
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand entry pathways control the chemical space recognized by GPR183†
Viktoria Madeline Skovgaard
Kjær‡
 a,
Tomasz Maciej
Stępniewski‡
bcd,
Brian
Medel-Lacruz
b,
Lisa
Reinmuth
a,
Marija
Ciba
e,
Elisabeth
Rexen Ulven
e,
Massimiliano
Bonomi
f,
Jana
Selent
*b and
Mette Marie
Rosenkilde
*a
a,
Tomasz Maciej
Stępniewski‡
bcd,
Brian
Medel-Lacruz
b,
Lisa
Reinmuth
a,
Marija
Ciba
e,
Elisabeth
Rexen Ulven
e,
Massimiliano
Bonomi
f,
Jana
Selent
*b and
Mette Marie
Rosenkilde
*a
aDepartment of Biomedical Sciences, Faculty of Health and Medical Sciences University of Copenhagen, Blegdamsvej 3B, 2200, København N, Denmark. E-mail: Rosenkilde@sund.ku.dk
bResearch Programme on Biomedical Informatics (GRIB), Hospital del Mar Research Institute (IMIM) & Pompeu Fabra University (UPF), Dr Aiguader 88, E-8003, Barcelona, Spain. E-mail: jana.selent@upf.edu
cInterAx Biotech AG, PARK innovAARE, 5234, Villigen, Switzerland
dBiological and Chemical Research Centre, Faculty of Chemistry, University of Warsaw, 02-089, Warsaw, Poland
eDepartment of Drug Design and Pharmacology, University of Copenhagen, Jagtvej 160, 2100, København Ø, Denmark
fInstitut Pasteur, Université Paris Cité, CNRS UMR3528, Structural Bioinformatics Unit, 75015, Paris, France
First published on 25th September 2023
Abstract
The G protein-coupled receptor GPR183 is a chemotactic receptor with an important function in the immune system and association with a variety of diseases. It recognizes ligands with diverse physicochemical properties as both the endogenous oxysterol ligand 7α,25-OHC and synthetic molecules can activate the G protein pathway of the receptor. To better understand the ligand promiscuity of GPR183, we utilized both molecular dynamics simulations and cell-based validation experiments. Our work reveals that the receptor possesses two ligand entry channels: one lateral between transmembrane helices 4 and 5 facing the membrane, and one facing the extracellular environment. Using enhanced sampling, we provide a detailed structural model of 7α,25-OHC entry through the lateral membrane channel. Importantly, the first ligand recognition point at the receptor surface has been captured in diverse experimentally solved structures of different GPCRs. The proposed ligand binding pathway is supported by in vitro data employing GPR183 mutants with a sterically blocked lateral entrance, which display diminished binding and signaling. In addition, computer simulations and experimental validation confirm the existence of a polar water channel which might serve as an alternative entrance gate for less lipophilic ligands from the extracellular milieu. Our study reveals knowledge to understand GPR183 functionality and ligand recognition with implications for the development of drugs for this receptor. Beyond, our work provides insights into a general mechanism GPCRs may use to respond to chemically diverse ligands.
Introduction
G protein-coupled receptors (GPCRs) represent the largest family of cell surface receptors in the human genome.1 Due to their expression in almost every type of human cell and involvement in most physiological processes, GPCRs are attractive drug targets, with over one-third of currently available drugs targeting this protein family.2 Designing novel molecules that can interact and modulate the activity of GPCRs thus remains a relevant research question. When designing GPCR binders, it is typically assumed that each receptor is biased towards a specific type of ligand (e.g., based on hydrophobicity or size), and this bias is primarily driven by the properties of the orthosteric ligand binding site. Ligand binding towards GPCRs, however, appears to be a multi-step process involving various meta-stable binding states,3–5 each of them requiring a specific fit between the molecule and the receptor. Thus, it is likely that the ligand entry pathway on its own is an important factor driving the selectivity of molecules toward GPCRs.6In this work, we focus on GPR183, alias Epstein-Barr Virus (EBV) Induced Gene 2 EBI2, which belongs to class A GPCRs.7,8 Even before its de-orphanization, GPR183 was established to signal via Gαi8 and to play an important role in humoral immunity, in the correct positioning of B cells within the follicle required for mounting of antibody responses.9–11 In 2011, two simultaneous papers presented the native ligands of GPR183 to be a group of hydroxylated cholesterol derivatives (oxysterols) with the most prominent agonist being 7α,25-dihydroxycholesterol (7α,25-OHC).12,13 The OH-groups at positions 7 and 25 were shown to be essential for binding to GPR183, as replacement of either of these for hydrogen (25-OHC and 7α-OHC respectively) resulted in a dramatically decreased GPR183 signaling. Aberrant expression and signaling by GPR183 have been associated with a variety of diseases ranging from inflammatory conditions to metabolic diseases and cancer to mention a few.14,15 These discoveries have sparked a pharmacological interest, which has led to the design of several GPR183 synthetic ligands comprising both agonists and antagonists.16–21 The variation in structure and hydrophobicity between the endogenous and synthetic ligands provokes the question of how one receptor can recognize such diverse ligands and whether they occupy the same binding site.
We and others previously conducted site-directed mutations followed by signaling and binding studies to identify 7α,25-OHC anchor points within GPR183. As such, multiple residues within the receptor core were found to be important for 7α,25-OHC binding (e.g. R872.60, Y1123.33, Y1163.37 and Y2606.51); however, substantially different ligand docking modes arose from the molecular modeling experiments.22,23 More recently though, a research group published two experimentally solved GPR183 structures and suggested that an opening between transmembrane domain (TM) 4 and TM5 serves as a gate for 7α,25-OHC.24 Although modulation of GPCR activity by membrane components has been described in multiple independent studies,25–27 the lateral entry of an orthosteric ligand from the membrane environment is still an uncommon and poorly understood process. It has, however, been proposed in a handful of GPCRs, primarily with hydrophobic ligands i.e., the sphingosine-1-phosphate receptor (S1P1)28 and cannabinoid receptor 1 (CB1)29 that all contain an entry point for their respective ligands between TM1 and TM7. Furthermore, ligand entry through a channel between TM4 and TM5 has been described for both the adenosine 2A (A2AR),30 lysophosphatidic acid 6 (LPA6)31 and the melatonin 1 (MT1R)32,33 receptors. In this study, we combine in silico and in vitro experiments to elucidate the entrance gates of different GPR183 agonists namely the endogenous oxysterol 7α,25-OHC and two synthetic agonists.17 Our study reveals important insights into how a receptor uses promiscuous entrance gates to recognize chemically diverse ligands, which has important implications for drug development endeavors.
Results and discussion
To better understand the overall properties of GPR183, we studied the recently published cryo-EM structure in complex with 7α,25-OHC (PDB ID: 7TUZ)24 using molecular dynamics simulation (Fig. 1), a technique which has provided robust insights into GPCR functionality.34 In the cryo-EM structure, the endogenous agonist establishes the following polar interactions: (i) the 25-OH group in the aliphatic tail is bound to R872.60 and Y1123.33, (ii) the 7α-OH group forms contacts with Y1163.37 and Y2606.51 and (iii) the 3-OH group of the sterol scaffold is found in vicinity to Q1624.56 (Fig. S1†). To our surprise, unbiased all-atom MD simulations with an accumulation simulation time of 12 μs (6 × 2 μs) reveal that the aliphatic tail of 7α,25-OHC is highly flexible adopting three main conformations (clusters 1 to 3, Fig. 1A). In fact, this is in line with the cryo-EM density data in the experimentally solved structure of GPR183 (PDB ID: 7TUZ). When visualizing the cryo-EM density over the ligand structure (Fig. 1B), we find high density for the sterol fragment but only low density for the aliphatic tail which even disappears at an isovalue of 0.017 indicating high flexibility. Cluster 1 (17.5%, blue) (Fig. 1C) overall resembles the crystallized ligand binding pose but lacks interaction between the 25-OH group of the aliphatic tail and R872.60 in TM2. Instead, the aliphatic tail establishes polar interactions with Q2877.32 in the top of TM7. In cluster 2 (16%, red) (Fig. 1D), we observe a major conformational change of the aliphatic tail losing contacts with Q2877.32 in TM7 while forming new contacts with R1053.26 in TM3 with its 25-OH group. Cluster 3 (14.5%, green) (Fig. 1E) is similar to cluster 2, however, it lacks contacts between the 3-OH group and Q1624.56 in TM4 indicating a slight shift of the sterol scaffold. Whereas cluster 1 mainly overlaps with the crystallized binding pose, it is tempting to speculate that cluster 2 and 3 reflect conformational states that are visited during the binding and unbinding process of 7α,25-OHC. Cluster 1 to 3 and their ligand receptor contacts with Y1123.33, Y1163.37, Y2606.51 go along with previous mutational data (Table S1†) as alteration of each of these residues results in a substantial reduction in ligand binding and potency in G protein signaling. However, the high flexibility of the aliphatic tail and the consequential loss of the direct contact between the 25-OH group and R872.60 is surprising as this residue has been implicated in ligand binding in previous studies (see Table S1†).22–24 Interestingly, there is evidence that the effect of R872.60 on 7α,25-OHC binding is linked to specific receptor conformations. For instance, its ligand binding impact appears to be negligible in a receptor conformation induced and stabilized by a D77R mutation.22 Most importantly, R872.60 has been also associated with the general mechanism of GPR183 activation as demonstrated by its relevance for constitutive activity.35 These results suggest that an R872.60A mutation shifts the receptor towards inactive conformations24 which typically goes along with a lower affinity for agonists. All in all, the structural (PDB ID: 7TUZ) and functional data indicate that the R872.60A mutation impedes 7α,25-OHC binding both directly (through contacts) and indirectly (through modifying the receptor conformation). Finally, these points would also explain why the oxysterol ligand can exist in binding modes that do not involve direct interaction with R872.60 as observed in our simulations (Fig. 1, cluster 1 to 3) even though in vitro mutation of this residue to alanine abolishes ligand binding and receptor signaling.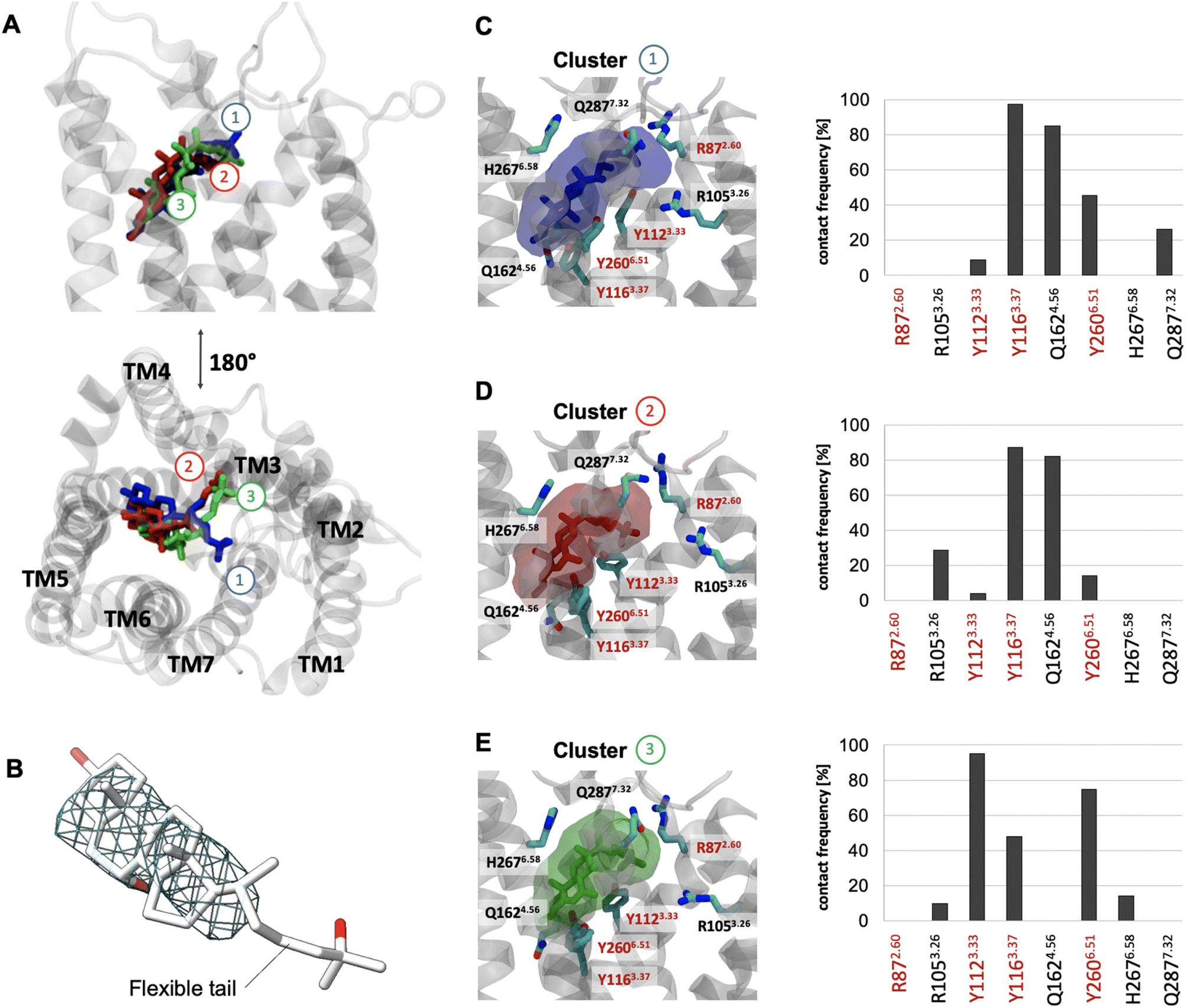 | ||
| Fig. 1 Binding mode sampling of 7α,25-OHC in the receptor interior using unbiased all-atom MD simulation. (A) Superimposition of cluster 1 (blue), 2 (red) and 3 (green) based on the positioning of 7α,25-OHC computed over an accumulated 12 μs simulation time (6 × 2 μs). (B) Cryo-EM density map (EMDB-26136) superimposed to the resolved structure (PDB ID: 7TUZ) and visualized at an isovalue of 0.017 in the region occupied by the 7α,25-OHC ligand. (C–E) Transparent surface reflects the conformational space of cluster 1 (C), cluster 2 (D) or cluster 3 (E) with associated contact frequencies computed over frames belonging to cluster 1, 2 or 3. Red labels are residues found to establish polar interaction with 7α,25-OHC in the experimental structure. Residues highlighted with a red frame have been shown to impact ligand binding in mutational experiments.22,23 | ||
In the experimentally solved structure of GPR18324 the authors observe a small channel between TM4 and TM5 and suggest this opening as a potential entrance gate for 7α,25-OHC. In support of this, oxysterols like 7α,25-OHC have been reported to be integral membrane components36 which would favor an entrance from the lipid bilayer through the proposed lateral channel between TM4 and TM5. To investigate the binding pathway of 7α,25-OHC, we carried out enhanced sampling using metadynamics by placing 7α,25-OHC in the intracellular or extracellular leaflet of the membrane at the TM4-TM5 receptor interface (Fig. 2A and S2†). The entrance pathway was monitored using the distance between the center of mass (COM) of the ligand and the COM of the orthosteric binding pocket within the receptor as reaction coordinates. Starting simulations with 7α,25-OHC located at the intracellular membrane leaflet, we observed that the ligand moved along a cleft on the TM4-5 receptor surface contacting diverse polar residues. In the initial configuration after structural relaxation, 7α,25-OHC interacted with R138 in the intracellular loop 2 (ICL2) via its 3-OH group and with N1203.41via its 25-OH group in the aliphatic tail (Fig. 2A). Thus, the computational data suggest that the interface formed between TM4 and TM5 in the intracellular membrane leaflet is a first anchoring point for 7α,25-OHC molecules. Interestingly, when studying other experimentally solved GPCR structures we found several examples of a cholesterol molecule (or a close derivative of cholesterol) bound within this initial anchoring point (Fig. S3†). This could suggest that the intercellular cleft between TM4 and TM5 represents a common interface between GPCRs and the lipid bilayer, where membrane components can bind to the receptor and either modify its function allosterically or afterwards enter the orthosteric ligand binding pocket. When proceeding the simulations of GPR183 and its ligand, we observed that 7α,25-OHC crawled further up along this TM4-TM5 cleft, contacting now N1203.41 with its 7α-OH group (Fig. 2B). Subsequently, an entrance gate flanked by residues Q1624.56, A2005.42 and G2045.56 opened, and the ligand started intercalating between TM4 and TM5 (Fig. 2C) before entirely penetrating the receptor (Fig. 2D). When simulating the 7α,25-OHC entry from the opposite side, the extracellular membrane leaflet, we observe that the ligand penetrates the receptor with its 3-OH group first (Fig. S2†). Such an entrance mode results in a completely inverted binding mode of 7α,25-OHC in respect to that observed in the crystal structure. When estimating the free energy difference between the bound and the unbound states (Fig. S4†), we find that the experimentally resolved binding mode of 7α,25-OHC (i.e. ligand enters from the lower leaflet) is preferred by 3.9 kcal mol−1 over the inverted mode (i.e. ligand enters from the upper leaflet). Of note, the inverted ligand would require a 180° flip directing its 25-OH into the receptor interior to adopt the experimentally solved ligand binding pose. Based on our simulation data, it appears highly improbable for such a transition to occur, as no ligand re-orientation event was observed throughout the entire sampled (un)binding pathways (Fig. S5†).
 | ||
| Fig. 2 Lateral membrane entrance pathway of 7α,25-OHC probed by metadynamics and site-directed mutagenesis experiments. (A–D) Metadynamics experiments were used to explore the (un)binding pathway of 7α,25-OHC. The ligand is shown in orange whereas the entrance gate flanked by Q1624.56, A2005.42 and G2045.46 is highlighted in green van der Waals representation. The entrance was observed in 3 replicates of metadynamics applying a bias with a Gaussian width of 1 nm and height of 2 kJ mol−1 to the reaction coordinate (see also Fig. S2†). (A) Initial recognition of 7α,25-OHC in a cavity formed between the intracellular parts of TM4 and TM5, 7α,25-OHC is stabilized by polar interaction with residues N1203.41 and R138. (B) Transient meta-stable binding mode of 7α,25-OHC between TM4 and TM5, the molecules is stabilized by polar interactions with N1203.41 as well as hydrophobic interactions with Q1624.56, A2005.42 and G2045.46. (C) Partial insertion of 7α,25-OHC into the GPR183 orthosteric ligand binding site, the ligand is stabilized by polar interactions with N1694.63, a cavity is formed between the extracellular parts of TM4 and TM5, allowing the ligand to intercalate between the helixes. (D) Insertion of 7α,25-OHC into the GPR183 orthosteric ligand binding site. (E–G) Experimental validation testing GPR183 mutants Q163W, A200W and G204W: (E) ELISA-based expression of receptor variants normalized to that of WT receptor in each experiment. (F) G protein signaling determined by BRET Gαi assay. An increase in BRET ratio reflects a decrease in forskolin induced cAMP levels. (G) Competition binding on membranes from stably transfected CHO Flp-In cells. 3[H]7α,25-OHC radioligand (10 nM) was added followed by increasing amounts of 7α,25-OHC. All data are mean ± SEM and experiments were performed in duplicates or triplicates in 3–7 independent experiments. * = p < 0.05 by unpaired t-test. | ||
To experimentally verify the observed lateral entrance gate, we generated three GPR183 mutants (Q162W, A200W & G204W) all designed to introduce steric hindrance at the gate in positions 4.52, 5.42, and 5.46, respectively (Fig. 2). All mutants were expressed at the cell surface, the first two at similar levels to wild-type (WT) receptor and the latter at slightly increased levels (Fig. 2E). Using a BRET-based real-time Gαi protein signaling assay, we determined the dose–response relationships for the mutants when adding increasing amounts of endogenous agonist 7α,25-OHC. The mutants displayed robustly diminished signaling regarding the potency of 7α,25-OHC, determined by the -logEC50-value, and/or the efficacy Emax (Table 1), when compared to the WT receptor that exhibited a −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) EC50-value of 7.9 (Fig. 2F) in accordance with previous studies.12
EC50-value of 7.9 (Fig. 2F) in accordance with previous studies.12
To clarify whether the decreased signaling of mutants Q162W, A200W and G204W was in fact due to obstruction of the TM4–TM5 opening, we investigated ligand binding to these receptor variants. Homologous competition binding using 3[H]7α,25-OHC as radioligand and increasing amounts of non-labeled 7α,25-OHC was implemented to determine the ligand concentration displacing 50% of the specifically bound radioligand (IC50-value) (Table 1). At the WT GPR183, 7α,25-OHC displayed a pIC50-value of 8.1 (Fig. 2G and Table 1). None of the three mutants displayed any measurable radioligand binding at the concentration used (10 nM) (Fig. 2G), as determined from the lack of radioligand binding in the absence of competing unlabeled ligand. That would suggest the steric hindrance mutations in the lateral receptor opening do in fact interfere with the binding of 7α,25-OHC and consequently severely impair signaling by the receptor. Given that the GPR183 mutants still maintain low levels of G protein signaling (Fig. 2F), it can be expected that 7α,25-OHC binds to some degree. However, due to the low sensitivity of the radioligand in the binding assays which is a consequence of the low specific activity of tritium and high unspecific binding of 7α,25-OHC to the membrane, we are not able to detect it. Structural data suggest that two of the mutations in the gate (Q162W and A200W) might also interfere with the experimentally solved binding mode of 7α,25-OHC.23 To dissect whether the designed mutants impede 7α,25-OHC binding by blocking the lateral entry channel only, or via the combined effect of interfering with the final ligand binding pose and blocking the ligand entry, we analyzed the GPR183/7α,25-OHC complex (PDB ID: 7TUZ). Our structural model with the introduction of each individual mutation (Fig. S6†) indicated that the observed impact of the Q162W (Fig. S6B†) and A200W (Fig. S6C†) mutations on 7α,25-OHC binding and signaling might be the consequence of both impaired ligand entry and steric interference with the final ligand binding pose. Contrarily, the G204W mutation flanks the bound ligand but does not induce direct clashes with the ligand binding mode observed in the cryo-EM structure (Fig. S6D†). This suggests that the functional impact of the mutation is primarily by impairing 7α,25-OHC entry from the membrane. While we initially designed this mutant to sterically block the 7α,25-OHC entry through TM4 and TM5, we cannot exclude that also additional polar contacts formed between the G204W mutant and the ligand at the entry gate aggravate ligand entrance. Another interesting observation is that the Q162W mutant retains the highest potency for oxysterol-mediated receptor activation among the studied mutants despite structurally interfering with the experimentally solved 7α,25-OHC binding mode (Fig. S6B†). In contrast, the G204W mutant shows a lower potency even though no ligand interference is observed in our structural models (Fig. S6D†). In this respect, it is worth noting that the potency of a ligand to activate the receptor depends on various factors, including the association rate of the ligand. It is tempting to speculate that a G204W mutant more efficiently closes the ligand entry channel and, by this, slows down the 7α,25-OHC association rate, which in turn reduces the receptor activation potency.
| Receptor variant | −logIC50 ± SEM |
−logEC50 ± SEM |
E max (% of 7a,25-OHC on WT ± SEM) |
|---|---|---|---|
| a N/D denotes that experiments were not conducted. N/A is written if the value could not be reliably determined due to lack of signaling or not reaching a maximum effect at the highest concentration of ligand used. | |||
| Homologous competition binding | G protein signaling 7a,25-OHC | ||
| WT | 8.1 ± 0.2 | 7.9 ± 0.1 | 100 |
| A108F | N/D | 7.6 ± 0.1 | 90 ± 5 |
| Q162W | N/A | 8.1 ± 0.1 | 46 ± 2 |
| A200W | N/A | 6.5 ± 0.2 | N/A |
| G204W | N/A | 6.9 ± 0.1 | N/A |
| L290F | N/D | 7.5 ± 0.1 | 89 ± 3 |
|
|||
| Heterologous competition binding | G protein signaling TUG-2201 | ||
| WT | 7.4 ± 0.2 | 7.1 ± 0.2 | 25 ± 2 |
| A108F | N/D | N/A | N/A |
| Q162W | N/D | N/A | N/A |
| A200W | N/D | 6.7 ± 0.5 | 10 ± 2 |
| G204W | N/D | 7.5 ± 0.3 | 21 ± 2 |
| L290F | N/D | N/A | N/A |
|
|||
| Heterologous competition binding | G protein signaling TUG-2202 | ||
| WT | 7.2 ± 0.2 | 6.3 ± 0.1 | 87 ± 2 |
| A108F | N/D | 5.3 ± 0.2 | 84 ± 14 |
| Q162W | N/D | 7.2 ± 0.4 | 18 ± 3 |
| A200W | N/D | 5.7 ± 0.3 | 34 ± 7 |
| G204W | N/D | 6.7 ± 0.1 | 65 ± 3 |
| L290F | N/D | N/A | N/A |
In addition to the lateral membrane channel, we found evidence in our MD simulations of a second entrance gate in GPR183, which connects the orthosteric site directly to the extracellular milieu. To structurally confirm the existence of such an alternative channel, we carried out additional unbiased simulations for the GPR183 apo-form by removing 7α,25-OHC from the complex (PDB ID: 7TUZ). Intriguingly, computing an occupancy map for water molecules over an accumulated 4.5 μs simulation time (3 × 1.5 μs) revealed the formation of a wide water channel flanked by TM2, TM1, and TM7 which is not described in the originally solved GPR183 structure (PDB ID: 7TUZ) (Fig. 3). To experimentally validate the existence and physiological relevance of this previously undescribed entrance gate, we applied the following strategy. We employed two synthetic compounds, TUG-2201 and TUG-2202 that have been reported to positively modulate GPR183 G protein signaling in previous studies (known as 91 and 92, respectively, in the original publication).17 We determined the experimental LogD7.4 (distribution constant at pH 7.4) for the two synthetic compounds as 3.15 ± 0.06 and 4.13 ± 0.08 for TUG-2201 and TUG-2202, respectively, whereas the endogenous ligand 7α,25-OHC exhibited a LogD7.4 of >5.3. This difference to the endogenous ligand demonstrates that a higher fraction of the synthetic ligands exists in the aqueous phase compared to 7α,25-OHC. Furthermore, we can conclude that the lower lipophilicity of TUG-2201 and TUG-2202 compared to 7α,25-OHC, makes it less likely for them to utilize the hydrophobic lateral entry pathway via the cell membrane, but rather an alternative and preferentially more polar entrance gate. We confirmed that both compounds enter the GPR183 orthosteric ligand binding site by 3[H]7α,25-OHC based heterologous competition binding experiments with increasing amounts of TUG-2201 or TUG-2202 (Fig. 4A, B and Table 1). We then used the GPR183 mutant G204W in which the lateral entry channel is obstructed (as demonstrated for the natural agonist 7α,25-OHC, Fig. 2G) and studied the functional response of the less lipophilic compounds TUG-2201 and TUG-2202. Remarkably, this mutation did not change the signaling profile of the two compounds, supporting the notion that TUG-2201 and TUG-2202 could use a different receptor entry pathway (Fig. 4C and D). In contrast, we found that the Q162W and A200W mutants reduced the signaling induced by TUG-2201 and TUG-2202 (Fig. 4C and D) which is not surprising as both mutations interfere with orthosteric binding according to our structural models (Fig. S6†). To further confirm the existence and relevance of the extracellular gate as the preferred entry pathway for TUG-2201 and TUG-2202, we carried out site directed mutagenesis. Based on the structural model, we hypothesized that mutation of L290 into the bulkier phenylalanine would obstruct the channel, thereby preventing the entry of ligand molecules from the extracellular side (Fig. 5). To investigate this potential blocking effect, we first performed molecular dynamics simulations on the L290F mutant. We estimated the degree of channel opening, and communication between the ligand binding site and the solvent, by computing the water occupancy in the WT and L290F receptors (Fig. 5A and B). Our findings confirm that the L290F mutation disrupts the water channel, both in terms of its structural integrity and the amount of solvent present in the orthosteric pocket (Fig. 5B) compared to the WT receptor (Fig. 5A). Experimental validation of the L290F mutant reveals a significant impairment in receptor signaling by the synthetic ligands TUG-2201 and TUG-2202 (Fig. 5D and E) suggesting that ligand entrance through the extracellular side is indeed impeded by this mutation. Intriguingly, when exposing GPR183 to the endogenous agonist 7α,25-OHC, receptor signaling is largely preserved (Fig. 5C), suggesting that this ligand utilizes a different entry channel than TUG-2201 and TUG-2202, even though both ligands occupy the same orthosteric binding pocket (Fig. 4A and B).
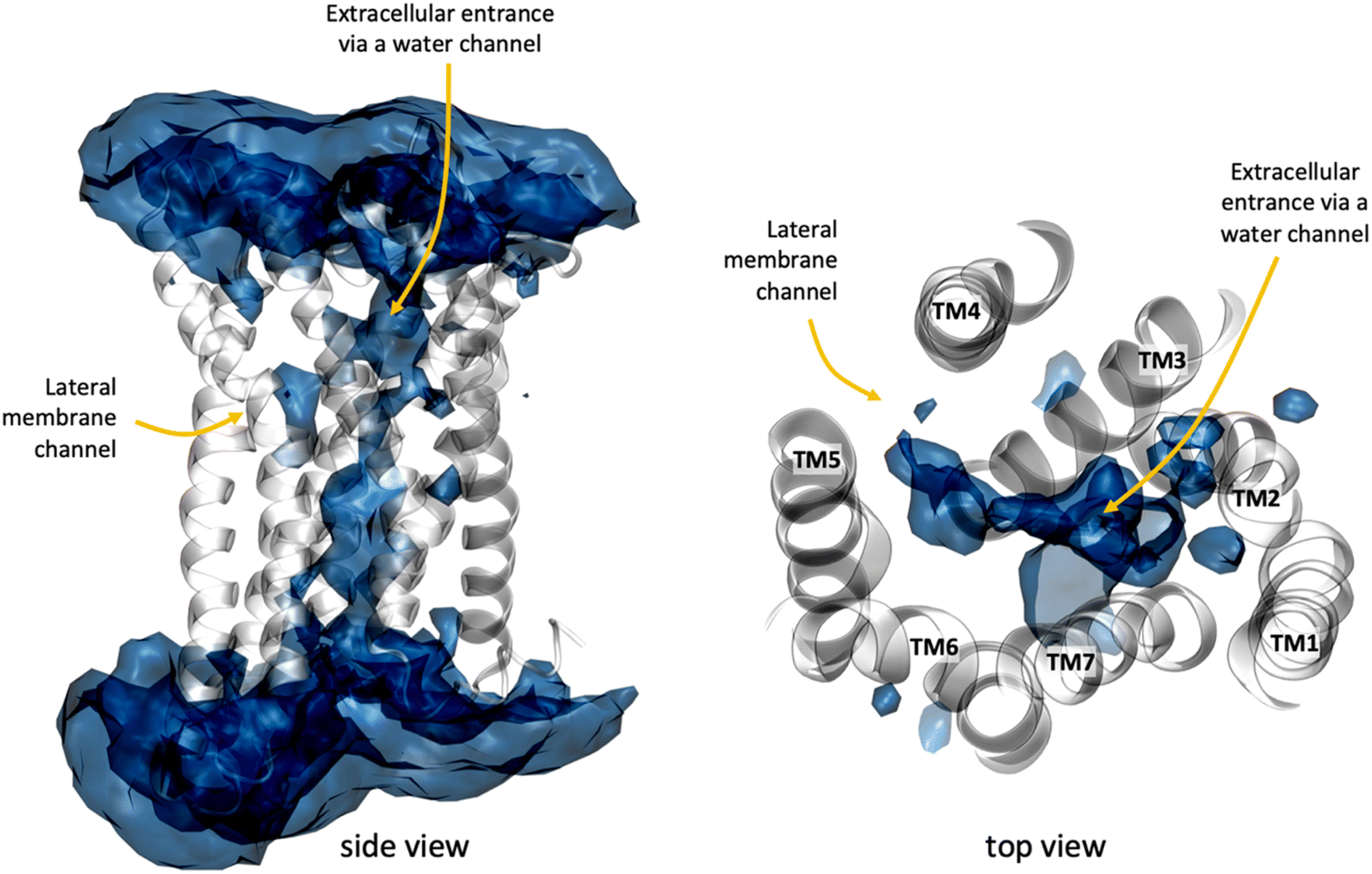 | ||
| Fig. 3 A water channel connects the extracellular side with the receptor interior in GPR183. The water map was computed as water occupancy over an accumulated simulation time of 3 μs (3 × 1 μs) for the apo GPR183 using the volmap plugin of VMD1.9.3. The water occupancy highlights regions where water is observed for at least 22% of the simulation time. GPR183 is depicted in white cartoon whereas the natural agonist 7α,25-OHC in cyan licorice. | ||
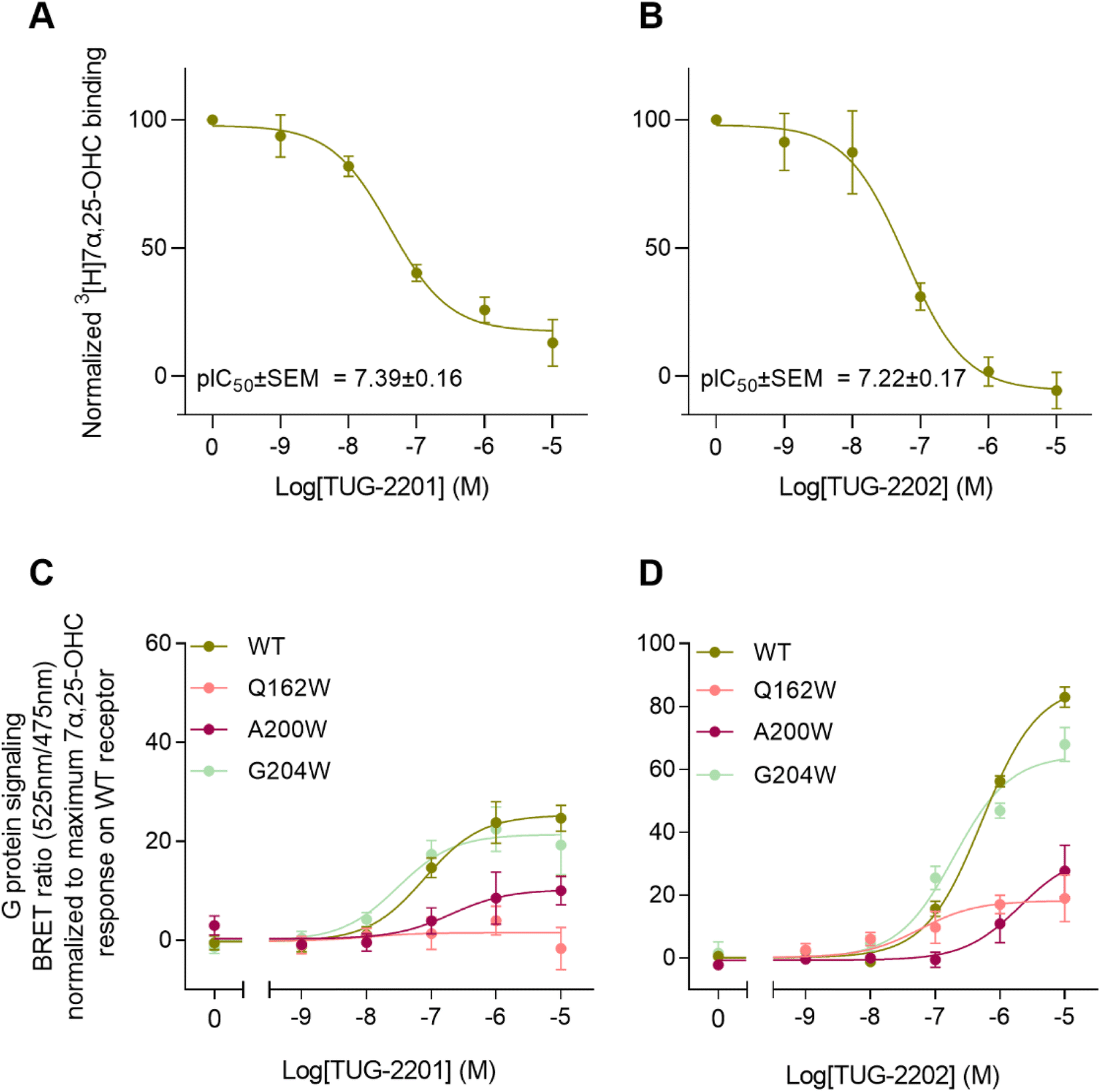 | ||
| Fig. 4 Activation of and binding to GPR183 by synthetic small molecule agonists TUG-2201 and TUG-2202. (A and B) Heterologous competition binding to GPR183 WT receptor with indicated amounts of TUG-2201 (A) or TUG-2202 (B) and a fixed (10 nM) amount of 3[H]7α,25-OHC radioligand. (C and D) G protein signaling (BRET assay) by GPR183 WT and steric hindrance mutants Q162W, A200W and G204W upon stimulation with synthetic compounds TUG-2201 (C) and TUG2202 (D). All data are mean ± SEM and experiments were performed in duplicates or triplicates in 3–5 independent experiments. | ||
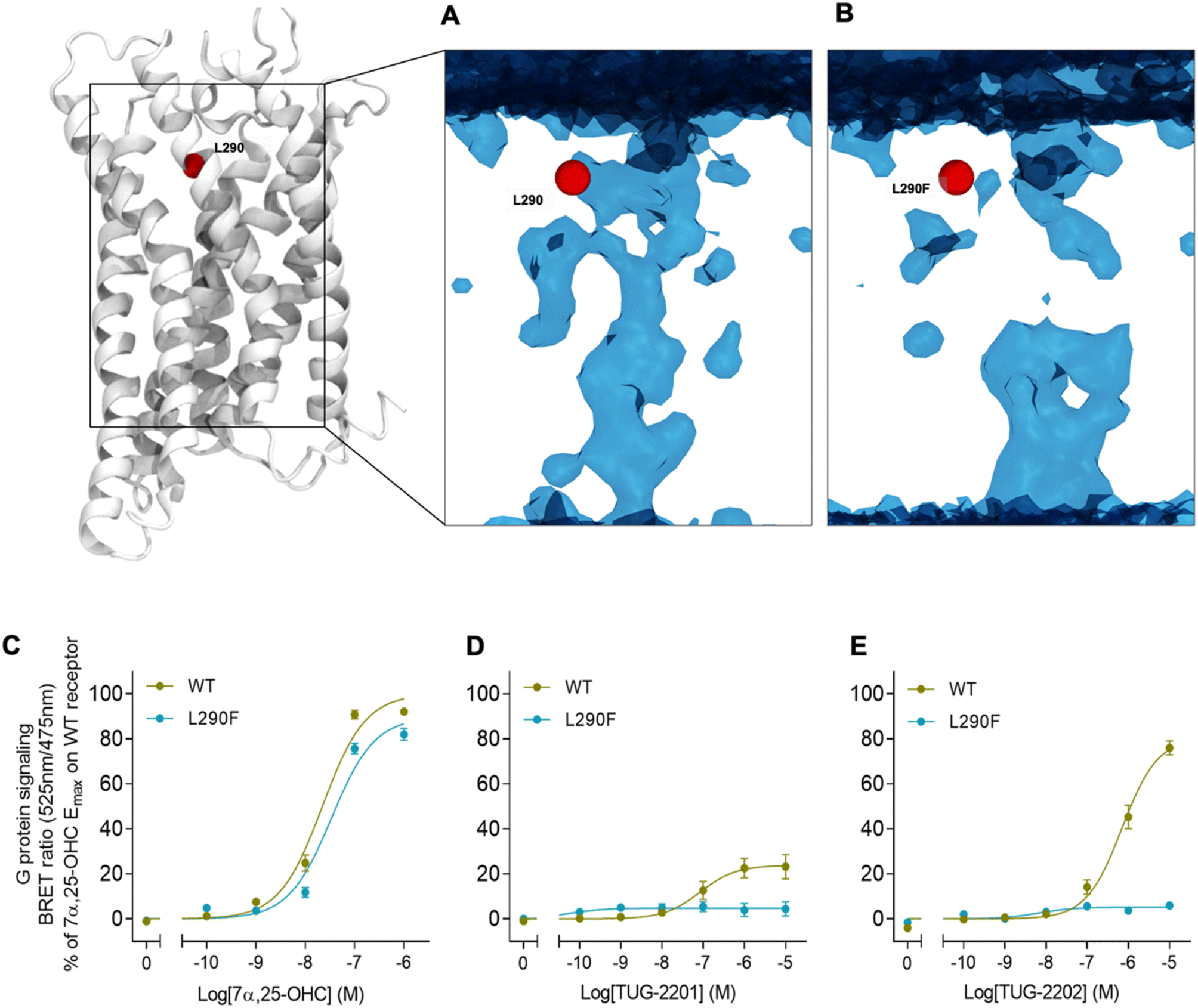 | ||
| Fig. 5 Impact on mutations on extracellular ligand entry into GPR183. (A) Structure of GPR183 with mutated position 290 highlighted in red. (B) A water map in GPR183 computer as occupancy over an accumulated simulation time of 4.5 μs (3 × 1.5 μs). The introduction of the L290F mutation narrows-down the extracellular channel, thus impairing water entry into the receptor, as it can be seen in the resulting map. (C–E) G protein signaling by GPR183 WT and L290F mutant upon stimulation with indicated concentrations of either 7α,25-OHC, TUG-2201 or TUG-2202. Data represent mean ± SEM and experiments were performed in triplicates in 4–8 independent experiments. | ||
All in all, we suggest that the synthetic agonists TUG-2201 and TUG-2202 can modulate GPR183 by entering the receptor directly from the extracellular milieu via a water channel based on the following findings: (i) TUG-2201 and TUG-2202 are less lipophilic than 7α,25-OHC which will aggravate receptor entrance from the hydrophobic membrane side, (ii) the existence of a wide water channel that connects the extracellular with the orthosteric binding site, (iii) the mutation G204W closes the lateral channel and impedes the function of the lipophilic 7α,25-OHC, but not the synthetic ligands TUG-2201 and TUG-2202 and lastly, (iv) obstructing the water channel by the L290F mutation reduces G protein signaling induced by TUG-2201 and TUG-2202 without affecting the 7α,25-OHC induced signal.
Conclusion
Our study provides evidence that a GPCR can utilize different ligand entry channels to recognize structurally diverse ligands. On one hand, our simulation data suggest that the endogenous agonist 7α,25-OHC preferentially enters the GPR183 orthosteric ligand binding pocket from the inner leaflet of the cell membrane. The existence of a lateral channel gate is supported by site-directed mutagenesis in which channel closure by the steric hindrance G204W mutation inhibits 7α,25-OHC binding and function. Importantly, our molecular simulations revealed an initial recognition site for 7α,25-OHC in the intracellular part of the cleft between TM4 and TM5 (Fig. 2A), and we find that this observed sterol binding site exists in numerous experimental GPCR structures (Fig. S3†) supporting our suggested entry pathway. Of note, oxysterols not only bind to GPR183 but are involved in the regulation of the immune system through interaction with multiple GPCRs.37 It is tempting to speculate that the common sterol anchoring point between TM4 and TM5 is of general relevance for membrane-induced effects on multiple GPCRs either via an allosteric action or, as shown in this study, as an initial point to enter the orthosteric ligand binding site.In addition to the membrane channel, we report a water channel connecting the binding pocket to the extracellular milieu (Fig. 3). Our data suggest that the two synthetic agonists TUG-2201 and TUG-2202 use this water-filled channel to enter the receptor as mutational closure of the lateral channel does not fully inhibit receptor activation and downstream signaling, whereas obstruction of the water channel does (Fig. 5). Altogether our findings indicate that GPR183 can respond to structurally diverse ligands thanks to the existence of different entrance channels. In theory, the chemotactic nature of GPR183 implies that its endogenous ligand 7α,25-OHC must exist in the extracellular milieu.38,39 The location in the extracellular milieu could allow a direct 7α,25-OHC entry through the observed water channel; however, our mutational data suggest that the lateral channel is the preferential entry gate for this ligand and that 7α,25-OHC integrates into the membrane to access the GPR183 interior.
Promiscuity in ligand entry pathways has previously been reported for other GPCRs, such as the melatonin receptor 2 (MT2)32 and the dopamine D3 receptor (DRD3)40 but it is still largely neglected in drug design efforts. In this respect, our findings have important implications for the development of drug candidates targeting GPR183. We find that the structural ligand properties will determine the way of ligand entrance (i.e. via the hydrophobic lateral or polar extracellular channel) which in turn could drive ligand binding kinetics. Ultimately, the detailed structural view into the binding pathway of the natural oxysterol agonist from the intracellular lipid leaflet and its entrance gate provides the opportunity to rationally design novel GPR183 orthosteric agonists and antagonists. In addition, it allows the design of compounds that target the meta-stable binding sites of the natural agonist along its entry pathway, and by this blocking physiological GPR183 activation. Considering this novel insight, several questions related to the role of the cell membrane as a ligand reservoir or its promotion of ligand–receptor interaction and finally ligand entrance will be an important focus of future work.
Methods
Experimental section
:9 ratio using Lipofectamine 2000 transfection method (Invitrogen). Post transfection (48 h), selection medium consisting of growth medium supplemented with 600 μg mL−1 Hygromycin B (Roche) was added to the cells and replenished every 3–4 days until foci were visible. Cells were grown in selection medium throughout the course of experiments and passaged every 3–5 days.
000 or 250000 per well in a 6-well plate in growth medium. The following day or two days after, respectively, cells were transfected using the Lipofectamine 2000 method (Invitrogen). A mixture of 160 ng M1-tagged receptor DNA in pcDNA3.1+ vector and 840 ng CAMYEL sensor DNA was made in 125 μL Opti-MEM™ while 6 μL Lipofectamine was mixed with 125 μL Opti-MEMTM in a separate tube. After 5 min incubation, the two solutions were mixed and incubated at RT for 20 min. Cell medium was changed to 1 mL Opti-MEM™ and the Lipofectamine 2000/DNA mixture was dripped on top of the cell medium and incubated for approximately 24 hours until assay was performed. On assay day cells were suspended in PBS + 5 mM glucose (3 mL per well). 84 μL cell suspension was added to each well in an opaque flat bottomed 96-well plate. Subsequently, 10 μL 50 μM coelenterazine luciferase substrate in PBS was added to each well (5 μM in well) followed by 1 μL 100× ligand in 100% DMSO. Five minutes post ligand addition, 5 μL 200 μM forskolin in PBS was added to each well (10 μM in well). At 40 minutes post ligand addition the BRET emissions at 525 nm (acceptor) and 485 nm (donor) were measured on a PerkinElmer EnVision plate reader and the BRET ratio was determined as acceptor counts/donor counts. Ligands TUG-2201 and TUG-2202 were kindly provided by Trond Ulven, Department of Drug Design and Pharmacology, University of Copenhagen.
446g for 45 minutes. Pellets were resuspended in 0.5–0.75 mL storage buffer (20 mM HEPES, 2 mM MgCl2, 0.4 mM CaCl2 and cOmplete™, EDTA-free Protease Inhibitor Cocktail 1 tablet/50 mL). The protein content of membrane preparations was determined using Pierce BCA Protein Assay Kit (ThermoFisher) according to the manufacturer's protocol.
:10 with MeOH (+0.1% TFA)/Milli-Q water (4:1, v/v) and analyzed by HPLC (Dionex UltiMate HPLC system, Gemini-NX C18 column (3 μm, 4.6 mm × 250 mm, 110 Å)). The interface was removed and the PBS7.4 phase was analyzed directly by HPLC or ESI-LCMS (Agilent 6130 Mass Spectrometer instrument using electron spray ionization (ESI) coupled to an Agilent 1200 HPLC system) in single ion mode, where [M + H+–2H2O]+ ion of 7α,25-OHC at m/z 383.41 (ref. 42) was used for quantification. LogD values were calculated from the peak areas and adjusted for difference in injection volume. All compounds were analyzed in triplicate.
Computational section
The systems were first relaxed for 100 ns under constant pressure and temperature (NPT) with a time step of 2 fs and gradually decreasing harmonic constraints applied to the protein backbone. Temperature was maintained at 310 K using the Langevin thermostat 50 and pressure was kept at 1 bar using the Berendsen barostat.50 The equilibration run was followed by production runs under constant volume and temperature (NVT) with a 4 fs time step. The temperature was maintained at 310 K using the Langevin thermostat. No harmonic constraints are applied in the NVT phase. For the complex with the ligand we amassed out 6 × 2 μs of simulation time, and for the apo system 3 × 1 μs. Simulation analysis was carried out in the Visual Molecular Dynamics package.51 Clustering was carried out in VMD. Simulations frames were aligned using the protein backbone, afterwards they were clustered based on the position of the ligand heavy atoms, using an RMSD cutoff of 2.2 Å. Subsequently the three most populated cluster were analyzed (amounting to 17.56%, 15.98% and 14.69% of simulation frames).
Data availability
Simulation data is available through the open access GPCRmd data respository (https://www.gpcrmd.org).Author contributions
Conceptualization: VMSK, TMS, JS, MMR. In vitro experiments: VMSK, MC, ERU, LR. Supervision of in vitro experiments: MMR. In silico experiments: TMS, BML, LR, MB. Supervision of in silico experiments: JS. Writing original draft: VMSK, TMS, JS, MMR. Writing, review and editing: VMSK, TMS, BML, LR, MC, ERU, MB, JS, MMR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thank you to Trond Ulven (Dep. of Drug Design and Pharmacology, University of Copenhagen) for providing synthetic agonists TUG-2201 and TUG-2202. The authors acknowledge support from the National Center of Science, Poland (2017/27/N/NZ2/02571), Sara Borrell grant CD22/00007 funded by the Institute of Health Carlos III (ISCIII) and resources of grant 2021 SGR 00046 funded by Agència de Gestió d'Ajuts Universitaris i de Recerca Generalitat de Catalunya (AGAUR) to TMS, Instituto de Salud Carlos III (ISCIII) (AC18/00030) as well as the Instituto de Salud Carlos III (ISCIII) and co-funded by the European Union (PI18/00094) to JS, the Danish Council for Independent Research | Medical Sciences (9039-00298B) to MMR and the Lundbeck Foundation (R307-2018-2950) to ERU. TMS, BML, JS and MMR participate in the European COST Action CA18133 (ERNEST).References
- J. Craig Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Yuan Wang, A. Wang, X. Wang, J. Wang, M. H. Wei, R. Wides, C. Xiao, C. Yan, A. Yao, J. Ye, M. Zhan, W. Zhang, H. Zhang, Q. Zhao, L. Zheng, F. Zhong, W. Zhong, S. C. Zhu, S. Zhao, D. Gilbert, S. Baumhueter, G. Spier, C. Carter, A. Cravchik, T. Woodage, F. Ali, H. An, A. Awe, D. Baldwin, H. Baden, M. Barnstead, I. Barrow, K. Beeson, D. Busam, A. Carver, A. Center, M. Lai Cheng, L. Curry, S. Danaher, L. Davenport, R. Desilets, S. Dietz, K. Dodson, L. Doup, S. Ferriera, N. Garg, A. Gluecksmann, B. Hart, J. Haynes, C. Haynes, C. Heiner, S. Hladun, D. Hostin, J. Houck, T. Howland, C. Ibegwam, J. Johnson, F. Kalush, L. Kline, S. Koduru, A. Love, F. Mann, D. May, S. McCawley, T. McIntosh, I. McMullen, M. Moy, L. Moy, B. Murphy, K. Nelson, C. Pfannkoch, E. Pratts, V. Puri, H. Qureshi, M. Reardon, R. Rodriguez, Y. H. Rogers, D. Romblad, B. Ruhfel, R. Scott, C. Sitter, M. Smallwood, E. Stewart, R. Strong, E. Suh, R. Thomas, N. Ni Tint, S. Tse, C. Vech, G. Wang, J. Wetter, S. Williams, M. Williams, S. Windsor, E. Winn-Deen, K. Wolfe, J. Zaveri, K. Zaveri, J. F. Abril, R. Guigo, M. J. Campbell, K. V. Sjolander, B. Karlak, A. Kejariwal, H. Mi, B. Lazareva, T. Hatton, A. Narechania, K. Diemer, A. Muruganujan, N. Guo, S. Sato, V. Bafna, S. Istrail, R. Lippert, R. Schwartz, B. Walenz, S. Yooseph, D. Allen, A. Basu, J. Baxendale, L. Blick, M. Caminha, J. Carnes-Stine, P. Caulk, Y. H. Chiang, M. Coyne, C. Dahlke, A. Deslattes Mays, M. Dombroski, M. Donnelly, D. Ely, S. Esparham, C. Fosler, H. Gire, S. Glanowski, K. Glasser, A. Glodek, M. Gorokhov, K. Graham, B. Gropman, M. Harris, J. Heil, S. Henderson, J. Hoover, D. Jennings, C. Jordan, J. Jordan, J. Kasha, L. Kagan, C. Kraft, A. Levitsky, M. Lewis, X. Liu, J. Lopez, D. Ma, W. Majoros, J. McDaniel, S. Murphy, M. Newman, T. Nguyen, N. Nguyen, M. Nodell, S. Pan, J. Peck, M. Peterson, W. Rowe, R. Sanders, J. Scott, M. Simpson, T. Smith, A. Sprague, T. Stockwell, R. Turner, E. Venter, M. Wang, M. Wen, D. Wu, M. Wu, A. Xia, A. Zandieh and X. Zhu, Science, 2001, 291, 1304–1351 CrossRef.
- R. Santos, O. Ursu, A. Gaulton, A. P. Bento, R. S. Donadi, C. G. Bologa, A. Karlsson, B. Al-Lazikani, A. Hersey, T. I. Oprea and J. P. Overington, Nat. Rev. Drug Discovery, 2017, 16, 19–34 CrossRef CAS.
- R. O. Dror, A. C. Pan, D. H. Arlow, D. W. Borhani, P. Maragakis, Y. Shan, H. Xu and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13118–13123 CrossRef CAS PubMed.
- T. Thomas, E. Yuriev and D. K. Chalmers, J. Chem. Theory Comput., 2020, 16, 3879–3888 CrossRef CAS PubMed.
- N. Stanley, L. Pardo and G. De Fabritiis, Sci. Rep., 2016, 22639 CrossRef CAS PubMed.
- P. Fronik, B. I. Gaiser and D. Sejer Pedersen, J. Med. Chem., 2017, 60, 4126–4134 CrossRef CAS PubMed.
- B. Mark, J. Knud, Y. Ramana, L. Gilbert and K. Elliott, J. Virol., 1993, 67, 2209–2220 CrossRef.
- M. M. Rosenkilde, T. Benned-Jensen, H. Andersen, P. J. Holst, T. N. Kledal, H. R. Lüttichau, J. K. Larsen, J. P. Christensen and T. W. Schwartz, J. Biol. Chem., 2006, 281, 13199–13208 CrossRef CAS.
- D. Gatto, D. Paus, A. Basten, C. R. Mackay and R. Brink, Immunity, 2009, 31, 259–269 CrossRef CAS.
- J. P. Pereira, L. M. Kelly, Y. Xu and J. G. Cyster, Nature, 2009, 460, 1122–1126 CrossRef CAS.
- K. N. Nobles, K. Xiao, S. Ahn, A. K. Shukla, C. M. Lam, S. Rajagopal, R. T. Strachan, T. Y. Huang, E. A. Bressler, M. R. Hara, S. K. Shenoy, S. P. Gygi and R. J. Lefkowitz, Sci. Signaling, 2011, 4(185), ra51 CrossRef CAS PubMed.
- S. Hannedouche, J. Zhang, T. Yi, W. Shen, D. Nguyen, J. P. Pereira, D. Guerini, B. U. Baumgarten, S. Roggo, B. Wen, R. Knochenmuss, S. Noël, F. Gessier, L. M. Kelly, M. Vanek, S. Laurent, I. Preuss, C. Miault, I. Christen, R. Karuna, W. Li, D. I. Koo, T. Suply, C. Schmedt, E. C. Peters, R. Falchetto, A. Katopodis, C. Spanka, M. O. Roy, M. Detheux, Y. A. Chen, P. G. Schultz, C. Y. Cho, K. Seuwen, J. G. Cyster and A. W. Sailer, Nature, 2011, 475, 524 CrossRef CAS PubMed.
- C. Liu, X. V. Yang, J. Wu, C. Kuei, N. S. Mani, L. Zhang, J. Yu, S. W. Sutton, N. Qin, H. Banie, L. Karlsson, S. Sun and T. W. Lovenberg, Nature, 2011, 475, 519–523 CrossRef CAS.
- S. Bartlett, A. T. Gemiarto, M. D. Ngo, H. Sajiir, S. Hailu, R. Sinha, C. X. Foo, L. Kleynhans, H. Tshivhula, T. Webber, H. Bielefeldt-Ohmann, N. P. West, A. M. Hiemstra, C. E. MacDonald, L. v. V. Christensen, L. S. Schlesinger, G. Walzl, M. M. Rosenkilde, T. Mandrup-Poulsen and K. Ronacher, Front. Immunol., 2020, 11, 2890 Search PubMed.
- S. Sun and C. Liu, Front. Pharmacol, 2015, 6, 60 Search PubMed.
- F. Gessier, I. Preuss, H. Yin, M. M. Rosenkilde, S. Laurent, R. Endres, Y. A. Chen, T. H. Marsilje, K. Seuwen, D. G. Nguyen and A. W. Sailer, J. Med. Chem., 2014, 57, 3358–3368 CrossRef CAS PubMed.
- V. M. S. Kjær, L. Ieremias, V. Daugvilaite, M. Lückmann, T. M. Frimurer, T. Ulven, M. M. Rosenkilde and J. Våbenø, ChemMedChem, 2021, 16, 2623–2627 CrossRef PubMed.
- T. Benned-Jensen, C. Smethurst, P. J. Holst, K. R. Page, H. Sauls, B. Sivertsen, T. W. Schwartz, A. Blanchard, R. Jepras and M. M. Rosenkilde, J. Biol. Chem., 2011, 286, 29292–29302 CrossRef CAS PubMed.
- K. Braden, L. A. Giancotti, Z. Chen, C. DeLeon, N. Latzo, T. Boehm, N. D'Cunha, B. M. Thompson, T. M. Doyle, J. G. McDonald, J. K. Walker, G. R. Kolar, C. K. Arnatt and D. Salvemini, J. Pharmacol. Exp. Ther., 2020, 375, 367–375 CrossRef CAS PubMed.
- R. Ardecky, E. Sergienko, J. Zou, S. Ganji, B. Brown, Q. Sun, C.-T. Ma, B. Hood, K. Nguyen, S. Vasile, E. Suyama, A. Mangravita-Novo, S. Salaniwal, P. Kung, L. H. Smith, T. D. Y. Chung, M. R. Jackson, A. B. Pinkerton and R. Rickert, Probe Reports from NIH Mol. Libr. Progr., 2015 Search PubMed.
- T. Benned-Jensen, C. M. Madsen, K. N. Arfelt, C. Smethurts, A. Blanchard, R. Jepras and M. M. Rosenkilde, FEBS Open Bio, 2013, 3, 156–160 CrossRef CAS.
- T. Benned-Jensen, C. Norn, S. Laurent, C. M. Madsen, H. M. Larsen, K. N. Arfelt, R. M. Wolf, T. Frimurer, A. W. Sailer and M. M. Rosenkilde, J. Biol. Chem., 2012, 287, 35470–35483 CrossRef CAS.
- L. Zhang, A. Y. Shih, X. V. Yang, C. Kuei, J. Wu, X. Deng, N. S. Mani, T. Mirzadegan, S. Sun, T. W. Lovenberg and C. Liu, Mol. Pharmacol., 2012, 82, 1094–1103 CrossRef CAS PubMed.
- H. Chen, W. Huang and X. Li, Structure, 2022, 30(7), 1016–1024 CrossRef CAS PubMed.
- G. A. Kumar, P. Sarkar, T. M. Stepniewski, M. Jafurulla, S. P. Singh, J. Selent and A. Chattopadhyay, Sci. Adv., 2021, 7(30), eabh2922 CrossRef CAS PubMed.
- S. Oddi, T. M. Stepniewski, A. Totaro, J. Selent, L. Scipioni, B. Dufrusine, F. Fezza, E. Dainese and M. Maccarrone, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 523–532 CrossRef CAS PubMed.
- J. M. Ramírez-Anguita, I. Rodríguez-Espigares, R. Guixà-González, A. Bruno, M. Torrens-Fontanals, A. Varela-Rial and J. Selent, Biotechnol. Appl. Biochem., 2018, 65, 29–37 CrossRef PubMed.
- M. A. Hanson, C. B. Roth, E. Jo, M. T. Griffith, F. L. Scott, G. Reinhart, H. Desale, B. Clemons, S. M. Cahalan, S. C. Schuerer, M. G. Sanna, G. W. Han, P. Kuhn, H. Rosen and R. C. Stevens, Science, 2012, 335, 851–855 CrossRef CAS PubMed.
- J. Jakowiecki, U. Orzeł, S. Chawananon, P. Miszta and S. Filipek, Molecules, 2021, 26(9), 2456 CrossRef CAS.
- R. Guixà-González, J. L. Albasanz, I. Rodriguez-Espigares, M. Pastor, F. Sanz, M. Martí-Solano, M. Manna, H. Martinez-Seara, P. W. Hildebrand, M. Martín and J. Selent, Nat. Commun., 2017, 8(1), 14505 CrossRef.
- R. Taniguchi, A. Inoue, M. Sayama, A. Uwamizu, K. Yamashita, K. Hirata, M. Yoshida, Y. Tanaka, H. E. Kato, Y. Nakada-Nakura, Y. Otani, T. Nishizawa, T. Doi, T. Ohwada, R. Ishitani, J. Aoki and O. Nureki, Nature, 2017, 548, 356–360 CrossRef CAS PubMed.
- L. C. Johansson, B. Stauch, J. D. McCorvy, G. W. Han, N. Patel, X. P. Huang, A. Batyuk, C. Gati, S. T. Slocum, C. Li, J. M. Grandner, S. Hao, R. H. J. Olsen, A. R. Tribo, S. Zaare, L. Zhu, N. A. Zatsepin, U. Weierstall, S. Yous, R. C. Stevens, W. Liu, B. L. Roth, V. Katritch and V. Cherezov, Nature, 2019, 569, 289 CrossRef CAS PubMed.
- B. Stauch, L. C. Johansson, J. D. McCorvy, N. Patel, G. W. Han, X. P. Huang, C. Gati, A. Batyuk, S. T. Slocum, A. Ishchenko, W. Brehm, T. A. White, N. Michaelian, C. Madsen, L. Zhu, T. D. Grant, J. M. Grandner, A. Shiriaeva, R. H. J. Olsen, A. R. Tribo, S. Yous, R. C. Stevens, U. Weierstall, V. Katritch, B. L. Roth, W. Liu and V. Cherezov, Nature, 2019, 569, 284 CrossRef CAS.
- I. Rodríguez-Espigares, M. Torrens-Fontanals, J. K. S. Tiemann, D. Aranda-García, J. M. Ramírez-Anguita, T. M. Stepniewski, N. Worp, A. Varela-Rial, A. Morales-Pastor, B. Medel-Lacruz, G. Pándy-Szekeres, E. Mayol, T. Giorgino, J. Carlsson, X. Deupi, S. Filipek, M. Filizola, J. C. Gómez-Tamayo, A. Gonzalez, H. Gutiérrez-de-Terán, M. Jiménez-Rosés, W. Jespers, J. Kapla, G. Khelashvili, P. Kolb, D. Latek, M. Marti-Solano, P. Matricon, M. T. Matsoukas, P. Miszta, M. Olivella, L. Perez-Benito, D. Provasi, S. Ríos, I. R. Torrecillas, J. Sallander, A. Sztyler, S. Vasile, H. Weinstein, U. Zachariae, P. W. Hildebrand, G. De Fabritiis, F. Sanz, D. E. Gloriam, A. Cordomi, R. Guixà-González and J. Selent, Nat. Methods, 2020, 17, 777–787 CrossRef PubMed.
- T. Benned-Jensen and M. M. Rosenkilde, Mol. Pharmacol., 2008, 74, 1008–1021 CrossRef CAS.
- A. J. Brown and W. Jessup, Mol. Aspects Med., 2009, 30, 111–122 CrossRef CAS.
- L. Reinmuth, C. C. Hsiao, J. Hamann, M. Rosenkilde and J. Mackrill, Cells, 2021, 10(8), 2078 CrossRef CAS PubMed.
- D. Gatto and R. Brink, Trends Immunol., 2013, 34, 336–341 CrossRef CAS PubMed.
- T. Yi, X. Wang, L. M. Kelly, J. An, Y. Xu, A. W. Sailer, J. A. Gustafsson, D. W. Russell and J. G. Cyster, Immunity, 2012, 37, 535–548 CrossRef CAS.
- F. Lolicato, H. Juhola, A. Zak, P. A. Postila, A. Saukko, S. Rissanen, G. Enkavi, I. Vattulainen, M. Kepczynski and T. Róg, ACS Chem. Neurosci., 2020, 11, 1914–1924 CrossRef CAS PubMed.
- L. I. Jiang, J. Collins, R. Davis, K. M. Lin, D. DeCamp, T. Roach, R. Hsueh, R. A. Rebres, E. M. Ross, R. Taussig, I. Fraser and P. C. Sternweis, J. Biol. Chem., 2007, 282, 10576–10584 CrossRef CAS PubMed.
- W. S. Choi, G. Lee, W. H. Song, J. T. Koh, J. Yang, J. S. Kwak, H. E. Kim, S. K. Kim, Y. O. Son, H. Nam, I. Jin, Z. Y. Park, J. Kim, I. Y. Park, J. I. Hong, H. A. Kim, C. H. Chun, J. H. Ryu and J. S. Chun, Nature, 2019, 566, 254–258 CrossRef CAS PubMed.
- G. Pándy-Szekeres, C. Munk, T. M. Tsonkov, S. Mordalski, K. Harpsøe, A. S. Hauser, A. J. Bojarski and D. E. Gloriam, Nucleic Acids Res., 2018, 46, D440–D446 CrossRef PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins: Struct., Funct., Genet., 2005, 61(4), 704–721 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2016, 14, 71–73 CrossRef PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CAS.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81(8), 3684–3690 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Banáš, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bottaro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. Chen, W. Chen, F. Colizzi, S. De, M. De La Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Ferguson, M. Filizola, J. S. Fraser, H. Fu, P. Gasparotto, F. L. Gervasio, F. Giberti, A. Gil-Ley, T. Giorgino, G. T. Heller, G. M. Hocky, M. Iannuzzi, M. Invernizzi, K. E. Jelfs, A. Jussupow, E. Kirilin, A. Laio, V. Limongelli, K. Lindorff-Larsen, T. Löhr, F. Marinelli, L. Martin-Samos, M. Masetti, R. Meyer, A. Michaelides, C. Molteni, T. Morishita, M. Nava, C. Paissoni, E. Papaleo, M. Parrinello, J. Pfaendtner, P. Piaggi, G. M. Piccini, A. Pietropaolo, F. Pietrucci, S. Pipolo, D. Provasi, D. Quigley, P. Raiteri, S. Raniolo, J. Rydzewski, M. Salvalaglio, G. C. Sosso, V. Spiwok, J. Šponer, D. W. H. Swenson, P. Tiwary, O. Valsson, M. Vendruscolo, G. A. Voth and A. White, Nat. Methods, 2019, 16(8), 670–673 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05962b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |