
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn organelle-specific photoactivation and dual-isotope labeling strategy reveals phosphatidylethanolamine metabolic flux†
Clémence
Simon
 *a,
Antonino
Asaro
a,
Suihan
Feng
b and
Howard
Riezman
a
*a,
Antonino
Asaro
a,
Suihan
Feng
b and
Howard
Riezman
a
aDepartment of Biochemistry, NCCR Chemical Biology, University of Geneva, Geneva 1205, Switzerland. E-mail: clemence.simon@univ-lille.fr
bUnit of Chemical Biology and Lipid Metabolism, Center for Microbes, Development and Health (CMDH), Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, 200031, China
First published on 16th January 2023
Abstract
Phosphatidylethanolamine metabolism plays essential roles in eukaryotic cells but has not been completely investigated due to its complexity. This is because lipid species, unlike proteins or nucleic acids, cannot be easily manipulated at the single molecule level or controlled with subcellular resolution, two of the key factors toward understanding their functions. Here, we use the organelle-targeting photoactivation method to study PE metabolism in living cells with a high spatiotemporal resolution. Containing predefined PE structures, probes which can be selectively introduced into the ER or mitochondria were designed to compare their metabolic products according to their subcellular localization. We combined photo-uncaging with dual stable isotopic labeling to track PE metabolism in living cells by mass spectrometry analysis. Our results reveal that both mitochondria- and ER-released PE participate in phospholipid remodeling, and that PE methylation can be detected only under particular conditions. Thus, our method provides a framework to study phospholipid metabolism at subcellular resolution.
Introduction
Despite the essential functions of lipids in health and disease, lipid metabolism is still less studied than the metabolism of other biomolecules. Unlike proteins or nucleic acids, the highly diverse lipid species are not directly genetically encoded and cannot be selectively overexpressed/reduced. Moreover, delivery of exogenous lipids into living cells is a great challenge in lipid biology as many of them are highly hydrophobic, resulting in low solubility in aqueous solutions. In addition, some of them also bear charges on their headgroup, preventing them from crossing the plasma membrane. Thus, it cannot be predicted if and where they will be transported into cells. Lastly, lipid metabolic processes are very fast and occur in precise subcellular locations, making them difficult to detect. Over the past twenty years, chemical tools have been developed and combined with bioanalytical techniques in order to address lipid chemical complexity (for example chemical reporters, and photocrosslinkable, photoswitchable or photocaged lipids).1,2 The photocaging method allows native species to be delivered on demand with light and is therefore ideal for studying lipid biological processes with high temporal resolution. It consists of adding a photolabile protecting group on the lipid headgroup to inhibit its activity. The caged lipid can be incorporated and accumulated in living cells in which it won't be recognized by enzymes. Then, a rapid UV illumination removes the chemical protecting group, and delivers the bioactive lipid in a non-invasive manner. Photocaging has been applied to a number of lipid messengers like phosphoinositides,3 diacylglycerides,4 ceramides5 and fatty acids.6,7 While these studies focused on the roles of lipids in signalling processes, examples of photocaging tools to trace their metabolism are rare. In the past few years, our group has developed a series of photocaged lipids targeting different subcellular compartments including lysosomes and mitochondria. We combined this approach with stable-isotope labeling to trace sphingosine metabolism, and found that its metabolic conversion to ceramide versus sphingosine-1-phosphate depends on its original location in subcellular compartments, showing a metabolic bias based on subcellular location.8,9 Here we extended this approach of tracking local lipid metabolic flux to phosphatidylethanolamine (PE).PE represents 25% of all phospholipids in mammals and corresponds to the second most abundant glycerophospholipid (GPL) behind phosphatidylcholine (PC).10,11 Enriched in the mitochondrial inner membrane, PE has a myriad of biological roles in cells.12–16 Given its biological importance, abnormal PE metabolism can lead to severe diseases like cancers or diabetes.17,18 PE metabolism holds a unique place in mammalian lipid cell biology and has not been completely investigated due to its complexity.10,11 First, PE can be synthesized by 2 major pathways localized either in the ER by the CDP–ethanolamine pathway starting with ethanolamine or in mitochondrial inner membranes by phosphatidylserine (PS) decarboxylation mediated by phosphatidylserine decarboxylase (PSD). Then, PE metabolism occurs, which involves its conversion into other GPL species and its remodeling into other PE species with different acyl chain compositions (Fig. S1†). Studies of PE metabolism in mammals, based for a long time on radioactive precursors19–22 and more recently on stable isotope-labeled precursors,23–25 suggest that its metabolism, transport and functions depend on its localization between the ER and mitochondria. However, little is known in terms of the quantitative and qualitative contribution of each pathway.
Here, we use an organelle-targeting photoactivation method to study PE metabolism in living cells with a high spatiotemporal resolution. We designed different PE probes which can be selectively introduced into the ER versus mitochondria to compare their metabolic products. We combined the photo-uncaging method with dual-stable isotopic labeling, which enabled us to track PE transformations in living cells with high resolution mass spectrometry (Fig. 1).
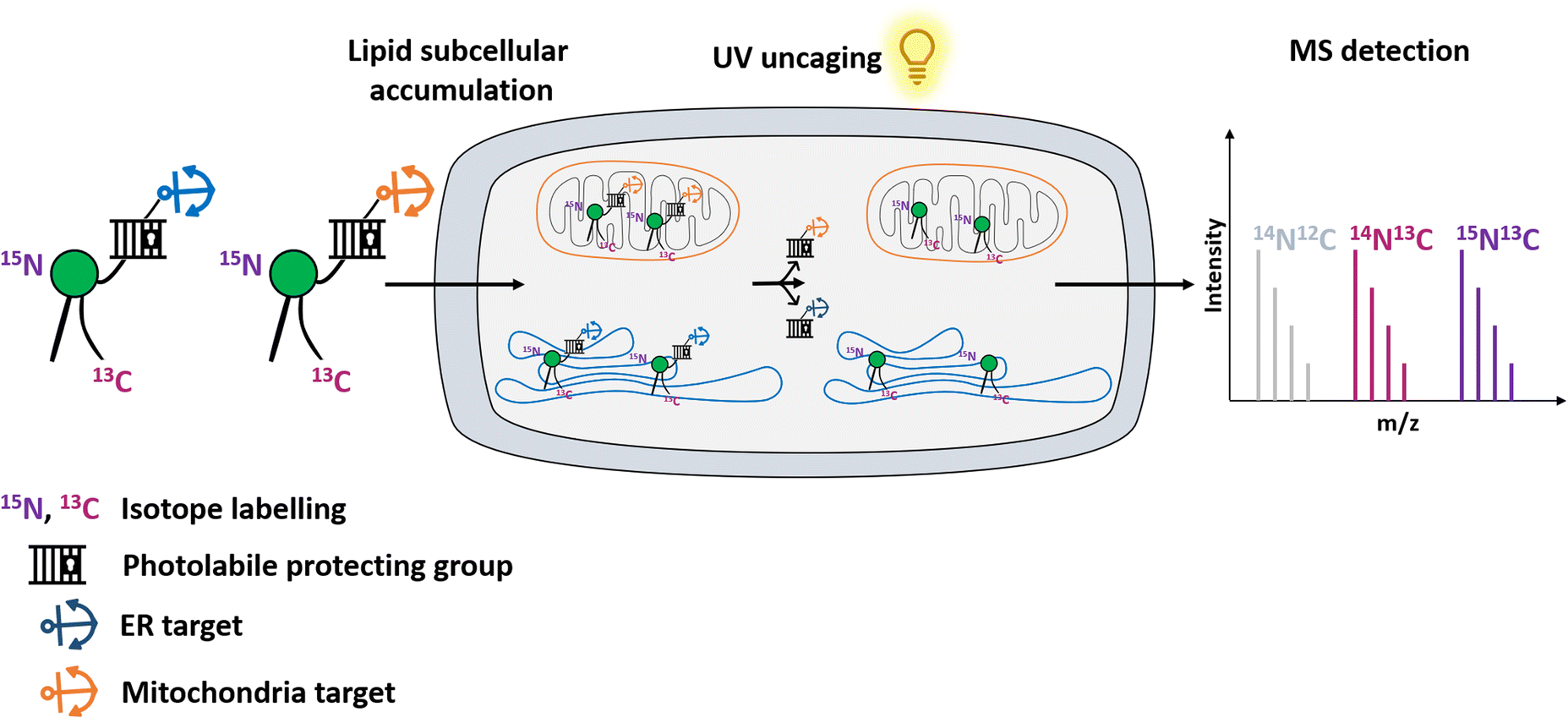 | ||
| Fig. 1 Schematic illustration of organelle-specific uncaging and MS-based metabolic tracking of PE probes. Isotope-labeled PE is equipped with a photocaging group functionalized with targeting groups that localize it to specific compartments (mitochondria or ER) in living cells, where the phospholipid can subsequently be photo-released. | ||
Results
The first step of the strategy was to design photocaged PE probes. Concerning its composition, the synthesized PE bears a palmitoyl and an oleoyl chain on the sn1 and sn2 positions respectively, giving PE34:1 (or PE16:0-18:1), one of the most abundant PE species in mammals. Most importantly, we labeled PE with two different heavy isotopes to ensure the tracing of different metabolic events using mass spectrometry: eighteen 13C labeled carbons on the oleoyl acyl chain and one 15N on the amine headgroup, giving 15N13C-PE34:1 (or 15N-PE16:0-13C18:1).PE synthesis is described in Scheme 1A and starts with the (R) enantiomer of protected glycerol. The first key step of the synthetic route is the esterification of the hydroxyl at the sn2 position of compound 1b with commercial oleic acid-13C18.4 The second key step is the nucleophilic addition of 15N-ammonia on the cyclic phosphotriester intermediate (1e) to get the final 15N13C-PE34:1 (compound 1f).26 The measured optical rotation of the isotope labeled PE was similar to that of its non-labeled naturally occurring commercial analog (R)-POPE (+4.5 and +5.1 respectively), confirming its high enantiospecificity.
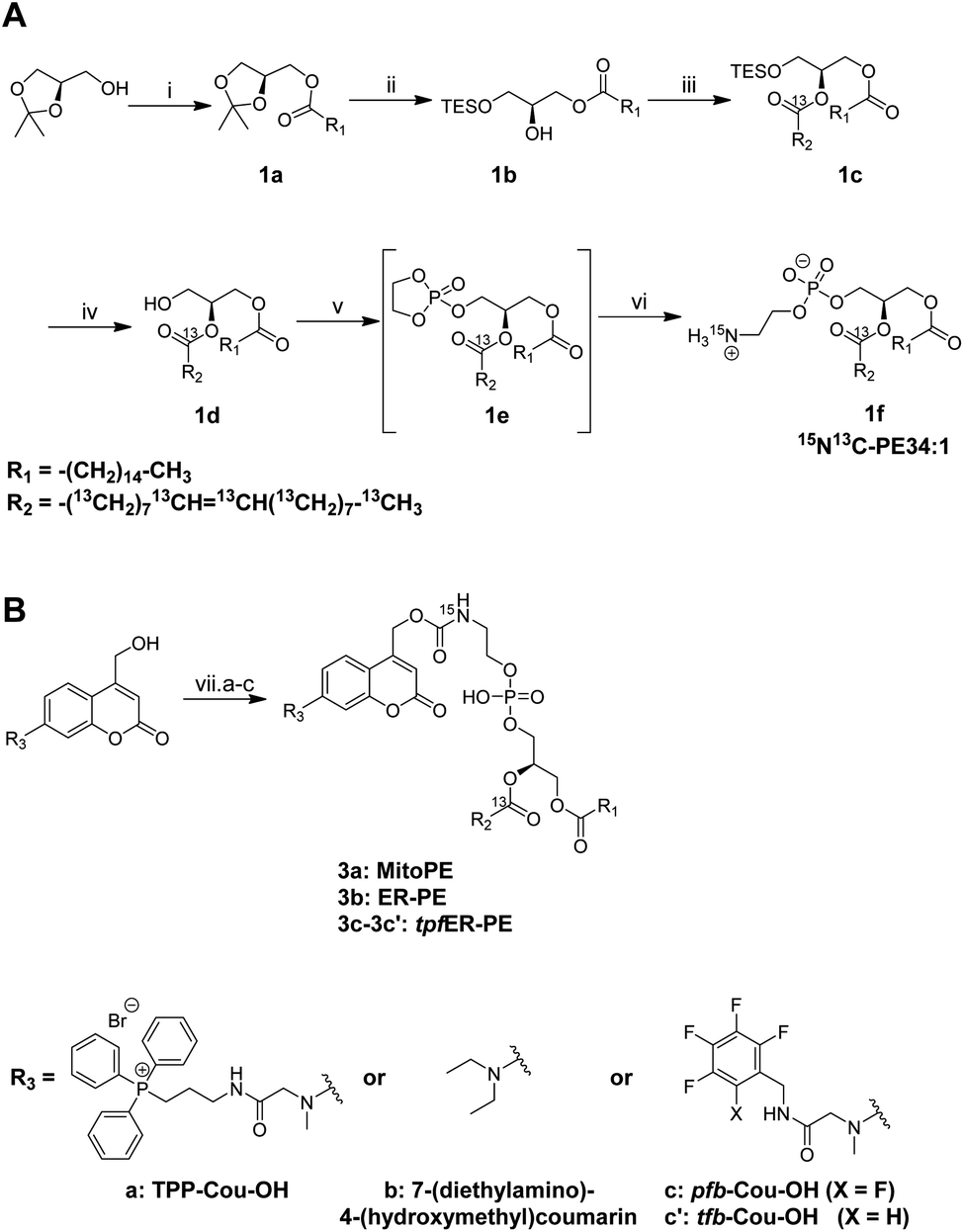 | ||
| Scheme 1 (A) Synthesis of 15N13C-PE34:1. Conditions: (i) palmitic acid, DCC, DMAP, DCM, rt, 18 h – 85%; (ii) (1) TESOTf, DIEA, DCE, 95 °C, 36 h; (2) I2, Na2CO3, H2O, THF, rt, 4 h – 54% over 2 steps; (iii) oleic acid-13C18, EDC, DMAP, DCM, rt, 12 h – 93%; (iv) FeCl3, MeOH, DCM, rt, 1 h – 92%; (v) 2-chloro-2-oxo-1,3,2-dioxaphospholane, TEA, ACN, 0 °C to rt, 12 h; (vi) 15NH3, ACN, 0 °C to 65 °C, 24 h – 6% over 2 steps. (B) Synthesis of MitoPE, ER-PE and tpfER-PE (compounds 3a, 3b and 3c respectively). Conditions: (vii-a) for MitoPE: (1) bis-(4-nitrophenyl)carbonate, DIEA, DCM, rt, 3 h; (2) 15N13C-PE34:1, DCM, 38 °C, 6 h – 41%. Conditions (vii-b) for ER-PE: (i) (1) triphosgene, DIEA, THF, 0 °C, 4 h; (2) 15N13C-PE34:1, DIEA, DCM, 0 °C to rt, 12 h – 22%. Conditions (vii-c) for tpfER-PE: (1) bis-(4-nitrophenyl)carbonate, DIEA, DCM, rt, 12 h; (2) 15N13C-PE34:1, DCM, 39 °C, 24 h – 43%. | ||
To capture the rapid turnover of PE, we synthesized different caged PE probes from 15N13C-PE34:1 to deliver it to specific subcellular compartments (Scheme 1B). Bioactive small molecules are usually caged with nitrophenyl or coumarin derivatives to inhibit their activity. Coumarin was used in our case because of its intrinsic fluorescence properties visible by optical microscopy and was added on the PE headgroup. First, we synthesized the triphenylphosphonium-labeled-coumarin-alcohol (TPP-Cou-OH) using a previously described protocol to deliver PE specifically into mitochondria.8 Compound 3a was synthesized by coupling TPP-Cou-OH with 15N13C-PE34:1 thought a carbamate linkage. In the same way, we combined the commercial 7-(diethylamino)-4-(hydroxymethyl)coumarin bearing no organelle tag to PE in order to see if this global-caged lipid (compound 3b) could be delivered into cells in a more diffuse way. Aiming to develop an ER-specific probe, we also coupled 15N13C-PE34:1 with a coumarin functionalized with a mixture of pentafluorobenzyl and tetrafluorobenzyl (pfb- and tfb-Cou-OH) to accumulate PE specifically in the ER (compounds 3c and 3c′).27
We first tested the photo-cleavage efficiency of the synthesized probes. Upon UV illumination, compounds 3a, 3b and 3c and 3c′ were quickly hydrolysed in chloroform/methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give free 15N13C-PE34:1 within 2 min (Fig. S2†). Next, we sought to determine the localization of each compound in living cells. In HeLa cells, we incubated compound 3a (25 μM), which was complexed with liposomes (POPC/cholesterol (1:1)) and methyl-β-cyclodextrin (mβCD), and compared its fluorescent signals with the ones from MitoTracker. The fluorescence signal of compound 3a showed a good colocalization with a MitoTracker, suggesting it is mainly introduced into mitochondria in living cells (Fig. 2A and B), and leading us to rename it “MitoPE” to highlight its subcellular localization. Importantly, optimization experiments showed that MitoPE needed to be complexed first with a correct ratio of mβCD/liposomes to be efficiently introduced into HeLa cells (Fig. S3D†). Indeed, previous studies showed that mβCD is a good donor to transfer long-chain fluorescent or natural phospholipids from vesicles (composed of POPC/cholesterol) to cultured cells.28,29 In the absence of mβCD or when there is a non-optimal donor/vesicles ratio, there is no specific staining inside cells, but blue dots representing insoluble aggregates apparently inside and outside cells (Fig. S3A–C†).
:1) to give free 15N13C-PE34:1 within 2 min (Fig. S2†). Next, we sought to determine the localization of each compound in living cells. In HeLa cells, we incubated compound 3a (25 μM), which was complexed with liposomes (POPC/cholesterol (1:1)) and methyl-β-cyclodextrin (mβCD), and compared its fluorescent signals with the ones from MitoTracker. The fluorescence signal of compound 3a showed a good colocalization with a MitoTracker, suggesting it is mainly introduced into mitochondria in living cells (Fig. 2A and B), and leading us to rename it “MitoPE” to highlight its subcellular localization. Importantly, optimization experiments showed that MitoPE needed to be complexed first with a correct ratio of mβCD/liposomes to be efficiently introduced into HeLa cells (Fig. S3D†). Indeed, previous studies showed that mβCD is a good donor to transfer long-chain fluorescent or natural phospholipids from vesicles (composed of POPC/cholesterol) to cultured cells.28,29 In the absence of mβCD or when there is a non-optimal donor/vesicles ratio, there is no specific staining inside cells, but blue dots representing insoluble aggregates apparently inside and outside cells (Fig. S3A–C†).
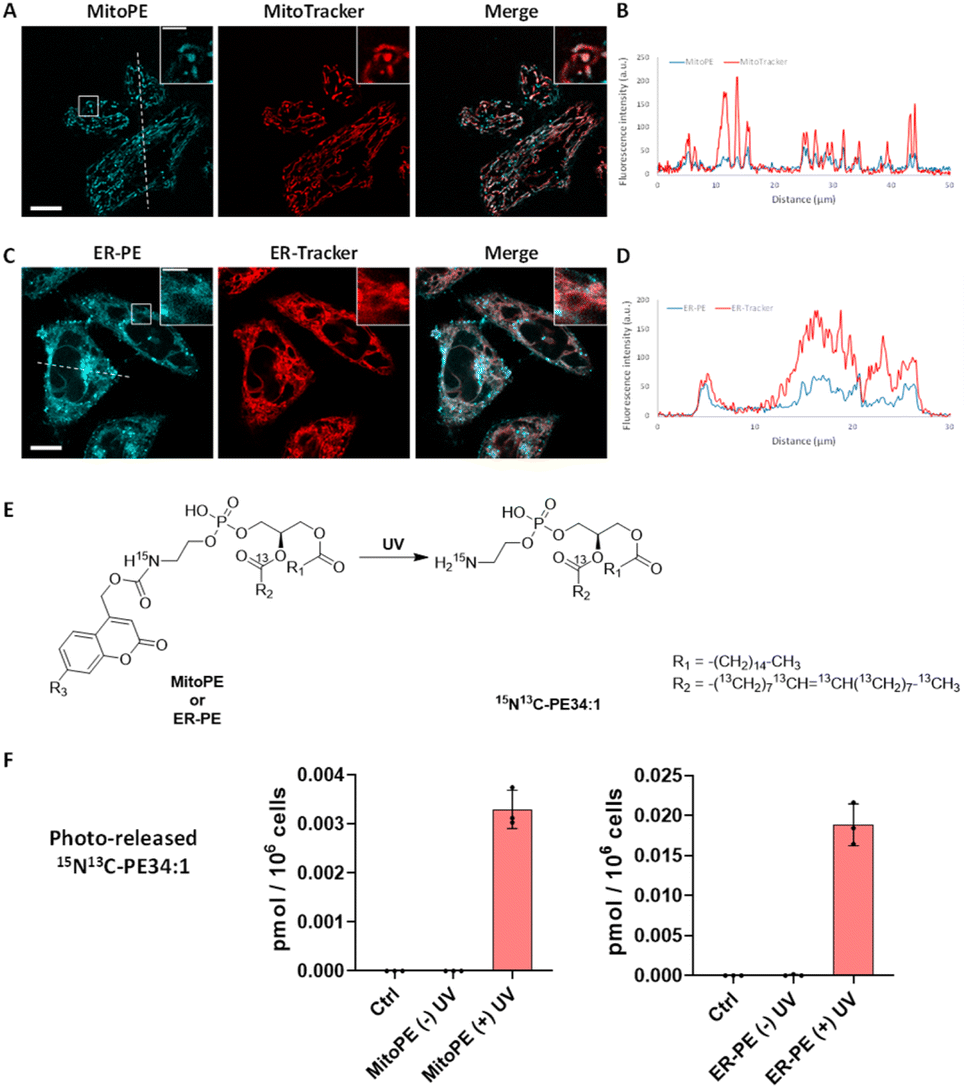 | ||
| Fig. 2 Localization and uncaging efficiency of organelle-targeting caged PE probes in living HeLa cells. (A and C) Representative fluorescence image of HeLa cells stained with (A) MitoPE (25 μM, previously complexed with cholesterol/POPC 1:1 and mβCD, see detailed description in the ESI†) and MitoTracker (50 nM) or (C) ER-PE (25 μM) and ER-Tracker (1 μM). Scale bar: 10 μm and 3 μm (inset). (B and D) Intensity profiles of the white dotted lines in (B) MitoPE and MitoTracker channels, or (D) ER-PE and ER-Tracker channels. (E) Illustration of photoactivation of PE probes. (F) Uncaging efficiency of MitoPE and ER-PE in living cells. Fully confluent cells in 35 mm culture dishes were incubated with the caged probes (i) MitoPE (5 μM), previously complexed with cholesterol/POPC (1:1) and mβCD or (ii) ER-PE (5 μM) for 15 min, washed, and irradiated under 350–450 nm UV light for 2 min on ice. Cells were collected immediately after UV irradiation. Lipids were extracted and the 15N13C-PE34:1 signal was measured by LC-MS. Values were normalized with respect to the amount of internal standards and cell numbers. Data represent the average of three independent experiments. Error bars represent SD. 15N13C-PE34:1 released from MitoPE and ER-PE corresponds to 0.12% and 0.81% of endogenous total PE species respectively. | ||
Concerning compound 3b, no vesicle carrier was necessary to introduce it into living HeLa cells. The absence of the three hydrophobic benzenes forming the TPP cation could explain its better solubility compared to MitoPE, even if the latter has a supplementary positive charge. Hence, HeLa cells were incubated with the global-caged PE (25 μM) for 15 min before washing. Despite the presence of some nonspecific dots (Fig. S4B†), its fluorescence staining mostly overlaps with an ER-Tracker, meaning that it is mainly localized in the ER (Fig. 2C and D). In the live cell images there is some signal that resembles labeling of the Golgi, however colocalization experiments on fixed cells with a Golgi marker did not confirm this localization (Fig. S4A†). We therefore renamed compound 3b “ER-PE” to highlight its subcellular localization. Similarly, compounds 3c and 3c′ complexed with liposomes/mβCD accumulate mainly in the ER (Fig. S5A and B†), so we renamed them “tpfER-PE”. However, tpfER-PE seemed to present more non-specific puncta than ER-PE, which can be explained by the low solubility of the fluorinated groups. So, we used the latter during our study. For each probe, we performed a colocalization analysis based on Mander's coefficient which confirms the strong colocalization with the organelle marker of interest (Fig. S6†).
Next, we sought to determine the efficiency of 15N13C-PE34:1 released into living cells. Isotope labeling allowed it to be distinguished from its endogenous analogue by mass spectrometry (Fig. 2E). HeLa cells were incubated with the probe of interest and exposed to UV. Directly after the uncaging, cells were collected for lipid extraction and analysis by LC-MS. For both MitoPE and ER-PE, data showed a robust appearance of the 15N13C-PE34:1 signal after UV irradiation compared to the cell treated with caged-lipid without UV exposure and control cells treated with no caged-molecule (Fig. 2F). The signal specificity of 15N13C-PE34:1 in cells and of all following labeled species was further confirmed by LC-MS/MS analysis (see the LC/MS-MS spectrum in the ESI†). Therefore, the uncaging was efficient for both probes. Interestingly, the level of released 15N13C-PE34:1 after UV uncaging is higher (approximately 6 times) in cells incubated with ER-PE than MitoPE (0.0188 versus 0.0033 pmol per 106 cells respectively). This could be explained by the difference in size of the organelles where the probe is localized (mitochondria vs. ER) and perhaps by differences in solubility between the two probes. Even though we treated the cells with equal amounts of probes, only MitoPE needs to be complexed beforehand with a donor carrier, meaning that it will not enter into the HeLa cells as easily as ER-PE. Taken together, these results show that MitoPE and ER-PE can selectively be introduced into mitochondria and the ER of HeLa cells respectively, and they can efficiently release 15N13C-PE34:1 under UV illumination. To our knowledge, this is the first time that a PE has been delivered with a high spatiotemporal resolution into living cells.
Immediately after photo-uncaging, 15N13C-PE34:1 can be released at specific places in living HeLa cells, recognized by enzymes and recycled, degraded or converted into different metabolites. These potential metabolites can be traced by high resolution mass spectrometry due to the 15N and 13C isotopic labeling on the amine headgroup and oleoyl sn2 acyl chain respectively, allowing us to study phospholipid remodeling. Also called the Lands cycle, this pathway consists of deacylation/reacylation steps of newly synthesized phospholipids performed by phospholipases and acyltransferases respectively to modulate their composition and maintain membrane diversity in mammalian cells.30 To monitor phospholipid remodeling, we treated cells either with MitoPE, or with ER-PE. After the uncaging steps cells were incubated at 37 °C for different times (0, 20 or 90 min) and then collected for lipid extraction and LC-MS analysis. To be able to compare the data coming from MitoPE and ER-PE photolysis, we normalized the signals of interest by the quantity of released 15N13C-PE. PE34:1 originating in the two locations was involved in sn2 remodeling, but this pathway was more prevalent for the ER localized lipid (Fig. 3A and B). For the ER localized lipid, a signal corresponding to 15N-LysoPE16:0-0:0 is visible from 0 min to 90 min with a slight increase at 20 min, meaning that phospholipase A2 enzymes are able to cleave the 13C18-oleic acid at the sn2 position and generate 15N-LysoPE 16:0-0:0. Then 13C18-oleic acid must be converted to oleoyl-CoA and used by lysophospholipid acyltransferases (LPLAT) to acylate the sn2 position of endogenous phospholipids like PC, giving 13C-labeled PC species. The composition of detected 13C-PC species follows the order of the more abundant species with an oleoyl chain at the sn2 position in HeLa cells. Five species are detected after ER-PE uncaging (13C-PC34:1, 36:2, 36:1, 34:1-O and 34:2) and seem to continue to increase up to 90 min. Three species are visible after MitoPE photolysis (13C-PC34:1, 36:2 and 34:2) and they seem to reach a plateau already at 20 min. Some species are already visible at 0 min, meaning that the remodeling can take place already during the photoactivation period which lasts 2 min.
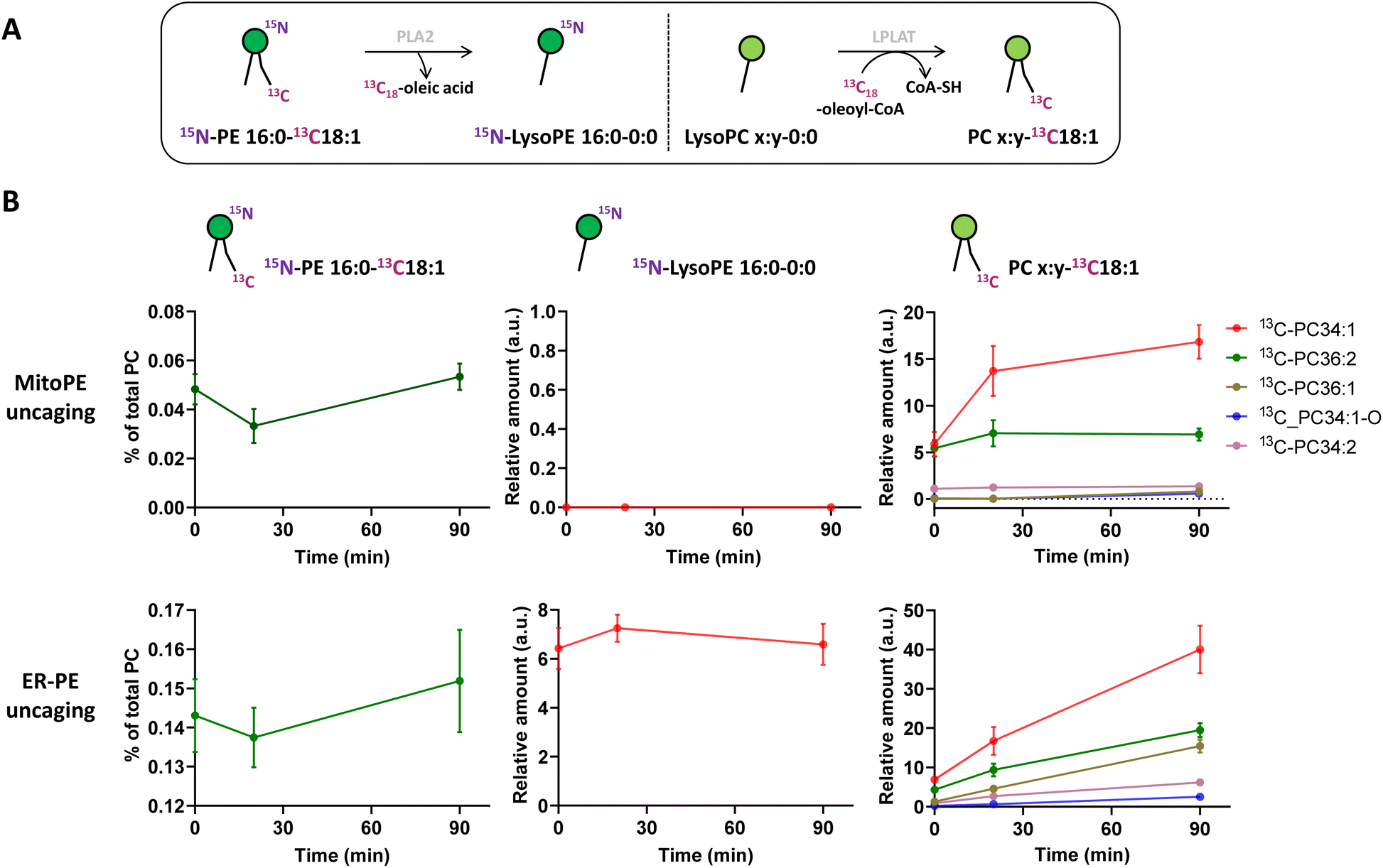 | ||
| Fig. 3 Released 15N13C-PE34:1 participates in sn2 remodeling of endogenous lysoPC species in living cells. MitoPE or ER-PE was introduced into HeLa cells for 15 min as outlined in the “experimental procedures”. Then the cells were washed, irradiated with UV light for 2 min on ice, incubated at 37 °C, and collected for lipid extraction at different time points (0, 20 or 90 min). (A) The 13C18-oleoyl acyl chain of released 15N13C-PE34:1 is cleaved by phospholipase A2 (PLA2) and used by lysophospholipid acyltransferase (LPLAT) to acetylate the sn2 position of endogenous lysoPC species. (B) LC-MS analysis of labeled species involved in sn2 remodeling of endogenous lysoPC species. Values were normalized with respect to the amount of internal standards and of (i) total endogenous PC species for photo-released 15N13C-PE34:1, or of (ii) photo-released 15N13C-PE34:1 for 15N-LysoPE 16:0-0:0 and 13C-PC species. Data represent the average of three independent experiments. Error bars represent SD. | ||
The longer incubation time of 15N13C-PE34:1 after probe photo-release leads to sn2 reacylation of other GPL classes with 13C-oleic acid. After 4 h and 16 h of ER-PE uncaging, we observed significant appearance of the more abundant PE species (13C-PE36:2, 34:1 and 36:1 after ER-PE uncaging; 13C-PE34:1 after MitoPE uncaging), PI species (13C-PI36:2, 36:1, 34:1, and 34:2 after ER-PE uncaging only) and the more abundant PS species as well (13C-PS36:1, 36:2 and 34:1 after ER-PE uncaging only) (Fig. S7†). Although 13C-PS 36:1 and 36:2 are necessarily generated by reacylation of their respective lysoPS forms, we cannot exclude the possibility that 13C-PS34:1 could also come from base exchange reaction between 15N-ethanolamine of 15N13C-PE34:1 and endogenous serine. The solution to verify this hypothesis would be to synthesize a new PE probe with labeling of the glycerol skeleton in addition to the oleoyl acyl chain. Interestingly, while we can detect sn2 remodeling of endogenous lysoPE species with the cleaved 13C-oleic acid, reacylation of 15N-LysoPE16:0 to give 15N-PE16:0-18:1 or 15N-PE16:0-18:2 as abundant PE species is not detectable (data not shown).
Tracing the metabolism of released 15N13C-PE34:1 after long incubation times allows its sn1 remodeling to be followed. After the uncaging of ER-PE or MitoPE, cells were incubated at 37 °C for 4 h or 16 h. First, for ER-PE photo-release, we can observe a signal specific to 15N-LysoPE0:0-13C18:1 at times 4 h and 16 h (Fig. 4B). We can also detect specific signals for 15N-PE18:0-13C18:1 and 15N-PE18:1-13C18:1. Together, these data suggest that phospholipase A1 has cleaved the palmitoyl acyl chain at the sn1 position and an LPLAT is able to reacetylate the 15N-LysoPE0:0-13C18:1 with stearic acid or oleic acid (Fig. 4A). In the case of MitoPE, a signal of 15N-LysoPE0:0-13C18:1 is not visible, most likely because it is below the detection limit. However, a signal of 15N-PE18:0-13C18:1 starts to appear and a signal of 15N-PE18:1-13C18:1 is significant at 16 h. As phospholipid reacylation by LPLAT usually occurs in the ER, this implies that 15N13C-PE34:1 released into mitochondria from MitoPE can be transported to the ER in HeLa cells. Together, these results show that 15N13C-PE34:1 is remodeled at the sn1 position and participates as well in the sn2 remodeling of endogenous lysoPL species.
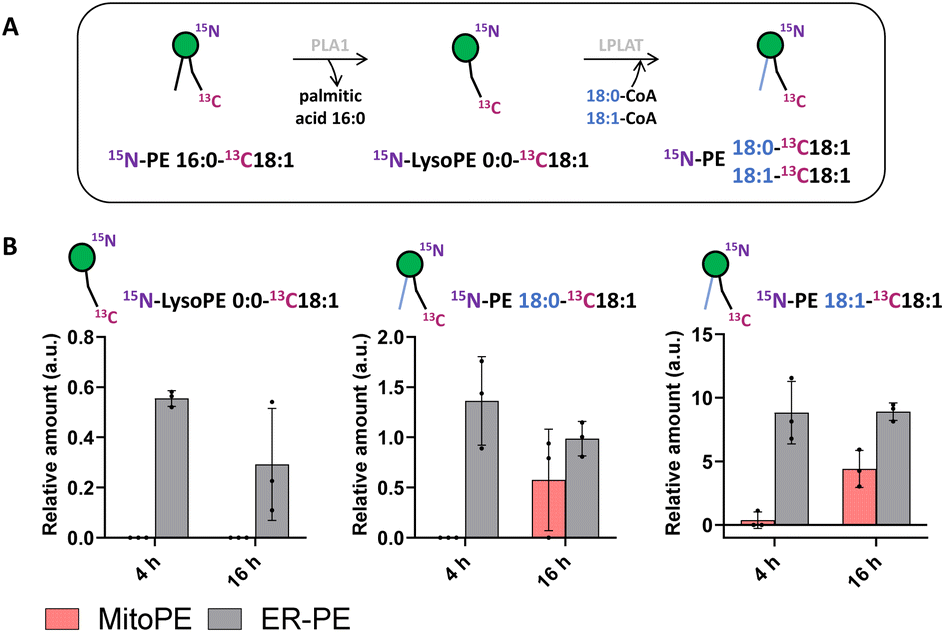 | ||
| Fig. 4 Sn1 remodeling of photo-released 15N13C-PE34:1 in living cells. MitoPE or ER-PE were introduced into HeLa cells for 15 min as outlined in the “experimental procedures”. Then cells were washed, irradiated with UV light for 2 min on ice, incubated at 37 °C, and collected for lipid extraction and LC-MS analysis at 4 or 16 h. (A) Illustration of sn1 remodeling of released 15N13C-PE34:1 by phospholipase A1 (PLA1) and lysophospholipid acyltransferase (LPLAT). (B) Analysis of labeled species involved in 15N13C-PE sn1 remodeling after its release from different sources as indicated. Values were normalized with respect to the amount of internal standards and photo-released 15N13C-PE34:1. Data represent the average of three independent experiments. Error bars represent SD. | ||
As expected, 15N13C-PE released from tpfER-PE after UV illumination is also involved in sn2 remodeling of endogenous PC species and undergoes its own sn1 remodeling (Fig. S5C and D†).
Another important aspect of PE metabolism is its transformation into PC by the phosphatidylethanolamine-N-methyl transferase (PEMT, Fig. S1†).31 PEMT is an integral membrane protein enriched in the ER and mitochondria-associated membrane (MAM) contact sites which converts PE into PC through 3 consecutive transmethylation reactions from S-adenosyl-methionine (SAM). This process is one of the three mammalian PC biosynthesis pathways and is essential in the liver where it accounts for 30% of total PC production. In contrast to yeast in which PEMT is a major pathway, its activity is less clear in mammals and seems to be organ-dependent. Moreover, the study of PE methylation is complex because of the lack of a suitable mammalian cell line that significantly expresses PEMT. Indeed, the photo-release of 15N13C-PE34:1 from different sources (mitochondria or ER) during different incubation times (from 0 min up to 16 h) in HeLa cells did not lead to detection of any 15N13C-PC34:1 (i.e.15N-PC16:0-13C18:1) signal which would come from PEMT methylation. This nevertheless demonstrates the importance of labeling PE at two different places in the chemical structure (here the oleoyl acyl chain versus the headgroup) to discriminate 13C-PC34:1 involving sn2 remodeling of endogenous lysoPC species from 15N13C-PC34:1 which would come from PEMT methylation.
PEMT is known to be liver specific, with the mRNA initial level very high initially in liver primary culture, even if it rapidly decreases within 16 h of isolation.32 We therefore decided to photo-release PE from the ER-PE probe in primary mouse hepatocytes suitable for metabolism studies with a maximum of 6 hours of incubation. However, after 20 min, 90 min or 4 h of incubation, we did not detect any signal of 15N13C-PC34:1 coming from PEMT activity, but we did detect some 13C-PC species showing activity of the remodeling pathway (Fig. S8†).
We therefore decided to go back to HeLa cells and to overexpress PEMT by transfecting a plasmid containing isoform 2 of PEMT under the control of a constitutive promoter. Immunofluorescence of PEMT and western blot analysis (Fig. 5A and B and S9†) confirmed a high rate of transfection. According to the immunofluorescence, overexpressed PEMT is localized in the ER and Golgi. To test for overexpression of PEMT activity, we incubated control cells and PEMT overexpressing cells with d4-ethanolamine (d4-Etn) for 1.5 h or 5 h before cell collection and lipid extraction. The obtained lipidomic data showed the incorporation of d4-Etn into d4-PE species, and a significant increase of d4-labeled PC species in overexpressed cells compared to control cells, confirming the increase of PEMT activity (Fig. S10† and 5C). After 5 h of incubation, the d4-PC34:1 signal increased approximately 114-fold in PEMT overexpressing cells compared to mock-transfected cells, corresponding approximately to 20% of the d4-PE34:1 signal (Fig. 5C).
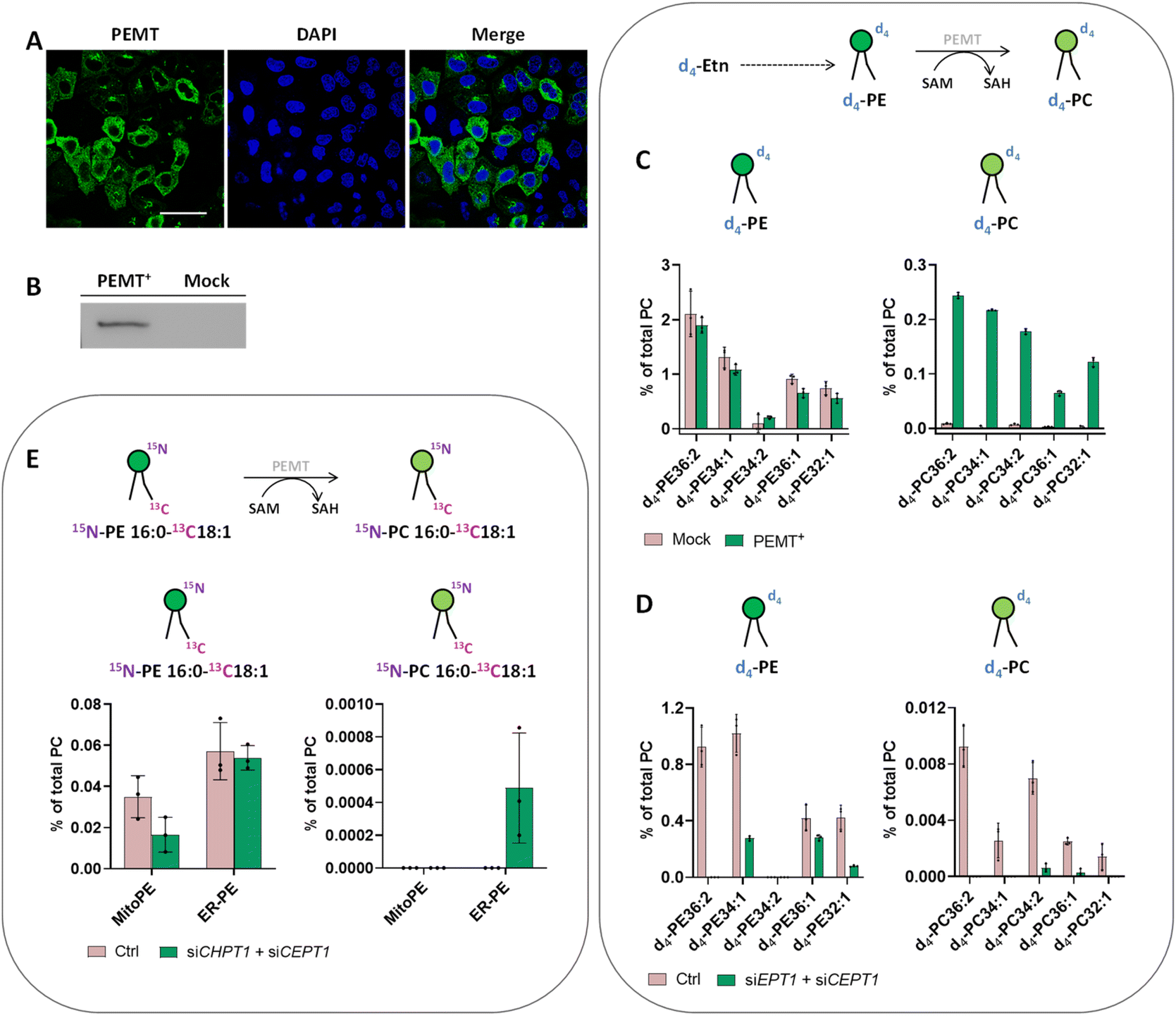 | ||
| Fig. 5 Analysis of PE methylation. (A and B) PEMT gene overexpression in HeLa cells. (A) HeLa cells were grown on glass dishes and transfected with PEMT-myc expression construct. 24 h after transfection, cells were fixed and stained with anti-Myc antibody (green). Cell nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (B) Western blot analysis of PEMT-myc levels in cell extracts from transiently transfected cells. (C) Incorporation of d4-Etn into lipid metabolites of HeLa cells transfected with PEMT2. HeLa control cells and cells transiently transfected with PEMT2 were washed once with PBS and incubated with d4-Etn-HCl (0.5 mM) for 5 h at 37 °C. Then the cells were washed three times with PBS and collected for lipid extraction. Top: Illustration of d4-PE methylation to d4-PC. Bottom: LC-MS analysis of labeled species involved in the PEMT pathway. (D) Incorporation of d4-Etn into lipid metabolites of HeLa cells presenting PEMT overexpression and CDP–ethanolamine pathway downregulation. PEMT overexpressing cells treated with negative siRNA (Ctrl) or siRNA for EPT1 (siEPT1) and CEPT1 (siCEPT1) were washed once with PBS and incubated with d4-Etn-HCl (0.5 mM) for 90 min at 37 °C. Then the cells were washed three times with PBS and collected for lipid extraction. (E) Formation of 15N13C-PC34:1 in HeLa cells presenting PEMT overexpression and CDP–choline pathway downregulation after photoactivation. PEMT overexpressing cells treated with negative siRNA (Ctrl) or siRNA for CPT1 (siCHPT1) and CEPT1 (siCEPT1) were washed once with PBS and incubated with MitoPE or ER-PE for 15 min as outlined in the “experimental procedures”. Then the cells were washed, irradiated with UV light for 2 min on ice, incubated for 4 h at 37 °C and collected for lipid extraction. Top: Illustration of 15N13C-PE34:1 methylation to 15N13C-PC34:1 by PEMT. Bottom: LC-MS analysis of labeled species involved in the PEMT pathway. For each LC-MS analysis, values were normalized with respect to the amount of internal standards and total PC species. Data represent the average of three independent experiments. Error bars represent SD. | ||
In some organisms including plants, the PEMT enzyme can methylate the ethanolamine headgroup not only when part of the phospholipid but also directly after its activation to phosphoethanolamine in the CDP–ethanolamine pathway.33 To prove that increased PEMT activity is working on PE and not one of its precursors in HeLa cells, the latter were transfected with two siRNA targeting EPT1 and CEPT1, afterwards PEMT was overexpressed and cells were incubated with d4-Etn for 1.5 h. EPT1 and CEPT1 encode respectively phosphotransferase EPT1 and CEPT1 that act at the last step of the CDP–ethanolamine pathway to generate PE from CDP–choline. Their effective silencing in transfected cells was confirmed by RT-qPCR (Fig. S11A†). Then, the lipidomic data confirmed a decrease of PE synthesis in cells presenting PEMT overexpression combined with CDP–ethanolamine pathway downregulation compared with control cells with no siRNA silencing (Fig. 5D). The signal of d4-PC species significantly decreased as well, meaning that in HeLa cells, PEMT enzymes seem to be mainly methylate PE phospholipids after their complete biosynthesis and they cannot act upstream at the start of the CDP–ethanolamine pathway.
Despite PEMT overexpression and demonstration of its activity with d4-Etn, the use of our probes to photo-release 15N13C-PE34:1 did not lead to any detectable 15N13C-PC34:1 signal (Fig. S12†). The major pathway for PC biosynthesis in mammalian cells is the CDP–choline pathway by analogy to the CDP–ethanolamine pathway for PE biosynthesis. We therefore decided to inhibit the CDP–choline pathway in addition to PEMT overexpression to further promote PE methylation. The final step of the CDP–choline pathway is mediated by two choline phosphotransferases (CPT1 and CEPT1 respectively encoded by CHPT1 and CEPT1) able to transfer phosphocholine from CDP–choline into a diacylglycerol backbone to form PC. Hence, HeLa cells were transfected with two siRNA targeting CHPT1 and CEPT1, afterwards they were transfected with PEMT2 plasmid. The effective silencing of targeted genes in transfected cells was confirmed by RT-qPCR (Fig. S11B†). Then, after MitoPE or ER-PE incubation and uncaging, transfected cells were incubated at 37 °C for 4 h (Fig. 5E). The 15N13C-PC34:1 signal was finally detectable when 15N13C-PE34:1 was released from the ER in cells presenting PEMT overexpression combined with CDP–choline pathway downregulation, meaning that the ER-PE probe can be used to specifically study PE methylation. The signal of 15N13C-PC34:1 corresponds to only 0.9% of the 15N13C-PE34:1 released signal. No signal was detected under the same conditions with MitoPE. This difference between both probes can be explained by the localization of overexpressed PEMT in the ER, directly accessible for ER-PE.
Discussion
PE metabolism holds a unique place in lipid cell biology and exemplifies the complexity of lipid biosynthesis and transport mechanisms. Although some studies suggest that its metabolism and function depend on its biosynthetic origin,17,22,23 the experimental means to directly address this issue are limited. Here, we developed novel photo-activation techniques, which enable the delivery of PE into different subcellular compartments, and thereafter the release of PE molecules with a high spatiotemporal resolution. We combined them with dual isotopic labeling to quantitatively analyze and compare PE metabolic conversion in HeLa cells. Installation of the labeling on two strategic positions of the lipid molecule is essential to discriminate the remodeling pathway from the PEMT pathway.Phospholipid remodeling was mainly studied in the past by using radioactively labeled or stable isotopically labeled precursors (choline, ethanolamine or glycerol). However, due to the lack of specificity, it needs details on the remodeling pathways and the kinetics. Only a few papers describe instead the use of isotopically labeled phospholipids in mammalian cells or in yeast.34,35 In mammalian cells, the authors showed that atypical phospholipids were rapidly remodeled to endogenous abundant species whereas the introduced species identical to endogenous ones were remodeled only to a limited degree. Our probes allow phospholipid remodeling to be studied with a high spatiotemporal resolution due to the presence of functionalized photolabile protecting groups on the PE. The use of two different stable isotopes at two strategic positions of the lipid allows the sn1 remodeling of the labeled lipid itself to be differentiated from sn2 remodeling of endogenous phospholipids. While sn2 remodeling has been well described,30,36 it is not clear how the composition of saturated fatty acid is controlled at the sn1 position. It has been recently shown that lysophosphatidylglycerol acyltransferase 1 (LPGAT1) is a key remodeling enzyme located in the ER and involved in the regulation of the stearate/palmitate ratio at the sn1 position of LysoPE species.37 It is possible that LPGAT1 is responsible for the 15N-PE16:0-13C18:1 (i.e.15N13C-PE34:1) remodeling into 15N-PE18:0-13C18:1. This could be investigated by the use of LPGAT1 knock-out cell lines.
The PEMT pathway represents the second biosynthetic pathway of PC via PE methylation in the ER. However, PEMT activity has not been clearly implicated in a number of tissues because this pathway is mainly liver specific, and cell culture models usually express only traces of PEMT activity. To our knowledge, PE methylation into PC in mammalian models was demonstrated with GPL precursors and never with complete GPLs.38,39 Some previous studies also postulate that only the newly made PE could be methylated.40 As HeLa cells only present a minor PEMT activity, we overexpressed the gene and confirmed the increase of d4-PE methylation into d4-PC using d4-Etn as PE precursors. However, overexpression alone was not sufficient to detect any signal of 15N13C-PC34:1 coming from 15N13C-PE34:1 methylation, suggesting that this pathway is very inefficient in consuming PE. We then combined PEMT overexpression with disruption of the CDP–choline pathway to further promote methylation of the PE probe. This condition allowed detection of the formation of 15N13C-PC34:1 when PE was photo-released from the ER, meaning that the ER-PE probe can be used to study PEMT activity. We also photo-released PE from the ER-PE probe in other cell lines including mouse primary hepatocytes, in which the PEMT initial level should be very high. However, we did not detect any signal of 15N13C-PC coming from PEMT activity. The striking inefficiency of this pathway suggests that only a very small portion of the PE in cells is the substrate for PEMT. When compared to our measurements of the efficiency of PE to PC conversion by labeling with d4-Etn, we must conclude that there are limitations on the metabolism of PE that we do not understand. Two possible explanations could be entertained as limitations for accessibility to PEMT. First, it could be that only newly synthesized PE can be converted to PC by PEMT. Second, it could be that not all PE can access the location where PEMT is localized, suggesting that PE cannot freely diffuse everywhere in the ER. The latter explanation is consistent with our previous data on sphingosine transport.9 Sphingosine coming from lysosomes can access ceramide synthases, but not sphingosine kinases, both of which are found in the ER. Our data show that PE metabolism is far more complex than originally thought, and can only be seen using tools like those we have prepared.
The diversity of PE metabolism implies its transport between the ER and mitochondria, probably involving the MAM and some lipid transfer proteins.41 The lipid probes we developed present a chemical tool that can be combined with genetic tools to investigate PE transport and its impact on PE metabolism.
Conclusions
To summarize, we developed organelle-targeting photoactivated probes to study PE metabolism in living cells with a high spatiotemporal resolution. We designed different PE probes which can be selectively introduced into the ER versus mitochondria to compare their metabolism according to their subcellular localization. LC-MS results revealed that both mitochondria- and ER-photo-released PE are involved in sn1 and sn2 phospholipid remodeling. They also showed that PEMT methylation was very inefficient but could be detected with ER-PE under specific conditions, suggesting unknown limitations on the PE methylation pathway. Our data thus provide a framework to study phospholipid metabolism at subcellular resolution.Data availability
Experimental data associated with this work have been provided in the ESI.†Author contributions
C. S. and H. R. designed all the experiments; C. S. and A. A. performed the research; C. S. analyzed the data; C. S. wrote the manuscript; C. S., A. A., S. F. and H. R. corrected the manuscript; C. S., S. F. and H. R. conceived and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Takeshi Harayama for technical support in lipidomic experiments. We thank the Bioimaging Center (University of Geneva) for technical assistance. This work was supported by the Human Frontier Science Program (LT000552/2020-L), the Leducq Foundation, the Swiss National Science Foundation (310030_184949), the NCCR Chemical Biology (51NF40-185898) and the Deutsche Forschungsgemeinschaft. S. F. was supported by the Shanghai Municipal Science and Technology Major Project (2019SHZDZX02) and Shanghai Pujiang Program (22PJ1414500).Notes and references
- T. W. Bumpus and J. M. Baskin, Trends Biochem. Sci., 2018, 43, 970–983 CrossRef CAS PubMed.
- A. Laguerre and C. Schultz, Curr. Opin. Cell Biol., 2018, 53, 97–104 CrossRef CAS PubMed.
- T. Bittner, C. Wittwer, S. Hauke, D. Wohlwend, S. Mundinger, A. K. Dutta, D. Bezold, T. Dürr, T. Friedrich, C. Schultz and H. J. Jessen, J. Am. Chem. Soc., 2020, 142, 10606–10611 CrossRef CAS PubMed.
- A. Nadler, G. Reither, S. Feng, F. Stein, S. Reither, R. Müller and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 6330–6334 CrossRef CAS PubMed.
- Y. A. Kim, D. M. C. Ramirez, W. J. Costain, L. J. Johnston and R. Bittman, Chem. Commun., 2011, 47, 9236–9238 RSC.
- P. Gaur, M. Galkin, S. Hauke, R. Redkin, C. Barnes, V. V. Shvadchak and D. A. Yushchenko, Chem. Commun., 2020, 56, 10646–10649 RSC.
- A. Poryvai, M. Galkin, V. Shvadchak and T. Slanina, Angew. Chem., Int. Ed., 2022, 61, e202205855 CAS.
- S. Feng, T. Harayama, S. Montessuit, F. P. David, N. Winssinger, J.-C. Martinou and H. Riezman, eLife, 2018, 7, e34555 CrossRef PubMed.
- S. Feng, T. Harayama, D. Chang, J. T. Hannich, N. Winssinger and H. Riezman, Chem. Sci., 2019, 10, 2253–2258 RSC.
- J. E. Vance and G. Tasseva, Biochim. Biophys. Acta, 2013, 1831, 543–554 CrossRef CAS PubMed.
- J. E. Vance, J. Lipid Res., 2018, 59, 923–944 CrossRef CAS PubMed.
- K. D'Herde and D. V. Krysko, Nat. Chem. Biol., 2017, 13, 4–5 CrossRef PubMed.
- D. P. Siegel and R. M. Epand, Biophys. J., 1997, 73, 3089–3111 CrossRef CAS PubMed.
- P. Rockenfeller, M. Koska, F. Pietrocola, N. Minois, O. Knittelfelder, V. Sica, J. Franz, D. Carmona-Gutierrez, G. Kroemer and F. Madeo, Cell Death Differ., 2015, 22, 499–508 CrossRef CAS PubMed.
- E. van den Brink-van der Laan, J. Antoinette Killian and B. de Kruijff, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 275–288 CrossRef CAS PubMed.
- S. Kumar, C. Chitraju, R. V. Farese, T. C. Walther and C. G. Burd, Biol. Open, 2021, 10, bio058516 CrossRef CAS PubMed.
- J. N. van der Veen, J. P. Kennelly, S. Wan, J. E. Vance, D. E. Vance and R. L. Jacobs, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1558–1572 CrossRef CAS PubMed.
- M. Tavasoli, S. Lahire, T. Reid, M. Brodovsky and C. R. McMaster, J. Biol. Chem., 2020, 295, 17877–17886 CrossRef CAS PubMed.
- R. G. Sleight and R. E. Pagano, J. Biol. Chem., 1983, 258, 9050–9058 CrossRef CAS PubMed.
- B. A. Lipton, E. P. Davidson, B. H. Ginsberg and M. A. Yorek, J. Biol. Chem., 1990, 265, 7195–7201 CrossRef CAS PubMed.
- L. Heikinheimo and P. Somerharju, J. Biol. Chem., 1998, 273, 3327–3335 CrossRef CAS PubMed.
- O. B. Bleijerveld, J. F. H. M. Brouwers, A. B. Vaandrager, J. B. Helms and M. Houweling, J. Biol. Chem., 2007, 282, 28362–28372 CrossRef CAS PubMed.
- V. Kainu, M. Hermansson, S. Hänninen, K. Hokynar and P. Somerharju, Biochim. Biophys. Acta, 2013, 1831, 429–437 CrossRef CAS PubMed.
- J. Montesinos, E. Area-Gomez and M. Schlame, Methods Cell Biol., 2020, 155, 321–335 CAS.
- Y. Horibata, H. Ando and H. Sugimoto, J. Lipid Res., 2020, 61, 1221–1231 CrossRef CAS PubMed.
- E. Altamura, A. Comte, A. D'Onofrio, C. Roussillon, D. Fayolle, R. Buchet, F. Mavelli, P. Stano, M. Fiore and P. Strazewski, Symmetry, 2020, 12, 1108 CrossRef CAS.
- N. Wagner, M. Stephan, D. Höglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339–13343 CrossRef CAS PubMed.
- L. Heikinheimo and P. Somerharju, Biochim. Biophys. Acta, 2002, 1591, 75–85 CrossRef CAS PubMed.
- V. Kainu, M. Hermansson and P. Somerharju, J. Lipid Res., 2010, 51, 3533–3541 CrossRef CAS PubMed.
- B. Wang and P. Tontonoz, Annu. Rev. Physiol., 2019, 81, 165–188 CrossRef PubMed.
- D. E. Vance, Biochim. Biophys. Acta, 2014, 1838, 1477–1487 CrossRef CAS PubMed.
- L. K. Cole and D. E. Vance, J. Biol. Chem., 2010, 285, 11880–11891 CrossRef CAS PubMed.
- F. Gibellini and T. K. Smith, IUBMB Life, 2010, 62, 414–428 CrossRef CAS.
- V. Kainu, M. Hermansson and P. Somerharju, J. Biol. Chem., 2008, 283, 3676–3687 CrossRef CAS PubMed.
- L. Deng, R. Fukuda, T. Kakihara, K. Narita and A. Ohta, Biochim. Biophys. Acta, 2010, 1801, 635–645 CrossRef CAS PubMed.
- H. Shindou, D. Hishikawa, T. Harayama, K. Yuki and T. Shimizu, J. Lipid Res., 2009, 50, S46–S51 CrossRef PubMed.
- Y. Xu, P. C. Miller, C. K. L. Phoon, M. Ren, T. Nargis, S. Rajan, M. M. Hussain and M. Schlame, J. Biol. Chem., 2022, 298, 101685 CrossRef CAS PubMed.
- Z. Cui, M. Houweling and D. E. Vance, Biochem. J., 1995, 312(3), 939–945 CrossRef CAS PubMed.
- M. W. Lee, M. Bakovic and D. E. Vance, Biochem. J., 1996, 320(3), 905–910 CrossRef CAS PubMed.
- D. E. Vance, C. J. Walkey and Z. Cui, Biochim. Biophys. Acta, 1997, 1348, 142–150 CrossRef CAS PubMed.
- L. H. Wong, A. T. Gatta and T. P. Levine, Nat. Rev. Mol. Cell Biol., 2019, 20, 85–101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06069h |
| This journal is © The Royal Society of Chemistry 2023 |