
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrostatically tuned phenols: a scalable organocatalyst for transfer hydrogenation and tandem reductive alkylation of N-heteroarenes†
Brijesh
Patel
ab,
Shilpa
Dabas
ab,
Parth
Patel
 ab and
Saravanan
Subramanian
*ab
ab and
Saravanan
Subramanian
*ab
aInorganic Materials and Catalysis Division, CSIR – Central Salt & Marine Chemicals Research Institute, Bhavnagar-364002, Gujarat, India. E-mail: saravanans@csmcri.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Gaziabad-201002, India
First published on 20th December 2022
Abstract
One of the fundamental aims in catalysis research is to understand what makes a certain scaffold perform better as a catalyst than another. For instance, in nature enzymes act as versatile catalysts, providing a starting point for researchers to understand how to achieve superior performance by positioning the substrate close to the catalyst using non-covalent interactions. However, translating this information to a non-biological catalyst is a challenging task. Here, we report a simple and scalable electrostatically tuned phenol (ETP) as an organocatalyst for transfer hydrogenation of N-arenes using the Hantzsch ester as a hydride source. The biomimetic catalyst (1–5 mol%) displays potential catalytic activity to prepare diverse tetrahydroquinoline derivatives with good to excellent conversion under ambient reaction conditions. Kinetic studies reveal that the ETP is 130-fold faster than the uncharged counterpart, towards completion of the reaction. Control experiments and NMR spectroscopic investigations elucidate the role of the charged environment in the catalytic transformation.
Introduction
It is often thought that compounds with complicated structural features could be the choice for better catalytic activity. This is a consequence of the fact that enzymes – natures ingenious complex machines1 – play a paramount role in performing most processes essential to life at high rates under mild conditions.2 One of the unique features of enzymes is the creation of a precise environment, created using electrostatic contributions that stabilize specific transition states, enabling the remarkable catalytic effect. Due to these inbuilt features, it should be stressed that enzymes are highly complex systems in every imaginable aspect.3 Thus, translating the tremendous potential of this extended environment, particularly the electrostatic effects, to simple and non-biological catalysts is a dream for developing next-generation catalyst systems.In our line of search for a simple and modular catalytic system,4,5 we were attracted to the possibility of using phenols, a low-price compound known for its catalytic activity, as a Brønsted acid and hydrogen bond donor system. Tuning the electronic nature of phenol not only alters the acidity and hydrogen bond donor strength, but also typically make it a more active catalytic component.6–8 Wassermann’s seminal work on Diels–Alder reactions in the presence of phenol9 has enabled the development of numerous phenolic systems for diverse catalytic applications. To date, a variety of strategies have been devised10–12 to enhance the catalytic profile of phenolic scaffolds (Fig. 1A). Hine et al. reported 1,8-biphenylenediol (BPD) that activates oxygen bearing reactants such as pyrone and epoxide by acting as a dual hydrogen bond donor.6 It was observed that BPD has 3 times improved activity due to its two hydroxyl groups, however, it lacks solubility in chlorinated solvents. Subsequently, Kelly et al. developed a soluble analog, 4,5-dinitro-2,7-dipropylbiphenylene-1,8-diol, for Diels–Alder reactions, and observed excellent catalytic activity over monoprotic phenol counterparts such as p-nitrophenol.10
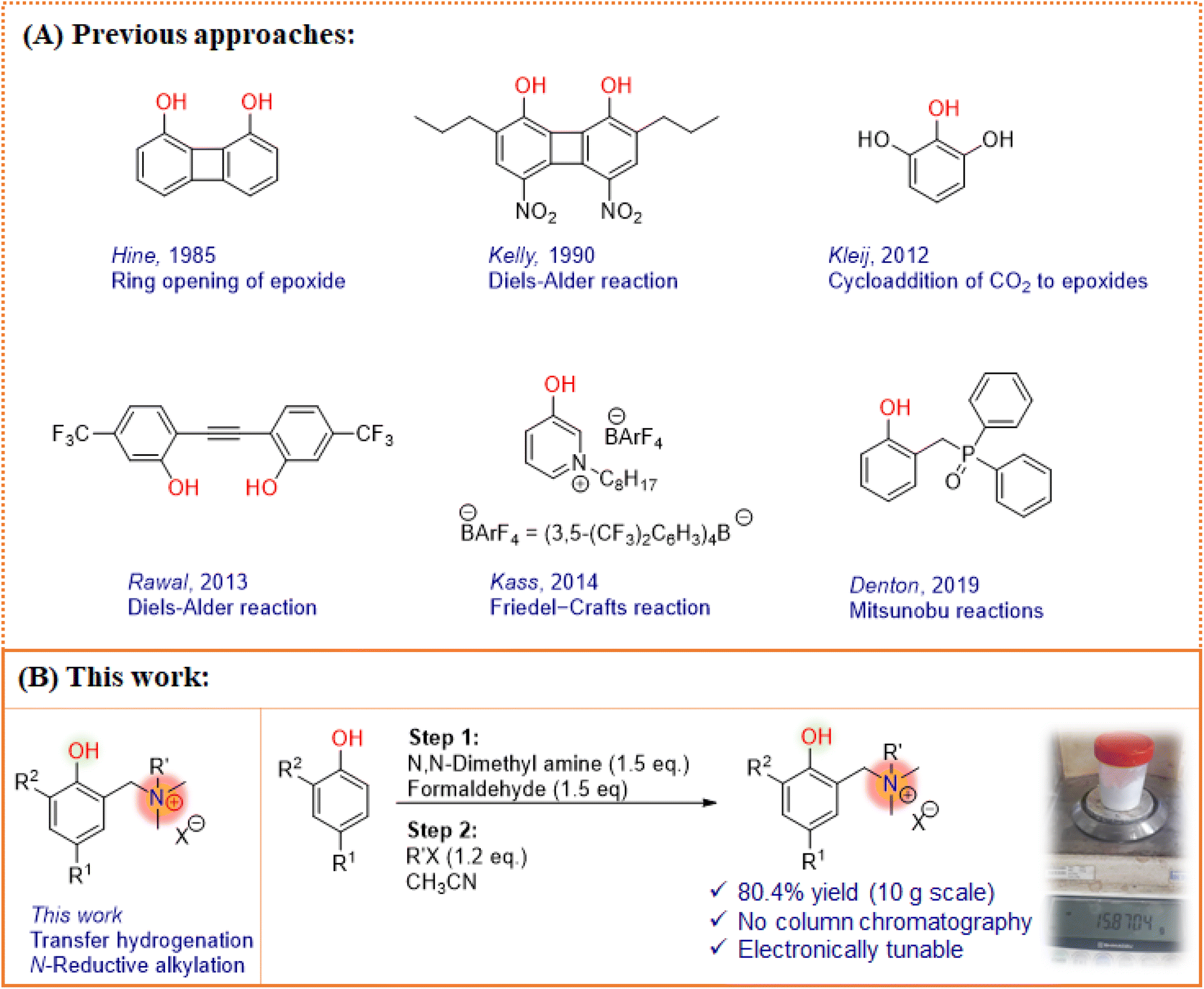 | ||
| Fig. 1 (A) Different phenol based catalytic system designs; (B) synthesis of electrostatically tuned phenols. | ||
Kleij et al. developed a binary catalytic system comprised of substituted phenols and ammonium salts for the cycloaddition of epoxides with carbon dioxide. The multiphenolic scaffold with intramolecular hydrogen-bond pattern demonstrated large stabilization effects which thereby leads to prominent synergistic activity towards the formation of organic carbonates.12 Rawal and co-workers explored the alkyne linked phenol (diarylacetylenediol) scaffolds for the reaction between cyclopentadiene and methyl vinyl ketone. The structurally rigid diol was found to be a more effective catalyst due to the two-point hydrogen bond activation of the carbonyl compound.7 In this line, a different approach was applied by Denton and co-workers.8 A redox-neutral phosphine oxide was introduced as a potential organocatalyst for Mitsunobu reactions where the phenolic –OH plays a crucial role to generate oxyphosphonium salt as the active species prior to SN2 reaction.13 In their elegant work, Kass and co-workers investigated the effect of charged substituents on the phenolic moiety with weakly coordinating anions in nonpolar media.14,15
Interestingly, a charged system provides several orders of rate enhancement and performs better for organic transformations,16–21 proton coupled electron transfer mechanisms,22 secondary coordination sphere approaches,23 and molecular electrocatalysis.24 However, in many cases, the charge effect was found to be beneficial when it resides on a complicated aromatic system or the heteroatoms of aromatic units. These findings on charge activated catalytic systems inspired us to identify a related but more simple, modular scaffold that could offer synthetically accessible and effective organocatalytic variants (Fig. 1B). Thus, we planned to synthesize a phenol based catalyst with the α-alkylammonium group as a charged arm that regulates the catalytic behavior of phenol, together represented as electrostatically tuned phenols (ETPs). We envisioned that the ETP dispenses the catalytic activity towards the transfer hydrogenation of N-arenes using Hantzsch 1,4-dihydropyridine esters (HE), a close analog of biological NAD(P)H. The choice of the reaction is inspired by the similar selective reduction in biological systems involving enzymes and cofactors with the influence of electrostatic environments.25,26
On the other hand, the transfer hydrogenation of N-arenes is an interesting and challenging transformation because of the associated resonance stability.27–30 Nevertheless, significant effort has been devoted to improve the reactivity profile of N-arenes such as quinoline for its selective partial hydrogenation to access 1,2,3,4-tetrahydroquinoline (THQ) derivatives.31–39 In pioneering work, Rueping and co-workers reported the organocatalytic reduction of N-arenes with a biomimetic reductant, HE, and also successfully extended the strategy to an asymmetric version using a chiral phosphoric acid catalyst.40–42 Subsequently, diverse catalytic systems have been explored for the transfer hydrogenation reaction of quinolines. In the context of a metal-free system, a few classes of compounds have been found to be successful, particularly, phosphoric acids,31,43,44 thioureas,45 chalcogen bond donor catalyst,46,47 and halogen bond catalyst.48 Despite these remarkable advances, most literature reports in the organocatalytic transfer hydrogenation, demand the use of strong acids, multi-step and expensive protocols for the catalyst synthesis which thereby limits their large-scale application.
Herein, we report ETPs derived from non-specialty chemicals which serves as an efficient metal-free catalyst for the transfer hydrogenation of N-arenes under mild conditions with high efficiency. The catalytic protocol of ETP was also successfully extended for the one-pot reductive alkylation of N-arenes using aromatic aldehydes, to prepare N-alkylated arenes. In this study, we have observed that the incorporation of a charged arm on the phenol results in a large increase in the activity compared to its uncharged counterpart. We also performed NMR studies to understand the mechanism of the activity.
Results and discussion
Catalyst synthesis
Developing a simple catalytic system that works under mild conditions is a challenging task and requires a well-designed chemical scaffold. Generally a strong acidic/basic system or bifunctional strategy could enhance the reactivity profile of substrates. On the otherhand, incorporation of a charged center as an additional control element has been identified as a new strategy to increase the catalytic activity and thereby influence the reaction rate.14 Also, the phenol–tyrosine combination has a significant role in a variety of biological systems.49 Inspired by these studies and continuing our interest in exploring organocatalytic scaffolds from commercially available sources,4,50–52 in this work, we focused on examining the ability of ETP moieties to catalyze the transfer hydrogenation reaction. With the literature support,49,53–55 we designed a catalyst having phenol as a core unit and an α-alkylammonium group as a side arm. The choice of α-alkylammonium group is due to the intervention of the alkyl group between the aromatic ring and ammonium group that might largely enhance the electrostatic effects by preventing conjugation. All the ETPs investigated (1–8, Fig. 2) were prepared by reacting substituted phenols (1 eq.) with formaldehyde (1.5 eq.) and their corresponding amines (1.5 eq.), by stirring at room temperature for 12 h. Subsequently, the crude product obtained by evaporation of solvent under reduced pressure was treated with quaternizing agents in acetonitrile. The resultant precipitate was filtered and washed with diethyl ether to yield the ETPs in good yields (up to 89%). Considering the low-priced starting materials and synthetic ease involved in the procedure, encouraged us to scale up the preparation of ETP-2 to 10 g, obtaining an isolated yield of 80.4% (Fig. 1B). Also, we calculated that ETP-2 can be prepared for about US$ 0.7 per g (cost estimate is presented in the ESI†). In the case of ETP-4, only the second step differs with the use of NaH (1 eq.) as the base followed by quaternization with methyl iodide (2.5 eq.). Overall, the ETP moieties differ with the aromatic substituents, alkylating sources and counter anions (Fig. 2). Further, to evaluate the effectiveness of ETPs as catalysts for the transfer hydrogenation reaction we chose quinoline as a model substrate with HE as the hydride donor using dichloroethane as the solvent at 60 °C. | ||
| Fig. 2 Evaluation of metal-free catalysts for the transfer hydrogenation of quinoline. aUnless otherwise mentioned all the reactions are carried out with catalyst (5 mol%), quinoline (0.5 mmol) with HE (2.5 mol eq.) using dry DCE (1 mL) as the solvent at 60 °C. bReaction was carried out at room temperature (28–30 °C). | ||
Catalyst optimization
To our delight, ETP-1 demonstrates good catalytic activity and provides the product 1,2,3,4-tetrahydroquinoline with 74% conversion at 3 h of reaction time. Encouraged by the preliminary results, next we studied the effects of substituents in the aromatic ring of phenol. ETP-2 and ETP-3 with bulky tert-butyl group substitution(s) resulted in the product tetrahydroquinoline with nearly quantitative conversion (Fig. 2A). To identify the role of phenolic O–H in the transfer hydrogenation reaction, the O–H group was masked with the methyl as a protecting group (ETP-4). The lack of the phenolic O–H disrupted the catalytic activity, leading to only trace conversions. This clearly shows its crucial role, and without the acidic hydroxyl group there is no driving force to activate the quinoline substrate. Further to elucidate the catalytic character and probe the effect of the charged arm, we then studied the uncharged counterpart of ETP-2 (i.e., DMAMP) and the chemical precursor DTBP under the same reaction conditions. The presence of uncharged amine and the lack of charged side arm, resulted in only trace conversions.In order to check the impact of Brønsted acidity on the transfer hydrogenation reaction, an electron withdrawing substituent on the phenolic ring was tested (ETP-5). Unexpectedly, only 7% conversion was achieved and even prolonging the reaction time led to no significant change. These results indicate that the catalytic activity of ETP-2 not only directly depends on the acidic parameter but could also be attributed to the incorporation of the charged arm in the phenolic moiety that renders the electrostatic environment. This prompted us to evaluate the effect of anions (Fig. 2C), and we observed that the association of the weakly coordinating tetrafluoroborate anion with the charged ammonium arm of phenol (ETP-6), outpaces its analogues (ETP-7 and 8) in terms of reaction rate – obtaining the quantitative conversion in just 10 min. An ∼130-fold increase in the rate is seen with ETP-6 over its uncharged counterpart DMAMP, and an ∼9 fold increase over ETP-2 (ESI, Fig. S2†), presumably because of the association with weakly coordinating anions leads to pronounced electrostatic effects. It is worth mentioning here that the transfer hydrogenation of quinoline using HE proceeds smoothly to completion in the presence of ETP-6 even at room temperature (28–30 °C, Fig. 2C). Alternatively, the reaction employed using choline chloride (CC), an aliphatic structural analogue of ETP, afforded only low conversion (Fig. 2D) even after prolonging the reaction time to 24 h. Similar results were observed with pyridinium (BMPTFB) and imidazolium tetrafluoroborate (BMIMTFB) ionic liquids as catalyst employed under similar reaction conditions. These reactions clearly depict that there is no significant impact of sole anion/charged ammonium centers. Overall, the intriguing observations imply the importance of the substituted charged arm that regulates the catalytic behaviour of phenol by electrostatic enhancement in the transfer hydrogenation of quinoline using HE as the hydride source.
With ETP-6 as the identified best choice of catalyst, we screened various reaction parameters to optimize the catalytic activity. Though ETP-6 is found to be efficient for catalyzing the transfer hydrogenation of quinoline using HE at room temperature, taking advantage of the reaction time at 60 °C, we performed further screening experiments at this temperature. Initially, the catalyst loading was explored, upon decreasing the catalyst loading to 2.5 mol% we observed a significant decrease in the conversion to 91% (Table 1, entry 2). Whereas, increasing the catalyst loading to 10 mol% resulted in no significant change in the conversion but with a change in the time of reaction (Table 1, entry 3). Thus, 5 mol% of catalyst is found to be optimal for the reaction. Furthermore, different solvents were screened for their suitability in the transfer hydrogenation reaction and we realized that 1,2-dichloroethane was the best (Table 1, entries 4–14). We tested the catalytic reaction using 2-methyl tetrahydrofuran, a successful neoteric bio-based solvent,56,57 and found benefits in terms of conversion. Also, we observed that 2.5 equivalents of hydride source is desirable to achieve maximum conversion of quinoline, as decreasing it to 2 equivalents resulted in a deleterious effect (Table 1, entry 15).
|
|
||
|---|---|---|
| Entry | Deviation | Conversionb (%) |
| a Unless otherwise mentioned all the reactions are carried out with catalyst (5 mol%), quinoline (0.5 mmol) and HE (2.5 mol eq.) using solvent (1 mL) at 60 °C for 10 min. b Determined using GC analysis. | ||
| 1 | None | 99 |
| 2 | 2.5 mol% | 91 |
| 3 | 10 mol% | 99 |
| 4 | Toluene | 91 |
| 5 | Xylene | 99 |
| 6 | Dioxane | 97 |
| 7 | Tetrahydrofuran (THF) | 98 |
| 8 | 2-Methyl tetrahydrofuran | 98 |
| 9 | Ethyl acetate | 89 |
| 10 | Dichloromethane | 93 |
| 11 | Dimethylformamide (DMF) | 32 |
| 12 | Dimethylsulfoxide (DMSO) | 02 |
| 13 | Acetonitrile | 74 |
| 14 | Methanol | 30 |
| 15 | 2 mol eq. HE | 82 |
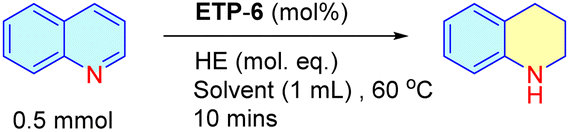
Next, using the optimal conditions, we evaluated the scope of the catalyst using N-arenes with different substitution patterns at both 60 °C and room temperature (Fig. 3). In general, N-heteroarenes bearing alkyl, aryl, nitro and halogen substituents proceeded smoothly and afforded the corresponding substituted 1,2,3,4-tetrahydroquinolines with good to excellent conversion. The substitution position of the methyl group had an impact on the yield of the transfer hydrogenation reaction. We observed that other than the 4-methyl substituted quinoline, methyl substitutions at the 2, 3, 6, 7 and 8 positions of quinoline are found to be suitable and resulted in the corresponding product with 80–94% conversion. We were also delighted to find chemoselective transfer hydrogenation of nitro- and halogen- substituted N-arenes that did not interfere with the reducing reactions. 8-Amino substituted quinoline resulted in the product with 65% conversion. Other N-arenes like benzo[h]quinoline were also investigated and we obtained the product with 70% conversion. Interestingly, (E)-2-(3,4-dimethoxystyryl)quinoline, the precursor of a natural alkaloid, also underwent transfer hydrogenation and resulted in the product with good conversion. To our surprise, only the 4-methyl substituted quinoline resulted in lower conversion (50%). We then attempted to increase the amount of hydride and also extended the reaction time to 24 h, but observed no significant improvement. Thus, we focused this study on the differently behaved substrate with the aim of understanding the mechanism behind the ETP for the transfer hydrogenation reaction.
 | ||
| Fig. 3 Chemoselective transfer hydrogenation of N-arenes using ETP-6 as catalyst. aUnless otherwise mentioned all the reactions are carried out with catalyst (5 mol%), substrate (0.5 mmol) with HE (2.5 mol eq.) using solvent (1 mL) at 60 °C for 10 min. The conversions mentioned in the parentheses are the reactions carried out at room temperature (28–30 °C) for 7 h, keeping all the other parameters same. bSubstrate: (E)-2-(3,4-dimethoxystyryl) quinoline has an additional olefinic bond and requires HE in 3.5 mol eq. with all other parameters kept the same. | ||
Mechanism
The optimization and scope of substrate studies revealed four significant points: (i) the necessity of phenolic moiety, (ii) the crucial role of the charged arm, (iii) the impact of C-4 substitution on the N-arene substrate, and (iv) the non-polar nature of the solvent. In order to gain further insight into the ETP-6 catalyzed transfer hydrogenation of N-arenes, we conducted additional control experiments. Thus, we first reviewed our substrate scope and attempted to extend it to different N-arenes such as isoquinoline, quinoxaline and acridine. Surprisingly, these electronically varied N-arenes are found to behave differently, wherein, the former two remain the same with no conversion observed. However, there is a striking difference with the acridine substrate which resulted in 99% conversion in the stipulated reaction time of 3 h (Fig. 4B). This observation somehow matches with the reactivity trend of alkyl substituted quinolines and suggest that the transfer hydrogenation of quinoline catalyzed by ETP-6 proceeds via the 1,4-pathway. Accordingly, we presume that the mechanism of transfer hydrogenation reaction is as follows: first, the quinoline gets activated by the interaction of nitrogen with ETP’s –OH which may possibly generate quinolinium and phenoxide ions as Int-I, leading to two ion-pairs. This gives rise to two instances for Int-I where (1) the generated phenoxide ion is counter balanced by the protonated quinolinium center or (2) it could also be counter balanced by its own alkyl ammonium arm. The 1H NMR spectra show two singlets for the alkyl ammonium protons [–C![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) 2- at 4.22 ppm and –N+(C3)3 at 2.89 ppm] of ETP-6 and found they shifted upfield (to 4.07 and 2.73 ppm, respectively) upon interaction with the quinoline. The observed shift might be due to the change in the environment of the generated phenoxide ion that could be counter balanced by its own alkylammonium arm, wherein the nitrogen faces relatively high electron density (more shielding than the one in ETP-6, associated with the weakly coordinating tetrafluroborate ion) and thereby influences the neighbouring protons. On the other hand, there is also a noticeable chemical shift of the hydrogen at the C4 position of quinoline from 8.15 ppm to 8.23 ppm (Fig. 4D), indicating interaction with ETP’s –OH. Consequently, this rules out the pathway for 1,2-addition. In the next step, the hydride from the HE transfer to Int-I and leads to the formation of enamine Int-II and furthermore the proton transfer regenerates the catalyst and completes Step-1. The Int-II undergoes a similar process in Step-2 again, resulting in the formation of the 1,2,3,4-tetrahydroquinoline product (Fig. 4A).
2- at 4.22 ppm and –N+(C3)3 at 2.89 ppm] of ETP-6 and found they shifted upfield (to 4.07 and 2.73 ppm, respectively) upon interaction with the quinoline. The observed shift might be due to the change in the environment of the generated phenoxide ion that could be counter balanced by its own alkylammonium arm, wherein the nitrogen faces relatively high electron density (more shielding than the one in ETP-6, associated with the weakly coordinating tetrafluroborate ion) and thereby influences the neighbouring protons. On the other hand, there is also a noticeable chemical shift of the hydrogen at the C4 position of quinoline from 8.15 ppm to 8.23 ppm (Fig. 4D), indicating interaction with ETP’s –OH. Consequently, this rules out the pathway for 1,2-addition. In the next step, the hydride from the HE transfer to Int-I and leads to the formation of enamine Int-II and furthermore the proton transfer regenerates the catalyst and completes Step-1. The Int-II undergoes a similar process in Step-2 again, resulting in the formation of the 1,2,3,4-tetrahydroquinoline product (Fig. 4A).
 | ||
| Fig. 4 (A) Proposed mechanism for ETP-6 catalyzed transfer hydrogenation of N-arenes, (B) control experiments for mechanistic investigations, (C) recycling process of the Hantzsch ester, (D and E) 1H NMR spectra for the interaction study of ETP-6 and quinoline. | ||
Scale-up studies of transfer hydrogenation of quinoline and its purification process
The most important critique associated with the Hantzsch ester based transfer hydrogenation reaction is the generation of a stoichiometric amount of its oxidized version (OHE) as by-product. This necessitates the use of conventional column chromatography to separate and purify the desired product from the reaction mixture and limits its practical applicability. To address this limitation and to enhance the practicality of our catalytic reaction from an academic and industrial viewpoint, we further investigated the post workup procedure with the aim of efficiently separating, recovering, regenerating and reusing the Hantzsch ester. It is worth mentioning here that there are reports that deal with the regeneration of HE, but only after its separation from the product using column chromatography.36 Ironically, to the best of our knowledge, without isolating the product THQ from the reaction mixture (containing catalyst, OHE and unutilized HE), separation and recovery of OHE from the crude reaction mixture using a simple procedure has not been widely studied.Thus, we believe that developing a simple recovery and regeneration process will add a significant impact on the transfer hydrogenation reactions. The rationale behind the workup based separation process is taking advantage of: (1) charged side arm in ETP-6 catalyst [easily precipitates out in non-polar solvent], (2) possible phase separation of product (THQ, liquid) and by-product (OHE, solid) using water to wash under vacuum filtration followed by any additional required chemical treatments.
To evaluate our hypothesis of workup based separation, we utilized the optimized conditions to a preparative-scale reaction up to 10 g to validate the simplicity and efficiency of the ETP catalytic system. In this particular case, the loading of the ETP-6 catalyst can be lowered to 1 mol% with an observed quantitative conversion (99.5%). The detailed experimental procedure is provided in the ESI.† The above mentioned two key properties motivated us to attempt the simple workup based separation of catalyst (ETP-6), HE and THQ individually, and accordingly we categorized the process involving three steps (Fig. 5). After completion of the reaction, the solvent was removed (∼90%) under reduced pressure leaving behind the crude reaction mixture in a minimum amount of solvent (THQ + OHE + catalyst, ETP-6 + some unutilized HE) and proceeded further with the following steps.
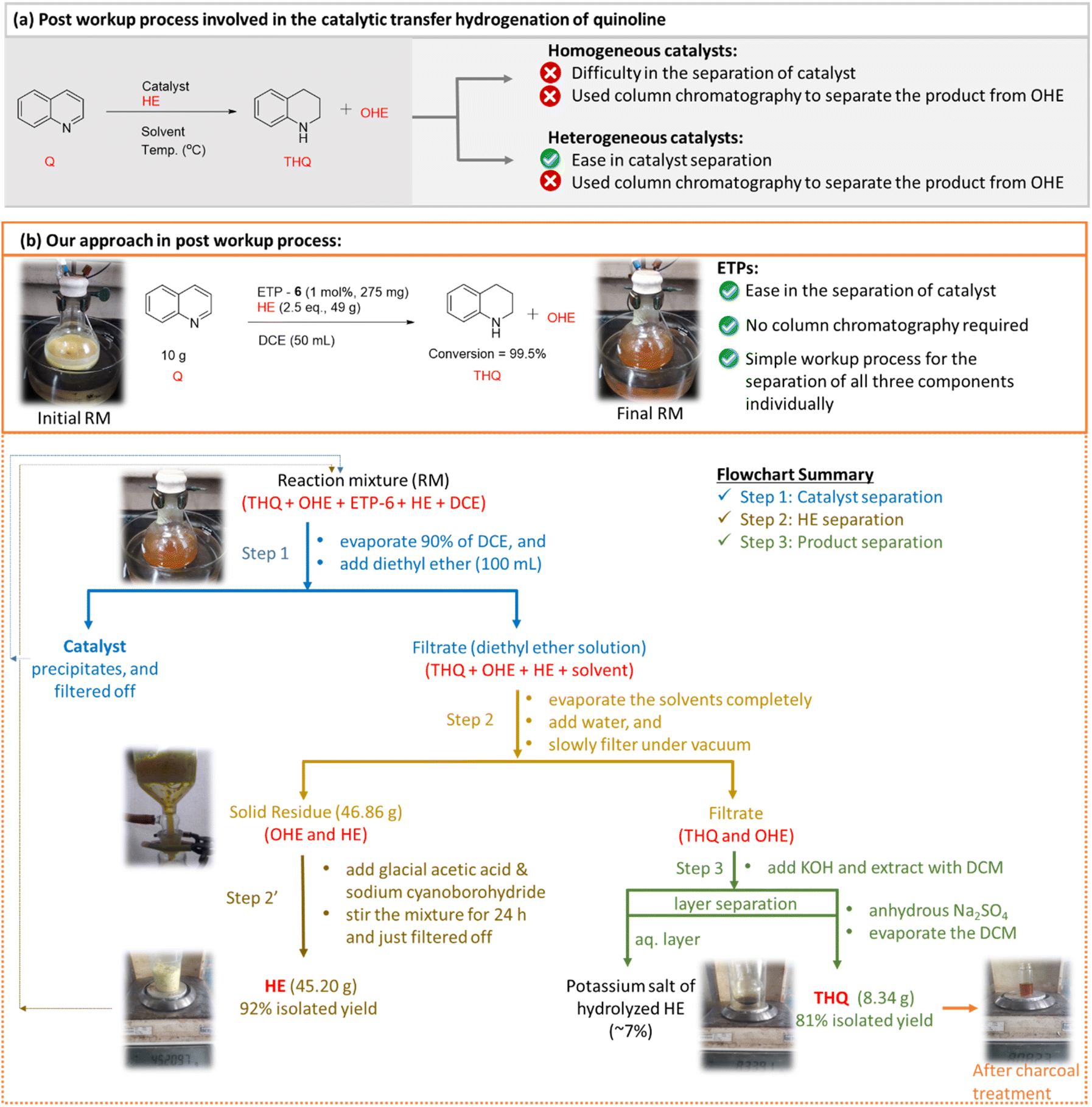 | ||
| Fig. 5 Scaling up, separation and purification process for the transfer hydrogenation of quinoline using ETP-6 as the catalyst in the presence of HE. | ||
In step 1, a non-polar solvent (preferably, diethyl ether in excess) was added to the crude reaction mixture and that resulted in the separation of catalyst by precipitation. Again, yet another advantage of the charged side arm in our ETP catalyst that makes it easier. The recovered catalyst has been characterized with 1H NMR spectroscopy and tested for its catalytic activity. The recovered catalyst demonstrated similar activity towards the transfer hydrogenation of quinoline. Upon the removal of the catalyst from the reaction mixture, the resultant filtrate containing diethyl ether and a minimum amount of reaction solvent was removed completely under reduced pressure. In step 2, to the obtained residual solid, water was added (5 × 20 mL) and subsequently filtered off slowly under vacuum conditions. In step 2′, the recovered OHE (off-white residue) was treated further with glacial acetic acid and sodium cyanoborohydride in the presence of water as a solvent36 (ESI-8†). The mixture was stirred at room temperature for 24 h to achieve the regenerated HE as a yellow solid (92%). In addition, we also tested the recovery and regeneration percentage of HE and reused it for the further catalytic transfer hydrogenation of quinoline [ESI-10†]. The regenerated HE worked very similarly to the freshly prepared HE which is evident by the conversion of quinoline achieved in the reusability studies. On the other hand, the aqueous filtrate contains the product (THQ) and residual OHE (∼5–8%). Step 3 involves the removal of the leftover OHE from the product, KOH was added which converts the OHE into the water soluble potassium salt of hydrolysed HE.58 Furthermore, upon addition of dichloromethane the product is extracted from the aqueous layer and subsequent removal of the organic layer results in the desired THQ with 81% yield.
One-pot reductive alkylation reaction
To demonstrate the general applicability of the ETP catalyst and to extend the scope of the process, we attempted to carry out further functionalization in one pot without the separation of the formed tetrahydroquinoline, to simplify the process. However, such strategies also involve additional challenges such as efficiency of catalyst towards the second step functionalization and the selectivity of the final product. Understanding these difficulties, we employed the ETP for the one-pot preparation of N-alkylated tetrahydroquinoline, due to the excellent bioactivity profile.In general, the N-alkylated tetrahydroquinoline is accessible via N-alkylation of partially reduced quinoline by treating it with alkyl halides, carboxylic acids and nitriles. These traditional methods require an additional reaction step, separation, purification, and are also limited by a narrow substrate scope and low yields.59,60 Our approach resulted in the direct synthesis of N-benzyltetrahydroquinolines by tandem reduction and N-alkylation of quinoline using the ETP-6 as an efficient organocatalyst (Fig. 6). We observed that both aromatic and aliphatic aldehyde derivatives underwent the tandem reaction smoothly and resulted the product with good conversion.
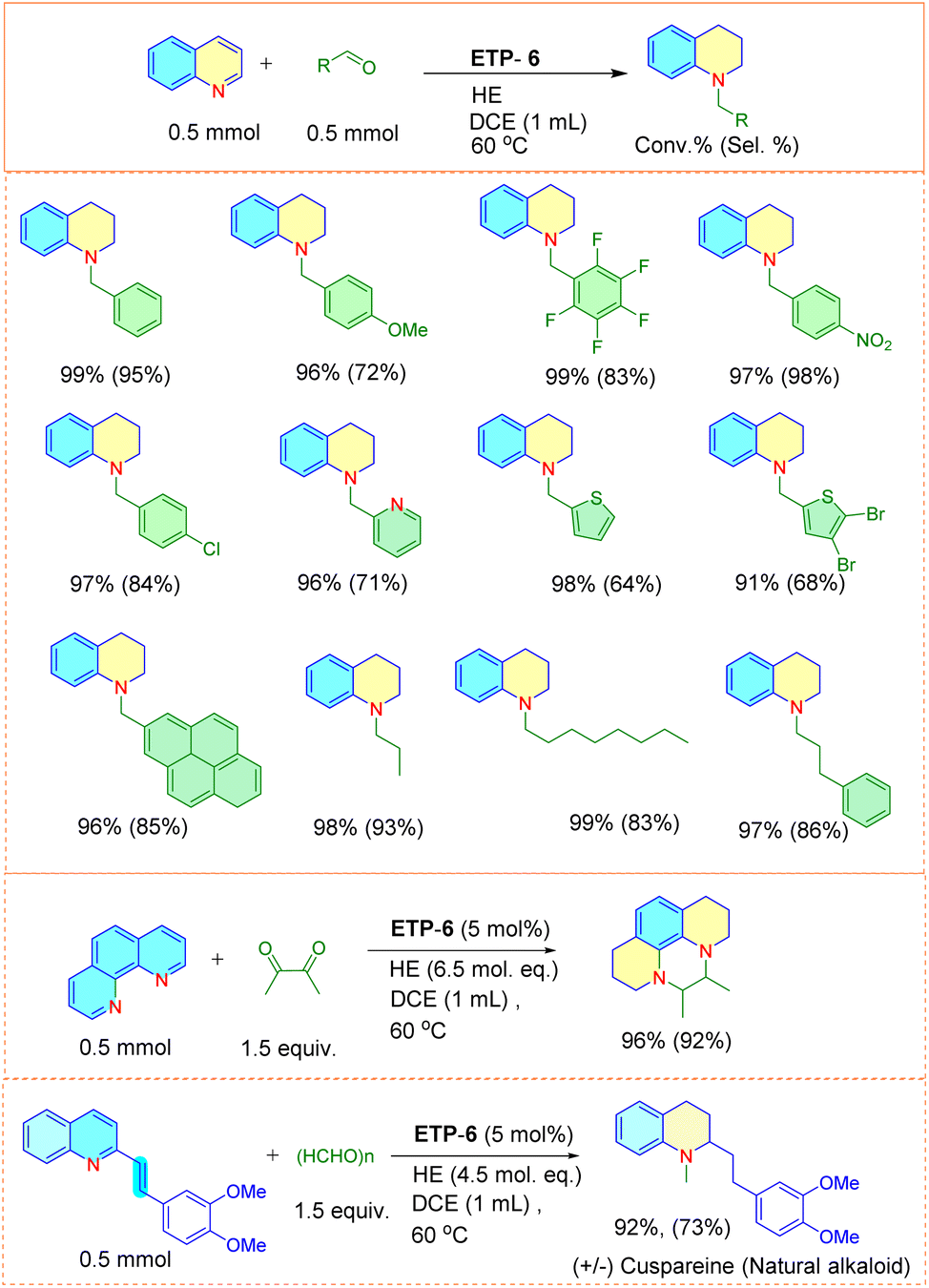 | ||
| Fig. 6 One-pot reductive alkylation of N-arenes using ETP-6 as the catalyst. aUnless otherwise mentioned all the reactions are carried out with catalyst (5 mol%), substrate quinoline (0.5 mmol), aldehyde (0.5 mmol) with HE (3.5 mol eq.) using DCE (1 mL) at 60 °C for 1 h. | ||
To our delight, we found that the catalyst is also capable of N-alkylating phenanthroline with diketone (diacetyl), resulting in the product 5,6-dimethyl-1,2,3,5,6,8,9,10-octahydropyrazino[1,2,3,4-lmn][1,10]phenanthroline in 96% conversion with 92% selectivity. Furthermore, we also extended the catalytic protocol for the synthesis of natural alkaloid, cuspareine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36via a tandem route. ETP-6 is efficient in selectively reducing the partial quinoline ring, olefinic double bond followed by N-alkylation using the paraformaldehyde. To the best of our knowledge this is the first report enabling the tandem preparation of cuspareine (92% yield and 73% selectivity) which involves hydrogenation followed by N-alkylation. This not only depicts the catalytic efficiency of ETP-6 but also may pave the path for step- and cost-economical preparation of N-alkylated tetrahydroquinoline products.
36via a tandem route. ETP-6 is efficient in selectively reducing the partial quinoline ring, olefinic double bond followed by N-alkylation using the paraformaldehyde. To the best of our knowledge this is the first report enabling the tandem preparation of cuspareine (92% yield and 73% selectivity) which involves hydrogenation followed by N-alkylation. This not only depicts the catalytic efficiency of ETP-6 but also may pave the path for step- and cost-economical preparation of N-alkylated tetrahydroquinoline products.
Conclusions
In summary, we have successfully developed electrostatically tuned phenol systems as efficient catalysts for the transfer hydrogenation of N-arenes using the Hantzsch ester. The easy-to-synthesise and scalable organocatalysts (ETPs-2 and 6) can effectively convert different N-arenes to partially saturated tetrahydroquinolines with good conversions under ambient reaction conditions. These results suggest that the associated charged arm regulates the catalytic behaviour of phenolic moiety. Efforts toward extension of this functional asymmetry for their straightforward application to enantioselective transformations are ongoing in our laboratory. We do believe this understanding may provide new means for producing more simple and scalable organocatalytic systems for application in diverse organic transformations.Data availability
All the experimental procedures and characterization details are available in the ESI.†Author contributions
B. P. synthesized and characterized the catalysts and carried out the catalytic experiments. S. D. and P. P. carried out a few optimization studies and substrate variations. S. S. conceived the project and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CSIR-CSMCRI Communication no. 106/2022. SS thanks DST for the INSPIRE grant (DST/INSPIRE/04-I/2017/000003). The authors thank CSIR (project no. MLP-0028 and SRF fellowship to PP) and UGC (for BP’s fellowship) for financial support. Analytical facilities from AESD and CIF of CSIR-CSMCRI are gratefully acknowledged. This article is dedicated to (the late) Dr Rukhsana I. Kureshy, Dr Noor-ul H. Khan and Dr Sayed H. R. Abdi for their significant contributions in the field of homogeneous catalysis.Notes and references
- L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9397–9404 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- A. Prah, E. Frančišković, J. Mavri and J. Stare, ACS Catal., 2019, 9, 1231–1240 CrossRef CAS.
- S. Subramanian, J. Oppenheim, D. Kim, T. S. Nguyen, W. M. H. Silo, B. Kim, W. A. Goddard and C. T. Yavuz, Chem, 2019, 5, 3232–3242 CAS.
- S. Subramanian, J. Park, J. Byun, Y. Jung and C. T. Yavuz, ACS Appl. Mater. Interfaces, 2018, 10, 9478–9484 CrossRef CAS.
- J. Hine, S. M. Linden and V. M. Kanagasabapathy, J. Am. Chem. Soc., 1985, 107, 1082–1083 CrossRef CAS.
- Y. E. Türkmen and V. H. Rawal, J. Org. Chem., 2013, 78, 8340–8353 CrossRef PubMed.
- R. H. Beddoe, K. G. Andrews, V. Magné, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon and R. M. Denton, Science, 2019, 365, 910–914 CrossRef.
- A. Wassermann, J. Chem. Soc., 1942, 618–621 RSC.
- T. R. Kelly, P. Meghani and V. S. Ekkundi, Tetrahedron Lett., 1990, 31, 3381–3384 CrossRef.
- C. Thomas, F. Peruch, A. Deffieux, A. Milet, J.-P. Desvergne and B. Bibal, Adv. Synth. Catal., 2011, 353, 1049–1054 CrossRef.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef PubMed.
- Y. Zou, J. J. Wong and K. N. Houk, J. Am. Chem. Soc., 2020, 142, 16403–16408 CrossRef PubMed.
- M. Samet, J. Buhle, Y. Zhou and S. R. Kass, J. Am. Chem. Soc., 2015, 137, 4678–4680 CrossRef.
- G. F. Riegel and S. R. Kass, J. Org. Chem., 2020, 85, 6017–6026 CrossRef PubMed.
- C. F. Gorin, E. S. Beh, Q. M. Bui, G. R. Dick and M. W. Kanan, J. Am. Chem. Soc., 2013, 135, 11257–11265 CrossRef PubMed.
- V. M. Lau, C. F. Gorin and M. W. Kanan, Chem. Sci., 2014, 5, 4975–4979 RSC.
- R. Zhang and J. J. Warren, J. Am. Chem. Soc., 2020, 142, 13426–13434 CrossRef.
- A. Rostami, A. Ebrahimi, N. Sakhaee, F. Golmohammadi and A. Al-Harrasi, J. Org. Chem., 2022, 87, 40–55 CrossRef PubMed.
- C.-X. Yan, R.-Z. Wu, K. Lu, F.-L. Yang, X.-S. Yang, R. Wang, X. Yang, P.-P. Zhou and X. Shao, Org. Chem. Front., 2019, 6, 1821–1831 RSC.
- J. Wang, T. A. Young, F. Duarte and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 17743–17750 CrossRef PubMed.
- R. E. Warburton, P. Hutchison, M. N. Jackson, M. L. Pegis, Y. Surendranath and S. Hammes-Schiffer, J. Am. Chem. Soc., 2020, 142, 20855–20864 CrossRef.
- N. D. Loewen, S. Pattanayak, R. Herber, J. C. Fettinger and L. A. Berben, J. Phys. Chem. Lett., 2021, 12, 3066–3073 CrossRef.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef PubMed.
- S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef PubMed.
- S. Banerjee and P. J. Sadler, RSC Chem. Biol., 2021, 2, 12–29 RSC.
- A. K. Kar and R. Srivastava, ACS Sustainable Chem. Eng., 2019, 7, 13136–13147 CrossRef.
- R. Adam, J. R. Cabrero-Antonino, A. Spannenberg, K. Junge, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 3216–3220 CrossRef PubMed.
- N. Gandhamsetty, S. Joung, S.-W. Park, S. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 16780–16783 CrossRef PubMed.
- N. Gandhamsetty, S. Park and S. Chang, Synlett, 2017, 28, 2396–2400 CrossRef.
- M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683–3686 CrossRef PubMed.
- A. M. Faísca Phillips and A. J. L. Pombeiro, Org. Biomol. Chem., 2017, 15, 2307–2340 RSC.
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS.
- I. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119, 5057–5191 CrossRef PubMed.
- M. El-Shahat, J. Heterocycl. Chem., 2022, 59, 399–421 CrossRef.
- D. Bhattacharyya, S. Nandi, P. Adhikari, B. K. Sarmah, M. Konwar and A. Das, Org. Biomol. Chem., 2020, 18, 1214–1220 RSC.
- V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef.
- X. Qiao, M. El-Shahat, B. Ullah, Z. Bao, H. Xing, L. Xiao, Q. Ren and Z. Zhang, Tetrahedron Lett., 2017, 58, 2050–2053 CrossRef.
- B. Chatterjee, D. Kalsi, A. Kaithal, A. Bordet, W. Leitner and C. Gunanathan, Catal. Sci. Technol., 2020, 10, 5163–5170 RSC.
- M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683–3686 CrossRef CAS PubMed.
- M. Rueping, T. Theissmann, M. Stoeckel and A. P. Antonchick, Org. Biomol. Chem., 2011, 9, 6844–6850 RSC.
- M. Rueping, T. Theissmann and A. P. Antonchick, Synlett, 2006, 2006, 1071–1074 CrossRef.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS.
- J. Greindl, J. Hioe, N. Sorgenfrei, F. Morana and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 15965–15971 CrossRef CAS PubMed.
- X. Qiao, Z. Zhang, Z. Bao, B. Su, H. Xing, Q. Yang and Q. Ren, RSC Adv., 2014, 4, 42566–42568 RSC.
- S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef CAS PubMed.
- P. Wonner, T. Steinke and S. M. Huber, Synlett, 2019, 30, 1673–1678 CrossRef CAS.
- C. T. Ser, H. Yang and M. W. Wong, J. Org. Chem., 2019, 84, 10338–10348 CrossRef CAS PubMed.
- T. Maki, Y. Araki, Y. Ishida, O. Onomura and Y. Matsumura, J. Am. Chem. Soc., 2001, 123, 3371–3372 CrossRef CAS PubMed.
- S. Subramanian, H. A. Patel, Y. Song and C. T. Yavuz, Adv. Sustainable Syst., 2017, 1, 1700089 CrossRef.
- S. Saravanan, A. Sadhukhan, N.-u. H. Khan, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, J. Org. Chem., 2012, 77, 4375–4384 CrossRef CAS PubMed.
- D. Ghosh, D. Sahu, S. Saravanan, S. H. R. Abdi, B. Ganguly, N.-u. H. Khan, R. I. Kureshy and H. C. Bajaj, Org. Biomol. Chem., 2013, 11, 3451–3460 RSC.
- Y. Fang, L. Liu, Y. Feng, X.-S. Li and Q.-X. Guo, J. Phys. Chem. A, 2002, 106, 4669–4678 CrossRef CAS.
- J. J. Warren and J. M. Mayer, Biochemistry, 2015, 54, 1863–1878 CrossRef CAS.
- L. Sun, M. Burkitt, M. Tamm, M. K. Raymond, M. Abrahamsson, D. LeGourriérec, Y. Frapart, A. Magnuson, P. H. Kenéz, P. Brandt, A. Tran, L. Hammarström, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1999, 121, 6834–6842 CrossRef CAS.
- A. Pellis, F. P. Byrne, J. Sherwood, M. Vastano, J. W. Comerford and T. J. Farmer, Green Chem., 2019, 21, 1686–1694 RSC.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef PubMed.
- V. N. Wakchaure, M. Nicoletti, L. Ratjen and B. List, Synlett, 2010, 2010, 2708–2710 CrossRef.
- K. Azizi, S. Akrami and R. Madsen, Chem.–Eur. J., 2019, 25, 6439–6446 CrossRef PubMed.
- Q. Xu, H. Xie, E.-L. Zhang, X. Ma, J. Chen, X.-C. Yu and H. Li, Green Chem., 2016, 18, 3940–3944 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05843j |
| This journal is © The Royal Society of Chemistry 2023 |