
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePanchromatic light harvesting and record power conversion efficiency for carboxylic/cyanoacrylic Fe(II) NHC co-sensitized FeSSCs†
Anil
Reddy-Marri
 a,
Edoardo
Marchini
b,
Valentin Diez
Cabanes
c,
Roberto
Argazzi
d,
Mariachiara
Pastore
*c,
Stefano
Caramori
*b and
Philippe C.
Gros
*e
a,
Edoardo
Marchini
b,
Valentin Diez
Cabanes
c,
Roberto
Argazzi
d,
Mariachiara
Pastore
*c,
Stefano
Caramori
*b and
Philippe C.
Gros
*e
aDepartment of Chemistry, North Carolina State University, 851 Main Campus Drive, Raleigh, North Carolina 27695-8204, USA
bDepartment of Chemical and Pharmaceutical Sciences, University of Ferrara, Via L. Borsari 46, 44121, Ferrara, Italy. E-mail: cte@unife.it
cUniversité de Lorraine, CNRS, LPCT, F-54000 Nancy, France. E-mail: mariachiara.pastore@univ-lorraine.fr
dCNR-ISOF c/o Department of Chemical, Pharmaceutical and Agricultural Sciences, University of Ferrara, Via L. Borsari 46, 44121, Ferrara, Italy
eUniversité de Lorraine, CNRS, L2CM, F-54000 Nancy, France. E-mail: philippe.gros@univ-lorraine.fr
First published on 24th March 2023
Abstract
Fe(II) pyridyl-NHC sensitizers bearing thienylcyanoacrylic (ThCA) anchoring groups have been designed and characterized with the aim of enhancing the metal to surface charge separation and the light harvesting window in iron-sensitized DSSCs (FeSSCs). In these new Fe(II) dyes, the introduction of the ThCA moiety remarkably extended the spectral response and the photocurrent, in comparison with their carboxylic analogues. The co-sensitization based on a mixture of a carboxylic and a ThCA–iron complex produced a panchromatic absorption, up to 800 nm and the best photocurrent and efficiency (Jsc: 9 mA cm−2 and PCE: 2%) ever reported for an FeSSC.
1. Introduction
Dye sensitized solar cells (DSSCs) are a credible contender in the search for a cost-effective alternative to conventional silicon based solar cells.1–4 Some of the best efficiencies have been achieved using Ru(II) complexes as sensitizers (up to 11%).5 The reason for such a success results from the combination of long-lived metal-to-ligand charge transfer (MLCT) states (ns regime) responsible for the electron transfer into the semiconductor conduction band (CB) and efficient electron–hole separation preventing the parasitic charge recombination processes at the dye/semiconductor interface.6–8 However, modern research is intended to integrate holistic approaches taking into account the availability, cost and strategic nature of raw materials. In this context, ruthenium complexes are not the best candidates due to the scarcity of ruthenium, and their high cost, tricky purifications, and environmental issues precluding potential large-scale applications.Thus, researchers have focused their attention on more cost-effective alternative sensitizers such as earth abundant first row transition metal complexes9,10 or metal-free organic conjugated compounds.11–14 Since the pioneering studies by Ferrere15,16 first investigating Fe(dcbpy)2(CN)2 as a sensitizer and despite the recent improvements achieved by McCusker,17 the maximum efficiency attained with iron(II) polypyridyl sensitizers is 0.35%. The origin of such a poor performance lies in the fact that, in contrast with Ru(II) polypyridyl complexes possessing long-lived MLCT states,18 ultrafast relaxation to metal centred (MC) states, and subsequent ground-state recovery, are observed for the iron analogues19–22 precluding their successful utilization in photochemical applications requiring stabilized MLCT states, such as DSSCs.23 The introduction of N-heterocyclic carbenes (NHCs) in the ligands surrounding the iron centre has been decisive in the quest for long-lived MLCTs. Especially tridentate pyridyl-N-heterocyclic carbenes (pyridyl-NHCs), due to their marked σ-donating strength, promoted an unprecedented stabilization of the MLCT states with respect to the MC ones.24–28 This strategy opened the way for the preparation of Fe(II) complexes with MLCT state lifetimes up to the picosecond.27,29–31 Complex C1 (Fig. 1) which combines σ-donating NHC ligands (showing MLCT lifetimes up to 16 ps) with π-accepting carboxylic groups for grafting to the TiO2 surface, was the first Fe(II)NHC prototype to be employed in an operating DSSC (FeSSC), albeit with poor photovoltaic performances.25 Very recently, some of us reported that this efficiency could be boosted up to 1% with a photocurrent reaching 3.3 mA cm−2 by introducing Mg2+ cations in the electrolyte composition.32 Mg2+ cations are capable of adsorption at the sensitizer/semiconductor interface and of improving the interfacial electron transfer kinetics (regeneration and/or electron injection), thus resulting in higher measured photocurrents.33 This result was very recently further improved by our group to reach the best efficiencies ever reported of 1.44% (Jsc = 4.8 mA cm−2) and 1.83% (Jsc = 4.8 mA cm−2) by sensitizing FeSSCs, respectively, with the heteroleptic complexes ARM13 (ref. 33) and ARM130 (ref. 34) (Fig. 1). The heteroleptic approach was also recently applied by Wärnmark which reported the introduction of diarylamino substituents on the central pyridine of the ancillary ligand resulting in 1.31% efficiency and Jsc = 3.5 mA cm−2.35
 | ||
| Fig. 1 Iron sensitizers reported and complexes studied in this work; the counterion is PF6− in every case. | ||
Besides the stereoelectronic tuning of the ancillary ligand, to our knowledge, the modification of the anchoring functionality has not been investigated yet in this series of sensitizers. Indeed, the nature and strength of the anchoring group's interaction with the surface of the DSSC's photoanode are critical to ensure an efficient injection and to prevent back electron transfer (recombination) processes.
When compared with the carboxylic function, the cyanoacrylic group, which has been used extensively with organic dyes,11 exhibits a bifunctionality in terms of enhanced π-conjugation via the acrylate double bond. Cole has reported that the coordination of CN with the titanium atoms of the TiO2 surface ensured the necessary electron withdrawal from the oxidized sensitizer, while the carboxylate group acts as an efficient anchor.36,37 The same group also disclosed a very interesting property of the cyanoacrylic function when linked to a thiophene unit in organic dyes. Indeed, a synchrotron-based X-ray analysis highlighted the creation of a very strong intramolecular interaction with an energy similar to that of a chemical bond between the thiophene's sulfur atom and the cyano group.37 The study revealed that this robust S⋯CN bond led to two intramolecular charge transfer (ICT) processes (Fig. 2), ICT1 (blue path) and ICT2 (red path). The summed contribution of these two ICTs greatly enhanced the cell performance.
 | ||
| Fig. 2 The S⋯CN bonding reported by Cole.37 | ||
These remarkable properties drew us to investigate the impact of thienylcyanoacrylic (ThCA) anchoring groups on the electronic properties of Fe(II) NHC complexes and on the performance of a DSSC sensitized by the same.
Herein we report the synthesis and characterization of the new iron sensitizers ARM12 and ARM242 decorated with ThCAs (Fig. 1) and the investigation of their photovoltaic properties. The homoleptic ARM12 promoted a remarkable improvement in the photocurrent compared to C1 bearing carboxylic groups, whereas co-sensitization of C1 and ARM12 led to the best efficiency and photocurrent ever reported for FeSSCs.
2. Results and discussion
2.1. Synthesis of ligand precursors and complexes
The synthesis of the target complexes first required the preparation of the pyridylimidazolium salts L1H and L2H, precursors of the pyridyl-NHC ligands to be coordinated to iron (Scheme 1). L1H was obtained in good yield by quaternization of the pyridylimidazole 3 with methyl iodide. 3 was the result of a palladium-catalyzed Suzuki coupling of the bromo derivative 1 (ref. 33) with 5-formyl-2-thienylboronic acid followed by a condensation reaction with cyanoacetic acid to generate the cyanoacrylic function. L2H was prepared according to previously reported studies.24 With the ligands in hand we turned to the preparation of the homoleptic and heteroleptic complexes ARM12 and ARM242, respectively. ARM12 was prepared by first reacting 2 equiv. of L1H with FeCl2 followed by addition of t-BuOK as the base to generate the NHC. The heteroleptic ARM242 was obtained using a statistical synthesis that we reported previously, consisting in reacting FeCl2 with an equimolar mixture of L1H and L2H ending with KHMDS addition.33 KHMDS was found to be more efficient than t-BuOK in this case. ARM242 was further cleanly separated by column chromatography from a mixture containing both the homoleptic complexes coming from each ligand concomitantly formed during the process.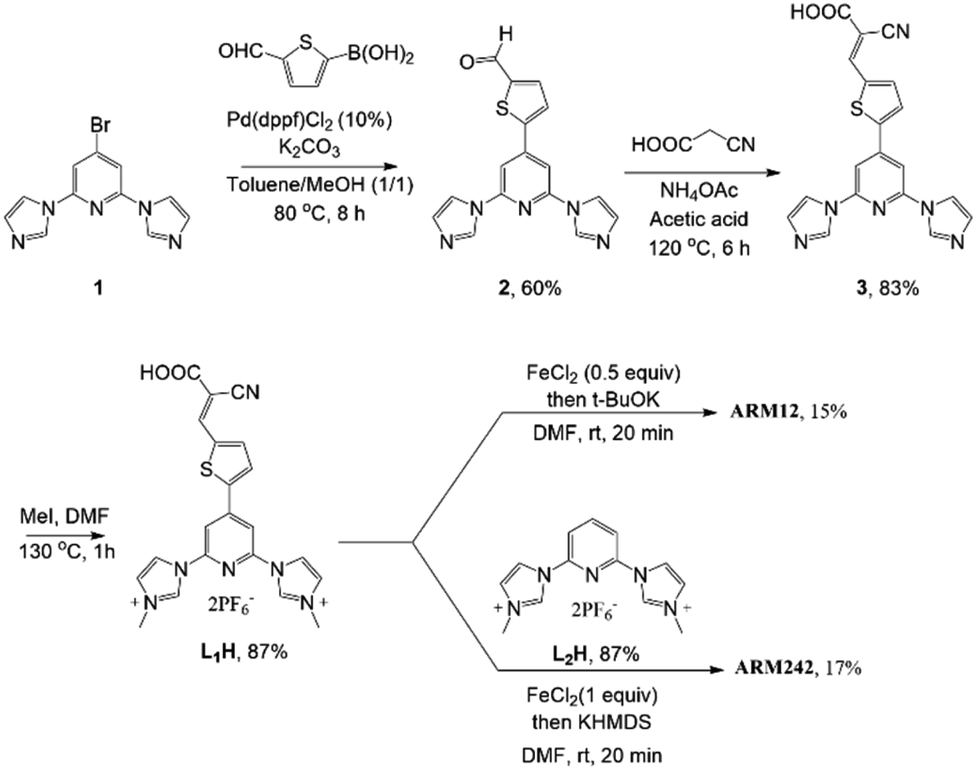 | ||
| Scheme 1 Synthesis of L1H and complexes ARM12 and AM242. | ||
2.2. Spectroscopic properties of the complexes
The UV-vis spectra recorded in methanol solution are shown in Fig. 3 and the corresponding absorption maxima, in Table 1.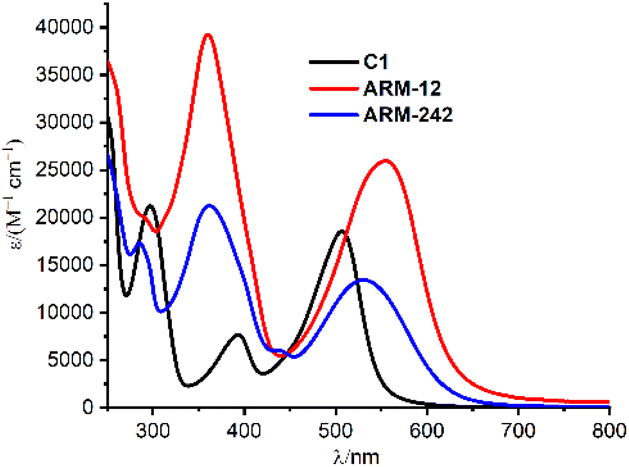 | ||
| Fig. 3 UV-vis spectra of C1, ARM12 and ARM242 in methanol. | ||
| Dye | Experiments | Theorya | ||
|---|---|---|---|---|
| λ max (104ε (M−1 cm−1)) | λ max (TiO2) | E max (osc. str.) | Transition (%) | |
| a All energy values are in nm. Calculated vertical excitation properties for C1, ARM242 and ARM12 computed at the TD-B3LYP* and TD-B3LYP levels of theory, respectively, within a 6-311G(d) basis set in their corresponding ground state (GS) geometries. | ||||
| C1 | 506 (1.85) | 504 | 464 (0.324) | H − 2 → L + 1 (45.4%) |
| H − 1 → L (45.1%) | ||||
| ARM12 | 555 (2.59) | 553 | 538 (1.193) | H − 2 → L + 1 (49.8%) |
| ARM242 | 531 (1.34) | 556 | 524 (0.566) | H − 1 → L (68.9%) |
As already reported, C1 displayed three distinct absorption bands in this energy region.25 The band at higher energy (250–350 nm) is assigned to π–π* transitions, while the two lower-energy bands correspond to the Fe–NHC MLCT transitions (340–420 nm) and Fe–pyridine MLCT transitions (410–650 nm).
Overall, the introduction of ThCA as an anchoring moiety induces a significant red shift of the Fe–pyridine MLCT bands, whose extent is directly proportional to the number of ThCA units in the complex, namely 0.22/0.11 eV for ARM12/ARM242 (Table 1). In ARM12 and ARM242, the Fe–NHC MLCT band was masked by the very broad π–π* character absorption band.
Focusing on the lowest-energy portion of the spectrum, the MLCT Fe–pyridine band, ARM12 and ARM242 present a broad absorption, ranging from 450 to 650 nm. Among them, the homoleptic ARM12 showed the broadest, most intense and largest red shifted absorption (λmax: 555 nm, ε: 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1). Similar absorption features were observed in the computed TD-DFT spectra which, indeed, were able to fairly reproduce the red-shift and intensity increase prompted by the ThCA units (Table 1 and Fig. S11†). The estimated red shifts for ARM12 and ARM242 are 0.37 and 0.31 eV, respectively, being slightly overestimated with respect to the experimental one due to the small blue shift of the calculated C1 MLCT band maximum when using the B3LYP* functional employed in our previous studies (see the ESI† for details).33
900 M−1 cm−1). Similar absorption features were observed in the computed TD-DFT spectra which, indeed, were able to fairly reproduce the red-shift and intensity increase prompted by the ThCA units (Table 1 and Fig. S11†). The estimated red shifts for ARM12 and ARM242 are 0.37 and 0.31 eV, respectively, being slightly overestimated with respect to the experimental one due to the small blue shift of the calculated C1 MLCT band maximum when using the B3LYP* functional employed in our previous studies (see the ESI† for details).33
As is apparent, on the other hand, B3LYP very accurately (within 0.1 eV) predicts the excited state energy of the new complexes that, due to the larger electron withdrawing effect of the cyanoacrylic functionality are characterized by a greater extent of intramolecular CT (0.8 for C1 and 0.9 for ARM242 and ARM12, as the CT index (0 < CT < 1)),38 thus requiring the use of a slightly larger fraction of non-local exchange to be properly treated (20% in B3LYP vs. 15% in B3LYP*). Regarding the nature of the MLCT bands, they are mainly composed of transitions between molecular orbitals (MOs) in the vicinity of the highest occupied MO (HOMO), such as HOMO−1 and HOMO−2 levels, and the degenerate lowest unoccupied MO (LUMO) levels of the dyes, in a similar way as has been reported for other Fe–NHC complexes.33,34 Fig. S12† displays the isosurfaces of the MOs involved in the main transitions composing the MLCT bands of the studied dyes. As expected, the nature of the frontier occupied MOs is barely influenced by the introduction of the ThCA moiety, whereas the degenerated LUMO levels of ARM242/ARM12 more largely delocalize over the ligands and toward the anchoring unit, resulting in a larger stabilization with respect to those of ARM13/C1 (see Tables S3 and S4†), and in the red-shift of the MLCT bands.
2.3. Spectroscopic and electrochemical properties of FeNHC-sensitized films
In order to gain information about the spectroscopic features of the new Fe(II)NHC series grafted onto TiO2, relatively thick 13 μm electrodes sensitized with ARM12, ARM242 and C1 have been prepared. Their relative absorption spectra are depicted in Fig. 4a and λmax values are reported in Table 1. The dyes bearing the ThCA functions are characterized by a broad and intense symmetric MLCT band, spanning the 500–700 nm interval with λmax ≈ 550 nm, while C1 exhibits a narrower and blue shifted main band (λmax ≈ 500 nm).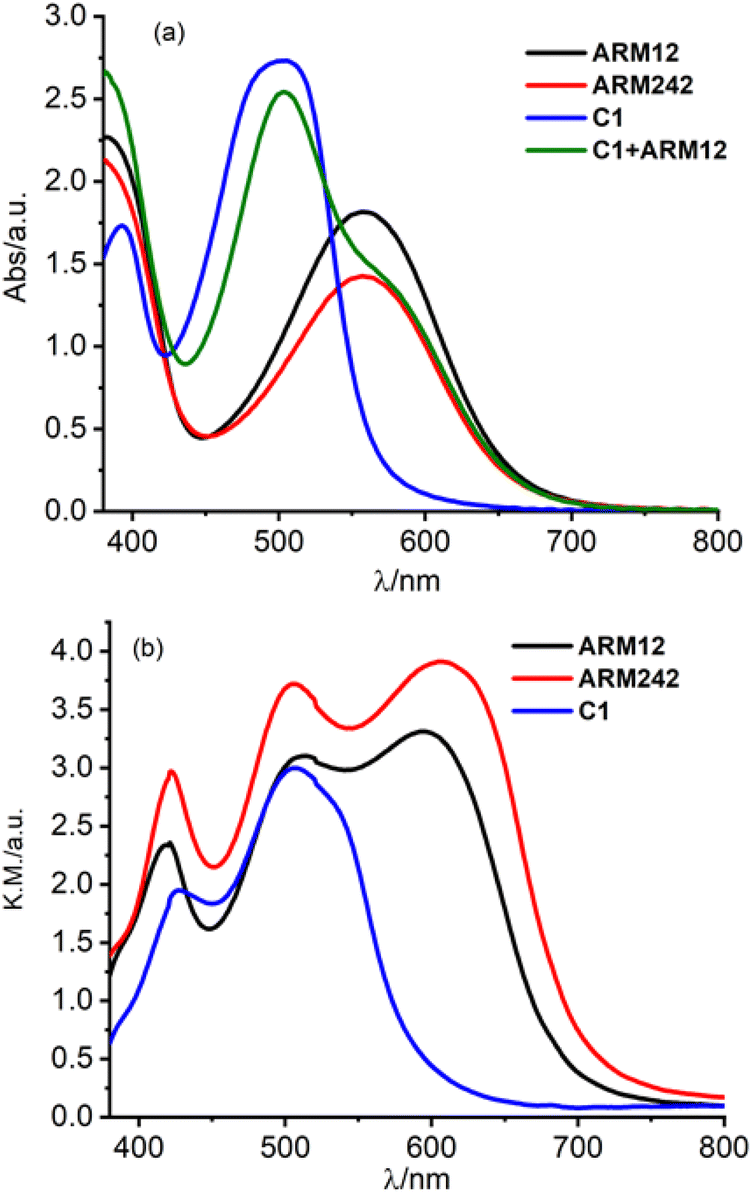 | ||
| Fig. 4 Absorption spectra (recorded against an identical undyed electrode as a reference) of the Fe(II)NHC series together with ARM12 + C1 loaded (a) on 13 μm TiO2 and (b) on the 20 μm + top scattering layer. In the latter case the C1 + ARM12 system saturated the spectrophotometer response at an absorbance higher than 4. | ||
The red shifted MLCT bands, with a strong Fe(II)–pyridine character are consistent with the reduced electrochemical gap that we measured by cyclic voltammetry on the dyed TiO2 or ZrO2 thin films in contact with 0.1 M LiClO4 in acetonitrile solution (Fig. 5 and Table 2). ZrO2 was used as an electrochemically inert porous support allowing the exploration of the energetics of dye reduction.39–41
 | ||
| Fig. 5 Cyclic voltammetry recorded for Fe(II)NHC on (a) TiO2 and on (b) ZrO2 (full scan) in 0.1 M LiClO4/ACN solution. | ||
| Dye | E ox (TiO2) (V vs. SCE) | E ox (ZrO2) (V vs. SCE) | E (peak)red (ZrO2) (V vs. SCE) | ΔEredoxb (ZrO2) (eV) |
|
E 00–Eox |
|
|---|---|---|---|---|---|---|---|
a Data taken from ref. 32.
b Calculated according to: |Eox(ZrO2)| + |Ered(ZrO2)|.
c The  energy was estimated from the onset of the absorption spectrum, by taking 5% absorbance relative to the MLCT maximum on TiO2.
d Calculated according to: energy was estimated from the onset of the absorption spectrum, by taking 5% absorbance relative to the MLCT maximum on TiO2.
d Calculated according to: 
|
|||||||
| C1 | 0.67a | 0.61 | −1.39 | 2.00 | 2.10 | 1.49 | −1.43 |
| ARM12 | 0.59 | 0.57 | −0.88 | 1.85 | 1.81 | 1.24 | −1.22 |
| −1.34 | |||||||
| ARM242 | 0.58 | 0.57 | −0.94 | 1.93 | 1.81 | 1.24 | −1.23 |
| −1.36 | |||||||
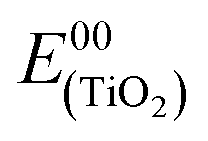

On the titania substrates (Fig. 5a), both the new complexes, ARM12 and ARM242, revealed a reversible Fe(II)/(III) oxidation potential (ΔE ≈ 59 mV) with an Eox of 0.59 V and 0.58 V, respectively, more cathodic (≈70 mV) than that recorded for our standard C1 (0.67 V vs. SCE32). All these complexes are characterized by irreversible cathodic waves. The complexes bearing ThCA, ARM12 and ARM242 highlight a first irreversible process (wave at −0.88 V and −0.94 V respectively), which is absent in C1. The same complexes display a second more intense polyelectronic wave peaking at −1.34 and −1.36 V vs. SCE, similar to that recorded in the case of C1 at −1.39 V. The estimation of the HOMO–LUMO gap, based on these potentials, provides values ranging between 650 and 833 nm, which embraces the onset of the absorption spectrum found at around 700 nm, confirming the MLCT character of the lowest energy transition. Indeed, on average, the manifold of electron acceptor states lying at the midpoint between the redox waves (−1.12 V) provides a gap value (732 nm) in excellent agreement with the onset of the experimental absorption spectrum of the thin film sensitized by these new dyes. Thus, both stabilization of the LUMOs and destabilization of the Fe(II) centered HOMO are responsible for the substantial red shift in the spectral response of the Fe complexes bearing cyanoacrylic (ThCA) moieties. The data obtained on ZrO2 could be safely extended to TiO2, where a marginal anodic shift (10–20 mV) of the Fe(II)//III wave is observed. Clearly, on TiO2, the observation of the cathodic features of the Fe(II) sensitizers is precluded by the interception of its CB states.
As for the excited state energetics, as expected,27 the iron(II) complexes did not emit in fluid solution nor loaded on the semiconductor, and thus, the spectroscopic E00 was estimated from the onset of the absorption spectra at ca. 5% of the maximum absorption. E00 was 1.81 eV for both chemisorbed ThCA substituted complexes, and this value was smaller than the energy calculated in the case of our standard C1 (2.10 eV). Such an estimate appears to be in acceptable agreement with the electrochemical gap discussed before, if the irreversible nature of the cathodic processes which precludes a rigorous thermodynamic information is taken into consideration. The excited state potential  where Eox is the Fe(III)/(II) ground state potential affords values of around −1.2 V for ARM12 and ARM242 and −1.4 V for C1. Thus, injection in the TiO2 semiconductor is an exergonic process whose driving force can be calculated according to ΔGinj = −e(|Eox| − |EFB|) where EFB represents the flat band potential of TiO2. Considering the flat band potential of TiO2 in an organic medium in the presence of lithium cations at ca. −0.7 V vs. SCE,42 we obtained ΔGinj ≈ 500 meV for ARM12 and ARM242 and around 700 meV for C1. The ground state potential also indicates that regeneration of the Fe(II) species by the iodide/triiodide-based electrolyte is an exergonic process by at least 200 meV.
where Eox is the Fe(III)/(II) ground state potential affords values of around −1.2 V for ARM12 and ARM242 and −1.4 V for C1. Thus, injection in the TiO2 semiconductor is an exergonic process whose driving force can be calculated according to ΔGinj = −e(|Eox| − |EFB|) where EFB represents the flat band potential of TiO2. Considering the flat band potential of TiO2 in an organic medium in the presence of lithium cations at ca. −0.7 V vs. SCE,42 we obtained ΔGinj ≈ 500 meV for ARM12 and ARM242 and around 700 meV for C1. The ground state potential also indicates that regeneration of the Fe(II) species by the iodide/triiodide-based electrolyte is an exergonic process by at least 200 meV.
From Fig. 4a, it can be seen that the enhanced red sensitivity of ARM12 and ARM242 leaves a “valley” in the absorption spectrum between 400 and 500 nm, where, in contrast with C1, their extinction coefficient is small. Thus, to further increase the spectral sensitivity of the thin films, we have realized the co-sensitization of C1 and ARM12 to obtain a panchromatic profile to collect both the relative blue (450–500 nm) and green-red (500–650 nm) energy portions of the visible region. Due to the stronger stability of the –CNCOO–Ti linkage36 of ARM12 with respect to the COO–Ti binding of C1, we first dipped the TiO2 electrodes for 1 h in ARM12 until a maximum MLCT absorbance of ca. 1 was reached, followed by 6 h of immersion in the C1 solution to complete the saturation of the remaining available adsorption sites of the TiO2 film. Fig. S20† shows the evolution of the absorption spectrum of the TiO2 film after 1 h of immersion in ARM12 and after treatment with C1. This co-sensitized electrode clearly exhibits both the spectroscopic fingerprints of the COOH- and ThCA-based complexes with the first band at around 500 nm and the second shoulder at 570 nm that, coherently with the presence of ARM12, collects photons up to 700 nm in the transparent film. The difference in the spectrum shows that spectral fingerprints of C1 are maintained in the C1 + ARM12 system, and that the interaction between both Fe(II) sensitizers is ostensibly weak. The average surface loading was estimated according to eqn (1):
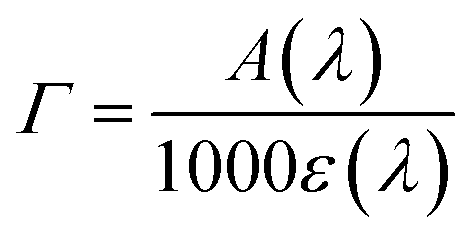 | (1) |
We have further improved the light harvesting capability of the sensitized TiO2 films by preparing ca. 20 μm transparent TiO2 equipped with a top scattering layer characterized by >100 nm nanoparticles. The increased optical path obtained with the scattering layer notably broadens the spectral features of the sensitized films (Fig. 4b): in particular C1 collects photons up to λ > 600 nm and ARM12 and ARM242, up to 750 nm. Under these conditions, C1 + ARM12 saturated the spectrophotometer response at an absorbance higher than 4.
2.4. Transient absorption spectroscopy
Regeneration and recombination dynamics were evaluated by means of transient absorption spectroscopy (TAS) in the μs to s timescale on dyed-TiO2 thin films in contact with 0.1 M LiClO4/ACN solutions. The absorption spectra of ARM12 and ARM242 are characterized by similar features with a strong band mirroring the MLCT manifold if the small bathochromic shift of the ground state spectrum occurring in the presence of small high-density cations such as Li+ and Mg2+ (Fig. S21†) interacting with the TiO2 film is considered.33 The transient absorption spectra of ARM12 and ARM242 (Fig. 6) exhibited a bleach centred at around 575 nm with a small shoulder at around 500 nm, a comparatively intense absorption band peaking at ca. 430 nm, and weaker and featureless broad absorption starting at ca. 670 nm and extending into the NIR region. These transient features are very long lived, showing a minor evolution during the first 4 μs after the laser pulse. The extremely long lived state and the bleaching of the ground state absorption is consistent with the photoinduced formation of Fe(III) following electron injection. The high (450 nm) and low energy absorption bands (>700 nm), whose time evolution parallels that of the bleaching are assigned to LMCT transitions, with weaker low energy contributions by the photoinjected electrons trapped inside TiO2. To confirm the interpretation of the TA spectra, we have performed spectroelectrochemical (SE) measurements on TiO2 (Fig. S22†). In general, we observed a remarkable agreement with the transient difference spectra on the same substrate, confirming the assignment we made to the photoinduced formation of the oxidized state of the iron complex following visible excitation (Fig. 6 and S23†). In detail, the spectra in Fig. S22† exhibited the deep bleach of the charge transfer bands at 500 nm in the case of C1 and at around 570 nm and 490 nm in the case of the ThCA-based systems. Consistent with the TAS analysis, the oxidized forms of ARM12 and ARM242 show distinctively higher energy (450 nm) and lower energy (>700 nm) absorption due to LMCT transitions, further corroborating our results from the TA spectra. It is interesting to observe that the SE changes for C1, upon electrochemical oxidation are quantitatively more significant (ΔAmin = −0.2) than those found with the cyanoacrylic dyes (ΔAmin ≈ −0.04), while during TAS measurements all complexes exhibit similar ΔA amplitudes corresponding to their main MLCT band. Considering that the electrochemical oxidation on TiO2 proceeds through charge hopping from molecules in proximity of the FTO back contact, this suggests that C1 is able to sensitize deeper layers of TiO2, closer to the FTO collector, than ARM12 or ARM242. This could be due to a better permeation of TiO2 by C1, due to its less hydrophobic and less sterically hindered structure. It is also interesting to note that the SE variations in the co-sensitized system are the most significant within the explored series, achieving a much more considerable bleaching of the ARM12 signature, i.e. more than 1 order of magnitude compared to that of ARM12@TiO2 (ΔA580 = −0.5, Fig. S22d†). This is indicative of an oxidation of ARM12 proceeding through hopping from oxidized C1 states. Notably, since the co-sensitized electrode was prepared by first dipping the TiO2 electrode in the ARM12 solution, the distribution of ARM12 within the co-sensitized TiO2 mesoporous substrate should be identical to that of the ARM12@TiO2 case.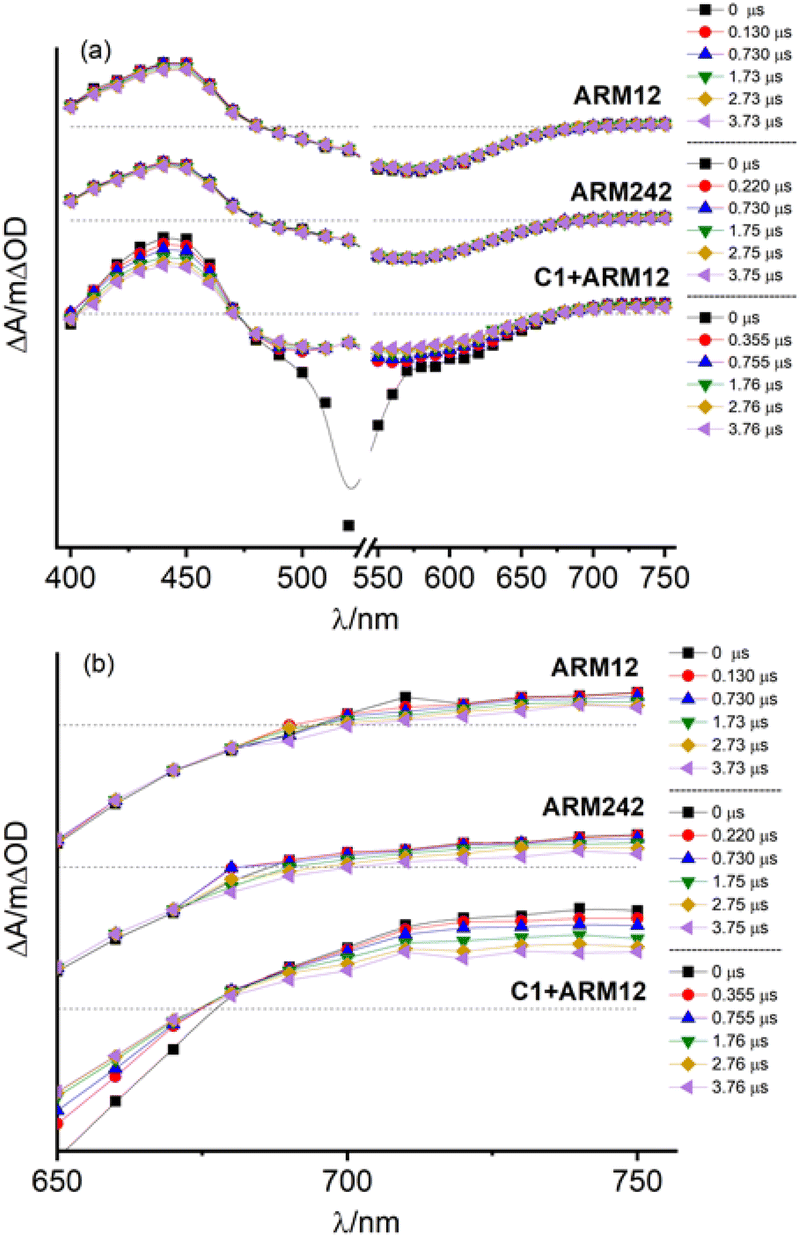 | ||
| Fig. 6 (a) Transient absorption spectra recorded for ARM12, ARM242 and C1 + ARM12 in contact with 0.1 M LiClO4/ACN solution. (b) Magnification of low energy flat absorption. Laser source: 532 nm. Laser power: 7 mJ per cm2 per pulse. Input impedance: 350 Ω. | ||
The TAS spectrum of C1 + ARM12 exhibited clear fingerprints of both components. While the formation of the charge separated state Fe(III)/e-(TiO2) is almost completely convolved with the instrumental time resolution of our apparatus (100 ns FWHM with 350 Ω impedance), we observed with our highest time resolution (50 Ω impedance and FWHM ≈ 7 ns), in agreement with a previous work,32 the residual of a very fast dynamics (relatively to the time scale of our apparatus) involving C1, which consists in the narrowing and a weak hypsochromic shift of the MLCT bleach, due to relaxation of the lowest triplet state (T1) which may also inject (Fig. S23a†), resulting in the increase of weak absorption bands for λ > 570 nm and λ ≈ 430 nm. A spectral evolution during the first 15 ns is clearly observed in the case of ARM12 (Fig. S23b†), where the increase of a well-defined absorption band centred at 450 nm occurs. After 100 ns, changes to the spectral shape almost completely ceased, and no further evolution in the spectral fingerprints reported in Fig. 6 are observed in the following hundreds of ms during which the amplitude of the transient signals simply decays by recombination according to non-monoexponential kinetics. Charge separated state recombination was probed at 500 nm in the case of C1, whereas the ground state recovery of ARM12 and ARM242 were both monitored at 570 nm. The transient spectrum of C1 + ARM12 (Fig. 6 and S23†) contains the contribution of the charge separated states of both C1 and ARM12, meaning that each dye is able to inject independently of each other, as indicated by the coexistence of two bleaching bands centred at 500 nm (C1) and at 570 nm (ARM12), which are well defined already at time scales convolved with the laser pulse (4–10 ns delay, Fig. S23c†). Note that in order to avoid the interference of 532 nm laser excitation with the ground state bleaching of the respective dyes, we performed experiments at short time scales by using 355 nm excitation. Similar to what is reported for ARM12 alone, we observe the formation of the 450 nm absorption band, assigned to the oxidized state of ARM12, within the first 40 ns after the excitation pulse. We confirmed that this latter spectral feature is absent on ZrO2 (Fig. S23d†) where, due to the lack of injection, we could observe the spectral contribution of the excited states of both ARM12 and C1, where the sharp and intense bleaching of C1 overlapped with the broader ground state bleaching of ARM12, extending up to 700 nm. This also confirmed that energy transfer from surface bound C1 to ARM12 was not a significant excited state deactivation process, at least compared to charge injection into TiO2. Regardless of the excitation wavelength, the shape of the transient spectrum of C1 + ARM12 after the first 100 ns, is very similar to that observed with a lower time resolution, at longer time scales (up to 3.2 μs delay (Fig. S24†)), where we only observe the recovery of the charge separated state by recombination.
In order to investigate in more detail the recombination processes underlying the solar cell device operation under relevant conditions, recombination kinetics were explored under a relatively low excitation power (480 μJ per cm2 per pulse) to avoid an excess of localized electron–hole pairs that could lead to an underestimation of the recombination lifetime. Fe(II) recovery was ca. 100% complete within a time window of 500 ms. Fig. 7 summarizes the evolution of the photoinduced bleaching of the dyed thin films in contact with the I−/I2 free electrolyte (0.1 M LiOTf, 0.6 M PMIOTf, 0.1 M GuNCS, 0.1 M TBAPF6, 0.1 Mg(OTf)2). The decay was fitted by the combination of a power law function (eqn (2)),43 owing to the fast bimolecular electron–hole recombination which contributes to ca. 50% of the decay of the initial amplitude, with a stretched exponential function (Kohlrausch–Williams–Watts (KWW), eqn (3)) accounting for the distribution of recombination rate constants motivated by the inherent inhomogeneity of the nanocrystalline titania film:44,45
| ΔAt<2 ms = A + bt−c | (2) |
 | (3) |
 | ||
| Fig. 7 (a) Recombination dynamics of the dyed TiO2 thin films in contact with the blank electrolyte 3 (0.1 M LiOTf, 0.6 M PMIOTf, 0.1 M GuNCS, 0.1 M TBAPF6, 0.1 MgOTf). The kinetics were obtained by joining traces sampled with 10 kΩ and 1 MΩ input impedances. The orange lines represent the fit of the decay. (b) Regeneration kinetics where the same films as in (a) are in contact with the reduced form of the electrolyte (deprived of I2). Laser excitation: 532 nm input impedance: 10 kΩ. Laser power for curves recorded in (a) and in (b): 480 μJ per cm2 per pulse. | ||
In eqn (3), β represents the stretch exponential parameter which was set to 0.25 in all cases. The weighted lifetime is defined from τ0 and β according to eqn (4):
 | (4) |
All the complexes exhibited 〈τ〉 values in the ms range (Table 3); in particular, 6 ms was obtained in the case of ARM12 and C1 and a shorter value of 1 ms in the case of the heteroleptic complex ARM242. Quite similar time constants values (1.6 and 2.9 ms probed at 500 and 570 nm) were also found in the C1 + ARM12 system. Nevertheless, considering that at 500 nm the spectral contribution of C1 is dominant, the shorter Fe(III) lifetime at 500 nm might be due to hole transfer to C1 from ARM12, consistent with the lower oxidation potential of the latter and with the observations from the steady state spectrochemistry discussed above. We note that the hole transfer from C1 to ARM12, even assuming slow kinetics, should be feasible, since the dyes' oxidized state on the surface is extremely long lived. This contrasts with the excited state lifetime, which, besides being short lived is further quenched by electron transfer to the semiconductor, making energy transfer unlikely to occur.
| Dye | τ 0 (s) | 〈τ〉 (ms) |
|---|---|---|
| ARM12 | 2.7 × 10−4 | 6.5 |
| ARM242 | 5.9 × 10−5 | 1.4 |
| C1 | 2.6 × 10−4 | 6.1 |
| C1 + ARM12 (500 nm) | 6.7 × 10−5 | 1.6 |
| C1 + ARM12 (570 nm) | 1.2 × 10−4 | 2.9 |
To better gain insight into the injection quantum yield in a shorter time scale, we have carried out comparative actinometry47 50 ns after 355 nm laser excitation and probed at the bleach of their respective MLCT states, to evaluate the injection of ARM12 relative to that of C1 according to eqn (5):
 | (5) |
When iodide was introduced into the electrolyte (electrolyte without I2), the bleach recovery of all Fe(III) was largely confined inside the instrumental response function of our spectrometer (10 kΩ input impedance), suggesting a regeneration process with nearly unitary efficiency ηreg expressed by eqn (6):
 | (6) |
2.5. Photovoltaic properties
The photovoltaic properties of the new Fe(II)NHC series have been evaluated in the presence of our previously reported customized electrolyte.33 The electrolyte contains Li+, Mg2+, TBAI and GuNCS. The role of the small high-density cations, able to interact with the titania surface, enhances the directionality of the excited state, via formation of a favorable surface dipole, leading to improved charge injection; TBAI contributes to decreasing the overall cell resistance helping to increase the concentration of iodide without further decreasing the open circuit photovoltage (Voc) as it would happen with potential determining ion containing salts such LiI; the mild base GuNCS also adsorbs on the titania surface and partially screens recombination with I3−, bringing a small improvement in the Voc and fill factor (FF). Unfortunately, the addition of stronger bases, like tBuPy proved detrimental in the case of all the Fe(II)NHC sensitizers explored so far causing a large drop in the resulting photocurrent.32,48 On transparent thin films (13 μm thick active layer), the incident photon to current conversion efficiency (IPCE) spectra (Fig. S25a†) clearly highlights the extended light harvesting capability provided by the introduction of the ThCA moieties generating photoconversion at the threshold of 800 nm. This is an unprecedented result for Fe(II) sensitizers, whose commonly narrow MLCT bands normally limit the conversion to the green/orange region of the visible spectrum. Among this series, C1 resulted in the highest IPCE value of 45% probably due to its higher injection driving force, but C1 was closely matched by ARM12 (max. 40%), which was superior to ARM242 (max. 35%).The integrated short circuit photocurrent density extracted from the IPCE spectra afforded maximum photocurrents in the order of 6 mA cm−2 with the ThCA-based dyes, in good agreement with the data obtained under AM 1.5 white light illumination: ARM12 indeed exhibited the best Jsc of 6.23 mA cm−2, a Voc of 0.37 V, a FF of 56% and the highest power conversion efficiency (PCE) among the series (1.28%); ARM242 exhibited a PCE of 1.14% owing to the lower photocurrent density of 5.30 mA cm−2 (Fig. S26 and Table S11†). In agreement with its narrowest spectral response, the standard C1 generates the lowest Jsc of 4.64 mA cm−2 but the highest Voc leading to a PCE of 1.11%. The computed absorbed photon to current conversion efficiency (APCE%) spectra confirm a slightly higher injection in the case of ARM12 compared to its heteroleptic analogue, providing a value of 40%, where the light harvesting efficiency is essentially unitary (Fig. S25b†). The panchromatic sensitization provided by the co-adsorption of C1 and ARM12 brought a further increase in photocurrent leading to ca. Jsc = 7 mA cm−2, a Voc = 0.42 V and a PCE of 1.56%. The favorable competition between dye regeneration and charge recombination leading to nearly unitary collection efficiency prompted us to further enhance the light harvesting capability of our series by increasing the thickness of the transparent TiO2 tasked with dye adsorption (up to ca. 20 μm) and by introducing a top scattering layer made of ca. 100 nm anatase nanoparticles. Under these conditions, the photocurrent reached 8 mA cm−2 for ARM12, ca. 7 mA cm−2 for ARM242 and 5 mA cm−2 to C1 associated with a PCE of 1.71%, 1.52% and 1.30%, respectively (Fig. 8 and Table 4).
 | ||
| Fig. 8 (a) IPCE spectra for the Fe(II)NHC complexes and their relative integrated photocurrent density. (b) J/V curves for the dyed 20 μm + scattering layer TiO2 electrodes. | ||
| Dye | J sc/(mA cm−2) | V oc/V | FF% | η% |
|---|---|---|---|---|
| ARM12 | 7.84 ± 0.38 | 0.39 ± 0.01 | 56 ± 1 | 1.71 ± 0.15 |
| ARM242 | 6.64 ± 0.12 | 0.39 ± 0.01 | 59 ± 1 | 1.52 ± 0.10 |
| C1 | 5.27 ± 0.14 | 0.45 ± 0.01 | 55 ± 1 | 1.30 ± 0.07 |
| C1 + ARM12 | 8.63 ± 0.24 (8.74) | 0.42 ± 0.01 (0.43) | 55 ± 1 (55) | 1.98 ± 0.10 (2.06) |
These are, to the best of our knowledge, the highest photocurrent densities ever reported for an iron sensitized solar cell. The lower performances obtained from ARM242 compared to ARM12 can be attributable both to the slightly lower injection quantum yield as shown by the APCE and by a faster recombination rate with I3− as pointed out by the less negative threshold of the dark current well visible in Fig. 8b. Finally, the C1 + ARM12 system generated Jsc ≈ 9 mA cm−2, Voc = 0.42 V and an average PCE of 2% that at the moment represents the best efficiency ever reported for n-type sensitization by Fe(II) dyes. ARM12 and ARM242 exhibited a lower Voc than the standard C1 by at least 30–60 mV.
Often a reduced Voc is obtained by metal complex-based sensitizers endowed with an increased red sensitivity, that is usually obtained by lowering the energy of the lowest unoccupied orbitals of the chromophoric ligands, in agreement with studies by Sayama49 in the case of Ru(II) complexes bearing biquinoline ligands.
This phenomenon was explained by the back-electron recombination of photoinjected electrons with the oxidized form of the electrolyte mediated through π delocalized ligands. The dark current related to the recombination of the photoinjected electrons with the I3− species (I3− + 2e−(CB(TiO2)) → 3I−) influences the Voc that can be written according to eqn (7), a simplified form of the diode equation:50
 | (7) |
According to eqn (7), the Voc logarithmically decreases with the increase in the back-recombination rate. To better quantify the recombination kinetics involving the electron transfer from ARM12 and ARM242 modified TiO2 films to I3− electron acceptors in the electrolyte, we have carried out a Tafel analysis on the polarization curves obtained from the dyed TiO2 films in contact with a simplified electrolyte where only 10−2 M LiI3 (10−2 M LiI + 10−2 M I2) was present. Fig. 9 shows the Tafel plots and their relative slopes obtained by linear fit on a current interval of ca. 2.5 decades.
 | ||
| Fig. 9 Tafel plots of the dark current of the Fe(II)NHC sensitized TiO2 films in contact with 10−2 M I−/10−2 I3−/CH3CN solution. Tafel slopes extrapolated by the plots are 126, 112, 99 and 139 mV dec−1 for C1, ARM12, ARM242 and bare TiO2 respectively. | ||
In general, we found Tafel slopes in the proximity of 120 mV dec−1, typical of an electrochemical process kinetically limited by the electron transfer kinetics, as it would be expected in the case of the generation of the I2(˙−) radical anion by a poorly electrocatalytic electrode, like TiO2. In general, the Tafel slope can be expressed as eqn (8):
 | (8) |
2.6. Computational analysis
Herein we discuss the results of DFT/TD-DFT calculations performed with the aim of shining light on the photovoltaic behaviour and the interfacial charge separation processes observed experimentally for the distinct Fe(II)NHC dye sensitizers. The TAS experiments in the μs to s timescale, discussed in Section 2.4, do not provide us with information on the ultrafast electron injection rates and thus, on the relative efficiency of the interfacial charge separation. By calculating the electronic structure of the dye@TiO2 interface and the electronic coupling between the dye and the metal oxide we can quantify the impact of the introduction of the ThCA anchoring group in terms of electron injection efficiency. Note that in this section we also considered the results of the unsubstituted heteroleptic Fe(II)NHC dye (ARM13) with the objective of having a reference system for ARM242.We first investigated the optoelectronic properties of the standalone dye sensitizers by focusing on (i) the efficiency of the intramolecular charge separation in the MLCT state, and (ii) the improvement of light harvesting capabilities in terms of the generated photocurrent. To quantify the hole/electron spatial delocalization in the MLCT excited state along the different molecular fragments conforming the dyes (see Fig. S13† for the fragment definitions), we used the transition density matrix analysis tools as implemented in the TheoDORE package.38 The fragment delocalization percentages for the heteroleptic and homoleptic dyes are summarized in Tables S5 and S6,† respectively. As is apparent in Fig. 10, when introducing the ThCA unit, the electron density is largely moving from the Fe–NHC core towards the carboxylic group, resulting in a larger electron delocalization around the anchoring moieties (almost a 16% increase) for ARM242 with respect to ARM13, as spatially represented by the electron natural transition orbitals (NTOs).51 It is important to stress here that the effect of the CA group on increasing the electron localization on the anchoring moiety is even larger than the one shown in Fig. 10 (about 24%), since we should compare it with the value of 8% calculated for the heteroleptic COOH-substituted dye bearing a thiophene bridge (ARM7 see ref. 33). On the other hand, the spatial delocalization of the hole density remained almost unaffected upon the chemical functionalization of the ThCA groups (Tables S5 and S6†). Identical results were, clearly, obtained for the homoleptic dye, but in this case the amount of delocalization on the fragments was halved due to the split of the charge density into two symmetric branches.
 | ||
| Fig. 10 Electron natural transition orbitals (NTOs) in ARM13 and ARM242 with the percentages of delocalization for each fragment. The colors employed to plot the isodensities correspond to the unbound pyridyl NHC ligand and iron (orange), anchoring ligand: pyridyl NHC (purple), thiophene spacer (green) and carboxylic group (red) moieties. The isovalue used for this plot was 0.02 a.u. | ||
Then, we evaluated the impact on the photogenerated current of the red-shift absorption prompted by the inclusion of the ThCA group, by following the methodology used in ref. 52, where the photocurrent (Jsc) generated by the dye's lowest energy absorption MLCT band was calculated by integrating the simulated absorption spectra against the ASTMG173 solar spectrum (Pin) applying eqn (9):
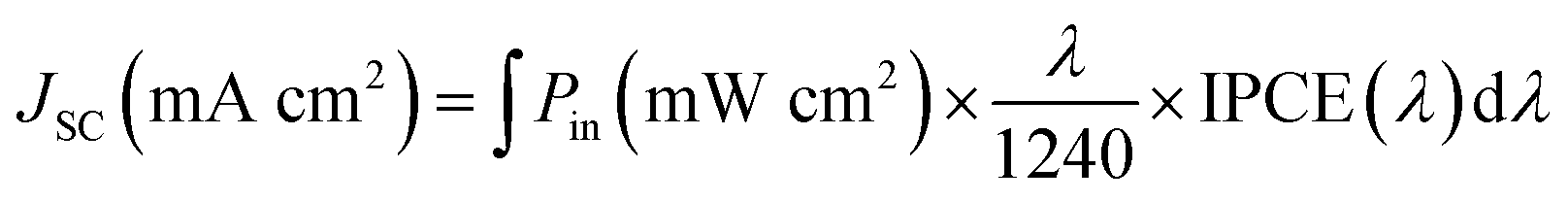 | (9) |
| LHE(λ) = 1 − 10A(λ) | (10) |
A(λ) is the reciprocal absorption of the electrode, given by the product of the molar extinction coefficients (ε) and the surface concentration (Γ) of the sensitizer. The values of Γ were extracted from the experimental absorption spectra (see Section 2.2), and amounted to 147, 107 and 70 nmol cm−2 for C1, ARM242 and ARM12; while for ARM13 we used a surface coverage of 120 nmol cm−2, as it was obtained in our previous work.33 Finally, in order to get consistent results which are not biased by the errors introduced by our theoretical models, the excited state vertical energies used to compute the simulated spectra have been rigidly shifted to make the main MLCT transition energies coincide with the experimental absorption maximum wavelengths, as reported in Table 1. The calculated Jsc curves as a function of the absorption in the vis region are represented in Fig. S14.†
Notably, the computed current intensities achieved by ARM242/ARM12 are about 1.8 and 1.5 higher than that of their previous heteroleptic/homoleptic homologues, respectively, which is in good qualitative agreement with the experiments ((Jsc)ARM242/(Jsc)ARM13 and (Jsc)ARM12/(Jsc)C1 ratios amounting to 1.2 and 1.4, respectively). This result confirms that the increase of the current intensities induced by the light harvesting enhancement originates from the effect of the acceptor ThCA moiety, even though the surface coverage by ARM242/ARM12 is lower with respect to that by the reference dyes.
Overall, based on the results discussed so far, the ThCA dyes should show high photocurrents and higher values of PCEs with respect to the reference ARM13/C1 by virtue of their red-shifted absorption and higher electron localization on the anchoring group. Till here, we are, however, neglecting the impact of the energetics, that is, the relative position between the donor (LUMO) dye's state and the acceptors CB states, and we already know from experiments that the introduction of ThCA translates into a reduction of the driving force for electron injection, estimated from experiments to be about 0.2 eV.
Thus, with the goal of extracting a more quantitative picture of the electron injection efficiency when the dyes are grafted to the TiO2 surface, we modelled the dye@TiO2–Mg interfaces as described in the Computational methods section of the ESI† (Fig. 11).
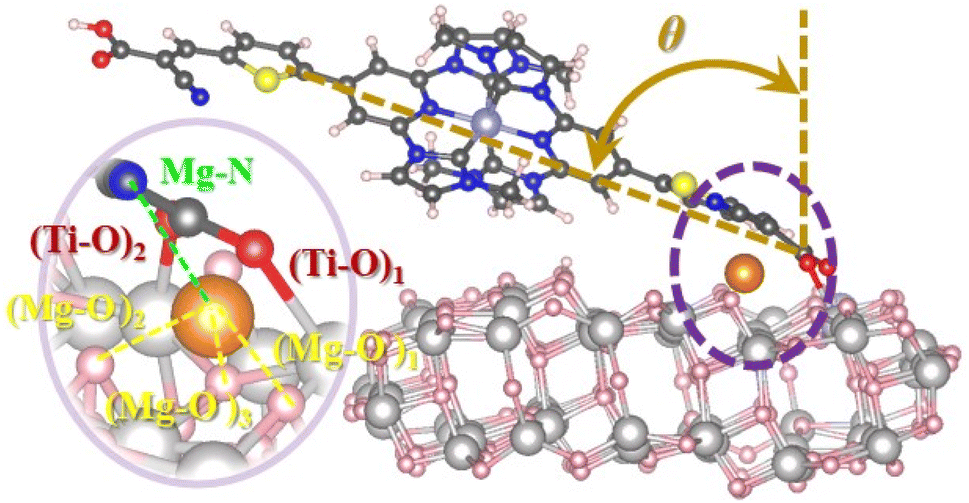 | ||
| Fig. 11 Optimized structure of the ARM12@Mg–TiO2 system representing tilted angles (θ) formed by the normal surface vector and the dye; whereas a zoomed image of the Mg2+ cation (orange color atom), its closest O neighbor atoms of the TiO2 surface and the N atom of the ThCA moiety is displayed in the onset of the lateral view as well. | ||
A first comment concerns the geometry of the dye@TiO2–Mg system: although all dyes exhibited similar O–Ti bond lengths (in the order of 2–2.3 Å) when attached to the surface, those bearing the ThCA units (ARM242/ARM12) resulted in being more bent toward the semiconductor surface by about 10° with respect to the C1/ARM13 dyes, see the relevant optimized structural parameters listed in Table S7;† this is the consequence of the extension of the π-electronic conjugation on the whole molecular skeleton, which results in the increase of the van der Waals interactions with the TiO2 surface.
Note that the “almost flat” adsorption configuration predicted by structural optimization of the dye@TiO2 surface slab is in line with the decreased surface coverage experimentally estimated for the two ThCA complexes. As one can expect, this particular adsorption configuration, leading to the breaking of the electronic conjugation between the dye and the surface, is, in principle, detrimental for the LUMO–CB state electronic coupling, despite the larger fraction of charge localized upon excitation on the anchoring group (Fig. 10).
Looking at the dye@TiO2–Mg electronic structure and energetic alignment at the interface, the projected density of states (PDOS) over the dye and the TiO2 components (Fig. 12) and the corresponding occupied/unoccupied dye and TiO2 Kohn–Sham (KS) levels listed in Table S9,† indicate that when introducing the ThCA group, the predicted stabilization of the dye's LUMO amounts to about 0.15–0.2 eV (from −3.79/−3.83 for ARM13/C1 to −3.98 eV for ARM242), in perfect agreement with the experimentally estimated value of 0.2 eV.
 | ||
| Fig. 12 Projected density of states (PDOS) of dye@TiO2–Mg systems along the ARM13 (bright blue) and ARM242 (red) dyes, TiO2 surface (purple) and Mg2+ cation (magenta), as calculated by Mulliken population analysis. Furthermore, a caption of the graph in the energy region where the dye's LUMO and TiO2 CB are located and delimited by the green dotted line, is also displayed in the right part. For visualization purposes, only the vertical bars of the dye's PDOS are represented here. In the same vein, the TiO2 DOS intensity has been divided by a factor of 14 in the main plot, whereas for the zoomed graph no DOS intensity renormalizations have been applied. The rest of PDOS plots are presented in Fig. S15.† | ||
In addition, in our simulations the LUMO vs. CB driving force is further reduced by a 0.1 eV upshift of the TiO2 CB states (Table S9†) driven by the presence of a larger interfacial dipole (Table S8†).53 As is apparent in Fig. 12, this change in the relative dye/TiO2 energetics places the dye LUMO at the edge of the semiconductor CB, in a lower density of states region, thus being detrimental for the electron injection process. Since, as we discussed before, the two families of dyes require different DFT functionals if one wants to accurately predict their optical properties, we checked if the larger extent of Hartree–Fock exchange going from B3LYP* to B3LYP may affect to some extent the dye/TiO2 relative energy lining up. Data listed in Table S10,† in line with previous results on organic sensitizers,54 show that the TiO2 and dye occupied/unoccupied levels are downshifted/upshifted by a comparable extent resulting in a similar LUMO vs. CBM alignment.
The bent adsorption configuration shown in Fig. 11 and the reduction in the electron injection driving force, discussed so far, qualitatively explain the similar maximum IPCE values with respect to C1, partially affecting the PCEs measured for the new ThCA-substituted dyes (Fig. 8a). By explicitly calculating the electronic coupling between the dye's LUMO and the TiO2 CB states as detailed in the Computational details section of the ESI,† we can quantitatively estimate the electron injection probability (Γinj) over the semiconductor manifold of unoccupied levels, as shown in Fig. 13. Evidently, the Γinj values estimated for the ThCA dye, ARM242, regardless of the LUMO position (driving force), are always lower with respect to the ones displayed by the COOH reference dye, indicating that the reduction in the electronic coupling due to the breaking of the dye–TiO2 electronic conjugation has quite a sizable impact on the injection efficiency.
 | ||
| Fig. 13 Probability distribution Γ(ε) for electron injection from the LUMO of ARM13 (bright blue) and ARM242 (red) dyes to the TiO2–Mg states within the energy region of the TiO2 CB edge. The probability distributions for all systems analysed here are plotted in Fig. S19.† | ||
Although these results nicely account for the measured PCE curves reported in Fig. 8a (note that the Γ(ε) curves for ARM13 and C1 are similar, as shown in Fig. S19†), the picture we are providing, based on the optimized bidentate adsorption configuration via the COO− deprotonated group, might only partially represent the actual working conditions.
Previous investigations have, indeed, evidenced the formation of bonds between the N atoms of the cyano (CN) groups and the Ti surface atoms (as schematised in Fig. 2), which can indeed be stabilized under certain conditions.55,56 In this regard, with the objective of evaluating the impact of the formation of such bonds on the interfacial electron injection properties, we carried out the diabatic analysis on ARM242 and ARM12 grafted in a bidentate mode to the surface by one Ti–N bond from the cyano moiety, and by one Ti–O bond from the carboxylic group (denoted in the following section as ARM242–CN and ARM12–CN), as represented in Fig. S18.† At this stage, it is important to stress that we relied on a bidentate CN/COO− mode to assess the modification of the interfacial electronic structure when changing one COO− by another CN-based bond, even though this configuration is less stable than the previous considered bidentate COO− type of anchoring, which is in line with the findings of previous theoretical studies.56 On the same footing, we are aware that to properly tackle the relative stability of the different anchoring groups that these dyes can form with the surface, one should rely on molecular dynamics (MD) simulations for all possible bidentate/tridentate and CN/COO−-based bond conformations. However, since this type of analysis has already been established for other organic dyes bearing similar anchoring groups,56 these stability issues are out of the scope of the present paper. Interestingly, the transition density matrix analysis of the isolated ARM242 revealed a high electron delocalization around the N atom of the CN moiety (4%) compared with the O atoms of the COOH group (2.8 and 0.6%), suggesting a strong coupling between the dye's LUMO and the TiO2 CB edge states for both ARM12–CN and ARM242–CN (see Fig. S16 and S17†). On top of that, the anchoring geometries achieved with CN/COO− moieties clearly enhance the electron injection due to a higher conjugation between the π-electrons of the dye and the surface, if one compares this with the bent geometries displayed by the bidentate COO− binding modes (see the comparison between Fig. 11 and S18†). As a result, the calculated injection probability, Γinj, over the TiO2 CB states is always higher with respect to the one calculated for both ARM242 and ARM12 in the COO− bidentate configurations (Fig. S19†). Notably, such an improvement in the electron injection rates driven by the formation of CN-based bonds has already been observed for other dyes containing CA anchoring groups.56 It is noteworthy to emphasize that the two considered systems correspond to the extreme scenario where only one type of anchoring group is formed, whereas under the experimental conditions both binding modes probably exist. Nonetheless, our calculations provided an interval for the injection probability values Γinj achieved by ARM242/ARM12 which, indeed, systematically underperformed those ones calculated for ARM13/C1. To sum up, based on the results of our atomistic simulations, we can conclude that the measured photocurrents come from a balance between the notable improvement of the light harvesting properties (Fig. 10) and the detrimental lowering of the driving force for electron injection, to different extents depending on the type of binding mode formed with the TiO2 surface.
3. Conclusions
Two new Fe(II) pyridylNHC sensitizers ARM242 and ARM12 bearing one and two thienylcyanoacrylic (ThCA) anchoring groups, respectively, have been prepared with the aim of promoting the metal to surface charge separation and the light harvesting window in iron-sensitized solar cells (FeSSCs). The complexes have been fully characterized in both the solution and chemisorbed on titanium dioxide photanodes by means of electrochemistry, transient absorption spectroscopy and TD-DFT calculations. Charge injection resulted in extremely long-lived charge separated states on TiO2 photoanodes which could be used to intercept the redox couple with nearly quantitative efficiency. The introduction of the ThCA moiety remarkably improved the photocurrent by comparison with the carboxylic analogue (C1) thanks to the unprecedented extension of spectral sensitization in the case of Fe(II) sensitizers. The use of a co-sensitizing mixture of a carboxylic and a ThCA–iron complex (C1 + ARM12) produced a panchromatic absorption intensely covering the 450–700 nm range and extending up to 800 nm. Co-sensitization allowed a further improvement of the photocurrent and efficiency of the FeSSC (Jsc: 9 mA cm−2 and PCE: 2%) to the best values reported so far. At this time, however, it has been impossible to obtain both reasonably high current density and high photovoltage which can only be obtained by using basic additives to shift the Fermi level of TiO2. A comparative ultrafast investigation to address the role of the electrolyte's composition in the excited state dynamics of these Fe(II) sensitizers grafted on TiO2 and ZrO2 semiconductors is currently in progress.Data availability
The data supporting this study is available within the main text and the ESI.† All original data generated during this work is available from the corresponding author upon request.Author contributions
A. R. M. performed the synthesis and ground state characterization studies of the ligands and complexes. E. M. assembled the DSSCs, characterized them and analysed the results with S. C. V. D.-C. performed DFT calculations and analysed the results with M. P. P. C. G. conceived the project and wrote the manuscript with S. C. and M. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The L2CM thanks the French Agence Nationale de Recherche (ANR-16-CE07-0013-02), Lorraine Université d'Excellence (IMPACT N4S) and the European Regional Development Funds (Programme opérationnel FEDER-FSE Lorraine et Massif des Vosges 2014–2020/“Fire Light” project: “Photo-bio-active molecules and nanoparticles”) for financial support and grants to A. R. M. The L2CM laboratory is also grateful to F. Dupire for mass spectrometry of the complexes. University of Ferrara acknowledges financial support from the Emilia Romagna Region. The LPCT thanks the European Regional Development Funds (Programme opérationnel FEDER-FSE Lorraine et Massif des Vosges 2014–2020/COMETE (COnception in silico de Matériaux pour l'EnvironnemenT et l'Energie) and Fire Light projects) for financial support to V. D.-C.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef
.
- M. Grätzel, Prog. Photovolt.: Res. Appl., 2000, 8, 171–185 CrossRef
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC
- A. B. Muñoz-García, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed
- J. R. Durrant, S. A. Haque and E. Palomares, Coord. Chem. Rev., 2004, 248, 1247–1257 CrossRef CAS
- A. Listorti, C. Creager, P. Sommeling, J. Kroon, E. Palomares, A. Fornelli, B. Breen, P. R. F. Barnes, J. R. Durrant, C. Law and B. O'Regan, Energy Environ. Sci., 2011, 4, 3494 RSC
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 RSC
- M. Sandroni, Y. Pellegrin and F. Odobel, C. R. Chim., 2016, 19, 79–93 CrossRef CAS
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915–1925 CrossRef CAS
- L. Zhang, X. Yang, W. Wang, G. G. Gurzadyan, J. Li, X. Li, J. An, Z. Yu, H. Wang, B. Cai, A. Hagfeldt and L. Sun, ACS Energy Lett., 2019, 4, 943–951 CrossRef CAS
- Y. Ren, D. Zhang, J. Suo, Y. Cao, F. T. Eickemeyer, N. Vlachopoulos, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Nature, 2023, 613, 60–65 CrossRef CAS PubMed
- S. Ferrere and B. A. Gregg, J. Am. Chem. Soc., 1998, 120, 843–844 CrossRef CAS
- S. Ferrere, Chem. Mater., 2000, 12, 1083–1089 CrossRef CAS
- C. R. Tichnell, J. N. Miller, C. Liu, S. Mukherjee, E. Jakubikova and J. K. McCusker, J. Phys. Chem. C, 2020, 124, 1794–1811 CrossRef CAS
- C. D. Clark, M. Z. Hoffman, D. Paul Rillema and Q. G. Mulazzani, J. Photochem. Photobiol., A, 1997, 110, 285–290 CrossRef CAS
- J. E. Monat and J. K. McCusker, J. Am. Chem. Soc., 2000, 122, 4092–4097 CrossRef CAS
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 CrossRef PubMed
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, J. Am. Chem. Soc., 2008, 130, 14105–14107 CrossRef CAS PubMed
- M. Cammarata, R. Bertoni, M. Lorenc, H. Cailleau, S. Di Matteo, C. Mauriac, S. F. Matar, H. Lemke, M. Chollet, S. Ravy, C. Laulhé, J.-F. Létard and E. Collet, Phys. Rev. Lett., 2014, 113, 227402 CrossRef PubMed
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed
- Y. Liu, T. Harlang, S. E. Canton, P. Chábera, K. Suárez-Alcántara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Chem. Commun., 2013, 49, 6412 RSC
- T. Duchanois, T. Etienne, C. Cebrián, L. Liu, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Eur. J. Inorg. Chem., 2015, 2015, 2469–2477 CrossRef CAS
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chábera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed
- L. Liu, T. Duchanois, T. Etienne, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Phys. Chem. Chem. Phys., 2016, 18, 12550–12556 RSC
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Acc. Chem. Res., 2016, 49, 1477–1485 CrossRef CAS PubMed
- M. Darari, E. Domenichini, A. Francés-Monerris, C. Cebrián, K. Magra, M. Beley, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Dalton Trans., 2019, 48, 10915–10926 RSC
- K. Magra, E. Domenichini, A. Francés-Monerris, C. Cebrián, M. Beley, M. Darari, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Inorg. Chem., 2019, 58, 5069–5081 CrossRef CAS PubMed
- K. Magra, M. Darari, E. Domenichini, A. Francés-Monerris, C. Cebrián, M. Beley, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, J. Phys. Chem. C, 2020, 124, 18379–18389 CrossRef CAS
- E. Marchini, M. Darari, L. Lazzarin, R. Boaretto, R. Argazzi, C. A. Bignozzi, P. C. Gros and S. Caramori, Chem. Commun., 2020, 56, 543–546 RSC
- A. R. Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori and P. C. Gros, J. Mater. Chem. A, 2021, 9, 3540–3554 RSC
- A. R. Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori, C. A. Bignozzi and P. C. Gros, Chem.–Eur. J., 2021, 27, 16260–16269 CrossRef CAS PubMed
- L. Lindh, O. Gordivska, S. Persson, H. Michaels, H. Fan, P. Chábera, N. W. Rosemann, A. K. Gupta, I. Benesperi, J. Uhlig, O. Prakash, E. Sheibani, K. S. Kjaer, G. Boschloo, A. Yartsev, M. Freitag, R. Lomoth, P. Persson and K. Wärnmark, Chem. Sci., 2021, 12, 16035–16053 RSC
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS PubMed
- J. M. Cole, M. A. Blood-Forsythe, T.-C. Lin, P. Pattison, Y. Gong, Á. Vázquez-Mayagoitia, P. G. Waddell, L. Zhang, N. Koumura and S. Mori, ACS Appl. Mater. Interfaces, 2017, 9, 25952–25961 CrossRef CAS PubMed
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed
- Y. Tachibana, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem., 1996, 100, 20056–20062 CrossRef CAS
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276–3279 CrossRef
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed
- Y. Ma, S. R. Pendlebury, A. Reynal, F. Le Formal and J. R. Durrant, Chem. Sci., 2014, 5, 2964–2973 RSC
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chábera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed
- J. R. Durrant, S. A. Haque and E. Palomares, Coord. Chem. Rev., 2004, 248, 1247–1257 CrossRef CAS
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2011, 115, 2439–2447 CrossRef CAS
- A. V. Müller, K. T. de Oliveira, G. J. Meyer and A. S. Polo, ACS Appl. Mater. Interfaces, 2019, 11, 43223–43234 CrossRef PubMed
- M. Karpacheva, C. E. Housecroft and E. C. Constable, Beilstein J. Nanotechnol., 2018, 9, 3069–3078 CrossRef CAS PubMed
- T. Funaki, H. Kusama, N. Onozawa-Komatsuzaki, K. Kasuga, K. Sayama and H. Sugihara, Eur. J. Inorg. Chem., 2014, 2014, 1303–1311 CrossRef CAS
- A. Hagfeldt and M. Graetzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS
- S. Fantacci, E. Ronca and F. De Angelis, J. Phys. Chem. Lett., 2014, 5, 375–380 CrossRef CAS PubMed
- E. Ronca, M. Pastore, L. Belpassi, F. Tarantelli and F. De Angelis, Energy Environ. Sci., 2013, 6, 183–193 RSC
- M. Pastore, S. Fantacci and F. De Angelis, J. Phys. Chem. C, 2013, 117, 3685–3700 CrossRef CAS
- F. Zhang, W. Ma, Y. Jiao, J. Wang, X. Shan, H. Li, X. Lu and S. Meng, ACS Appl. Mater. Interfaces, 2014, 6, 22359–22369 CrossRef CAS PubMed
- Y. Jiao, F. Zhang, M. Grätzel and S. Meng, Adv. Funct. Mater., 2013, 23, 424–429 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, additional computational details, and electrochemical and photovoltaic data. See DOI: https://doi.org/10.1039/d2sc05971a |
| This journal is © The Royal Society of Chemistry 2023 |