
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidation of styrene to benzaldehyde by discrimination of spin-paired π electrons†
Xiaoxue
Luo
a,
Xiaoxia
Tang
a,
Jingtian
Ni
a,
Baijing
Wu
a,
Cunpu
Li
 *a,
Minhua
Shao
b and
Zidong
Wei
*a
*a,
Minhua
Shao
b and
Zidong
Wei
*a
aThe State Key Laboratory of Power Transmission Equipment & System Security and New Technology, Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, College of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400044, China. E-mail: lcp@cqu.edu.cn; zdwei@cqu.edu.cn
bDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
First published on 27th January 2023
Abstract
The oxidation of styrene to benzaldehyde has been a considerable challenge in the electrochemical synthesis of organic compounds because styrene is more easily oxidized to benzoic acid. In this work, MnO2 with an asymmetric electronic configuration is designed to discriminate the spin-paired π electrons of styrene. One of these discriminated π electrons combined with reactive oxygen species (ROS), ˙OH, ˙OOH, etc., produced simultaneously on a MnO2/(Ru0.3Ti0.7)O2/Ti bifunctional anode, to form benzaldehyde via Grob fragmentation, rather than benzoic acid. However, only benzoic acid is obtained from the oxidation of styrene on the anodes MOs/(Ru0.3Ti0.7)O2/Ti, where MOs are other metal oxides with symmetric electronic configurations.
1 Introduction
Electrochemical synthesis is widely used in the development of chemical products.1–7 As electrons can be injected into or ejected from raw molecules by regulating the applied electric potentials, electrochemical synthesis therefore possesses many advantages, e.g., stoichiometric oxidants or reductants can be avoided, flow reactors can be used instead of batch reactors, and the reaction dynamics can be easily accelerated by adjusting the applied current density.8,9 Among the developed electrochemical synthesis methods, organic selective electrochemical oxidation is both intriguing and challenging from a catalysis perspective due to its complexity: the reaction either fails to proceed or proceeds in excess, making the aim of achieving intermediate products difficult.10–17 On the other hand, developing organic selective oxidation is important not only for the environment but also for the economies of energy and raw materials, for example, the production of the simplest aromatic aldehyde, benzaldehyde.18 Benzaldehyde is commonly used in medicine, pesticides, spices, dyes, etc. The conventional synthesis of benzaldehyde from styrene always requires massive amounts of oxidants (H2O2, TBHP, etc.) and catalysts (Au, Ag, TiO2, TS-1, etc.) and harsh reaction conditions (high temperature, high pressure, etc).19–23 Also, benzaldehyde is highly reactive, therefore, the inexpensive but hard-to-separate byproduct benzoic acid is inevitably generated, which decreases the utilization efficiency of styrene and increases the separation costs of benzaldehyde.24,25 Therefore, it is a not commonly used method to produce benzaldehyde from styrene, due to the superior stability of styrene and the poor separation efficiency. To overcome these obstacles, the electrochemical oxidation of styrene to benzaldehyde with green oxidants, can therefore be achieved as long as the reactivity of styrene and overoxidation problems can be solved.The vinyl bond of styrene is conjugated with the benzene ring, which forms an ultrastable aromatic system. Therefore, it is not easy to oxidize styrene, and it is even more difficult to selectively oxidize styrene because of its stable conjugated bonds, as mentioned before, resulting in either no reaction or overreaction. To achieve the selective oxidation of the vinyl bond, its conjugation with the aromatic ring must be weakened. Fortunately, the presence of MnO2 makes it possible. MnO2 has an asymmetric electronic density of states (DOS) distribution. This means that the energy distributions of the upper and lower electrons for MnO2 are not symmetric.26 Hence, as shown in Scheme 1, when styrene is adsorbed on the MnO2 surface via its vinyl group, the asymmetric distributed electrons in MnO2 will form different interactions with the paired two electrons in the vinyl group. Therefore, the two paired π electrons in the vinyl group that were previously degenerate in energy, will differ in energy and density distribution because of the asymmetric electronic interaction with MnO2 electrons. We call this phenomenon as the “discrimination” of the vinyl electrons. Therefore, after the vinyl group adsorbed onto MnO2, the electrons in the vinyl group will be discriminated to two energy distinct π electrons, accompanied by the weakening of the conjugative effect with aromatic rings. In other words, the styrene is selectively activated. This makes the styrene serve as a “diradical” (ph–CH˙–CH2˙), the two “singly occupied” electrons can be therefore selectively oxidized by different oxidants with different activities and energies, respectively. Reactive oxygen species (ROS), such as ˙OH and ˙OOH, can be easily generated on the anode via the oxygen evolution reaction (OER) during water electrolysis, where ROS are further converted to oxygen gas and emitted directly to the atmosphere.27–30 Therefore, coupling these ROS radicals with the styrene “diradical” to selectively oxidize styrene is not only a novel green approach to produce benzaldehyde from styrene but also a promising strategy to increase the energy efficiency of hydrogen production.
 | ||
| Scheme 1 Electrochemical oxidation of styrene with a MnO2/(Ru0.3Ti0.7)O2/Ti electrode in acid media coupled with a water splitting reaction. MnO2 and RuO2 were used to activate styrene and produce ROS, respectively. | ||
In this work, we developed an electrochemical oxidation system to produce benzaldehyde from styrene. Styrene was activated by discriminating its vinyl π electrons with the aid of the asymmetric electronic configuration of MnO2. ROS from RuO2-catalyzed water splitting were used as clean oxygen sources. Therefore, bifunctional catalytic electrodes consisting of MnO2 and RuO2 were developed to discriminate the spin-paired π electrons of styrene and produce ROS. As shown in Scheme 1, the asymmetric electronic configuration of MnO2 was employed to discriminate the paired π electrons of styrene to form the “diradical”. In addition, the traditional water electrolysis catalyst RuO2–TiO2 was used to generate ROS, thereby realizing the selective oxidation of styrene coupled with water splitting via Grob fragmentation.31
2 Results and discussion
2.1 Electrode design and preparation
All electrode preparation processes are described in the ESI.† Rutile-type RuO2 and TiO2 were simultaneously introduced to Ti mesh with a thermal chemical deposition method. Specifically, RuO2 was used to catalyze the splitting of water to obtain abundant ROS. TiO2 was used to maintain excellent adhesion between the metal oxide and the Ti mesh and to stabilize the crystallinity via the solid solution structure.32,33 The molar ratios of the Ru and Ti precursors were controlled at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, 2:8, 1:9 and 0.5:9.5, which was examined using the electro-probe microanalyzer (EPMA) images (Fig. S1†). Therefore, the obtained electrodes were named (Ru0.3Ti0.7)O2/Ti, (Ru0.2Ti0.8)O2/Ti, (Ru0.1Ti0.9)O2/Ti, and (Ru0.05Ti0.95)O2/Ti. γ-MnO2 was further introduced to the presynthesized (RuxTi1−x)O2/Ti (where x = 0.3, 0.2, 0.1, and 0.05) electrodes via an electrochemical oxidation method. The resultant bifunctional electrodes were named MnO2/(RuxTi1−x)O2/Ti (where x = 0.3, 0.2, 0.1, 0.05). Additionally, an electrode in which MnO2 was deposited directly on Ti mesh (MnO2/Ti) was prepared for comparison.
:7, 2:8, 1:9 and 0.5:9.5, which was examined using the electro-probe microanalyzer (EPMA) images (Fig. S1†). Therefore, the obtained electrodes were named (Ru0.3Ti0.7)O2/Ti, (Ru0.2Ti0.8)O2/Ti, (Ru0.1Ti0.9)O2/Ti, and (Ru0.05Ti0.95)O2/Ti. γ-MnO2 was further introduced to the presynthesized (RuxTi1−x)O2/Ti (where x = 0.3, 0.2, 0.1, and 0.05) electrodes via an electrochemical oxidation method. The resultant bifunctional electrodes were named MnO2/(RuxTi1−x)O2/Ti (where x = 0.3, 0.2, 0.1, 0.05). Additionally, an electrode in which MnO2 was deposited directly on Ti mesh (MnO2/Ti) was prepared for comparison.
To understand the effect of the asymmetric electronic configuration of MnO2 on the selective oxidation of styrene, rutile-type TiO2/(Ru0.3Ti0.7)O2/Ti and MoO3/(Ru0.3Ti0.7)O2/Ti electrodes were prepared by the same thermal treatment method by using TiO2 and MoO3 with perfectly symmetric electronic configurations. The precursors were tetrabutyl titanate and ammonium molybdate, respectively. For comparison, a MoO2/(Ru0.3Ti0.7)O2/Ti electrode with a slightly asymmetric electronic configuration of MoO2 was produced by the electrodeposition reduction method. Fig. S2–S5† show that all the corresponding materials were successfully synthesized by these procedures. The crystal form and exposed crystal plane of these materials were obtained from the corresponding XRD spectra and TEM images. Density functional theory (DFT) calculations were then performed according to the above obtained crystalline information.
2.2 Theoretical discussion
As discussed previously, MnO2 can potentially discriminate the vinyl π electrons of styrene molecules by its asymmetric electronic configuration. The (131) lattice plane of γ-MnO2, which was extracted from XRD, TEM and HAADF-STEM (Fig. S2 and S4†), was applied to perform the DFT calculation. As shown in Fig. 1(a), the DOS of the upper and lower spin electrons is highly asymmetric. Styrene itself has a closed shell configuration and spin-symmetric electronic orbitals (Fig. 1(b)). The completely symmetrically distributed DOS makes it difficult to selectively oxidize styrene to benzaldehyde. From the DOS of styrene, we can summarize the following reasons why styrene is too stable to be selectively oxidized. First, the DOS of each orbital of styrene is dissociated, which means that the electrons are strictly confined within a certain energy range (molecular orbitals). Second, the energy of the electrons in the highest occupied molecular orbital (HOMO) is much lower than the Fermi level (ca. 2 eV energy gap), which makes them difficult to be activated or removed. Third, the DOS of styrene is perfectly symmetric, indicating that the paired π electrons in the vinyl group of styrene can only be simultaneously activated. | ||
| Fig. 1 The total DOS of MnO2 and the partial DOS of styrene before and after the adsorption on MnO2 (131) with two adsorption patterns. | ||
In contrast, after being adsorbed onto the MnO2 surface, as shown in Fig. 1(c and d) and S6,† styrene underwent two changes. First, the DOS of styrene adsorbed on MnO2 was delocalized and merged with adjacent orbitals/energy bands. Second, the upper and lower DOS lost symmetry. The former facilitated the jump/transfer of the electrons of the adsorbed styrene between adjacent orbitals/energy bands. The latter enabled the selective activation of the electrons in the upper or lower orbitals because of their energy difference. All these factors facilitated the removal of only one electron from the vinyl moiety of the adsorbed styrene to produce the “diradical”. In addition, the DOS of the upper spin electrons of styrene became more spread out than that of the lower spin electrons because MnO2 has a more spread DOS of the upper electrons. Therefore, the introduction of MnO2 caused the paired π electrons in the vinyl group of styrene to be more easily discriminated and activated.
To further confirm our hypothesis, the asymmetrical electronic configuration of an electrode is essential for activating one of the double bonds of styrene to produce benzaldehyde. We chose electrode materials with different degrees of electronic configuration symmetries: TiO2 (perfectly symmetric), MoO3 (slightly symmetric), MoO2 (slightly asymmetric), and MnO2 (extremely asymmetric, as discussed above). The corresponding electrode materials were grown on the synthesized (Ru0.3Ti0.7)O2/Ti. Their exposed lattice faces were examined with XRD and TEM (Fig. S2 and S5†), and DFT calculations were performed (Fig. S7†). The corresponding DOS of the exposed material surfaces and PDOS for the adsorbed styrene on these surfaces were calculated and illustrated in Fig. 2. The DOS of TiO2 (101), MoO3 (110) and MoO2 (110) exhibited the expected symmetricities. The PDOS of the adsorbed styrene is consistent with the DOS symmetries of TiO2 (101), MoO3 (110), and MoO2 (110). All the synthesized electrodes with different DOS symmetries were used for the oxidation of styrene coupled with electric water splitting.
 | ||
| Fig. 2 The DOS of MoO2 (110), MoO3 (110) and TiO2 (101) and the PDOS of styrene before and after adsorption on the corresponding electrode materials, (a) for MoO2 (110), (b) for MoO3 (110) and (c) for TiO2 (101), respectively. | ||
2.3 Oxidation of styrene coupled with electric water splitting
The electrochemical oxidation of styrene was conducted in an H-type three-electrode system electrolytic cell (see Scheme 2), where the cathode was a Pt sheet, the anode was the studied electrode, and the electrolyte was 0.5 M sulfuric acid (pH = 0) with 1,4-dioxane. 1,4-Dioxane was introduced to increase the solubility of styrene in water. The volume ratio of electrolyte and 1,4-dioxane was controlled to be 25:5.
 | ||
| Scheme 2 The electrode design and electrolyte composition. | ||
As shown in Fig. 3, in the case of Ti mesh and MnO2/Ti, there is no anodic current observed on them regardless of styrene. This means that these two electrodes are not active for water oxidation and styrene oxidation. In the case of (Ru0.3Ti0.7)O2/Ti, the current density without styrene addition is greater than that with styrene. This means that (Ru0.3Ti0.7)O2/Ti is only active for water oxidation but not for styrene oxidation. Even worse, styrene inhibits water oxidation on (Ru0.3Ti0.7)O2/Ti. Fig. 3 shows that styrene oxidation only takes place on the (Ru0.3Ti0.7)O2/Ti electrode, where the current density with styrene addition becomes greater than that without styrene after the electrode potential passes through 1.78 V (vs. RHE). Below 1.78 V (vs. RHE), styrene adsorption on the electrode only weakens water adsorption and inhibits water oxidation. However, above 1.78 V (vs. RHE), the orbital energy of styrene is regulated to match the energy of ROS. In other words, some of the paired π electrons in the vinyl group can consume the produced ROS. In this case, the current density with styrene addition is greater than that without styrene. However, an electrode potential that is too high should be avoided in the case of styrene overoxidation to benzoic acid rather than to benzaldehyde, as shown in Fig. S9(b and c).† Therefore, we take 1.78 V (vs. RHE) as the styrene activation potential in the studied system. The styrene oxidation products were checked after 3 hours at 1.78 V (vs. RHE), as summarized in Table 1. Additionally, the Ru amount in RuO2/TiO2 is crucial for ROS generation. We further screened the Ru:Ti ratio to obtain the electrode with the highest catalytic efficiency, which is summarized in Fig. S8(a) and S9(a).† A Ru:Ti ratio of 3:7 is thought to be the best composition for styrene electro-oxidation.
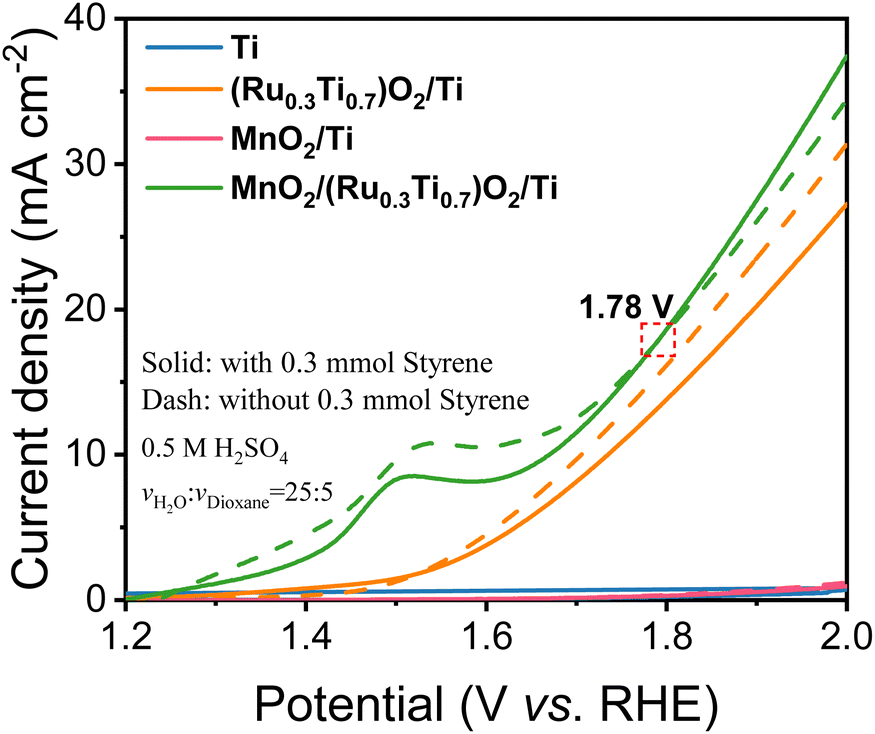 | ||
| Fig. 3 LSV curves of the Ti mesh electrodes catalyzed by different materials at a scan rate of 5 mV s−1 in 0.5 M H2SO4 and 1,4-dioxane cosolvents (v1:v2 = 25:5) with (solid lines) and without (dashed lines) 0.3 mmol styrene. | ||
| Entry | Electrode | Symmetry | ROS | Con./% | Sel./% |
|---|---|---|---|---|---|
| 1a | Ti | N.A. | N.A. | 0 | 0 |
| 1b | (Ru0.3Ti0.7O2)/Ti | N.A. | ✓ | 49.34 | 0 |
| 1c | MnO2/Ti | Asymmetric | N.A. | 0 | 0 |
| 1d | MnO2/(Ru0.3Ti0.7)O2/Ti | Asymmetric | ✓ | 79.78 | 65.02 |
| 1e | MoO2/(Ru0.3Ti0.7)O2/Ti | Slightly asymmetric | ✓ | 58.34 | 6.59 |
| 1f | MoO3/(Ru0.3Ti0.7O2)/Ti | Symmetric | ✓ | 35.07 | 0 |
| 1g | TiO2/(Ru0.3Ti0.7O2)/Ti | Perfectly symmetric | ✓ | 63.34 | 0 |
The onset potential for the optimized MnO2/(Ru0.3Ti0.7)O2/Ti electrode was evaluated with the potential step method. The potential was gradually stepped up from 1.28 V to 1.88 V (vs. RHE). From the obtained I–t curves as illustrated in Fig. S10,† we can find that when the potential increased to 1.33 V (vs. RHE), the current can be detected, and the benzaldehyde product can be obtained. Therefore, we believe that 1.33 V (vs. RHE) is the onset potential for the styrene electro-oxidation reaction. The amount of the styrene was also optimized to be 0.3 mmol in 30 mL electrolyte (Fig. S11†), where the amounts of ROS and adsorbed styrene can be made to strike a balance.
As expected from the theoretical discussion, Fig. 3 and Table 1 show that only the electrodes with an asymmetric DOS and with the participation of the ROS can selectively oxidize styrene to benzaldehyde, in which MnO2/(Ru0.3Ti0.7)O2/Ti with a grossly asymmetric DOS has 79.78% conversion and 65.02% selectivity for benzaldehyde (entry 1d), MoO2/(Ru0.3Ti0.7)O2/Ti with a slightly asymmetric DOS has 58.34% conversion and 6.59% (entry 1e). The electrodes with a symmetric DOS and even with the participation of the ROS cannot selectively oxidize styrene to benzaldehyde but directly to benzoic acid (entries 1f and 1g), which means that the paired π electrons in the vinyl group of styrene are simultaneously activated rather than only one electron being selectively activated. In the bifunctional electrodes, RuO2 was used to produce ROS. Therefore, the electrode without ROS (without the RuO2 catalyst) cannot produce benzaldehyde either (entries 1a and 1c).
2.4 Mechanism discussion
Commonly, water electrolysis can be performed in acidic or basic electrolytes to achieve high OER efficiency. Therefore, we performed our coupled electric oxidation of styrene with water splitting in multiple pH environments. However, as summarized in Table 2, we found that the selective oxidation of styrene can only be realized in rigorous acidic environments (pH = 0). To eliminate the possibility that the styrene was directly oxidized on the electrode, the electric oxidation reaction of styrene under anoxic and anhydrous conditions was performed. As shown in Fig. S12,† there was no obvious direct electron transfer process after the adsorption of styrene on the electrode surface. Additionally, no benzaldehyde product can be obtained when pure oxygen gas is pumped into the styrene–water system, which suggests that styrene is not directly oxidized by the oxygen gas either.|
|
|||
|---|---|---|---|
| Entry | pH | Con./% | Sel./% |
| 2a | 0 | 78.78 | 65.02 |
| 2b | 2 | N.A. | N.A. |
| 2c | 4 | N.A. | N.A. |
| 2d | 7 | N.A. | N.A. |
| 2e | 14 | N.A. | N.A. |
To delve into the mechanism of the reaction, electron paramagnetic resonance (EPR) and in situ attenuated total reflectance FTIR (ATR-FTIR) experiments were performed. As shown in the EPR spectra (Fig. 4(a)), both ˙OH and ˙OOH radicals can be observed in the electrolyte during the styrene electro-oxidation.34 Also, as shown in the ATR-IR spectra (Fig. 4(a)), three distinct absorption peaks at 1089 cm−1, 1128 cm−1 and 3729 cm−1 for ˙OO˙ and ˙OH radicals can be observed after the electro-oxidation happened.35,36 The benzaldehyde peaks (1702 cm−1 and 1980 cm−1) can be identified, as well as the formaldehyde byproduct peaks (1471 cm−1 and 1626 cm−1).37–39 The formed formaldehyde is extremely reactive, which will be further oxidized to formic acid (HCOOH, detected with HPLC, Fig. S13†).
 | ||
| Fig. 4 (a) The electron paramagnetic resonance (EPR) spectra of the reaction solution at different reaction times. The DMPO trapping agent was used to trap the radicals. In situ attenuated total reflectance FTIR (ATR-FTIR) spectra (b), EPR spectra (c), and in situ electrochemical XPS spectra (d) of the electrode–solution interface at different reaction times, respectively. MnO2/(Ru0.3Ti0.7)O2/Ti was used as the electrode, potential: 1.78 V (vs. RHE). | ||
The absorption peaks at 2857 cm−1 and 2968 cm−1 can be ascribed to styrene.40 It can be found that as the reaction time lapsed, the amount of the adsorbed styrene first increased then decreased. The increase can be attributed to the spontaneous enrichment of styrene on the electrode, when the electric potential is applied; the further decrease corresponds to the gradual drain of styrene, which is accompanied by the consumption of the ˙OH and ˙OOH radicals (decrease of the ˙OO˙ signal and formation of the negative peak for ˙OH). It is interesting that the absorption peaks for styrene somewhat red shifted to the region of phenylethane, which can be ascribed the formation of the “diradical” that the conjugated system between the vinyl group and aromatic ring is weakened.40 The interaction between styrene and MnO2 was further evaluated with EPR measurements (Fig. 4(c)) and in situ electrochemical XPS (Fig. 4(d)) at the electrode–electrolyte interface. We can find that after the activation of the electrode with cyclic voltammetry, some oxygen vacancies can be formed (g = 2.003), corresponding to the formation of Mn(II) and Mn(III) (Fig. 4(c and d), 0 h).41,42 However, when the electro-oxidation occurred, the formed ROS will decrease the amount of oxygen vacancies, and therefore increase the ratio of Mn(IV) (Fig. 4(c and d), 1 h).43 Then, as the reaction time lapsed, the amount of the oxygen vacancy will increase and then decrease, corresponding to the enrichment and consumption of styrene (Fig. 4(c), 2–3 h). This is consistent with the Mn oxidation state changes, as illustrated in Fig. 4(d).
In this regard, the mechanism of our designed coupled selective oxidation of styrene with water splitting can be suggested. As shown in Scheme 3, RuO2 will produce abundant ROS (˙OH, ˙OOH, ˙O, etc.). In the meantime, styrene will be activated on the MnO2 surface to act as a “diradical”. The discriminated π electrons that are far from the benzene ring will couple with ˙OOH to produce a stable benzyl radical. As discussed previously, the acidic environment is essential, where the proton will be gained by R–OOH. Therefore, Grob fragmentation will finally be boosted to produce benzaldehyde, formaldehyde, and H2O. The reactive formaldehyde will be further oxidized to HCOOH or CO2. Additionally, as the aldehyde group in benzaldehyde is relatively sluggish toward the radical reaction, the selectivity for our electric oxidation can be guaranteed to be high.
 | ||
| Scheme 3 Proposed mechanism for the oxidation of styrene coupled with electric water splitting. | ||
2.5 Reaction scope
To monitor the universality of the developed coupled selective oxidation of styrene with the water splitting method, we further evaluated the oxidation of other aromatic vinyl compounds (Fig. S14–16†). The results are summarized in Table 3. Moderate selectivity can be achieved for all substrate molecules. Vinylnaphthalene exhibited the best selectivity (86.84%), which can be attributed to the high reactivity nature of the substrate. Generally, our developed method can be used to synthesize multiple types of aromatic aldehydes coupled with water splitting.

|
3 Conclusion
In summary, a brand-new strategy to electrochemically oxidize aromatic vinyl compounds to aldehydes was developed. This strategy came out of the process by which the designed bifunctional electrode MnO2/(Ru0.3Ti0.7) O2/Ti can discriminate the paired π electrons in the aromatic vinyl group by MnO2 and produce ROS as the oxygen source on RuO2. Styrene is therefore activated as a “diradical”, couples with ROS radicals, and finally undergoes acid-catalyzed Grob fragmentation to form benzaldehyde. When coupled with electric hydrogen production, this method paves a novel way to increase the energy and material utilization rate of water splitting, and conventional abandoned ROS can be used as green oxidants to synthesize organic compounds, therefore demonstrating its significant advance in organic synthesis, electrochemical catalysis, and green chemistry.Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Also, other relevant data for this article are available from the corresponding authors, upon reasonable request.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research work was financially sponsored by the National Natural Science Foundation of China (Grant No. 22090030, 52021004, U21A20312, and 22075033).Notes and references
- S. Wu, H. Zhang, X. Huang, Q. Liao and Z. Wei, Ind. Eng. Chem. Res., 2021, 60, 8324–8330 CrossRef CAS.
- Y.-X. Tan, Z.-M. Chai, B.-H. Wang, S. Tian, X.-X. Deng, Z.-J. Bai, L. Chen, S. Shen, J.-K. Guo and M.-Q. Cai, et al. , ACS Catal., 2021, 11, 2492–2503 CrossRef CAS.
- S. Wu, X. Huang, H. Zhang, Z. Wei and M. Wang, ACS Catal., 2021, 12, 58–65 CrossRef.
- A. S. Fajardo, H. F. Seca, R. C. Martins, V. N. Corceiro, I. F. Freitas, M. E. Quinta-Ferreira and R. M. Quinta-Ferreira, J. Electroanal. Chem., 2017, 785, 180–189 CrossRef CAS.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed.
- S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed.
- T. Zheng, J. Jiang, J. Wang, S. Hu, W. Ding and Z. Wei, Acta Phys.-Chim. Sin., 2021, 37, 2011027 Search PubMed.
- H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed.
- J.-Y. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- B. You, G. Han and Y. Sun, Chem. Commun., 2018, 54, 5943–5955 RSC.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- S. R. Kubota and K.-S. Choi, ACS Sustainable Chem. Eng., 2018, 6, 9596–9600 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 1–5 CrossRef PubMed.
- X. Liu, J. He, S. Zhao, Y. Liu, Z. Zhao, J. Luo, G. Hu, X. Sun and Y. Ding, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338–1345 CrossRef CAS PubMed.
- K. Dang, H. Dong, L. Wang, M. Jiang, S. Jiang, W. Sun, D. Wang and Y. Tian, Adv. Mater., 2022, 2200302 CrossRef CAS PubMed.
- B. Liu, P. Wang, A. Lopes, L. Jin, W. Zhong, Y. Pei, S. L. Suib and J. He, ACS Catal., 2017, 7, 3483–3488 CrossRef CAS.
- Y. Gao, C. Xing, S. Hu and S. Zhang, J. Mater. Chem. A, 2021, 9, 10374–10384 RSC.
- M. S. Batra, R. Dwivedi and R. Prasad, ChemistrySelect, 2019, 4, 11636–11673 CrossRef CAS.
- X. Deng and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 17178–17179 CrossRef CAS PubMed.
- H. Fu, K. Huang, G. Yang, Y. Cao, H. Wang, F. Peng, Q. Wang and H. Yu, ACS Catal., 2019, 10, 129–137 CrossRef.
- N. Li, Y. Gao, X. Zhang, Z. Yu, L. Shi and Q. Sun, Chin. J. Catal., 2015, 36, 721–727 CrossRef CAS.
- X. Bao, H. Li, Z. Wang, F. Tong, M. Liu, Z. Zheng, P. Wang, H. Cheng, Y. Liu and Y. Dai, et al. , Appl. Catal., B, 2021, 286, 119885 CrossRef CAS.
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef.
- J. Huang and Y. Wang, Cell Rep. Phys. Sci., 2020, 1, 100138 CrossRef CAS.
- J. Deng, S. Chen, N. Yao, Q. Wang, J. Li and Z. Wei, Appl. Catal., B, 2020, 277, 119175 CrossRef CAS.
- S. Han, C. Wang, Y. Shi, C. Liu, Y. Yu, S. Lu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100462 CrossRef CAS.
- Y. Wang, X. Huang and Z. Wei, Chin. J. Catal., 2021, 42, 1269–1286 CrossRef CAS.
- C. Grob and W. Baumann, Helv. Chim. Acta, 1955, 38, 594–610 CrossRef CAS.
- V. M. Jovanović, A. Dekanski, P. Despotov, B. Ž. Nikolić and R. T. Atanasoski, J. Electroanal. Chem., 1992, 339, 147–165 CrossRef.
- L. Atanasoska, R. Atanasoski, F. Pollak and W. O'Grady, Surf. Sci., 1990, 230, 95–112 CrossRef CAS.
- L. Chen, J. Duan, P. Du, W. Sun, B. Lai and W. Liu, Water Res., 2022, 221, 118747 CrossRef CAS PubMed.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- V. Briega-Martos, W. Cheuquepán and J. M. Feliu, J. Phys. Chem. Lett., 2021, 12, 1588–1592 CrossRef CAS PubMed.
- G. A. Planes, E. Moran, J. L. Rodriguez, C. Barbero and E. Pastor, Langmuir, 2003, 19, 8899–8906 CrossRef CAS.
- Y. Li, H. Zhang and Q. Liu, Spectrochim. Acta, Part A, 2012, 86, 51–55 CrossRef CAS PubMed.
- H. Yue, X. Wang and C. Li, Chin. J. Catal., 2002, 23, 276–280 CAS.
- S. E. Wiberley, S. C. Bunce and W. H. Bauer, Anal. Chem., 1960, 32, 217–221 CrossRef CAS.
- L. Chen, Y. Liu, X. Fang and Y. Cheng, J. Hazard. Mater., 2021, 409, 125020 CrossRef CAS PubMed.
- W. Hong, M. Shao, T. Zhu, H. Wang, Y. Sun, F. Shen and X. Li, Appl. Catal., B, 2020, 274, 119088 CrossRef CAS.
- C. Yang, Z. Yang, K. Yang, Z. Yu, Y. Zuo, L. Cheng, Y. Wang, H. Sun, G. Yu, C. Zhang and X. Li, Sep. Purif. Technol., 2022, 301, 122022 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05913d |
| This journal is © The Royal Society of Chemistry 2023 |