
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEndogenous and exogenous wireless multimodal light-emitting chemical devices†
Miaoxia
Liu‡
 a,
Gerardo
Salinas‡
a,
Jing
Yu
a,
Antoine
Cornet
a,
Haidong
Li
b,
Alexander
Kuhn
*a and
Neso
Sojic
*a
a,
Gerardo
Salinas‡
a,
Jing
Yu
a,
Antoine
Cornet
a,
Haidong
Li
b,
Alexander
Kuhn
*a and
Neso
Sojic
*a
aUniv. Bordeaux, Bordeaux INP, ISM, UMR 5255 CNRS, Site ENSMAC, 33607 Pessac, France. E-mail: kuhn@enscbp.fr; sojic@u-bordeaux.fr
bCollege of Chemistry and Chemical Engineering, Yangzhou University, 225002 Yangzhou, China
First published on 5th September 2023
Abstract
Multimodal imaging is a powerful and versatile approach that integrates and correlates multiple optical modalities within a single device. This concept has gained considerable attention due to its potential applications ranging from sensing to medicine. Herein, we develop several wireless multimodal light-emitting chemical systems by coupling two light sources based on different physical principles: electrochemiluminescence (ECL) occurring at the electrode interface and a light-emitting diode (LED) switched on by an electrochemically triggered electron flow. Endogenous (thermodynamically spontaneous redox process) and exogenous (requiring an external power source) bipolar electrochemistry acts as a driving force to trigger both light emissions at different wavelengths. The results presented here interconnect optical imaging and electrochemical reactions, providing a novel and so far unexplored alternative to design autonomous hybrid systems with multimodal and multicolor optical readouts for complex bio-chemical systems.
Introduction
Multimodal imaging is based on the combination and correlation of two or more optical readouts of biological or chemical information, commonly within a single testing platform or device.1–12 The design of such complex systems is of crucial importance for many applications ranging from sensing to dynamic readout e.g. using self-propelled micro/nano-objects. It is a fast developing research area at the interface of several disciplines. In particular, light-emitting approaches based on fluorescence, electrochemiluminescence (ECL) or light emitting diodes (LEDs), are interesting alternatives to develop novel imaging systems.13–22 With these approaches, the physico-chemical information is encoded by light emission, allowing a direct visualization. This provides an easy and straightforward alternative to study complex dynamics with high spatial and temporal resolution. In this context, light-emitting imaging methods, based on ECL or LEDs, present rather strong light intensity, easily captured with simple commercial cameras, in comparison with fluorescence which requires expensive and more sophisticated optical microscopy set-ups. However, the majority of the available light-emitting devices require a direct electric connection to a power source.In recent years, bipolar electrochemistry (BE) has evolved into a powerful technique to induce light emission in a wireless manner.16,23–32 Briefly, under the influence of an electric field (ε), applied across an electrolyte solution, a polarization potential difference (ΔV) is generated at the extremities of a conducting object, the so-called bipolar electrode (BPE).33–36 In the presence of electroactive species, oxidation and reduction reactions take place at the extremities of the BPE, only when the ΔV overcomes the thermodynamic threshold potential (ΔVmin). The synergy between BE and light-emitting systems, based on ECL or by incorporating electronic devices within the BPE, has been extensively used for a broad range of applications, from sensing and optical mapping/imaging to the study of dynamic behaviors.37–52 Recently, a sophisticated and so far unexplored different type of BE, based on thermodynamically spontaneous redox reactions, has been developed. In such an approach a highly favored redox reaction i.e. the oxidation of Mg, is used as a source of electrons to trigger the light emission of a diode.53,54 However, the majority of the designed wireless light-emitting systems exhibit only a single color. A promising alternative to design wireless multimodal optical systems is to couple two different light sources based on different physical principles and powered by BE.
In this work, we design wireless multimodal light-emitting systems by taking advantage of the synergy between two light sources: the ECL emissions occurring at the anode and cathode of a hybrid BPE and the switching-on of a LED, triggered by the concomitant electron flow from the anodic to the cathodic extremities of the device. The ECL/LED systems are designed by combining three main ingredients: (i) the proper luminophore/co-reactant couple, (ii) a micro-LED, and (iii) bare metallic wires connected to both extremities of the LED acting as BPEs, where the interfacial ECL occurs (Scheme 1). The resulting hybrid devices emit light by exploiting the concept of BE in two different scenarios: when the driving force is exogenous (i.e. non-spontaneous, Scheme 1a and b), an external electric field is required, and endogenous (i.e. spontaneous, Scheme 1c and d), where thermodynamically favored redox reactions act as the source of electrons to trigger both light emissions. In other words, in the endogenous configuration, no external power source is required and the device is self-powered. Such light-emitting wireless devices offer a novel multimodal platform for the straightforward optical readout of chemical information in solution.
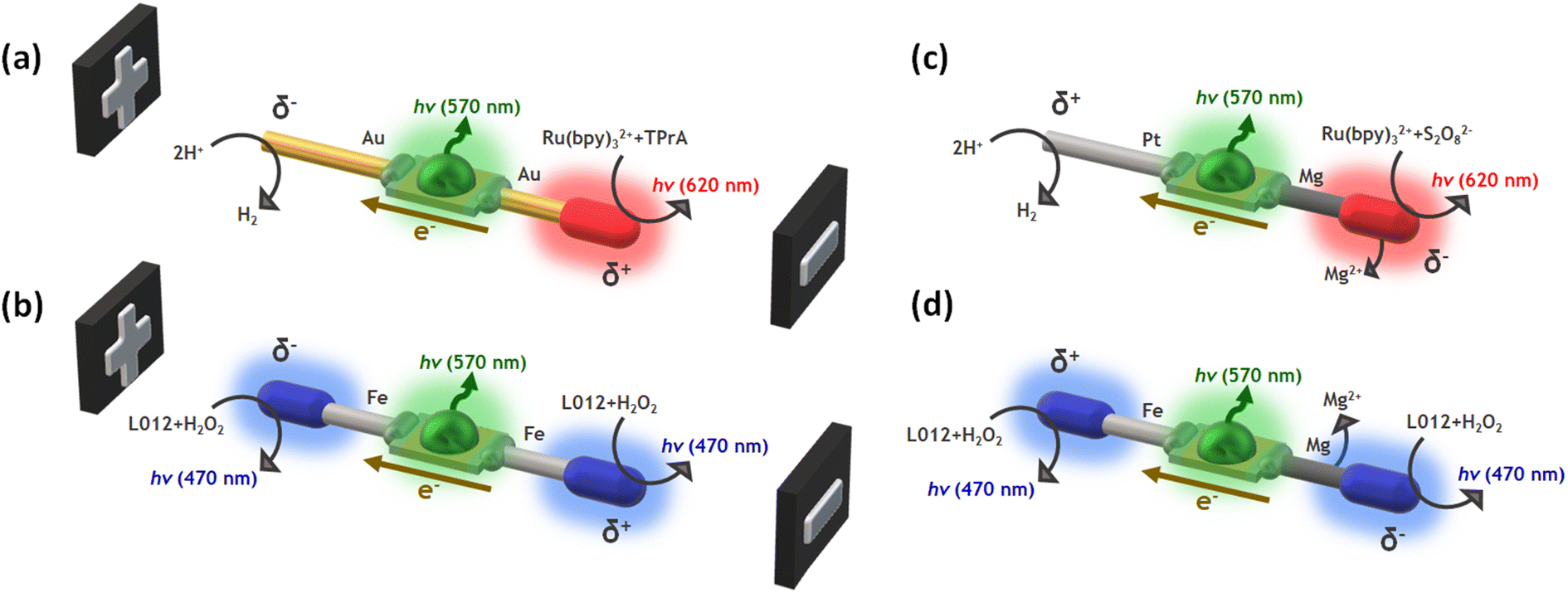 | ||
| Scheme 1 Schematic illustration of the different hybrid wireless light-emitting systems used for (a and b) the exogenous and (c and d) endogenous bipolar electrochemistry approach, with a representation of the associated chemical reactions, the electron flux and the color of the resulting light emissions. | ||
Results and discussion
Hybrid light-emitting bipolar electrodes
To demonstrate the versatility of our multimodal wireless approach, we developed different designs of hybrid light-emitting devices that were powered either exogenously by an external electric field, as in a classic BE configuration, or endogenously by exploiting the redox activity of spontaneous Mg oxidation (Scheme 1). The hybrid bipolar systems were elaborated by connecting two metal wires to the anode and cathode of a miniaturized green LED. Four different systems were designed and produced according to the nature of the wireless power source: (i) Au-LED-Au and Fe-LED-Fe for the exogenous devices (Scheme 1a and b); (ii) Pt-LED-Mg and Fe-LED-Mg for the endogenous ones (Scheme 1c and d). To generate different colors, we selected two typical ECL luminophores, Ru(bpy)32+ and L012 (8-amino-5-chloro-2,3-dihydro-7-phenyl-pyrido[3,4-d]pyri-dazine-1,4-dione), a luminol analog.55–58 They generate photons in the red (620 nm) and in the blue (470 nm) spectral regions, respectively. Both can operate in oxidation or in reduction, depending on the chosen co-reactant. Thus, the proper selection of the luminophore and its sacrificial co-reactant allows tuning the color of the ECL emission at the surface of the metallic extremities of the hybrid device. By coupling the oxidation of Ru(bpy)32+/TPrA with the reduction of protons, a characteristic red emission can be induced at the anode of the Au-LED-Au device (Scheme 1a). The flux of electrons from the anode to the cathode, through the LED, simultaneously also powers the green emission of the diode (570 nm). It is important to highlight the presence of dissolved dioxygen during the BE measurements, thus a fraction of the electrons passing through the BPE might be also associated to the reduction of dioxygen. With the same philosophy, the anodic and cathodic blue ECL emission of the L012/H2O2 system can be coupled to the green light produced from the LED by using the Fe-LED-Fe BPE (Scheme 1b). Indeed, blue ECL emission of L012 can be induced simultaneously in oxidation and in reduction on both anodic and cathodic poles, respectively.26On the other hand, as stated above, the endogenous bipolar approach requires a thermodynamically favourable source of electrons, in the present case the spontaneous oxidation of Mg at the anode of the device. Since the oxidation of Mg and the reduction of protons take place spontaneously with a standard redox potential difference of 2.34 V, in theory, a fraction of the released electrons passes from the anodic to the cathodic pole through the LED, triggering the emission of the diode. To favour this spontaneous flux of electrons along the endogenous BPE device, it is necessary to use the current, originating from the anodic reaction, for a kinetically favoured cathodic reaction. Furthermore, Mg can act as a strong reductant causing the spontaneous reduction of the luminophore/co-reactant system. Thus, in the case of the Pt-LED-Mg hybrid system, the ECL emission based on the reductive–oxidative mechanism of the Ru(bpy)32+/S2O82− system, occurs exclusively at the anode (Mg).59–61 In theory, such reactions lead to a sufficient flux of electrons that passes through the BPE towards the cathode where the kinetically favoured reduction of protons occurs on Pt, switching on the green light emission of the LED (Scheme 1c). The same concept can be extended to the L012/H2O2 system, where the blue ECL emission can be triggered at the anode and cathode of the BPE, via the spontaneous oxidation of Mg (Scheme 1d). Thus, in a first order approximation, such a spontaneous system can be considered as a short-circuited battery and this explains the polarity of the electrodes and the direction of the current indicated in Scheme 1c and d.
Exogenous bipolar electrochemical approach
To test the exogenous bipolar electrochemical approach, the intensity of the light emission produced by the Au-LED-Au and Fe-LED-Fe BPEs, as a function of the applied electric field was evaluated. The optical behavior of the devices was studied by placing the BPEs independently at the center of a classic bipolar electrochemical cell containing a 5 mM PBS (pH 7.4) solution in the presence of the corresponding luminophore/co-reactant system. For the Au-LED-Au device, the Ru(bpy)32+/TPrA couple (1 mM/100 mM ratio) was used. Under these conditions and at low ε values (below 2.1 V cm−1), there is not enough driving force to induce redox reactions at the anode and cathode of the device; thus, no light emission from the diode or the electrodes was observed (Fig. 1a). Once the electric field reaches the threshold value of around 2.2 V cm−1, the high enough ΔV triggers two coupled redox reactions, producing a flux of electrons from the anodic to the cathodic side of the diode and thus its concomitant light emission. However, at this point no ECL emission is generated, hence, the reduction of protons is probably coupled to the oxidation of TPrA. This was corroborated by evaluating the potentiodynamic profile of Ru(bpy)32+/TPrA on Au (Fig. S1†). As expected, during the anodic sweep, the oxidation of TPrA takes place at lower potentials than Ru(bpy)32+ (0.8 V vs. Ag/AgCl and 1.1 V vs. Ag/AgCl, respectively) (Fig. S1a†). However, at ε values above 2.6 V cm−1, the characteristic red ECL emission of the Ru(bpy)32+/TPrA at the anode occurs and is easily visible with naked eyes (Fig. 1a). In addition, the light intensity of the diode increases as a function of the applied electric field, whereas the intensity of the ECL emission reaches a maximum value at 3.4 V cm−1 (Fig. 1a bottom). This is expected since, according to the BE principles, the length of the anodic and cathodic regions, with the proper polarization to trigger the redox reactions, increases as a function of the applied electric field.62,63 Such an enlargement of the polarization regions is visualized by the continuous displacement of the red ECL emission towards the center of the BPE (Fig. 1a top). Thus above 3.4 V cm−1, the reduction of protons is coupled not only to the oxidation of the luminophore and co-reactant, but also to the oxygen evolution reaction at the anode, causing the continuous increase of the light intensity of the diode.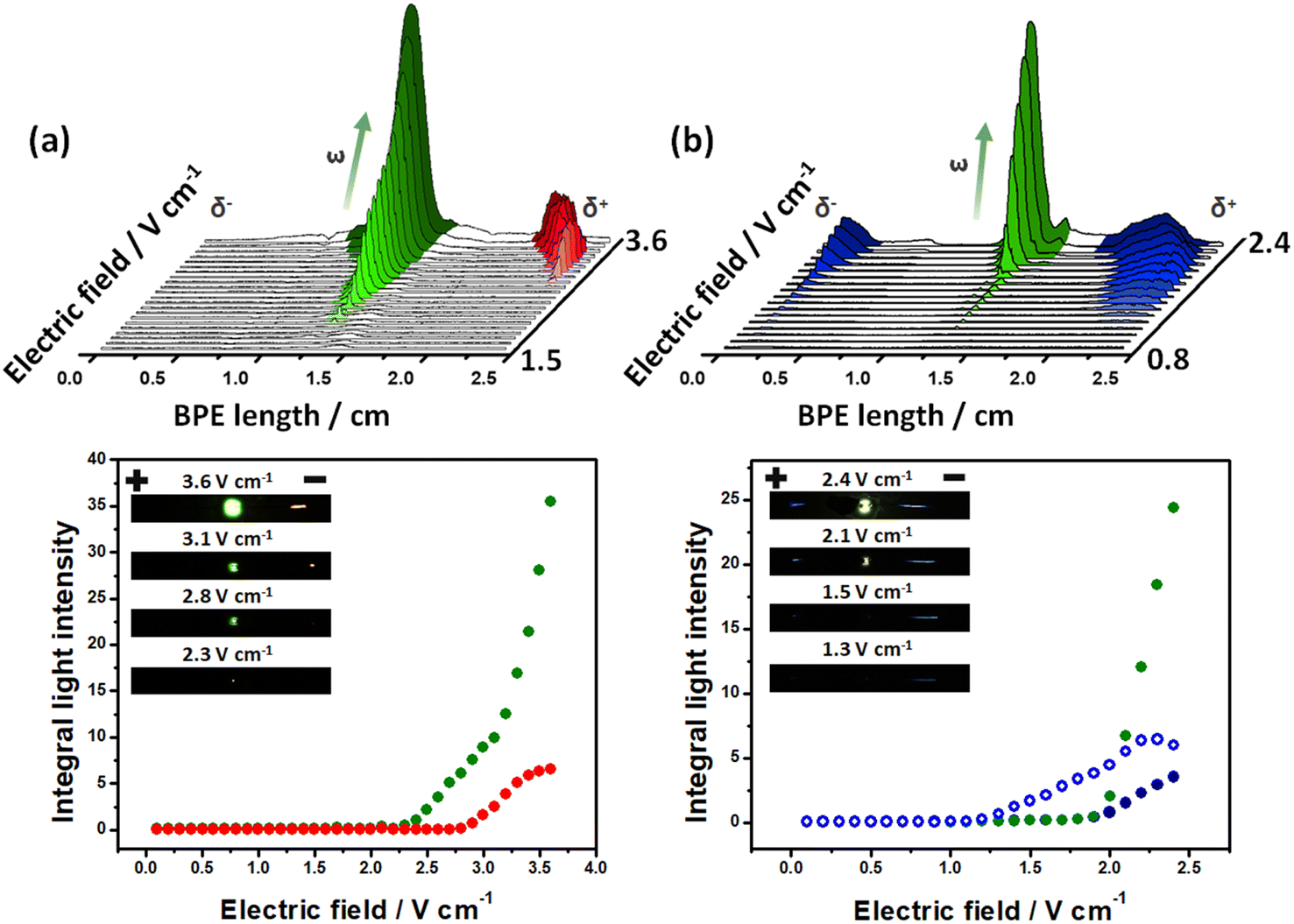 | ||
| Fig. 1 ECL and LED light generated by the exogenous multimodal devices. 3D representation of the light intensity profiles (top) and integrals of the light intensity (bottom) as a function of the imposed electric field obtained by using (a) an Au-LED-Au device in a 5 mM PBS (pH 7.4), 1 mM Ru(bpy)32+, 100 mM TPrA solution and (b) a Fe-LED-Fe device in a 5 mM PBS (pH 7.4), 1 mM L012, 100 mM H2O2 solution. Inset: optical pictures of the corresponding exogenous multimodal light-emitting devices at different applied electric fields (indicated in the figure). Plain and empty blue dots represent the ECL emissions of L012 occurring at the cathodic and anodic poles, respectively. | ||
To expand this concept to another model ECL luminophore, the light-emitting behavior of the Fe-LED-Fe device was evaluated in the presence of the L012/H2O2 system (1 mM/100 mM). Once again, at low ε values (below 1.1 V cm−1) there is not enough driving force to induce redox reactions, which is reflected by the absence of light emission from the diode or the electrodes (Fig. 1b). Above 1.2 V cm−1, a high enough ΔV triggers the characteristic blue ECL emission at the anode of the device. In this case, the oxidative and reductive mechanisms of the L012/H2O2 take place simultaneously at the surface of the Fe electrodes.26 The potentiodynamic measurements of the system present the anodic and cathodic ECL emission around 0.64 V vs. Ag/AgCl and −0.14 V vs. Ag/AgCl, respectively (Fig. S1b†), confirming the coupled redox reactions at the extremities of the BPE. However, under these conditions, cathodic ECL and the emission of the diode are not detected. This can be attributed to a relatively low current produced by the associated redox reactions for such a low electric field. However, as it can be seen, by applying higher ε values (above 1.9 V cm−1), it is possible to trigger the characteristic blue ECL emission at both the anodic and cathodic side of the BPE together with green light emission from the LED (Fig. 1b). Once again, the light intensity of the diode and the ECL at the cathode, increases as a function of the applied electric field, whereas the intensity of the anodic ECL emission reaches a maximum value at 2.2 V cm−1. It is important to highlight that additional resistive effects such as the interfacial potential drop, as well as the intrinsic resistance of the electrolyte and the LED, cause a considerable increase of the electric field required to trigger the LED and/or ECL emission. However, the possible fine-tuning of the applied electric field gives the exogenous system the advantage of wirelessly inducing a wide range of redox reactions.
Endogenous bipolar electrochemical approach
After this first set of experiments, demonstrating the exogenous induction of both ECL/LED emissions, we investigated the possibility to use endogenous BE for the design of multimodal autonomous light-emitting devices. As stated above, this is based on the efficient coupling of thermodynamically spontaneous redox reactions. Thus, the main driving force is the highly favored oxidation of Mg, at the anode of the LED, as a source of electrons. As stated above, this spontaneous system acts as a short-circuited battery, explaining the polarity of the electrodes and the direction of the current indicated in Scheme 1c and d. Therefore, the endogenous systems do not require any external power supply to generate both types of light. In theory, by connecting a Pt or Fe wire at the cathodic extremity of the diode, a fraction of the electrons produced by the oxidation of Mg can be directed to the cathode of the device, inducing the green light emission of the LED.Two different hybrid bipolar electrodes were designed: Pt-LED-Mg and Fe-LED-Mg. The light emission of these devices was tested independently as a function of the co-reactant concentration, containing the proper luminophore. For the Pt-LED-Mg device, the ECL intensity of Ru(bpy)32+ (1 mM) and the spontaneous emission of the diode were evaluated as a function of the concentration of S2O82− in a 0.1 M PBS (pH 7.4), 10 mM H2SO4 solution. As expected, under these conditions, in the absence of co-reactant, only the diode emits light, due to the spontaneous oxidation of Mg and reduction of protons at the Pt electrode, respectively (Fig. 2a). In the presence of the S2O82−, the reductive–oxidative ECL mechanism takes place spontaneously at the interface of the Mg anode, acting as a reductant, producing the characteristic red emission of Ru(bpy)32+ (Fig. 2a). Such reactions cause an increase of current passing through the LED, with a concomitant increase of the light intensity of the diode. As can be seen, both light emissions increase with the co-reactant concentration until reaching a maximum intensity at 5 mM (Fig. 2a and S2†).
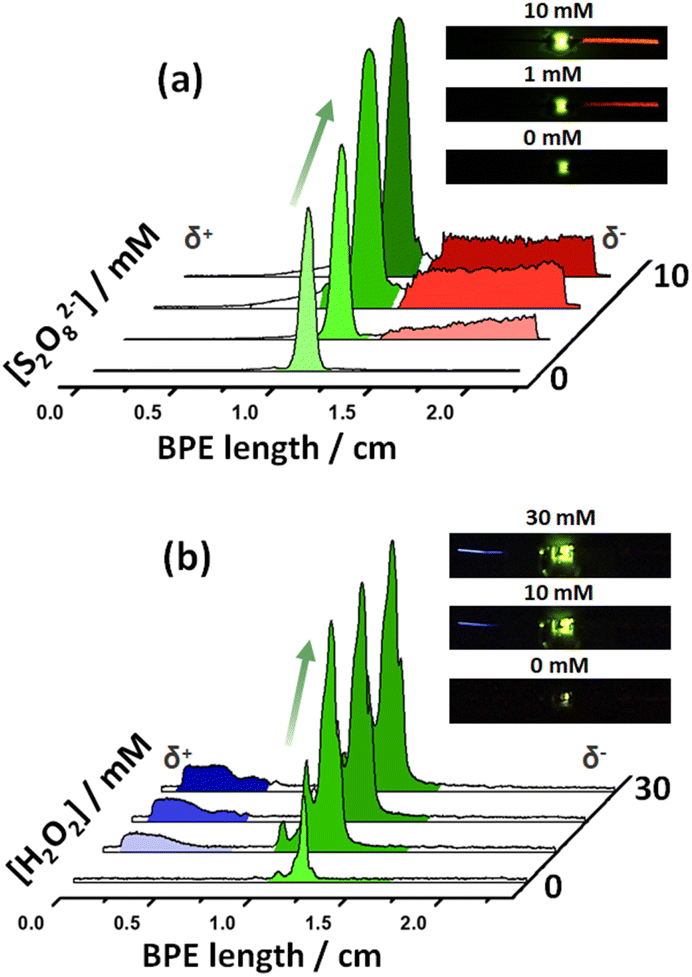 | ||
| Fig. 2 Endogenous light-emitting multimodal devices. 3D representation of the light intensity profiles as a function of the (a) S2O82− concentration obtained by using a Pt-LED-Mg device in a 0.1 M PBS (pH 7.4), 1 mM Ru(bpy)32+, 10 mM H2SO4 solution or (b) H2O2 concentration obtained by using a divided Fe-LED-Mg device in a 0.1 M PBS (pH 7.4) solution, in the presence of L012 (1 mM) and H2SO4 (10 mM) in the cathodic and anodic compartments, respectively. The insets show the optical pictures of the corresponding endogenous multimodal light-emitting devices at different co-reactant concentrations (indicated in the figure). | ||
With a similar philosophy, we studied the optical behavior of the Fe-LED-Mg device as a function of the H2O2 concentration in a 0.1 M PBS (pH 7.4), 1 mM L012 solution. It is important to highlight the absence of H2SO4 in this solution, due to the low solubility of the luminophore at low pH values. However, once again due to the thermodynamically favored oxidation and reduction of Mg and H+, respectively, only the diode emits light in the absence of the co-reactant (Fig. S3a†). Moreover, the intensity of the green emission increases as a function of the H2O2 concentration, whereas the characteristic blue ECL emission of L012 occurs, at the cathode and anode at concentrations of co-reactant above 20 mM and 50 mM, respectively (Fig. S3a and b†). The barely visible ECL emission at the cathodic and anodic extremities is associated with the relatively low amount of electrons produced during the oxidation of Mg in the absence of acid. Thus, under these conditions, high concentrations of co-reactant are required to trigger the oxidation of Mg and induce the ECL emission.
In order to corroborate this hypothesis and enhance the cathodic ECL emission, a divided Fe-LED-Mg endogenous bipolar system was designed. This consists in placing a polydimethylsiloxane (PDMS) barrier to separate physically the anodic and cathodic compartments (Fig. S4a†). In addition, a porous glass connector was integrated into the PDMS barrier as an ionic membrane in order to keep electroneutrality. This configuration is reminiscent of a closed configuration in BE, but without the feeder electrodes. The optical behavior of the divided Fe-LED-Mg device as a function of H2O2 concentration was tested, in a 0.1 M PBS (pH 7.4) solution, in the presence of L012 (1 mM) and H2SO4 (10 mM) in the cathodic and anodic compartments, respectively. Under these conditions, the light intensity of the diode increases by around one order of magnitude in the absence of H2O2, in comparison with the undivided system (Fig. 2b, and S4b†). Furthermore, the characteristic blue ECL emission of L012 at the cathode appears after the initial addition of H2O2, exhibiting an increase of the light intensity as a function of the co-reactant concentration (Fig. 2b and S4b†). Such an enhancement of the light intensity of both, the ECL and the LED emissions, is attributed to the presence of acid in the anodic compartment. This favors the oxidation of Mg, which translates into a considerable increase of current passing from the anode to the cathode of the hybrid BPE.
Endogenous multicolor light-emitting device
Finally, we tested whether the same compartmentalized device can be used to design a multimodal light-emitting platform composed of 3 different light sources. As stated above, in the presence of Ru(bpy)32+ and S2O82−, a reductive–oxidative ECL-like mechanism occurs spontaneously at the Mg anode. Thus, it is relatively straightforward to assume that by using such a luminophore/co-reactant system in the anodic compartment of the divided Fe-LED-Mg device, the characteristic red ECL emission can be produced (Scheme 2). Consequently, a flux of electrons from the anode to the cathode of the LED takes place, inducing the green emission of the diode and, in the presence of the L012/H2O2 system in the cathodic compartment, the blue ECL emission on the surface of the Fe wire (Scheme 2).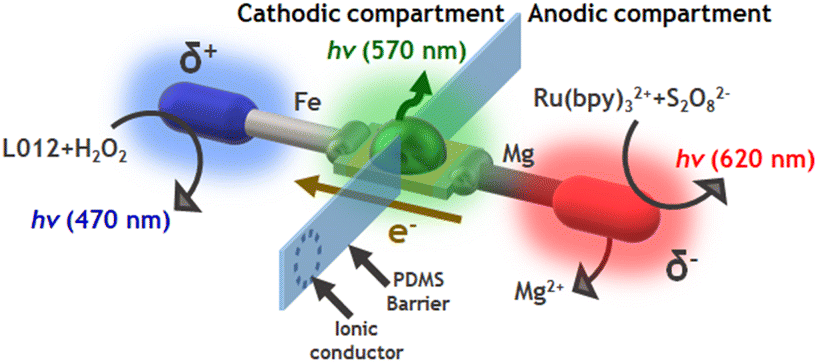 | ||
| Scheme 2 Schematic illustration of the divided light-emitting bipolar system used for the endogenous multimodal approach, with a representation of the associated chemical reactions, the electron flux, the physical separation, the ionic conductor (dot lines) and the cathodic (blue)/LED (green)/anodic (red) light emissions. | ||
First, we evaluated the optical behaviour of the divided hybrid systems as a function of the H2O2 concentration at the cathodic side in a 0.1 M PBS (pH 7.4), 1 mM L012 solution. For this set of experiments, we kept constant the composition of the anodic compartment, hence, a 0.1 M PBS (pH 7.4), 1 mM Ru(bpy)32+, 20 mM S2O82−, 10 mM H2SO4 solution was used. Under these conditions, in the absence of H2O2, only the red ECL emission of Ru(bpy)32+ at the anode was observed (Fig. 3a and S5a†). However, as expected, the addition of H2O2 to the cathodic compartment induces the generation of the reductive ECL of L012 with the characteristic blue ECL color. Furthermore, the kinetically favoured reaction leads to a faster consumption of electrons at the cathode, which causes an increase of current passing through the LED and a concomitant enhancement of light intensity (Fig. 3a and S5a†). Similarly, we evaluated the optical responses of the device as a function of the S2O82− concentration in the anodic compartment in a 0.1 M PBS (pH 7.4), 1 mM Ru(bpy)32+, 10 mM H2SO4 solution. In this case, the cathodic solution was composed of a 0.1 M PBS (pH 7.4), 1 mM L012, 100 mM H2O2 solution. As can be seen in Fig. 3b, the green emission of the LED and the blue ECL at the cathode were generated spontaneously in the absence of S2O82−. The addition of the S2O82− co-reactant to the anodic compartment enables the reductive–oxidative pathway of the Ru(bpy)32+/S2O82−system, which leads to the characteristic red ECL emission. Furthermore, the continuous addition of S2O82− causes an increase in the light emission of the LED and the cathodic ECL (Fig. 3b and S5b†). In this configuration, we generated spontaneously 3 light emissions: (i) blue cathodic ECL of L012, (ii) green LED light, and (iii) red anodic ECL.
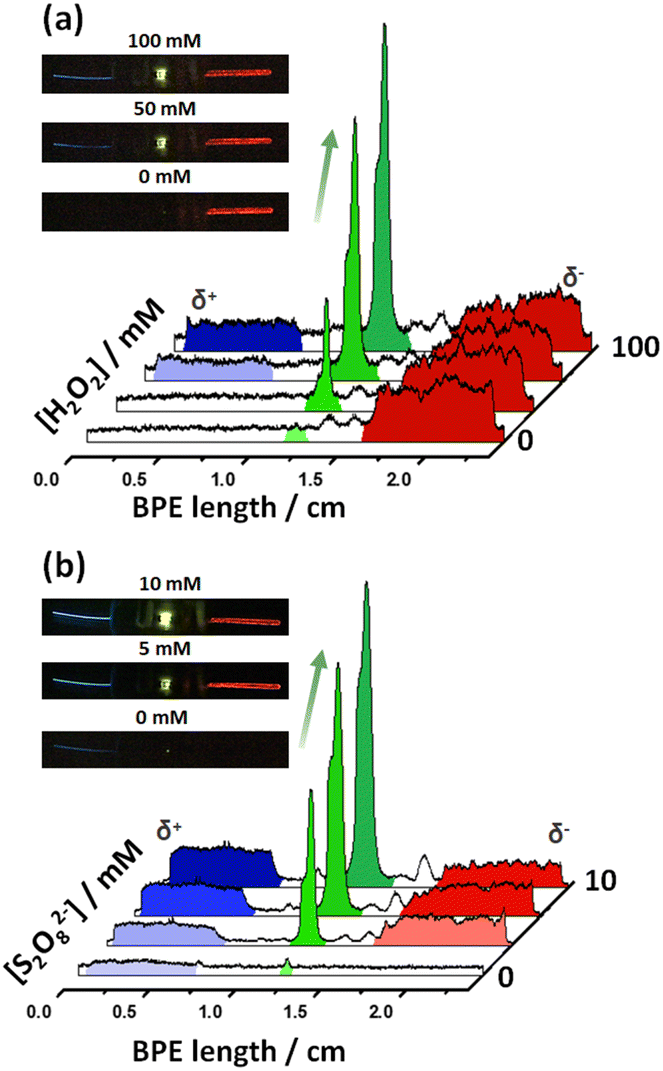 | ||
| Fig. 3 3D light intensity profiles obtained with a divided Fe-LED-Mg as a function of the co-reactant concentration, in a 0.1 M PBS (pH 7.4) solution in the presence of (a) L012 (1 mM) and increasing H2O2 concentrations (indicated in the figure) in the cathodic compartment and Ru(bpy)32+ (1 mM), H2SO4 (10 mM) and S2O82− (20 mM) in the anodic part, and (b) L012 (1 mM) and H2O2 (100 mM) in the cathodic compartment and Ru(bpy)32+ (1 mM), H2SO4 (10 mM) and increasing S2O82− concentrations (indicated in the figure) at the anodic side. Insets show optical pictures of the corresponding endogenous multimodal light-emitting devices at different co-reactant concentrations. | ||
Conclusions
In this work, we designed wireless multimodal light-emitting chemical devices by coupling two different light sources, ECL and LED, which are based on different physical principles. We used the concept of bipolar electrochemistry in exogenous and endogenous configurations. A pallet of different hybrid bipolar systems has been developed, depending on the nature of the wireless polarization in the presence of the proper luminophore/co-reactant couples. Both ECL/LED emissions were triggered by exogenous and endogenous bipolar electrochemistry. For the exogenous approach, both ECL/LED emission intensities directly depend on the applied electric field, whereas for the endogenous system, light emission is a function of the co-reactant concentration. In addition, a compartmentalized endogenous approach was developed, which significantly improves the ECL emission. Finally, we reported an original multimodal light-emitting platform, generating three different colors based on the same compartmentalized device. The direct interplay between the ECL/LED emission and the chemical composition in each compartment should allow using these so far unexplored multimodal optical readouts for the development of a new class of dynamic experiments with multifunctional objects. For example, it is possible to imagine that by using thermodynamically favored enzymatic reactions in one compartment, these devices can efficiently transduce information related to enzyme kinetics via multimodal optical readouts.Data availability
Additional data can be obtained from the corresponding author upon request.Author contributions
M. Liu: investigation, methodology, analysis, writing – original draft. G. Salinas: investigation, methodology, analysis, writing – original draft. J. Yu: investigation, methodology, analysis. A. Cornet: investigation, methodology, analysis. H. Li: conceptualization, writing – review & editing. A. Kuhn: conceptualization, supervision, writing – review & editing. N. Sojic: conceptualization, supervision, writing –review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no 741251, ERC Advanced grant ELECTRA). The authors thank the International Research Network ELECTROSENS funded by the CNRS. M. Liu acknowledges the China Scholarship Council for her PhD scholarship.Notes and references
- L. Guan, H. Sun, J. Xiong, W. Hu, M. Ding and Q. Liang, Sens. Actuators, B, 2022, 373, 132694 CrossRef CAS.
- Y. Kim, H. Kim, J. B. Son, M. Filatov, C. Ho, N. K. Lee and D. Lee, Angew. Chem., Int. Ed., 2023, 62, e202302107 CrossRef CAS PubMed.
- M. Nuriya, S. Fukushima, A. Momotake, T. Shinotsuka, M. Yasui and T. Arai, Nat. Commun., 2016, 7, 11557 CrossRef CAS PubMed.
- M. Tuck, L. Blanc, R. Touti, N. H. Patterson, S. V. Nuffel, S. Vilette, J. C. Taveau, A. Römpp, A. Brunelle, S. Lecomte and N. Desbenoit, Anal. Chem., 2021, 93, 445–477 CrossRef CAS PubMed.
- P. de Boer, J. P. Hoogenboom and B. N. G. Giepmans, Nat. Methods, 2015, 12, 503–513 CrossRef CAS PubMed.
- A. Louie, Chem. Rev., 2010, 110, 3146–3195 CrossRef CAS PubMed.
- D. E. Lee, H. Koo, I. C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- F. Du, Y. Chen, C. Meng, B. Lou, W. Zhang and G. Xu, Curr. Opin. Electrochem., 2021, 28, 100725 CrossRef CAS.
- C. de la Encarnación, E. Lenzi, M. Henriksen-Lacey, B. Molina, K. Jenkinson, A. Herrero, L. Colas, P. Ramos-Cabrer, J. Toro-Mendoza, I. Orue, J. Langer, S. Bals, D. Jimenez de Aberasturi and L. M. Liz-Marzan, J. Phys. Chem. C, 2022, 126, 19519–19531 CrossRef.
- L. Zhang, S. Tong, Q. Zhang and G. Bao, ACS Appl. Nano Mater., 2020, 3, 6785–6797 CrossRef CAS.
- M. Hou, X. Lu, Z. Zhang, Q. Xia, C. Yan, Z. Yu, Y. Xu and R. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 44316–44323 CrossRef CAS PubMed.
- H. F. Schmitthenner, T. M. Barrett, S. A. Beach, L. E. Heese, C. Weidman, D. E. Dobson, E. R. Mahoney, N. C. Schug, K. G. Jones, C. Durmaz, O. Otasowie, S. Aronow, Y. P. Lee, H. D. Ophardt, A. E. Becker, J. P. Hornak, I. M. Evans and M. C. Ferran, ACS Appl. Bio Mater., 2021, 4, 5435–5448 CrossRef CAS PubMed.
- N. Sojic and L. Bouffier, Curr. Opin. Electrochem., 2022, 34, 101007 CrossRef CAS.
- H. Xing, X. Zhang, Q. Zhai, J. Li and E. Wang, Anal. Chem., 2017, 89, 3867–3872 CrossRef CAS PubMed.
- X. Qin, J. Gao, H. J. Jin, Z. Q. Li and X. H. Xia, Chem. - Eur. J., 2023, 29, e2022026 Search PubMed.
- T. J. Anderson, P. A. Defnet and B. Zhang, Anal. Chem., 2020, 92, 6748–6755 CrossRef CAS PubMed.
- T. Iwama, K. Y. Inoue and H. Shiku, Anal. Chem., 2022, 94, 8857–8866 CrossRef CAS PubMed.
- A. De Poulpiquet, B. Diez-Buitrago, M. Milutinovic, B. Goudeau, L. Bouffier, S. Arbault, A. Kuhn and N. Sojic, ChemElectroChem, 2016, 3, 404–409 CrossRef CAS.
- J. Zhai, L. Yang, X. Du and X. Xie, Anal. Chem., 2018, 90, 12791–12795 CrossRef CAS PubMed.
- K. T. Lau, R. Shepherd, D. Diamond and D. Diamond, Sensors, 2006, 6, 848–859 CrossRef CAS.
- X. Zhang, Y. Han, J. Li, L. Zhang, X. Jia and E. Wang, Anal. Chem., 2014, 86, 1380–1384 CrossRef CAS PubMed.
- X. Ma, W. Gao, F. Du, F. Yuan, J. Yu, Y. Guan, N. Sojic and G. Xu, Acc. Chem. Res., 2021, 54, 2936–2945 CrossRef CAS PubMed.
- S. E. Fosdick, J. A. Crooks, B. Y. Chang and R. M. Crooks, J. Am. Chem. Soc., 2010, 132, 9226–9227 CrossRef CAS PubMed.
- H. Li, L. Bouffier, S. Arbault, A. Kuhn, C. F. Hogan and N. Sojic, Electrochem. Commun., 2017, 77, 10–13 CrossRef CAS.
- E. Villani, N. Shida and S. Inagi, Electrochim. Acta, 2021, 389, 138718 CrossRef CAS.
- M. Feng, A. L. Dauphin, L. Bouffier, F. Zhang, Z. Wang and N. Sojic, Anal. Chem., 2021, 93, 16425–16431 CrossRef CAS PubMed.
- V. Eβmann, J. Clausmeyer and W. Schuhmann, Electrochem. Commun., 2017, 75, 82–85 CrossRef.
- A. L. Dauphin, A. Akchach, S. Voci, A. Kuhn, G. Xu, L. Bouffier and N. Sojic, J. Phys. Chem. Lett., 2019, 10, 5318–5324 CrossRef CAS PubMed.
- A. L. Dauphin, S. Arbault, A. Kuhn, N. Sojic and L. Bouffier, ChemPhysChem, 2020, 21, 600–604 CrossRef CAS PubMed.
- P. A. Defnet and B. Zhang, ChemElectroChem, 2020, 7, 252–259 CrossRef CAS.
- J. S. Borchers, O. Riusech, E. Rasmussen and R. K. Anand, J. Anal. Test., 2019, 3, 150–159 CrossRef.
- J. S. Borchers, C. R. Campbell, S. B. Van Scoy, M. J. Clark and R. K. Anand, ChemElectroChem, 2021, 8, 3482–3491 CrossRef CAS.
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 10438–10456 CrossRef CAS PubMed.
- L. Koefoed, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2017, 2, 13–17 CrossRef CAS.
- K. L. Rahn and R. K. Anand, Anal. Chem., 2021, 93, 103–123 CrossRef CAS PubMed.
- N. Shida, Y. Zhou and S. Inagi, Acc. Chem. Res., 2019, 52, 2598–2608 CrossRef CAS.
- S. Cauteruccio, V. Pelliccioli, S. Grecchi, R. Cirilli, E. Licandro and S. Arnaboldi, Chemosensors, 2023, 11, 131 CrossRef CAS.
- Y. Liu, N. Zhang, J. B. Pan, J. Song, W. Zhao, H. Y. Chen and J. J. Xu, Anal. Chem., 2022, 94, 3005–3012 CrossRef CAS PubMed.
- A. J. Hsueh, N. A. A. Mutalib, Y. Shirato and H. Suzuki, ACS Omega, 2022, 7, 20298–20305 CrossRef CAS PubMed.
- E. Villani and S. Inagi, Anal. Chem., 2021, 93, 8152–8160 CrossRef CAS PubMed.
- M. Sentic, S. Arbault, B. Goudeau, D. Manojlovic, A. Kuhn, L. Bouffier and N. Sojic, Chem. Commun., 2014, 50, 10202–10205 RSC.
- B. Gupta, P. Suchomski, A. A. Melvin, S. Linfield, M. Opallo and W. Nogala, Electrochem. commun., 2022, 140, 107329 CrossRef CAS.
- G. Salinas, S. Arnaboldi, G. Bonetti, R. Cirilli, T. Benincori and A. Kuhn, Chirality, 2021, 33, 875–882 CrossRef CAS PubMed.
- G. Salinas, G. Bonetti, R. Cirilli, T. Benincori, A. Kuhn and S. Arnaboldi, Electrochim. Acta, 2022, 421, 140494 CrossRef CAS.
- G. Salinas, S. M. Beladi-Mousavi, L. Gerasimova, L. Bouffier and A. Kuhn, Anal. Chem., 2022, 94, 14317–14321 CrossRef CAS PubMed.
- G. Salinas, I. A. Pavel, N. Sojic and A. Kuhn, ChemElectroChem, 2020, 7, 4853–4862 CrossRef CAS.
- X. Zhang, C. Chen, J. Yin, Y. Han, J. Li and E. Wang, Anal. Chem., 2015, 87, 461264616 Search PubMed.
- J. Roche, S. Carrara, J. Sanchez, J. Lannelongue, G. Loget, L. Bouffier, P. Fischer and A. Kuhn, Sci. Rep., 2014, 4, 6705 CrossRef CAS PubMed.
- M. Sentic, G. Loget, D. Manojlovic, A. Kuhn and N. Sojic, Angew. Chem., Int. Ed., 2012, 51, 11284–11288 CrossRef CAS PubMed.
- W. Gao, K. Muzyka, X. Ma, B. Lou and G. Xu, Chem. Sci., 2018, 9, 3911–3916 RSC.
- W. Qi, J. Lai, W. Gao, S. Li, S. Hanif and G. Xu, Anal. Chem., 2014, 86, 8927–8931 CrossRef CAS PubMed.
- L. Qi, Y. Xia, W. Qi, W. Gao, F. Wu and G. Xu, Anal. Chem., 2016, 88, 1123–1127 CrossRef CAS PubMed.
- G. Salinas, A. L. Dauphin, C. Colin, E. Villani, S. Arbault, L. Bouffier and A. Kuhn, Angew. Chem., Int. Ed., 2020, 59, 7508–7513 CrossRef CAS PubMed.
- I. A. Pavel, G. Salinas, A. Perro and A. Kuhn, Adv. Intell. Syst., 2021, 3, 2000217 CrossRef.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- S. Rebeccani, A. Zanut, C. I. Santo, G. Valenti and F. Paolucci, Anal. Chem., 2021, 94, 336–348 CrossRef PubMed.
- C. Cui, Y. Chen, D. Jiang, J. J. Zhu and H. Y. Chen, Anal. Chem., 2017, 89, 2418–2423 CrossRef CAS PubMed.
- K. Hiramoto, K. Ino, K. Komatsu, Y. Nashimoto and H. Shiku, Biosens. Bioelectron., 2021, 181, 113123 CrossRef CAS PubMed.
- G. Salinas, A. L. Dauphin, S. Voci, L. Bouffier, N. Sojic and A. Kuhn, Chem. Sci., 2020, 11, 7438 RSC.
- H. S. White and A. J. Bard, J. Am. Chem. Soc., 1982, 104, 6891–6895 CrossRef CAS.
- J. Suk, Z. Wu, L. Wang and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 14675–14685 CrossRef CAS PubMed.
- J. F. L. Duval, M. Minor, J. Cecilia and H. P. van Leeuwen, J. Phys. Chem. B, 2003, 107, 4143–4155 CrossRef CAS.
- J. F. L. Duval, J. Buffle and H. P. van Leeuwen, J. Phys. Chem. B, 2006, 110, 6081–6094 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03678b |
| ‡ Authors contributed in an equal way to the work. |
| This journal is © The Royal Society of Chemistry 2023 |