DOI:
10.1039/D2SC06016G
(Edge Article)
Chem. Sci., 2023,
14, 533-539
Dative B←N bonds based crystalline organic framework with permanent porosity for acetylene storage and separation†
Received
1st November 2022
, Accepted 2nd December 2022
First published on 6th December 2022
Abstract
The utilization of dative B←N bonds for the creation of crystalline organic framework (BNOF) has increasingly received intensive interest; however, the shortage of permanent porosity is an obstacle that must be overcome to guarantee their application as porous materials. Here, we report the first microporous crystalline framework, BNOF-1, that is assembled through sole monomers, which can be scalably synthesized by the cheap 4-pyridine boronic acid. The 2D networks of BNOF-1 were stacked in parallel to generate a highly porous supramolecular open framework, which possessed not only the highest BET surface area of 1345 m2 g−1 amongst all of the BNOFs but also features a record-high uptake of C2H2 and CO2 in covalent organic framework (COF) materials to date. Dynamic breakthrough experiments demonstrated that BNOF-1 material can efficiently separate C2H2/CO2 mixtures. In addition, the network can be regenerated in organic solvents with no loss in performance, making its solution processable. We believe that BNOF-1 would greatly diversify the reticular chemistry and open new avenues for the application of BNOFs.
Introduction
Since the first crystalline porous materials were reported in the late 1980s and early 1990s, they have provided an ideal platform for a deep understanding of the structure–performance relationships of materials due to their clear and adjustable structures and controllable properties.1–3 Metal–organic frameworks (MOFs)4–6 and covalent organic frameworks (COFs)7–9 are the most representative of crystalline porous materials, and thus, they are known for their impressive material properties. However, insolubility and poor processability are the limitations of MOF and COF materials, which are related to the inherent strong bonding interactions (coordination/covalent bonds) used to aggregate their building blocks.10,11 Relatively weak intermolecular supramolecular interactions permit the framework to readily dissolve and regenerate in solvents, endowing the materials with processability to a certain extent.12–14 Various supramolecular interactions, such as hydrogen bonds,15,16 halogen bonds,17,18 and π–π interactions19 play important roles in producing crystalline supramolecular frameworks. However, the stability of these frameworks is usually not enough, for instance, the removal of lattice molecules often results in framework collapse.20–22 The few supramolecular networks, which show extrinsic porosity, are mostly based on hydrogen bonds.23–25 In addition, the large-scale and industrial usage of such materials is still difficult owing to prohibitive production costs.26–29
Dative boron–nitrogen bonds (B←N bonds) are very unique in having a strong covalent character, directionality, and reversible nature.30–32 The utilization of dative B←N bonds in structural supramolecular chemistry has become increasingly popular in recent years.33–35 Since Kay Severin pioneered the synthesis of the first crystalline organic framework based on the B←N bonds (BNOF) by using triboronate esters and bipyridyl ligands in 2011,36 other 1D and 2D crystalline BNOFs have been designed and synthesized by combining polytopic N-donor ligands with polyboronate esters.37–39 It is now known that the robustness of B←N bonds is significantly dependent upon the steric and electronic properties of the interacting components,33,40 as these properties can prevent the sensitive boronate ester moieties from hydrolyzing.41–43 Thus B←N bonds stabilize the resulting organic frameworks. In 2019, Severin's group explored the porosity of BNOFs with sheet-like structures by combining tetrahedral borates with bent diimidazole ligands.44 However, because these BNOFs exhibit an amorphous state after activation, the reported specific surface area is low (≤314 m2 g−1). The synthesis of BNOFs with permanent porosity is still in the initial stages.
When the dative B←N bond is formed, the Lewis basic N-donor strut is of a monodentate fashion; the configuration of the B atom is converted from trigonal-planar (sp2) to tetrahedral geometry (sp3), therefore BNOFs can be controlled and assembled.45–47 Previously reported the synthesis of supramolecular assemblies based on dative B←N bonds usually included two steps: (i) boric acid condensation with diols and/or self-condensation to form borate esters; (ii) adduct formation of borate esters with N-donor ligand (Scheme 1a and b).37,38 It was found that the formation of borate esters often increases the complexity of the monomers, leading to more flexible linkers, and forming unstable or interpenetrated frameworks with low void space.44,48 In contrast, the one-step reaction utilizing a rigid single component not only simplifies the formation procedures but also helps avoid by-products.49,50 Therefore, the single bifunctional component may be a promising alternative to building BNOFs. This strategy has however only rarely been studied.31 In this work, we employed an easily obtained and cheap bifunctional monomer, 4-pyridine boronic acid (PBA), to construct microporous BNOFs (Scheme 1c). It can be speculated that a 0D macrocyclic multimer could be formed through bridging pyridine, once boronic acid is condensed into a boroxine ring (B3O3). Nevertheless, isoreticular expansion needs to connect two B atoms in a stereoscopic configuration on the boroxine (one boroxine cannot exist with three tetrahedral B atoms due to the steric hindrance effect51). As we expected, a 2D crystalline B←N organic framework, BNOF-1, was successfully synthesized by PBA in one step, which is the first BNOF with permanent porosity based on the sole monomer. BNOF-1 can be easily prepared in large amounts and possesses the highest specific surface area compared to previous BNOFs. It also establishes a new benchmark organic framework material for C2H2 storage. According to the breakthrough curve, the high dynamic C2H2 productivity and selectivity allow BNOF-1 to be potentially applied to the separation of C2H2/CO2 mixtures under actual conditions.
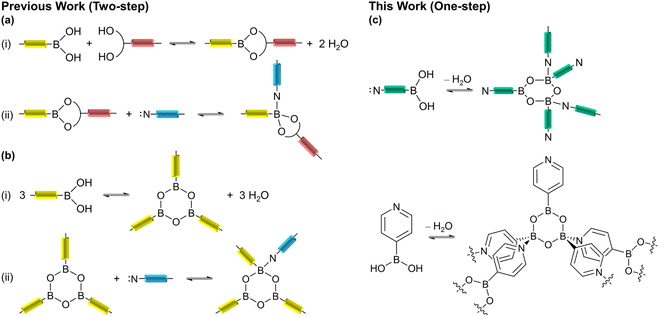 |
| | Scheme 1 (a) and (b) Supramolecular assemblies based on dative B←N bonds that were synthesized in two steps according to previous reports. (c) The incorporated bifunctional monomer used in this work and a fragment of the 2D network in BNOF-1. | |
Results and discussion
Crystal structure
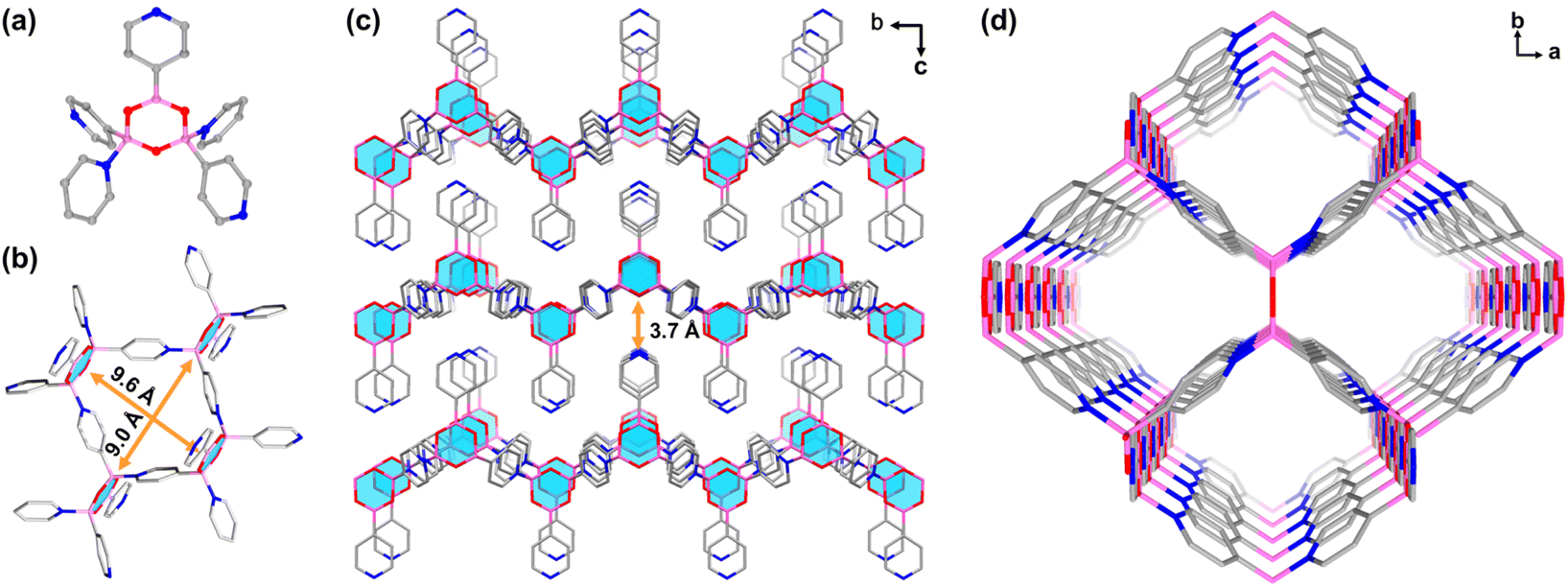
Colourless rhombic single crystals of BNOF-1 were obtained under a solvothermal reaction by heating PBA in a mixture of N-methylformamide (NMF), MeCN, and HCOOH at 120 °C for 12 h. The single-crystal structure shows that BNOF-1 crystallizes in the orthorhombic space group Pmmn. Three PBAs were condensed into a boroxine after removing trimolecular water, and the other two PBAs coordinated with two B atoms in the tetrahedral configuration on the boroxine through dative B←N bonds (Scheme 1c). The B–N bond length of 1.627 Å is similar to the previously reported values.40,47,52 Every boroxine is connected to four bridged pyridines in the trans conformation and one coplanar pyridine (Fig. 1a); the 4-c boroxine node is further extended to construct a 2D layer (Fig. 1b) with sra topology. From the a-axis, these layers are stacked and overlapped to give rise to a supramolecular framework through weak intermolecular forces, with an interlayer distance of about 3.7 Å (Fig. 1c). A square 1D open channel of ∼9.0 × 9.6 Å2 is formed along the c direction (Fig. 1b and d), with accessible free voids covering 49.0%. The end-capped pyridines were staggered and modified on the upper and lower surfaces of the layers (Fig. 1c).
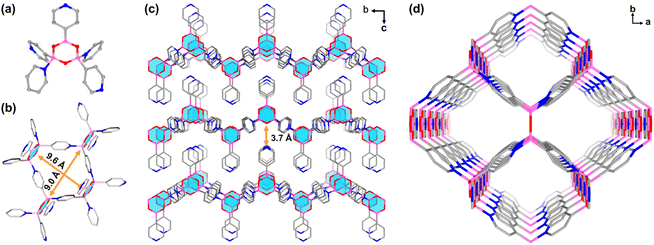 |
| | Fig. 1 (a) Connection mode of boroxine in BNOF-1. (b) Fragment of the tetrameric ring in BNOF-1. (c) 2D layers in BNOF-1 stacked along the a-axis. (d) The 3D supramolecular framework of BNOF-1 is viewed along the c axis. C gray, N navy, B pink, O red (H atoms are omitted for clarity). | |
Stability
The crystalline phase was characterized by powder X-ray diffraction (PXRD) that shows sharp and strong peaks at 2θ equaling 8.1 and 12.0° for synthesized crystals of BNOF-1 (Fig. S1†). Thermogravimetric analysis (TGA) of BNOF-1 showed a 23.7% weight loss between 30 and 260 °C, and then a platform was displayed at 260–380 °C (Fig. S2†), demonstrating high thermal stability. Although BNOF-1 exhibits thermostability at high temperatures, it quickly decomposed into the monomer PBA in water and acidic/basic solutions (Fig. S3†), resulting from the nature of the boroxine formation as a reversible dehydration process.42 The decomposition product, PBA, can be reused to synthesize BNOF-1 (Fig. S3†). However, BNOF-1 can retain its crystallinity in common organic solvents, such as EtOH, CH2Cl2, MeCN, n-hexane, N,N-dimethylformamide, methylbenzene (MB), and acetone (Fig. S4†).
Large-scale production and regeneration
Notably, the highly crystalline BNOF-1 material can be facily synthesized with gram-scale and a high yield, which is imperative from a view of industrial application. In a laboratory attempt, 0.73 g of BNOF-1 could be produced from 1 g of PBA by simply improving the raw material amount (Fig. 2a). Interestingly, the crystals of BNOF-1 can be dissolved in a hot organic solution, and the high-quality crystalline samples can be obtained again by slowly evaporating (Fig. 2b). This process is predominant due to the good reversibility of B←N bonds that enables recrystallization to the original framework after breakage and reassembly,38,44 indicating the excellent regenerability and solution processability of BNOF-1. The measured PXRD results are also consistent with the as-synthesized pattern, indicating phase purity (Fig. S1†).
 |
| | Fig. 2 (a) Large-scale synthesis and (b) regeneration process of BNOF-1. | |
Gas adsorption
Guest-free BNOF-1 can be readily produced by heating under a vacuum at 433 K for 10 h, where BNOF-1 maintains crystallinity after activation (Fig. S1†). The activated sample does not contain guest molecules, as confirmed by TGA (Fig. S2†). The permanent porosity of BNOF-1 was proven by N2 adsorption at 77 K. At low pressure (P/P0 < 0.01), the adsorption amount increased rapidly, exhibiting the I-type adsorption curve of microporous materials with a saturated loading of 340.7 cm3 g−1 (Fig. 3a). By fitting the adsorption data, Brunauer–Emmett–Teller (BET) and Langmuir specific surface areas were 1345 and 1450 m2 g−1, respectively. BNOF-1 represents the highest BET surface areas reported for B–N-based compounds (≤314 m2 g−1) to date.44 According to the Horvath–Kawazoe (H–K) model, the pore size of BNOF-1 is approximately 4.5–9.0 Å (Fig. 3a inset), which is close to the pore aperture metrics from the structure. The experimental pore volume of BNOF-1 is ∼0.50 cm3 g−1 (P/P0 = 0.12), agreeing with the theoretical pore volume (0.55 cm3 g−1). Furthermore, the porosity and BET surface areas can be well maintained for the scale-up and regenerated crystals (Fig. 3f and S5†).
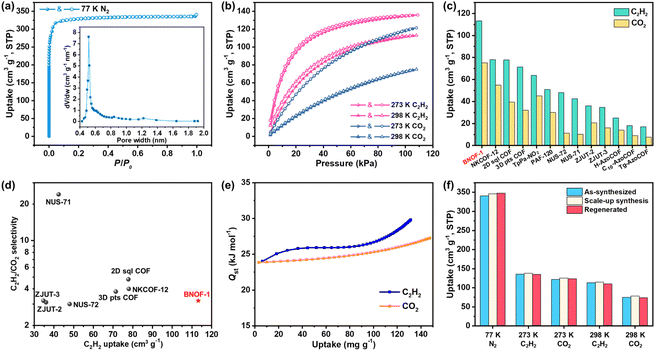 |
| | Fig. 3 (a) N2 sorption isotherm and pore size distribution of BNOF-1 at 77 K. (b) C2H2 and CO2 sorption isotherms of BNOF-1 at 273 and 298 K. (c) Comparison of C2H2 and CO2 uptakes by BNOF-1 with other C2H2-selective COF materials at 298 K. (d) Comparison of selectivity and C2H2 adsorption uptake among COFs at 298 K and 100 kPa. (e) Qst of BNOF-1 for C2H2 and CO2. (f) Comparison of adsorption amounts for N2, C2H2, and CO2 in different samples of BNOF-1. | |
C2H2 is an important raw material widely used in the production of chemical products.53 In industry, trace CO2 is inevitably mixed in the process of producing C2H2 through partial combustion of methane or steam cracking,54 which seriously affects the purity of C2H2 in applications. However, as they have identical kinetic diameters (3.3 Å) and similar boiling points (C2H2, 189.3 K; CO2, 194.7 K, Scheme S1†),55,56 C2H2–CO2 separation is extremely challenging. However, BNOFs have not been explored in the field of gas separation. Inspired by the high porosity and large specific surface area of BNOF-1, we further investigated the adsorption properties of C2H2 and CO2. As shown in Fig. 3b, the adsorption capacities of C2H2 at 273 and 298 K at 1 bar are 135.9 and 113.2 cm3 g−1, respectively. The corresponding uptakes of CO2 are 121.6 and 75.2 cm3 g−1, respectively. From the crystal density and uptake, the C2H2 storage density in BNOF-1 at 298 K was calculated to be 1.48 g cm−3, which is far beyond the safe compression limit at 2 bar (∼704 times, 0.0021 g cm−3).57 In addition, the uptake amounts of BNOF-1 for large-scale prepared and regenerated samples remain unchanged, demonstrating the feasibility of large-scale synthesis and the intactness of the restored framework (Fig. 3f and S6–S9†). Under ambient conditions, BNOF-1 exhibits the highest volumetric adsorption capacity for C2H2 and CO2 (113.2, 75.2 cm3 g−1) as compared to the reported COF materials, such as NKCOF-12 (78, 55 cm3 g−1),58 2D sql COF (77.8, 39.4 cm3 g−1),59 3D pts COF (71.4, 32 cm3 g−1),59 TpPa-NO2 (63.73, 45.18 cm3 g−1),60 PAF-120 (50.8, 30 cm3 g−1),61 NUS-72 (48, 11.2 cm3 g−1),62 NUS-71 (42.4, 10.3 cm3 g−1),62 ZJUT-2 (36, 20.7 cm3 g−1),63 ZJUT-3 (34.7, 16.1 cm3 g−1),63et al. (Fig. 3c and Table S1†), which could be a new benchmark organic framework for C2H2 and CO2 storage.
Gas selectivity
Given the significant C2H2 loading of BNOF-1, the ideal adsorbed solution theory (IAST) was used to evaluate the separation performance of BNOF-1 on C2H2 and CO2 mixtures. According to the fitting results through the single-point Langmuir–Freundlich equation (Fig. S10†), the selectivity of 2.8–3.2 at 298 K (Fig. S11†) is comparable to other COFs, such as ZJUT-2 (3.2),63 2D sql COF (4.8),59 and NKCOF-12 (5.8)58 (Fig. 3d). Subsequently, the Virial II equation was used to estimate the interactions of BNOF-1 with C2H2 and CO2 (Fig. S12†). As shown in Fig. 3e, the adsorption heats (Qst) of CO2 and C2H2 at 298 K were in the range of 23.8–27.3 and 24.0–29.8 kJ mol−1, respectively. The Qst for both C2H2 and CO2 increased with the uptake, which may be because the adsorption process benefits from molecular interactions between the adsorbates.64,65 The overall higher Qst of BNOF-1 for C2H2 than CO2, indicates a higher affinity. The zero-loading Qst (24.0 kJ mol−1) of BNOF-1 for C2H2 is lower than some known COFs (Table S1†). Such a moderate Qst implies that the regeneration of BNOF-1 material is more energy-saving for the uptake and release of C2H2.
Molecular simulations
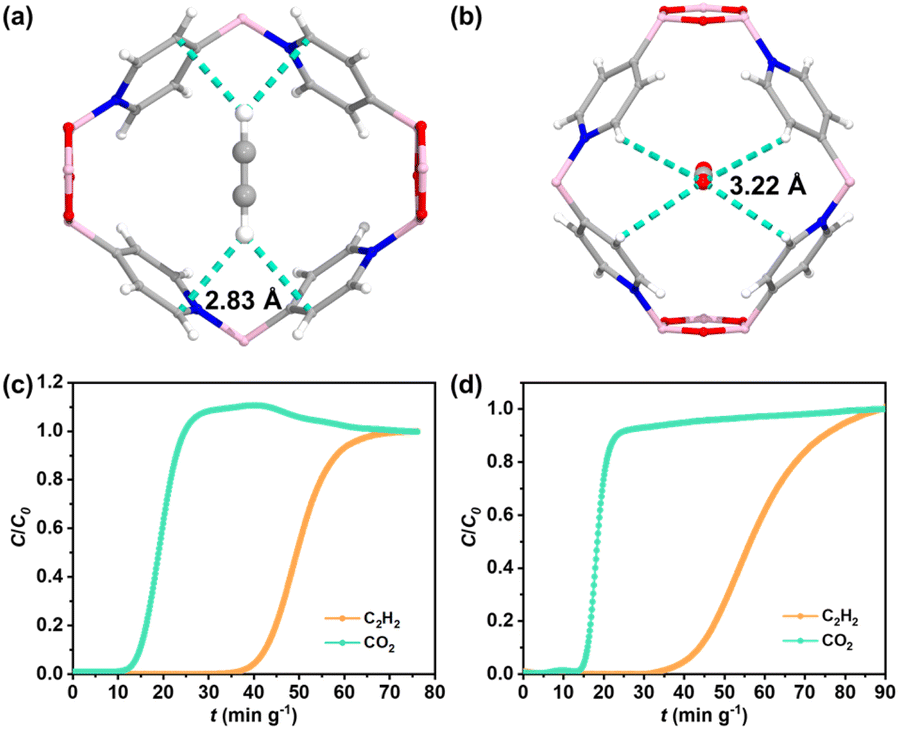
To accurately illustrate the interactions between BNOF-1 and gas molecules, the preferential combined sites of C2H2 and CO2 in the framework were simulated by using the Grand Canonical Monte Carlo (GCMC). As described in Fig. 4a and b, both CO2 and C2H2 molecules are located in the centre of the rhombic channel of BNOF-1. The adsorbed CO2 molecule shows a side-on orientation, while C2H2 has an end-on orientation in terms of the local pore environment, which has also been observed in previous reports on energy reduction.66 One O atom of CO2 molecule forms four C–H⋯O hydrogen bonds (∼3.22 Å) with the C–H units of four pyridine rings. For the C2H2 molecule, two H atoms form four C–H⋯C hydrogen bonds with the C atoms of four pyridine rings (∼2.83 Å), as reported in other MOF materials.67 These similar weak interactions explain the small difference in Qst between C2H2 and CO2, but as the hydrogen bonds formed by the framework with C2H2 are shorter than CO2, it leads to stronger interactions for C2H2 from the framework.
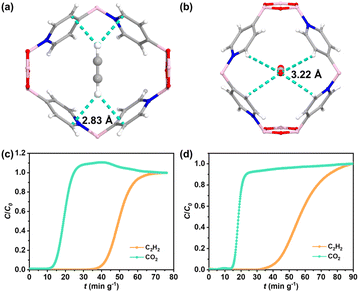 |
| | Fig. 4 Preferential adsorption sites for (a) C2H2 and (b) CO2. Breakthrough plots for the separation of C2H2/CO2 mixtures at 298 K: (c) C2H2–CO2–Ar = 5%–5%–90%, (d) C2H2–CO2–Ar = 5%–10%–85%. | |
Breakthrough experiment
To verify the ability of BNOF-1 to capture C2H2 and separate C2H2/CO2 in gas mixtures, the activated BNOF-1 (0.6 g) was filled into the packed bed for dynamic breakthrough experiments. At a flow rate of 5 mL min−1, CO2 was detected at 15 min g−1 for the equimolar C2H2 and CO2 mixture (C2H2–CO2–Ar = 5%–5%–90%), while C2H2 could be retained at 40.4 min g−1. Therefore, the penetration time interval was 25.3 min g−1 (Fig. 4c), exceeding COF material NKCOF-12 (14 min g−1)58 and MOF material FJU-22 (11 min g−1)68 under similar conditions. Based on the working capacity, 1 kg of BNOF-1 could adsorb 12.5 L of C2H2 in BNOF-1 and directly produce 4.0 L of CO2 with high purity (>99.0%). The yield of C2H2 was higher than that of NKCOF-12 (10.04 L),58 but lower than the COF materials, NUS-71 (31.4 L)62 and NUS-72 (35.8 L),62 as well as several benchmark MOFs for C2H2/CO2 separation supported by open metal sites and/or polar functional groups, such as TIFSIX-2-Cu-i (73.9 L),69 FJU-90 (41.9 L),70 CPL-1-NH2 (30.9 L),71 and UTSA-300 (17.2 L).72 The dynamic separation ratio could be determined to be 3.1, which was consistent with the IAST selectivity (3.2) and surpassed the values in some MOFs, such as FJU-22 (1.9),68 SNNU-45 (2.9),73 and NKMOF-1-Ni (2.6).74 When the ratio of C2H2 and CO2 is changed to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (C2H2–CO2–Ar = 5%–10%–85%), BNOF-1 can still completely separate C2H2 and CO2, with the breakthrough time interval of 25 min g−1 (Fig. 4d). The excellent C2H2/CO2 separation ability of BNOF-1 may be attributed to high C2H2 adsorption capacity and significant C2H2/CO2 selectivity. In addition, after the breakthrough experiments, the samples of BNOF-1 are still intact, as confirmed by PXRD (Fig. S1†).
:2 (C2H2–CO2–Ar = 5%–10%–85%), BNOF-1 can still completely separate C2H2 and CO2, with the breakthrough time interval of 25 min g−1 (Fig. 4d). The excellent C2H2/CO2 separation ability of BNOF-1 may be attributed to high C2H2 adsorption capacity and significant C2H2/CO2 selectivity. In addition, after the breakthrough experiments, the samples of BNOF-1 are still intact, as confirmed by PXRD (Fig. S1†).
Conclusions
In conclusion, we present the first example of crystalline microporous 2D BNOF with permanent porosity, which was constructed through single-component monomers. BNOF-1 with high porosity shows the highest surface area amongst known BNOFs and a benchmark C2H2 adsorption capacity in COFs. BNOF-1 can be conveniently prepared on a large scale by a one-step reaction and regenerated under mild conditions. The breakthrough experiment proves that BNOF-1 can efficiently separate CO2 and C2H2 mixtures with a long C2H2 retention time and high dynamic selectivity. The synthesis of BNOF-1 would enrich supramolecular chemistry and reticular chemistry. It is expected that more innovative B←N bonds-based organic frameworks will be produced for their broad applications in the future. Notably, a newly published study reported an example of BNOF with permanent porosity during the peer review stage of this study, which was based on two different monomers with a relatively low surface area (255 m2 g−1) compared to BNOF-1.75
Data availability
All experimental supporting data are provided in the ESI.†
Author contributions
W. Wang and L. Wang conceived the idea and designed the project, and they contributed equally to this work. L. Wang, F. Du and G.-D. Wang worked on the synthesis and characterization. W. Wang, B. Liu, and L. Hou analyzed the results and wrote the manuscript. B. Liu, L. Hou, Y.-Y. Wang, and Z. Zhu provided reagents, materials, and analysis tools, as well as supervised the work. All authors contributed to the discussion and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the support of this work by the Natural Science Foundation of Shaanxi Province (2021JM-084), and the National Natural Science Foundation of China (21871220).
Notes and references
- M. E. Davis, C. Saldarriaga, C. Montes, J. Garces and C. Crowdert, Nature, 1988, 331, 698–699 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- G. Maurin, C. Serre, A. Cooper and G. Férey, Chem. Soc. Rev., 2017, 46, 3104–3107 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef PubMed.
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef PubMed.
- X. Li, S. Cai, B. Sun, C. Yang, J. Zhang and Y. Liu, Matter, 2020, 3, 1507–1540 CrossRef.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2017, 345, 54–72 CrossRef.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- S. Subramanian and M. J. Zaworotko, Coord. Chem. Rev., 1994, 137, 357–401 CrossRef.
- G. Berger, J. Soubhye and F. Meyer, Polym. Chem., 2015, 6, 3559–3580 RSC.
- B. Li, S.-Q. Zang, L.-Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Cryst. Growth Des., 2018, 18, 7–15 CrossRef.
- L. J. Barbour, Chem. Commun., 2006, 1163–1168 RSC.
- T. Adachi and M. D. Ward, Acc. Chem. Res., 2016, 49, 2669–2679 CrossRef.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef PubMed.
- P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- U. Ryu, S. Jee, P. C. Rao, J. Shin, C. Ko, M. Yoon, K. S. Park and K. M. Choi, Coord. Chem. Rev., 2021, 426, 213544 CrossRef.
- L. Hashemi, M. Y. Masoomi and H. Garcia, Coord. Chem. Rev., 2022, 472, 214776 CrossRef.
- Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef PubMed.
- D. Salazar-Mendoza, J. Guerrero-Alvarez and H. Höpfl, Chem. Commun., 2008, 6543–6545 RSC.
- A. Dhara and F. Beuerle, Chem.–Eur. J., 2015, 21, 17391–17396 CrossRef.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- K. Ono, S. Shimo, K. Takahashi, N. Yasuda, H. Uekusa and N. Iwasawa, Angew. Chem., Int. Ed., 2018, 57, 3113–3117 CrossRef PubMed.
- E. C. Vargas-Olvera, F. J. Salas-Sánchez, A. Colin-Molina, S. Pérez-Estrada, B. Rodríguez-Molina, J. Alejandre, G. Campillo-Alvarado, L. R. MacGillivray and H. Höpfl, Cryst. Growth Des., 2022, 22, 570–584 CrossRef.
- E. Sheepwash, V. Krampl, R. Scopelliti, O. Sereda, A. Neels and K. Severin, Angew. Chem., Int. Ed., 2011, 50, 3034–3037 CrossRef.
- K. Severin, Dalton Trans., 2009, 5254–5264 RSC.
- B. Icli, E. Sheepwash, T. Riis-Johannessen, K. Schenk, Y. Filinchuk, R. Scopelliti and K. Severin, Chem. Sci., 2011, 2, 1719–1721 RSC.
- N. Luisier, K. Bally, R. Scopelliti, F. T. Fadaei, K. Schenk, P. Pattison, E. Solari and K. Severin, Cryst. Growth Des., 2016, 16, 6600–6604 CrossRef CAS.
- Y. Kubo, R. Nishiyabu and T. D. James, Chem. Commun., 2015, 51, 2005–2020 RSC.
- D. Kim, D. H. Jung, K.-H. Kim, H. Guk, S. S. Han, K. Choi and S.-H. Choi, J. Phys. Chem. C, 2012, 116, 1479–1484 CrossRef CAS.
- Y. Du, K. Mao, P. Kamakoti, P. Ravikovitch, C. Paur, S. Cundy, Q. Li and D. Calabro, Chem. Commun., 2012, 48, 4606–4608 RSC.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 RSC.
- A.
J. Stephens, R. Scopelliti, F. F. Tirani, E. Solari and K. Severin, ACS Mater. Lett., 2019, 1, 3–7 CrossRef.
- H. Höpfl, J. Organomet. Chem., 1999, 581, 129–149 CrossRef.
- A. L. Korich and P. M. Iovine, Dalton Trans., 2010, 39, 1423–1431 RSC.
- G. Campillo-Alvarado, K. P. D’mello, D. C. Swenson, S. V. Santhana Mariappan, H. Höpfl, H. Morales-Rojas and L. R. MacGillivray, Angew. Chem., Int. Ed., 2019, 58, 5413–5416 CrossRef PubMed.
- B. Icli, E. Solari, B. Kilbas, R. Scopelliti and K. Severin, Chem.–Eur. J., 2012, 18, 14867–14874 CrossRef PubMed.
- J. Cruz-Huerta, D. Salazar-Mendoza, J. Hernández-Paredes, I. F. Hernández Ahuactzi and H. Höpfl, Chem. Commun., 2012, 48, 4241–4243 RSC.
- D. Salazar-Mendoza, J. Cruz-Huerta, H. Höpfl, I. F. Hernández-Ahuactzi and M. Sanchez, Cryst. Growth Des., 2013, 13, 2441–2454 CrossRef CAS.
- C. Bao, Y.-J. Jiang, H. Zhang, X. Lu and J. Sun, Adv. Funct. Mater., 2018, 28, 1800560 CrossRef.
- A. Torres-Huerta, M. J. Velásquez-Hernández, D. Martínez-Otero, H. Höpfl and V. Jancik, Cryst. Growth Des., 2017, 17, 2438–2452 CrossRef CAS.
-
P. J. Stang and F. Diederich, Modern Acetylene Chemistry, VCH-Weinheim, 1995, pp. 1–499 Search PubMed.
- A. Corma, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Al-Bogami, M. S. Al-Ghrami and A. Bourane, Catal. Sci. Technol., 2017, 7, 12–46 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- C. R. Reid and K. M. Thomas, J. Phys. Chem. B, 2001, 105, 10619–10629 CrossRef CAS.
- W. Gong, H. Cui, Y. Xie, Y. Li, X. Tang, Y. Liu, Y. Cui and B. Chen, J. Am. Chem. Soc., 2021, 143, 14869–14876 CrossRef CAS PubMed.
- P. Zhang, Z. Wang, Y. Yang, S. Wang, T. Wang, J. Liu, P. Cheng, Y. Chen and Z. Zhang, Sci. China Chem., 2022, 65, 1173–1184 CrossRef CAS.
- L. Chen, C. Gong, X. Wang, F. Dai, M. Huang, X. Wu, C.-Z. Lu and Y. Peng, J. Am. Chem. Soc., 2021, 143, 10243–10249 CrossRef CAS.
- X.-H. Xiong, L. Zhang, W. Wang, N.-X. Zhu, L.-Z. Qin, H.-F. Huang, L.-L. Meng, Y.-Y. Xiong, M. Barboiu, D. Fenske, P. Hu and Z.-W. Wei, ACS Appl. Mater. Interfaces, 2022, 14, 32105–32111 CrossRef CAS.
- L. Jiang, P. Wang, M. Li, P. Zhang, J. Li, J. Liu, Y. Ma, H. Ren and G. Zhu, Chem.–Eur. J., 2019, 25, 9045–9051 CrossRef CAS PubMed.
- Z. Zhang, C. Kang, S. B. Peh, D. Shi, F. Yang, Q. Liu and D. Zhao, J. Am. Chem. Soc., 2022, 144, 14992–14996 CrossRef CAS PubMed.
- C. Gong, H. Wang, G. Sheng, X. Wang, X. Xu, J. Wang, X. Miao, Y. Liu, Y. Zhang, F. Dai, L. Chen, N. Li, G. Xu, J. Jia, Y. Zhu and Y. Peng, Angew. Chem., Int. Ed., 2022, 61, e202204899 CAS.
- S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS.
- H. Zeng, M. Xie, Y.-L. Huang, Y. Zhao, X.-J. Xie, J.-P. Bai, M.-Y. Wan, R. Krishna, W. Lu and D. Li, Angew. Chem., Int. Ed., 2019, 58, 8515–8519 CrossRef CAS PubMed.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS.
- S.-Q. Yang, L. Zhou, Y. He, R. Krishna, Q. Zhang, Y.-F. An, B. Xing, Y.-H. Zhang and T.-L. Hu, ACS Appl. Mater. Interfaces, 2022, 14, 33429–33437 CrossRef.
- Z. Yao, Z. Zhang, L. Liu, Z. Li, W. Zhou, Y. Zhao, Y. Han, B. Chen, R. Krishna and S. Xiang, Chem.–Eur. J., 2016, 22, 5676–5683 CrossRef PubMed.
- K.-J. Chen, H. S. Scott, D. G. Madden, T. Pham, A. Kumar, A. Bajpai, M. Lusi, K. A. Forrest, B. Space, J. J. Perry and M. J. Zaworotko, Chem, 2016, 1, 753–765 Search PubMed.
- Y. Ye, Z. Ma, R.-B. Lin, R. Krishna, W. Zhou, Q. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2019, 141, 4130–4136 CrossRef PubMed.
- L. Yang, L. Yan, Y. Wang, Z. Liu, J. He, Q. Fu, D. Liu, X. Gu, P. Dai, L. Li and X. Zhao, Angew. Chem., Int. Ed., 2021, 60, 4570–4574 CrossRef PubMed.
- R.-B. Lin, L. Li, H. Wu, H. Arman, B. Li, R.-G. Lin, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 8022–8028 CrossRef PubMed.
- Y.-P. Li, Y. Wang, Y.-Y. Xue, H.-P. Li, Q.-G. Zhai, S.-N. Li, Y.-C. Jiang, M.-C. Hu and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 13590–13595 CrossRef PubMed.
- Y.-L. Peng, T. Pham, P. Li, T. Wang, Y. Chen, K.-J. Chen, K. A. Forrest, B. Space, P. Cheng, M. J. Zaworotko and Z. Zhang, Angew. Chem., Int. Ed., 2018, 57, 10971–10975 CrossRef CAS PubMed.
- H. Zhang, Y. Li, L. Chen, Y. Yang, H. Lin, S. Xiang, B. Chen and Z. Zhang, Chem, 2022 DOI:10.1016/j.chempr.2022.10.023.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, PXRD analysis, TGA curves, additional adsorption isotherms, calculation adsorption selectivity and heat, IR spectra, comparison of adsorption performances and BET, X-ray crystallography details and other data. CCDC 2207247. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06016g |
| ‡ These authors have contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Zhonghua
Zhu
*b,
Zhonghua
Zhu