmC12 (9) demonstrate potency against vancomycin-resistant Gram-positive bacteria and were potential lead candidates. VanQAmC10 shows activity similar to telavancin and is better than dalbavancin. The MIC of semi-synthetic glycopeptides such as oritavancin, telavancin and dalbavancin against VRE (VanA phenotype) were reported as 0.14 μM, 4 μM and 18 μM respectively.4,20,41
Eradication of exponentially growing bacteria
To examine the antibacterial properties of the two leads, VanQbiph (4) and VanQAmC10 (8), the kinetics of bactericidal activity against exponentially growing, log-phase cells of MRSA was evaluated (Fig. 1B). The minimum bactericidal concentration (MBC) of VanQbiph (4) and VanQAmC10 (8) was at 2×MIC. VanQAmC10 (8) showed faster killing than both VanQbiph (4) and vancomycin. At 2×MIC, both the compounds exhibited complete eradication in 24 h like vancomycin. At 8×MIC, VanQAmC10 (8) showed complete eradication within 6 h while VanQbiph (4) showed a similar effect at 24 h. VanQAmC10 (8) showed a time and concentration-dependent activity while VanQbiph (4) and vancomycin showed only a time-dependent activity.Against VRSA, VanQAmC10 (8) exhibited better efficacy than VanQbiph (4) and was therefore tested for kinetics of killing. At MIC, it reduced the bacterial titer by 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log CFU mL−1 in 24 h (Fig. 1C). At 2×MIC, an initial reduction was observed, followed by a 1log CFU mL−1 increase in bacterial titer between 6 h and 24 h. At 6×MIC VanQAmC10 resulted in complete eradication after 24 h. Treatment with vancomycin at 20 μM (sub-MIC), showed an initial growth inhibitory effect, followed by resumption of bacterial growth. Resistance in VRSA results from a combination of target modification as well as thickening of the cell wall. The additional mechanisms of VanQAmC10 possibly contribute to its bactericidal activity against VRSA.
log CFU mL−1 in 24 h (Fig. 1C). At 2×MIC, an initial reduction was observed, followed by a 1log CFU mL−1 increase in bacterial titer between 6 h and 24 h. At 6×MIC VanQAmC10 resulted in complete eradication after 24 h. Treatment with vancomycin at 20 μM (sub-MIC), showed an initial growth inhibitory effect, followed by resumption of bacterial growth. Resistance in VRSA results from a combination of target modification as well as thickening of the cell wall. The additional mechanisms of VanQAmC10 possibly contribute to its bactericidal activity against VRSA.
Eradication of metabolically inactive bacteria
The stationary phase cells and persister cells are metabolically repressed and phenotypically different from the log-phase cells.27 For activity, the commonly used drugs, vancomycin, and linezolid require actively ongoing cellular processes. They are therefore ineffective against non-dividing bacteria. Even at the concentration of 40 μM, vancomycin did not reduce the number of viable stationary phase cells of MRSA in 24 h (Fig. 1D). The integrity of the bacterial membrane is imperative to survival irrespective of metabolic state. VanQbiph (4) and VanQAmC10 (8) were designed to incorporate membrane activity and therefore expected to be effective against the metabolically inactive cells of MRSA. VanQAmC10 (8) showed better activity than VanQbiph (4) at the same concentration, similar to that observed against log-phase cells of MRSA. Irrespective of treatment concentration, VanQbiph (4), exhibited slow killing with no significant reduction in viability up to 6 h. Complete eradication was observed at a higher concentration of 20 μM in 24 h. VanQAmC10 (8) showed rapid bactericidal activity that increased with concentration. It completely eradicated cells in 24 h at 10 μM and within 6 h at 20 μM.Persister cells are a subpopulation of bacterial cultures that are refractory to antibiotic treatment.28 Since VanQAmC10 (8) was more effective than VanQbiph (4) against stationary phase cultures, its activity against persister cells was evaluated. VanQAmC10 (8) exhibited a concentration-dependent bactericidal activity against the persister cells of MRSA (Fig. 1D). This activity was more rapid than that observed against stationary phase cells.
Another growing challenge in treating bacterial infections is the formation of biofilms, which are recalcitrant to antibiotic treatment.29 Most antibiotics including vancomycin are rendered ineffective against biofilms consisting of both dividing and non-dividing cells, majorly due to their inability to penetrate through the extracellular polymeric matrix. Thus, it is important to develop compounds that eradicate biofilms as well as planktonic cells. Untreated biofilms of MRSA grew to a thickness of 11.8 μm (Fig. S3A†). The vancomycin-treated biofilms remained intact with thickness similar to the untreated control. VanQAmC10 (2) was found to reduce the thickness of biofilms of MRSA by about 40%, showing a thickness of 7.4 μm as opposed to 11.8 and 10.1 μm for untreated and vancomycin treated cases respectively (Fig. S3B and C†).
Mechanism of action against Gram-positive bacteria
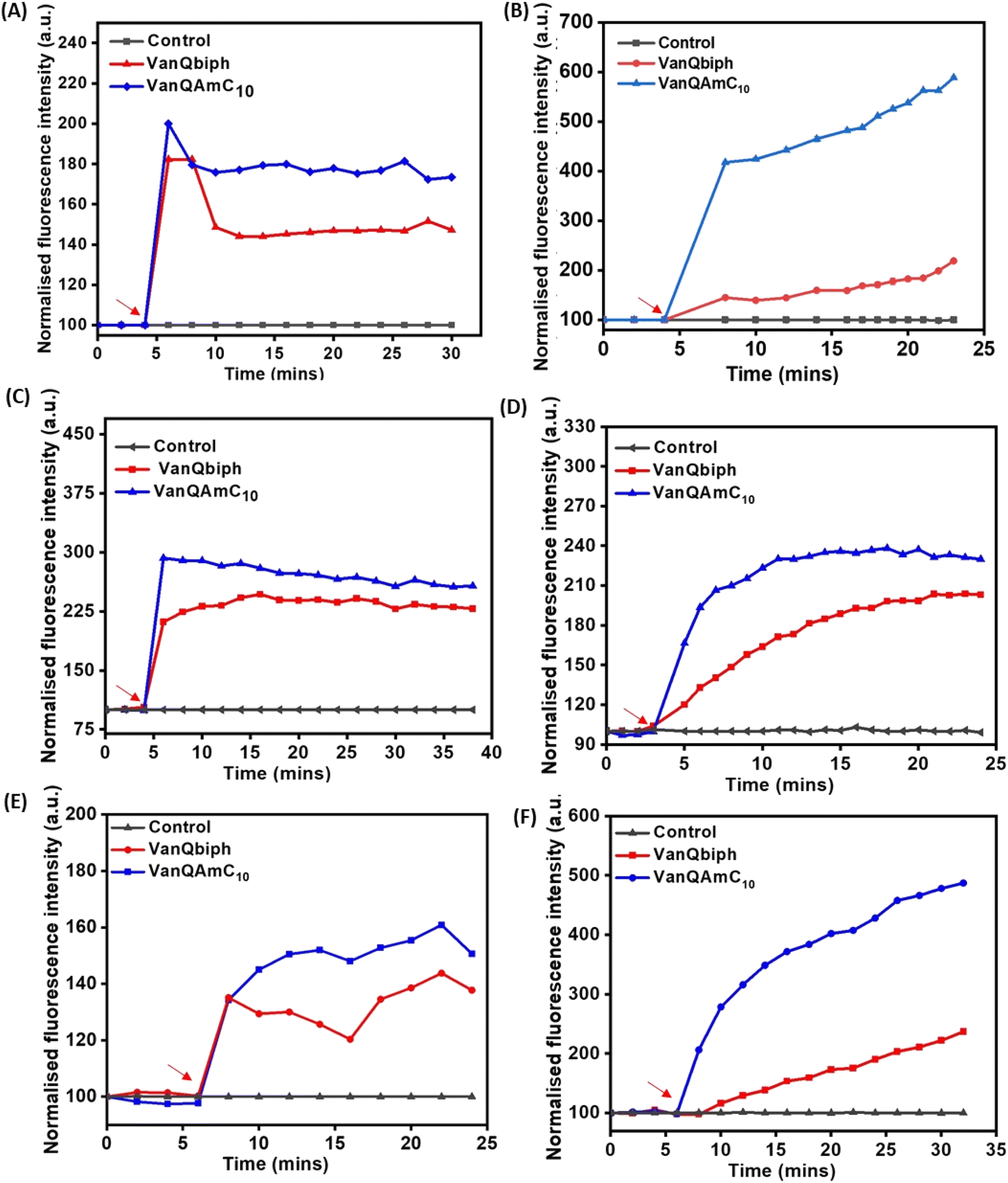 | ||
| Fig. 2 Membrane–perturbation properties of VanQbiph and VanQAmC10. Membrane depolarization against, (A) exponentially growing MRSA, (B) exponentially growing B. subtilis, upon treatment at 10 μM, and (C) stationary phase cells of MRSA upon treatment at 20 μM. Membrane permeabilization against, (D) exponentially growing MRSA, (E) exponentially growing B. subtilis, upon treatment at 10 μM, and (F) stationary phase cells of MRSA upon treatment at 20 μM. Red arrows indicate compound addition. The error percentage between replicates of an experiment was lesser than 5%. The images are representative of results from three independent experiments. | ||
To confirm that membrane perturbation contributed to cell death, the bacterial viability was evaluated under conditions similar to the mechanistic studies. A decrease in the number of viable cells was observed with an increase in the concentration of both VanQbiph (4) and VanQAmC10 (8) (Fig. 3A). The number of viable cells was lower upon treatment with VanQAmC10 (8) than when treated with VanQbiph (4) at the same concentration. At 5 μM and 10 μM, VanQbiph (4) showed 0.8log CFU mL−1 and 2.2log CFU mL−1 reduction respectively. VanQAmC10 (8) showed 2log CFU mL−1 and 4log CFU mL−1 reduction upon treatment at 5 μM and 10 μM respectively. Treatment with both the compounds at 20 μM resulted in complete eradication of the bacteria. Vancomycin did not reduce the viability of bacteria even at 40 μM, under the same conditions. The stronger membrane–perturbation properties of VanQAmC10 (8) therefore contribute to higher activity against both exponentially growing and stationary phase bacteria.
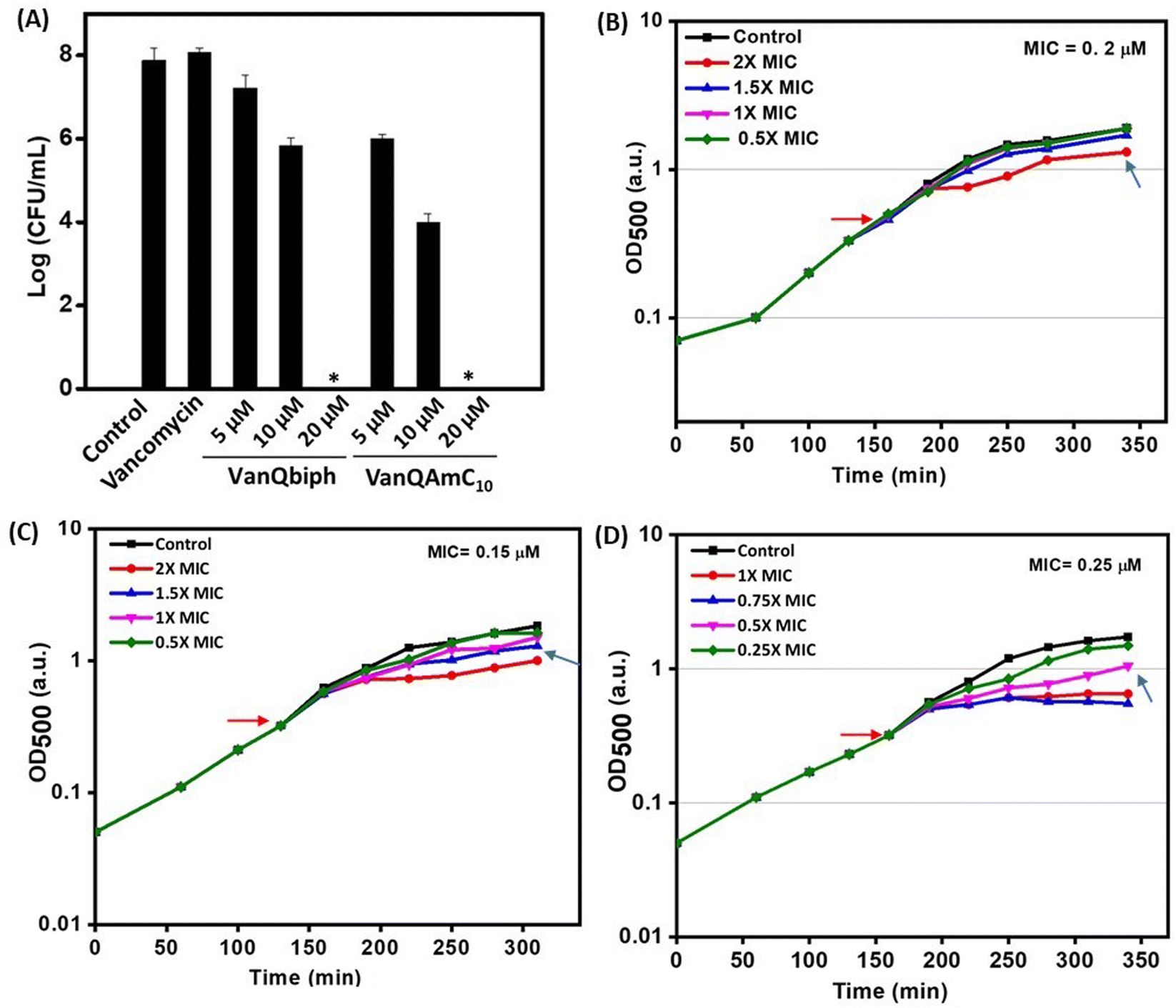 | ||
| Fig. 3 (A) Viability of planktonic cells post-treatment in HEPES-glucose; acute growth retardation upon treatment with (B) vancomycin, (C) VanQbiph, (D) VanQAmC10. ‘*’ indicates <50 CFU mL−1. Red arrow indicates compound addition and blue arrow indicates PEC. | ||
:3 mixture of acetic acid and methanol.30 This phenomenon is exhibited by compounds that inhibit cell wall biosyntheses like vancomycin and not agents that only perturb the membrane integrity such as gramicidin-A and valinomycin.31 Like vancomycin, VanQbiph (4) and VanQAmC10 (8) showed bubbles on the surface, indicating that they inhibit cell wall biosynthesis and compromise the integrity of the cell wall (Fig. 4A).
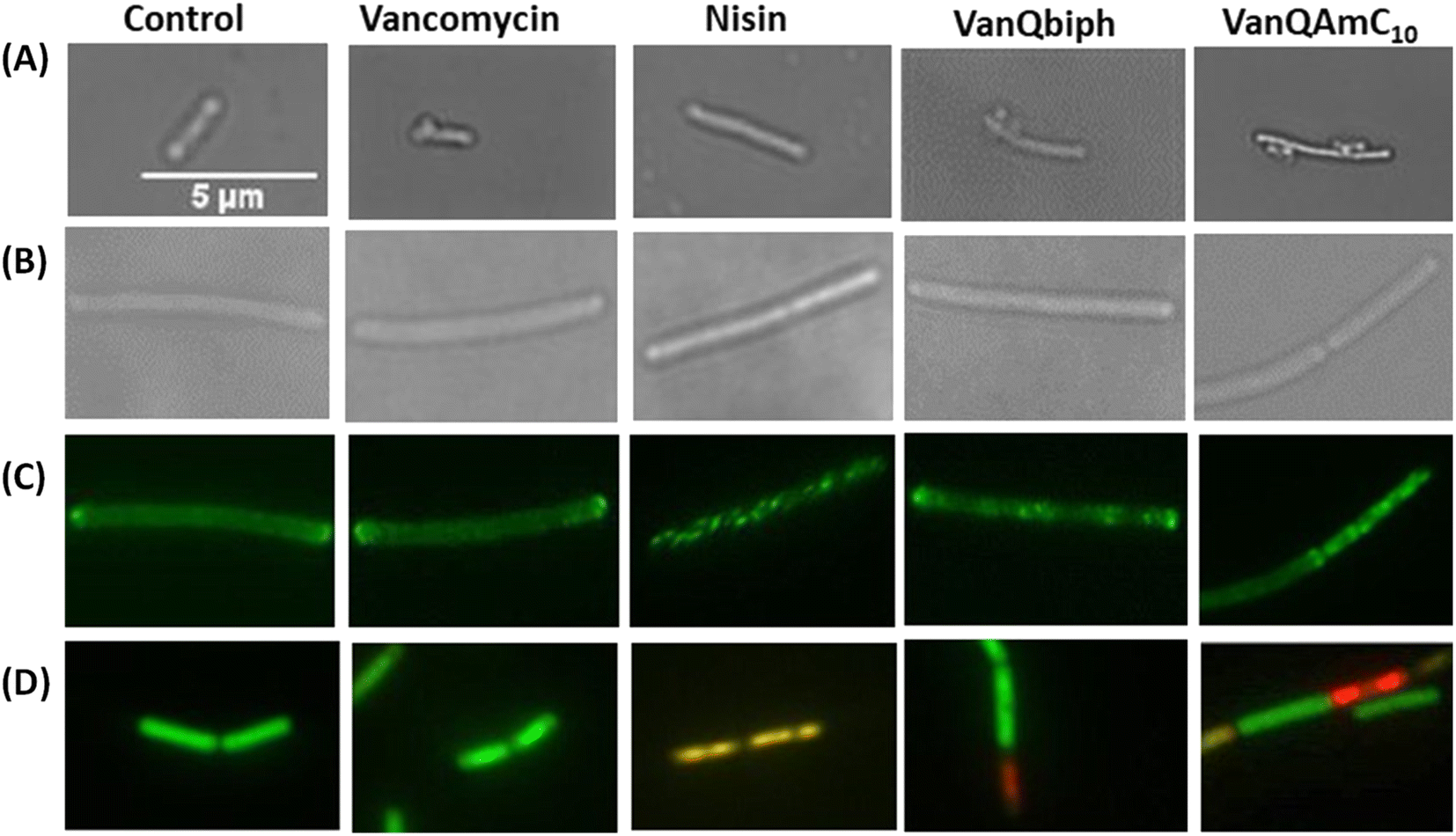 | ||
| Fig. 4 Examination of the effect of VanQbiph, VanQAmC10, vancomycin and nisin on cell membrane and wall integrity of B. subtilis upon treatment at respective PECs. (A) Inhibition of cell wall biosynthesis and compromise of cell wall integrity by visualization of ‘bubbles’ on the bacterial surface post acetic acid/methanol fixation through light microscopy; (B) light microscopy to assess the morphology of GFP-MinD expressing B. subtilis cells; (C) delocalization of GFP-MinD through fluorescence microscopy; (D) membrane permeabilization and pore formation monitored through fluorescence with the BacLight bacterial viability kit. Scale of all images is the same (5 μm). | ||
Mechanism of action in Gram-negative bacteria (E. coli)
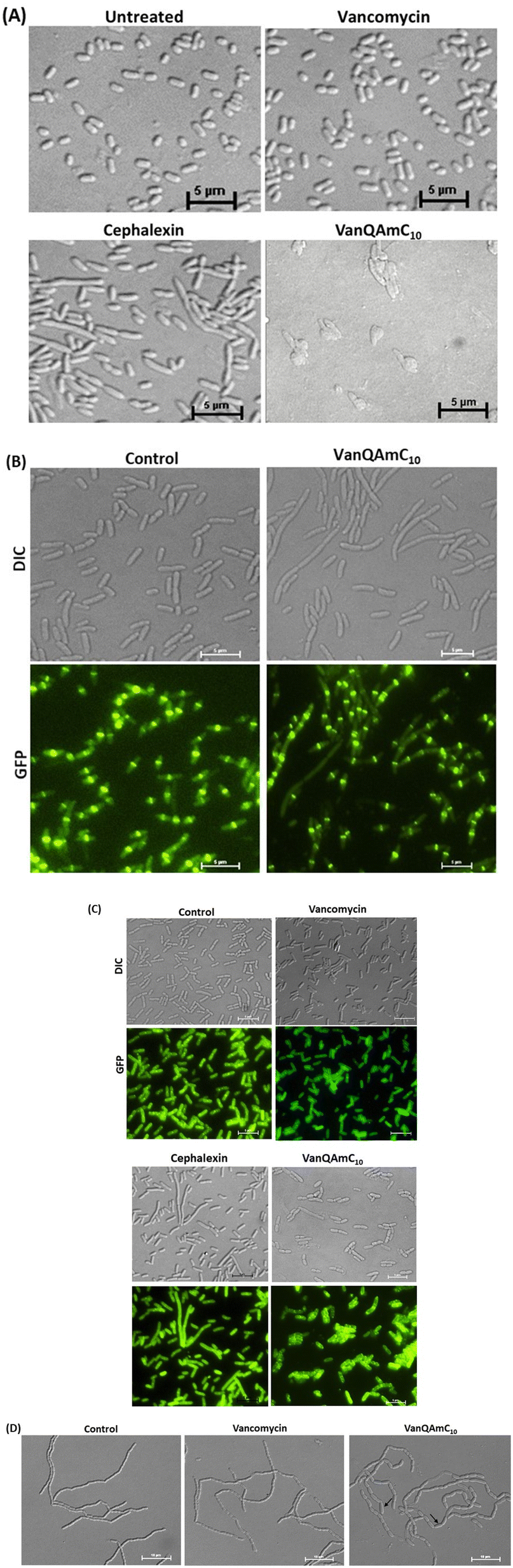 | ||
| Fig. 5 Examination of E. coli cells treated with vancomycin, cephalexin and VanQAmC10 to study the effect on bacterial membrane integrity and cell division. (A) Light microscopy of wild-type E. coli upon treatment with vancomycin (34 μM), and VanQAmC10 (25 μM); (B) light and fluorescence microscopy in GFP-FtsZ expressing E. coli to assess morphological changes and localization of FtsZ post-treatment with VanQAmC10 (15 μM) for 40 min; (C) delocalization of GFP-FtsI in GFP-FtsI expressing E. coli through fluorescence microscopy and morphological changes through light microscopy upon treatment with vancomycin (34 μM), cephalexin (22 μM) and VanQAmC10 (10 μM); (D) light microscopy to assess morphological changes in E. coli MG1655 ΔamiAC mutant, when untreated and treated with vancomycin (3 μM) and VanQAmC10 (2 μM). Black arrows indicate morphological aberrations. | ||
In vivo activity of VanQAmC10
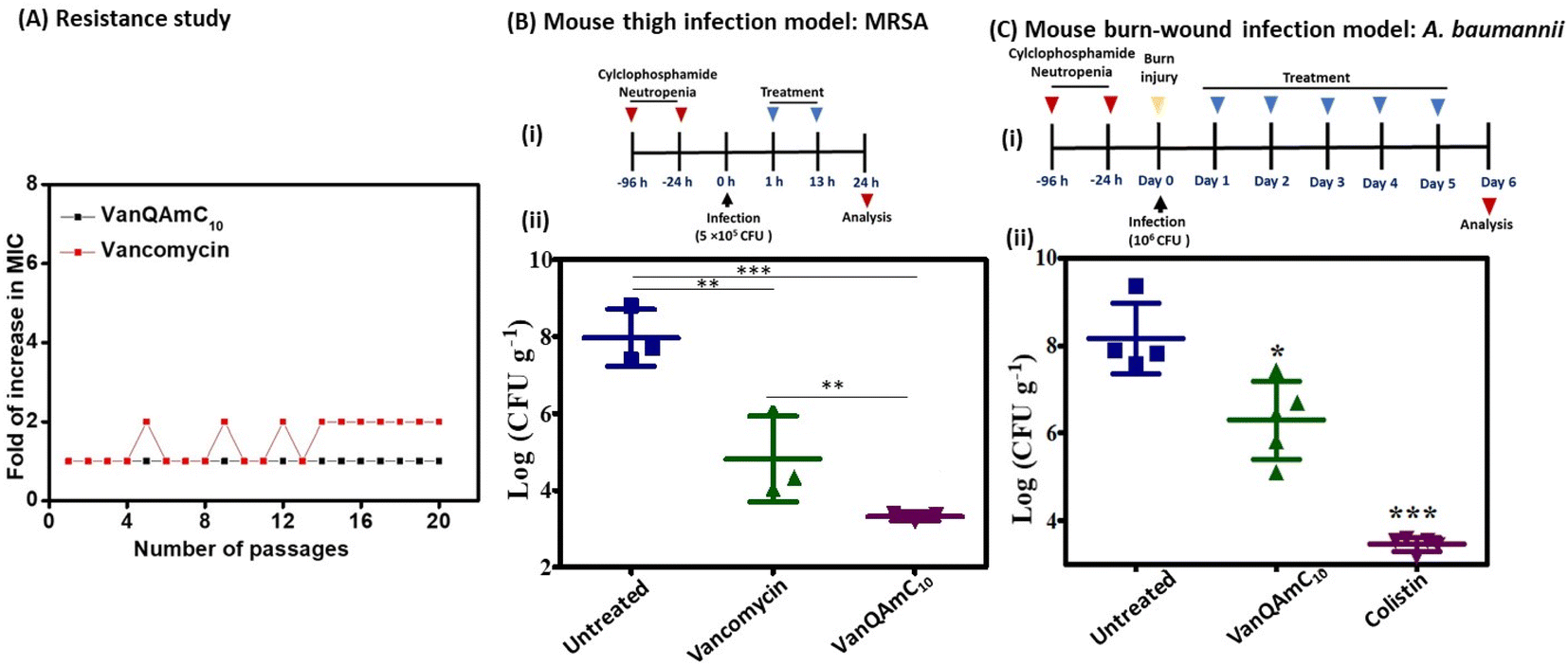 | ||
| Fig. 6 (A) Propensity of vancomycin and VanQAmC10 to induce resistance in MRSA upon serial exposure to sub-MIC concentrations; (B) (i) experimental design for in vivo efficacy study in a murine thigh infection model, (ii) in vivo efficacy of VanQAmC10 and vancomycin against the multidrug-resistant methicillin-resistant S. aureus (MRSA) (n = 3/dose). Antibiotics were administered intraperitoneally twice at 12 h intervals at a dose of 12 mg kg−1. (C) (i) Experimental design for in vivo efficacy study in murine burn wound infection model, (ii) in vivo efficacy of VanQAmC10 and colistin against the carbapenem-resistant A. baumannii (n = 5/dose, 1 mouse died in untreated control). VanQAmC10 and colistin were treated topically at 30 mg kg−1 for 5 days (‘***’ indicates p < 0.0001, ‘**’ indicates p < 0.001, ‘*’ p < 0.05, p = 0.08 (n.s.) for VanQAmC10 with respect to vancomycin). | ||
log CFU mL−1. In the untreated group, the bacterial load increased to 8log CFU g−1. The vancomycin treated group showed a bacterial load of 4.8log CFU g−1 of tissue. However, treatment with VanQAmC10 reduced the bacterial load to 3.3log CFU g−1, which is a 1.5log CFU g−1 lower bacterial load than observed against vancomycin. The results highlight the superior in vivo efficacy of VanQAmC10 as compared to that of the parent drug vancomycin.
Further, the efficacy of VanQAmC10 (8) was tested in a chronic burn-wound infection model of A. baumannii.40 Chronic burn wounds on the back of mice were infected with A. baumannii. 24 h post-infection, before initiation of treatment, the bacterial load was found to be 7.5log CFU g−1. Infected mice were treated with 30 mg kg−1 of VanQAmC10 and colistin for five days consecutively. The bacterial load in the untreated group increased to 8log CFU g−1 six days post-infection. Treatment with VanQAmC10 resulted in a 2.2log CFU g−1 lower bacterial load as compared to the untreated mice, while colistin showed a 4.8log CFU g−1 load lower bacterial load as compared to the untreated mice at the same dose (Fig. 6C). The use of colistin is however limited due to its toxicity and bacteria easily develop resistance to it. Therefore, VanQAmC10 presents a promising alternative for both Gram-positive and Gram-negative bacteria.
Discussion
Vancomycin served as a life-saving drug against MDR Gram-positive bacterial infections for over thirty years until the report of resistance. This makes it an attractive drug for further development against the escalating incidences of resistant bacteria. The more difficult to treat Gram-negative bacteria are inherently resistant to vancomycin. We have shown that the conjugation of cationic lipophilic moieties to vancomycin results in broad-spectrum activity against both Gram-negative bacteria and Gram-positive bacteria. The cationic lipophilic moiety incorporates interaction with the negatively charged bacterial membrane, therefore, perturbing membrane integrity. Through comparison of alkyl and aryl-substituted derivatives, VanQAmC10 and VanQbiph respectively, we demonstrate that the alkyl substitutions exhibit better membrane activity as well as bactericidal activity against both metabolically active and inactive bacteria (Fig. 1). Vancomycin, on the other hand, is ineffective against the metabolically inactive bacterial cells. The lead compound, VanQAmC10 shows remarkable improvement in antibacterial efficacy in a mouse thigh infection model as compared to vancomycin against MRSA (Fig. 6). It was also effective against burn wound infections in mice caused by A. baumannii.Scientific interest toward membrane-active vancomycin derivatives has been growing due to their ability to overcome both inherited and non-inherited resistance.3 Therefore, a holistic understanding of how membrane-active vancomycin derivatives affect the bacteria would be imperative for the further development of new agents. In a step toward this, we used a variety of biological assays to provide new insights into the mechanism of action of VanQAmC10. It acts through mechanisms in addition to cell wall synthesis inhibition by binding to the D-Ala–D-Ala terminus which is known for the parent drug, vancomycin. To rule out the secondary effects as a result of cell death, mechanisms of action were studied at sub-inhibitory concentrations. Systematic investigations in B. subtilis revealed that it acts by simultaneously, (i) inhibiting cell wall biosynthesis, (ii) permeabilizing cells through non-specific interactions with membrane, and (iii) dissipation of membrane-potential leading to delocalization of the cell division protein, MinD (Fig. 4). The dissipation of membrane potential by VanQAmC10 possibly results in malfunctioning of the bacterial cell division machinery.42 To comprehend how VanQAmC10 affects Gram-negative bacteria, studies were conducted on the model Gram-negative bacterium, E. coli. Treatment with VanQAmC10 results in the lysis of the cells and therefore cell death. Treatment of the various strains of E. coli (wild-type, GFP-FtsI, GFP-FtsZ, and ΔamiAC) at subinhibitory concentrations resulted in cells of variable sizes and distorted morphology (Fig. 5). It did not impede the formation of the Z-ring in the first stage of cell division. It delocalized the FtsI protein required for the synthesis of the cell wall during septum formation in the second stage of cell division. Mutants lacking AmiA and AmiC enzymes were more sensitive to VanQAmC10 which is consistent with reports that these mutants have increased susceptibility to surfactants. These confirm that VanQAmC10 inhibits cell division at sub-inhibitory concentrations through mechanisms different from the FtsZ inhibitors and the β-lactam, cephalexin. Our findings suggest that the membrane perturbing properties of VanQAmC10 affect the localization of proteins involved in cell division, leading to severe cell wall and membrane defects. This possibly ultimately leads to a breach in the cell membrane and cell death.
Overall, here we demonstrate a new vancomycin derivative, VanQAmC10 which is highly effective against Gram-positive as well as Gram-negative bacteria both in vitro and in vivo. The multiple mechanisms of action contribute to the negligible resistance induction and superior antibacterial properties of VanQAmC10 as compared to the parent drug. It represents a new class of multi-target, multi-effect glycopeptide antibiotics. The findings augment a new dimension to the understanding of the mechanism of such membrane-active glycopeptide derivatives.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.The ESI† includes materials and methods; experimental protocols for in vitro microbiological assays (activity, biofilms, time-kill kinetics, and mechanistic studies) and toxicity, and in vivo activity and toxicity; supplementary figures; details of characterization of intermediates and final compounds through 1H NMR, IR, and HR-MS.Author contributions
PS, project design, synthesis, characterisation, performing in vitro microbiological assays (activity and mechanistic studies) and in vivo infection studies, data curation, formal analysis, manuscript writing and editing; KD, synthesis and characterisation; GD, cytotoxicity testing and in vivo activity studies, manuscript editing; JEB, experimental design and analysis for mechanisms of action against B. subtilis, manuscript editing; MM and RP, experimental design and analysis for mechanisms of action against E. coli, manuscript editing; JH, project design, research supervision, manuscript writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge DST-DAAD bilateral cooperation project (INT/FRG/DAAD/P-15/2018) and JNCASR for funding. We thank Dr Sandip Samaddar (JNCASR), Dr Riya Mukherjee (JNCASR) and Pascal Dietze (RUB) for excellent technical support and scientific discussions. We thank Dr Chandradhish Ghosh and Dr Venkateswarlu Yarlagadda for fruitful scientific discussions. RP acknowledges funding from CSIR (37(1701)/17/EMR-II) and Shiv Nadar University. MM acknowledges support by DST Inspire fellowship. JEB acknowledges funding from the DAAD Program DST 2018 (57389759). We thank Dr Sidharth Chopra (Central Drug Research Institute, Lucknow) for the gift of drug-resistant clinical isolates.References
- https://www.who.int/foodsafety/cia/en/, 2019.
- E. Binda, F. Marinelli and G. L. Marcone, Antibiotics, 2014, 3, 572–594 CrossRef PubMed.
- G. Dhanda, P. Sarkar, S. Samaddar and J. Haldar, J. Med. Chem., 2019, 62, 3184–3205 CrossRef CAS PubMed.
- D. Zeng, D. Debabov, T. L. Hartsell, R. J. Cano, S. Adams, J. A. Schuyler, R. McMillan and J. L. Pace, Cold Spring Harb Perspect. Med., 2016, 6, a026989 CrossRef PubMed.
- M. A. T. Blaskovich, K. A. Hansford, M. S. Butler, Z. Jia, A. E. Mark and M. A. Cooper, ACS Infect. Dis., 2018, 4, 715–735 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5052–E5061 CrossRef CAS PubMed.
- V. Yarlagadda, P. Sarkar, G. B. Manjunath and J. Haldar, Bioorg. Med. Chem. Lett., 2015, 25, 5477–5480 CrossRef CAS PubMed.
- V. Yarlagadda, P. Sarkar, S. Samaddar and J. Haldar, Angew. Chem., Int. Ed. Engl., 2016, 55, 7836–7840 CrossRef CAS PubMed.
- D. Guan, F. Chen, Y. Qiu, B. Jiang, L. Gong, L. Lan and W. Huang, Angew. Chem., Int. Ed. Engl., 2019, 58, 6678–6682 CrossRef CAS PubMed.
- A. Antonoplis, X. Zang, M. A. Huttner, K. K. L. Chong, Y. B. Lee, J. Y. Co, M. R. Amieva, K. A. Kline, P. A. Wender and L. Cegelski, J. Am. Chem. Soc., 2018, 140, 16140–16151 CrossRef CAS PubMed.
- M. M. Fernandes, K. Ivanova, J. Hoyo, S. Perez-Rafael, A. Francesko and T. Tzanov, ACS Appl. Mater. Interfaces, 2017, 9, 15022–15030 CrossRef CAS PubMed.
- V. Yarlagadda, P. Sarkar, S. Samaddar, G. B. Manjunath, S. D. Mitra, K. Paramanandham, B. R. Shome and J. Haldar, ACS Infect. Dis., 2018, 4, 1093–1101 CrossRef CAS PubMed.
- A. Antonoplis, X. Zang, T. Wegner, P. A. Wender and L. Cegelski, ACS Chem. Biol., 2019, 14, 2065–2070 CrossRef CAS PubMed.
- E. van Groesen, C. J. Slingerland, P. Innocenti, M. Mihajlovic, R. Masereeuw and N. I. Martin, ACS Infect. Dis., 2021, 7, 2746–2754 CrossRef CAS PubMed.
- E. Lei, H. Tao, S. Jiao, A. Yang, Y. Zhou, M. Wang, K. Wen, Y. Wang, Z. Chen, X. Chen, J. Song, C. Zhou, W. Huang, L. Xu, D. Guan, C. Tan, H. Liu, Q. Cai, K. Zhou, J. Modica, S. Y. Huang, W. Huang and X. Feng, J. Am. Chem. Soc., 2022, 144, 10622–10639 CrossRef CAS PubMed.
- R. E. Impey, D. A. Hawkins, J. M. Sutton and T. P. Soares da Costa, Antibiotics, 2020, 9 CrossRef CAS PubMed.
- M. A. T. Blaskovich, K. A. Hansford, Y. Gong, M. S. Butler, C. Muldoon, J. X. Huang, S. Ramu, A. B. Silva, M. Cheng, A. M. Kavanagh, Z. Ziora, R. Premraj, F. Lindahl, T. A. Bradford, J. C. Lee, T. Karoli, R. Pelingon, D. J. Edwards, M. Amado, A. G. Elliott, W. Phetsang, N. H. Daud, J. E. Deecke, H. E. Sidjabat, S. Ramaologa, J. Zuegg, J. R. Betley, A. P. G. Beevers, R. A. G. Smith, J. A. Roberts, D. L. Paterson and M. A. Cooper, Nat. Commun., 2018, 9, 22 CrossRef PubMed.
- P. Sarkar, S. Samaddar, V. Ammanathan, V. Yarlagadda, C. Ghosh, M. Shukla, G. Kaul, R. Manjithaya, S. Chopra and J. Haldar, ACS Chem. Biol., 2020, 15, 884–889 CrossRef CAS PubMed.
- V. Yarlagadda, G. B. Manjunath, P. Sarkar, P. Akkapeddi, K. Paramanandham, B. R. Shome, R. Ravikumar and J. Haldar, ACS Infect. Dis., 2016, 2, 132–139 CrossRef CAS PubMed.
- V. Yarlagadda, P. Akkapeddi, G. B. Manjunath and J. Haldar, J. Med. Chem., 2014, 57, 4558–4568 CrossRef CAS PubMed.
- J. A. Karlowsky, K. Nichol and G. G. Zhanel, Clin. Infect. Dis., 2015, 61(Suppl 2), S58–S68 CrossRef CAS PubMed.
- M. T. Guskey and B. T. Tsuji, Pharmacotherapy, 2010, 30, 80–94 CrossRef CAS PubMed.
- G. G. Zhanel, D. Calic, F. Schweizer, S. Zelenitsky, H. Adam, P. R. Lagace-Wiens, E. Rubinstein, A. S. Gin, D. J. Hoban and J. A. Karlowsky, Drugs, 2010, 70, 859–886 CrossRef CAS PubMed.
- D. S. Uppu, P. Akkapeddi, G. B. Manjunath, V. Yarlagadda, J. Hoque and J. Haldar, Chem. Comm., 2013, 49, 9389–9391 RSC.
- J. Hoque, S. Gonuguntla, V. Yarlagadda, V. K. Aswal and J. Haldar, Phys. Chem. Chem. Phys., 2014, 16, 11279–11288 RSC.
- D. S. S. M. Uppu, M. M. Konai, U. Baul, P. Singh, T. K. Siersma, S. Samaddar, S. Vemparala, L. W. Hamoen, C. Narayana and J. Haldar, Chem. Sci., 2016, 7, 4613–4623 RSC.
- X. Zhou and L. Cegelski, Biochemistry, 2012, 51, 8143–8153 CrossRef CAS PubMed.
- I. Keren, N. Kaldalu, A. Spoering, Y. Wang and K. Lewis, FEMS Microbiol. Lett., 2004, 230, 13–18 CrossRef CAS PubMed.
- D. E. Moormeier and K. W. Bayles, Mol. Microbiol., 2017, 104, 365–376 CrossRef CAS PubMed.
- T. Schneider, T. Kruse, R. Wimmer, I. Wiedemann, V. Sass, U. Pag, A. Jansen, A. K. Nielsen, P. H. Mygind, D. S. Raventos, S. Neve, B. Ravn, A. M. Bonvin, L. De Maria, A. S. Andersen, L. K. Gammelgaard, H. G. Sahl and H. H. Kristensen, Science, 2010, 328, 1168–1172 CrossRef CAS PubMed.
- M. Wenzel, B. Kohl, D. Munch, N. Raatschen, H. B. Albada, L. Hamoen, N. Metzler-Nolte, H. G. Sahl and J. E. Bandow, Antimicrob. Agents Chemother., 2012, 56, 5749–5757 CrossRef CAS PubMed.
- Z. C. Wu, N. A. Isley, A. Okano, W. J. Weiss and D. L. Boger, J. Org. Chem., 2020, 85, 1365–1375 CrossRef CAS PubMed.
- L. I. Rothfield and S. S. Justice, Cell, 1997, 88, 581–584 CrossRef CAS PubMed.
- W. Margolin, Nat. Rev. Mol. Cell Biol., 2005, 6, 862–871 CrossRef CAS PubMed.
- J. E. Ward Jr and J. Lutkenhaus, Cell, 1985, 42, 941–949 CrossRef CAS PubMed.
- L. Araujo-Bazan, L. B. Ruiz-Avila, D. Andreu, S. Huecas and J. M. Andreu, Front. Microbiol., 2016, 7, 1558 Search PubMed.
- M. C. Wissel and D. S. Weiss, J. Bacteriol., 2004, 186, 490–502 CrossRef CAS PubMed.
- H. S. Chung, Z. Yao, N. W. Goehring, R. Kishony, J. Beckwith and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21872–21877 CrossRef CAS PubMed.
- R. Priyadarshini, M. A. de Pedro and K. D. Young, J. Bacteriol., 2007, 189, 5334–5347 CrossRef CAS PubMed.
- M. G. Thompson, C. C. Black, R. L. Pavlicek, C. L. Honnold, M. C. Wise, Y. A. Alamneh, J. K. Moon, J. L. Kessler, Y. Si, R. Williams, S. Yildirim, B. C. Kirkup Jr, R. K. Green, E. R. Hall, T. J. Palys and D. V. Zurawski, Antimicrob. Agents Chemother., 2014, 58, 1332–1342 CrossRef PubMed.
- J. M. Streit, Diagn. Microbiol. Infect. Dis., 2004, 48, 137–143 CrossRef CAS PubMed.
- P. Sarkar, J. Med. Chem., 2021, 10185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05600c |
| This journal is © The Royal Society of Chemistry 2023 |