
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStatistical copolymer metal organic nanotubes†
Jacob A.
Barrett
a,
Nathan D.
Rosenmann
b,
Karthikeyan
Gnanasekaran
 b,
Xian B.
Carroll
a,
Nathan C.
Gianneschi
*b and
David M.
Jenkins
*a
b,
Xian B.
Carroll
a,
Nathan C.
Gianneschi
*b and
David M.
Jenkins
*a
aDepartment of Chemistry, University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: jenkins@ion.chem.utk.edu
bDepartments of Chemistry, Materials Science & Engineering, Biomedical Engineering, Pharmacology, International Institute for Nanotechnology, Simpson-Querrey Institute, Chemistry of Life Processes Institute, Lurie Cancer Center, Northwestern University, Evanston, Illinois 60208, USA. E-mail: nathan.gianneschi@northwestern.edu
First published on 6th January 2023
Abstract
Metal–organic nanotubes (MONTs) are 1-dimensional crystalline porous materials that are formed from ligands and metals in a manner identical to more typical 3-dimensional metal–organic frameworks (MOFs). MONTs form anisotropically in one dimension making them excellent candidates for linker engineering for control of chemical composition and spacing. A novel series of MONTs was synthesized utilizing a mixture of 1,2,4-ditriazole ligands containing both a fully protonated aryl moiety and its tetrafluorinated analog in ratios of, 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:4, 1:1, 4:1, and 1:0, respectively. All MONTs were characterized by both bulk and nanoscale measurements, including SCXRD, PXRD, ssNMR and TEM, to determine the resulting co-polymer architecture (alternating, block, or statistical) and the ligand ratios in the solid materials. All characterization methods point towards statistical copolymerization of the materials in a manner analogous to 3D MOFs, all of which notably could be achieved without destructive analytical methods.
:1, 1:4, 1:1, 4:1, and 1:0, respectively. All MONTs were characterized by both bulk and nanoscale measurements, including SCXRD, PXRD, ssNMR and TEM, to determine the resulting co-polymer architecture (alternating, block, or statistical) and the ligand ratios in the solid materials. All characterization methods point towards statistical copolymerization of the materials in a manner analogous to 3D MOFs, all of which notably could be achieved without destructive analytical methods.
Introduction
Metal–organic nanotubes (MONTs) are 1-dimensional, tunable, porous materials synthesized from organic linkers and metal salts (Fig. 1).1 They share the properties of porosity and tunability with their more common 3D relation, metal–organic frameworks (MOFs) (Fig. 1A).2–4 Indeed, MONTs have already demonstrated a similar range of applications to MOFs that exploit their porosity including gas storage and liquid separations.5–8 Likewise, MONTs are tunable in that the design of the pore shape and size are controlled using modular approaches (e.g. 2-pillar, 4-pillar, and 6-pillar) reminiscent of MOFs' secondary building units (SBUs).8–10 However, a key aspect of tunability for MOFs that has yet to be fostered in MONTs is ligand multivariate functionality.11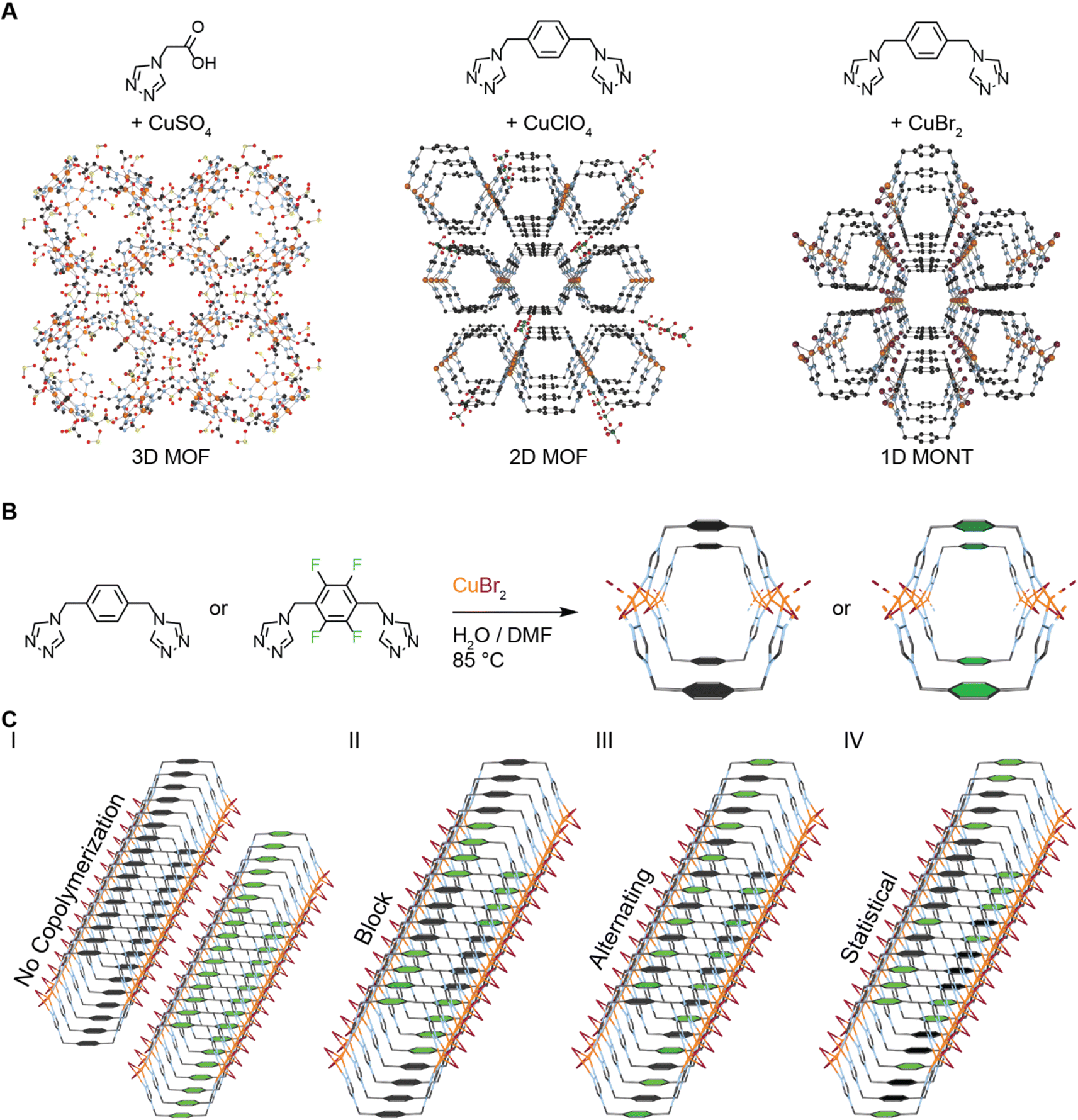 | ||
| Fig. 1 Strategies for preparing copolymerized MONTs from di-triazole ligands. (A) Different 1,2,4-triazole ligands combined with copper salts have led to distinct dimensionalities for materials. A typical example is shown for a 3D MOF with a mono-triazole ligand.26 Di-triazole ligands either form 2D sheet MOFs if non-coordination anions are employed or 1D metal–organic nanotubes (MONTs) if capping anions such as halides are used.23,27 (B) In this study, two similar di-1,2,4-triazole ligands, with and without fluorine, were reacted with copper(II) bromide to make MONTs under identical conditions. (C) Four distinct hypotheses for co-polymerization of MONTs shown. First, the ligands could not co-polymerize. Next, the copolymerization could be driven by the association of the previous ligand yielding block or alternating polymerization. Finally, the previous ligand insertion does not affect the copolymerization and it is a statistical copolymerization. | ||
Ligand multivariate functionality is the ability to control the mixing of different ligands within the same crystalline MOF.11 In the last fifteen years, numerous examples have been developed which showcase multiple functional groups within one MOF.11–15 These mixed linker MOFs have been employed for tunable catalysts, waste remediation, and fluorescent probes.16–18 While a few cases of ligand multivariance have allowed for quantification of ligands,19,20 with limited exception, the destruction of the MOF is required to determine the ratio of the ligands.4,13
Applications for MONTs that exploit their unique anisotropy, such as nanostraws and nanowires, could benefit considerably from the ability to mix ligands that are on the exterior surface of a finite bundle of these tubes.6,10,21,22 However, for the goal of ligand multivariate functionality in MONTs to be achieved, two conditions must be met. First, a robust isoreticular approach must be developed for a 1D system, ideally employing simple solvothermal synthesis.23,24 Second, the anisotropic materials must be synthesized on the nanoscale to achieve high external surface areas.24,25
In this manuscript, we report ligand multivariate functionality in MONTs that is coherent at the macroscale and nanoscale size regimes. The reaction of (4,4′-(1,4-(xylene)-diyl)bis(1,2,4-triazole)) or its tetrafluorinated analogue with copper bromide lead to isostructural MONTs (Fig. 1B). In addition, ligand ratios of the di-triazoles of 1:4, 1:1, and 4:1 reacting with copper bromide also lead to crystalline MONTs. A host of macro and nanoscale measurements were required to both quantitate the amount of each ligand present in the MONTs, as well as determine the type of copolymerization (Fig. 1C). The MONTs could copolymerize as block, alternating, or statistical (or even not at all). To this end, all five MONTs were characterized by single-crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD), 13C solid-state nuclear magnetic resonance (13C ssNMR), energy-dispersive X-ray spectroscopy (EDX), selected area electron diffraction (SAED), and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). These combined non-destructive measurements demonstrate that the ligand quantities in each MONT roughly match the ratios used in the solvothermal reactions and that the MONTs form statistical copolymers.
Results and discussion
We have pioneered a general method for isoreticular synthesis of MONTs with di-triazole ligands and with either copper or silver.23 Our two-column pillared MONTs feature di-1,2,4-triazoles in a syn conformation attached to the group 11 metals in a tetrahedral coordination.23 These MONTs have a (2,4) topology where 2 represents the number of links between copper and the organic ligand, while 4 represents the number of copper atoms linked to each organic ligand (Fig. S5†).1 We previously reported the synthesis of L1 by the method of Hórvath (Fig. 2A).27,28 The fluorinated version of the ligand, L2, is prepared in three steps starting from 2,3,5,6-tetrafluoro-1,4-benzenedimethanol (Fig. S1†) that are similar to the synthesis of L1.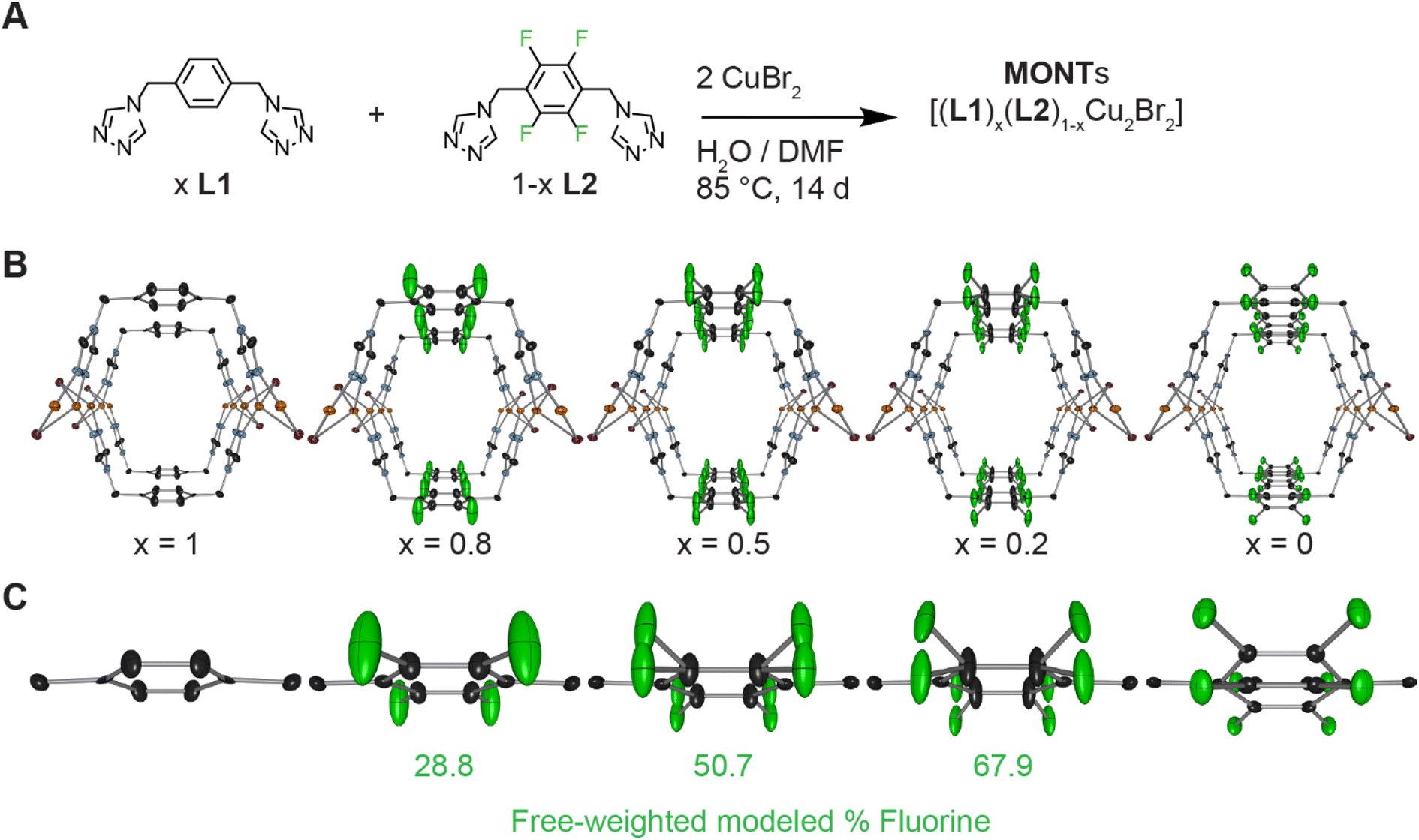 | ||
| Fig. 2 Synthesis and associated single crystal X-ray diffraction studies on copolymerized MONTs. (A) Synthesis of copolymerized MONTs employing L1 and L2 in different ratios. Values for x are based on the stoichiometry of the reactions. (B) Solid state structure of [(L1)x(L2)1−xCu2Br2] drawn at the 50% probability level. Hydrogen atoms are omitted. Orange, light blue, purple, black, and green ellipsoids represent copper, nitrogen, bromine, carbon, and fluorine atoms, respectively. The view is shown down the tubes to highlight the MONT pores. X-ray structure of x = 1 was previously reported.23 (C) Highlight of the aryl ring section of the solid state structure demonstrating the changes to the aryl orientation as percentage of fluorine increases with modeled fluorine percentage shown. | ||
The solution NMR of L2 is critical since assignments for carbons must be made prior to ssNMR studies on MONTs (vide supra). Only two resonances are observed for L2 in the 1H NMR at 8.58 and 5.49 ppm (Fig. S13†). Collecting two 13C NMR spectra with either typical 1H-decoupling or 19F-decoupling allowed us to assign the carbon resonances (Fig. S14 and S15†). The triazole carbon resonances are at 143.2 ppm, while the aryl carbons are at 144.4 ppm (carbons with F's) and 115.1 ppm (quaternary position). The solid-state structure for L2 was also determined with single crystal X-ray diffraction (Fig. S2†).
Reaction of L2 with copper(II) bromide in a DMF and water mixture heated to 85 °C for 14 days yielded colorless crystals (Fig. 2A). The crystals were isolated, rinsed with water and methanol followed by Soxhlet extraction overnight with methanol, and removal of residual solvent under reduced pressure to yield a white powder in 99% yield. The solid-state structure for [(L2)Cu2Br2] determined by SCXRD revealed the formation of a MONT that is isomorphous to L1 (Fig. 2B). However, there are subtle changes in the pore size, tube alignment, and position of the aryl ring compared to [(L1)Cu2Br2].23 The pore width for [(L2)Cu2Br2] is 9.456 Å while for [(L1)Cu2Br2] is 9.254 Å, which is an increase of just over 2% (Table S1†). Likewise, the pore height decreases by 0.137 Å (1% smaller). The non-covalent interactions between tubes are also affected by replacing the hydrogens on the aryl ring with fluorines. The largest effect is the vertical packing between the tubes as measured by the Cu⋯Cu distance and the aryl ring π–π stacking distance both decrease by 2–3% when switching to from L1 to L2. Finally, the aryl ring rotates out of plane by 16° (Fig. 2C, right) for [(L2)Cu2Br2] and is disordered over two positions (half pointing up and half down). The larger fluorine atoms do not have space to lay parallel to the tube as is seen in [(L1)Cu2Br2].
Other physical measurements for [(L2)Cu2Br2] were consistent with MONTs that were previously prepared by our group. The [(L2)Cu2Br2] MONT adsorps CO2 and CH4 is similar quantities to [(L1)Cu2Br2] (Fig. S22†).23 TGA results show excellent thermal stability until close to 300 °C (Fig. S23†). PXRD data is consistent with the simulated diffraction pattern (Fig. S24†).
With the completed characterization of [(L2)Cu2Br2], we turned towards synthesizing copolymerized MONTs. We accomplished this by mixing L1 and L2 together in ratios of 1:4, 1:1, and 4:1. The mixed ligand solutions were then combined with copper(II) bromide in a DMF and water mixture heated to 85 °C for 14 days (see ESI† for details). As in [(L2)Cu2Br2], these reactions yielded colorless needle crystals.
Single crystal X-ray diffraction for the copolymerized MONTs yielded similar results to [(L2)Cu2Br2] as all four crystals occupied the same space group (Imma) and had similar unit cell volumes. Notably, all three [(L1)x(L2)1−xCu2Br2] had distinct solutions which required modeling the fluorine differently in each case (Fig. 2C). For the [(L1)0.8(L2)0.2Cu2Br2] MONT, the fluorine was modeled in a single position with a floating free weight of 29%. The [(L1)0.5(L2)0.5Cu2Br2] MONT required splitting the fluorine into two positions with a floating free weight of 51%. Finally, the [(L1)0.2(L2)0.8Cu2Br2] came closer to the [(L2)Cu2Br2] with two very distinct fluorine positions and a free weighting of 68%. If alternating ligands for a co-polymerized MONT was the most favorable product (Fig. 1C), we would expect to have a distinct asymmetric unit cell with alternation between the two di-triazole ligands which is distinct from [(L1)Cu2Br2] or [(L2)Cu2Br2]. Furthermore, if the alternating model is correct, for a single crystal measurement, each ligand should have equivalent weighting no matter the starting ligand ratio.
To test the statistical copolymerization model in the bulk materials, we collected 13C ssNMR and PXRD measurements on all five MONTs. The 13C ssNMR for [(L1)Cu2Br2] contains four resonances at 53, 129, 135 and 146 ppm (Fig. 3A). These correspond to the B, D, C, and A carbons shown in the figure. The 13C ssNMR for [(L2)Cu2Br2] only contains three resonances at 41, 115, and 145 ppm (Fig. 3A). These correspond to the B′, C′, and A′ carbons shown in the figure. Notably, there is no signal for the aryl carbons bound to fluorine (D′), presumably due to the splitting of the F-atoms (which is seen in the solution state NMR, Fig. S14†).
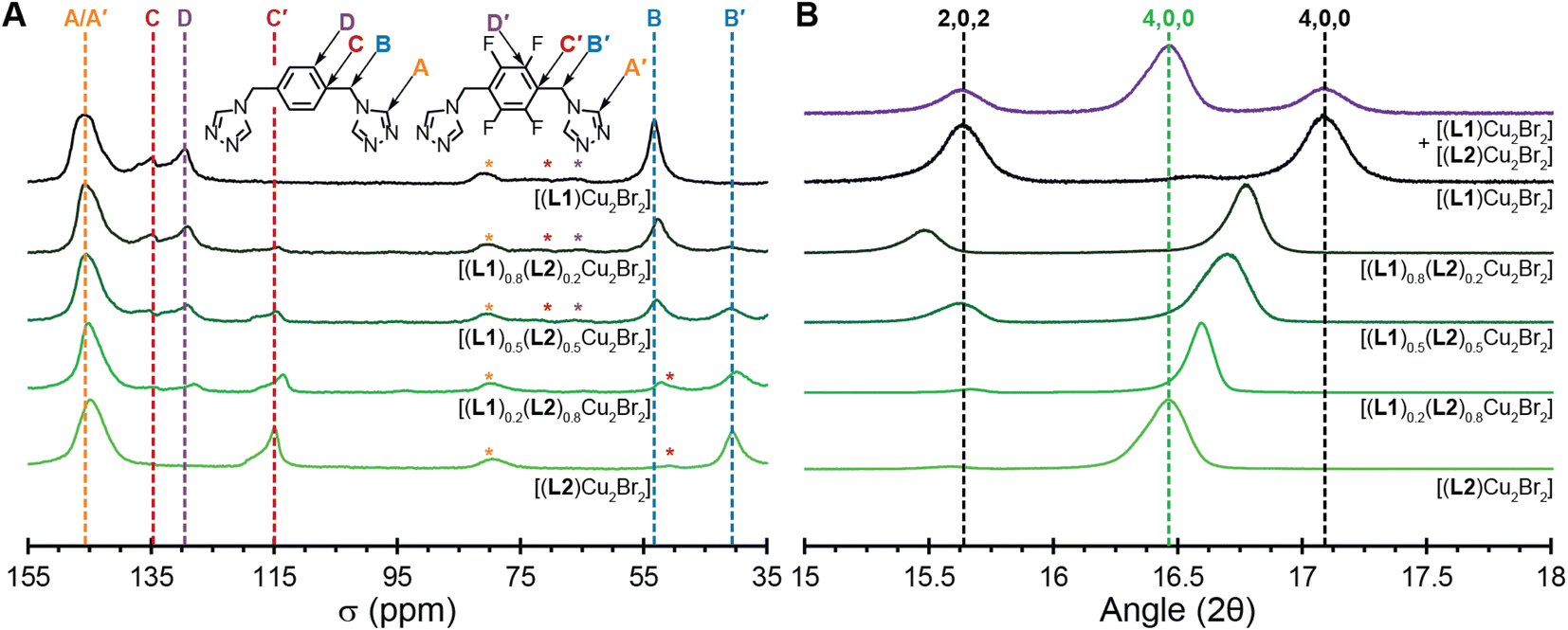 | ||
| Fig. 3 Characterization of copolymerized MONTs on the macroscale. (A) 13C ssNMR for [(L1)Cu2Br2] (black line) to [(L2)Cu2Br2] (bright green line). The copolymerized MONTs are between them with central dark green line representing [(L1)0.5(L2)0.5Cu2Br2]. Vertical dotted lines denote the resonances for each assigned carbon atom of the ligands. * Represent spinning sidebands. (B) Highlight of PXRD diffraction patterns for [(L1)Cu2Br2] (black line) to [(L2)Cu2Br2] (bright green line). Dotted vertical lines refer to hkl reflection values from calculated patterns based on SCXRD for [(L1)Cu2Br2] (black line) and [(L2)Cu2Br2] (bright green line), respectively. The copolymerized MONTs are between them with dark green line representing [(L1)0.5(L2)0.5Cu2Br2]. Purple line represents a 50/50 mix of [(L1)Cu2Br2] and [(L2)Cu2Br2] which is distinct from the copolymerized MONTs. | ||
The copolymerized MONTs [(L1)0.8(L2)0.2Cu2Br2], [(L1)0.5(L2)0.5Cu2Br2], and [(L1)0.2(L2)0.8Cu2Br2] show all the A–D and A′–C′ resonances. A and A′ overlap, but all of the other resonances are well separated in the spectrum. While 13C ssNMR are not typically integrated due to differences in structure or relaxation time concerns, these co-polymerized MONTs have very similar structures and the resonances are compared within the same spectra.19,29,30 Indeed, since we are measuring the same position on the ligand, the relative intensity is determined purely by the relative amount of the corresponding ligand.12,29 We integrated the B versus B′ resonances to give a bulk measurement for the quantity of L1versusL2 that is present in the MONTs. These comparative integrations revealed 20% L2 for [(L1)0.8(L2)0.2Cu2Br2], 45% L2 for [(L1)0.5(L2)0.5Cu2Br2], and 73% L2 for [(L1)0.2(L2)0.8Cu2Br2]. These values are 9%, 5%, and 5% different from the ratios measured by SCXRD and are in line with measured stoichiometry of each chemical reaction, which is additional evidence that the MONTs are copolymerized in a statistical manner.
While the five MONTs have similar PXRD diffraction patterns owing to similar unit cells, there are sufficient differences to determine effective ligand copolymerization. We highlight the 2θ region between 15–18° in Fig. 3B. MONT [(L1)Cu2Br2] (black line) has two reflections 2,0,2 (15.6°) and 4,0,0 (17.1°) that match the simulated powder pattern. MONT [(L2)Cu2Br2] (light green line) has only one visible reflection, 4,0,0, at 16.5°. Notably, the three copolymerized MONTs each have a 4,0,0 reflection that moves closer towards 17° as less fluorine is incorporated. Indeed, this reflection follows Vegard's law for solid-solutions (Fig. S26†).31,32 The [(L1)0.5(L2)0.5Cu2Br2] has two reflections at 15.6 and 16.7°. We compared this diffraction pattern to one that contained a 50/50 mixture of [(L1)Cu2Br2] and [(L2)Cu2Br2] (purple line), which shows that both MONT crystalline phases can clearly be seen and are discrete even when the MONTs are mixed together. This analysis of PXRD measurements prove conclusively that we have copolymerization of the ligands as the bulk materials.
To confirm the statistical copolymerization of the bulk MONTs with high spatial resolution we employed scanning transmission electron microscopy (STEM) combined with elemental composition analytics (Fig. 4). For this analysis, we prepared samples following the same procedure as for the bulk, except only heating the solutions for 15 minutes to generate nanoscale MONT structures. We observed under the same synthesis conditions, the average size of the MONTs increased upon increasing the amount of L2. Selected area electron diffraction (SAED) further confirm the crystal structure of the nanoscale materials and we observed that these materials display identical structures to the bulk MONTs (Fig. S27 and Table S2†).
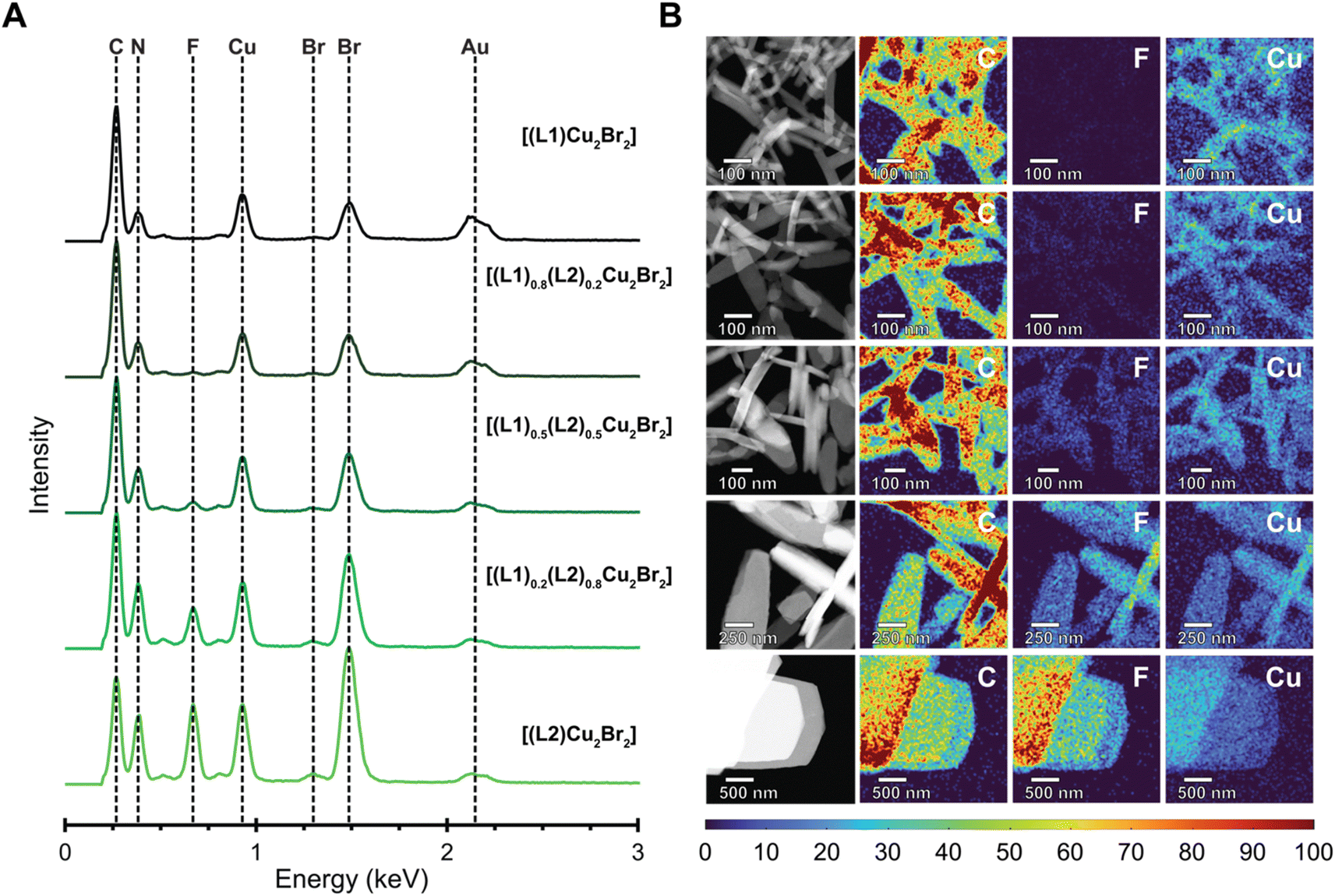 | ||
| Fig. 4 STEM EDX spectroscopy to analyze the nanoscale distribution of fluorine in the MONTs. (A) EDX spectra confirming the formation of nanoscale [(L1)Cu2Br2], [(L1)0.8(L2)0.2Cu2Br2], [(L1)0.5(L2)0.5Cu2Br2], [(L1)0.2(L2)0.8Cu2Br2], and [(L2)Cu2Br2]. The spectra display a noticeable increase in the intensity of the fluorene peak corresponding to the incorporation of L2 into the MONT bundles. The gold signal is retained throughout each spectrum due to the signal resulting from the gold TEM grid. (B) EDX maps of nanoscale MONT bundles grown at 85 °C. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and corresponding elemental maps confirming the formation of [(L1)Cu2Br2], [(L1)0.8(L2)0.2Cu2Br2], [(L1)0.5(L2)0.5Cu2Br2], [(L1)0.2(L2)0.8Cu2Br2], and [(L2)Cu2Br2] nanoscale MONT bundles. | ||
To determine whether the MONTs were block co-polymerized or statistically co-polymerized, we performed energy dispersive X-ray (EDX) spectroscopy of the nanoscale MONTs (Fig. 4A). This type of analysis has previously been employed to confirm the mixing of ligands or metals in a variety of MOFs.33–35 To minimize the degradation of the MONTs upon irradiation by the electron beam while ensuring a sufficient X-ray signal to construct 2D elemental maps, we chose to acquire maps at a relatively low resolution of 20 nm. To better distinguish the change in intensity between the MONT structures, we normalized the elemental maps and display the maps as intensity profiles. The resulting EDX maps demonstrate a similar strong and uniform signal for Cu, Br, C, and N for all CuBr MONT bundles (Fig. 4B). EDX mapping also shows that a uniform F signal that becomes visible after the composition of L2 becomes greater than 50%. As the F elemental map displays no clear bands in any of the MONT bundles across all compositions, these maps indicate that these structure are statistically copolymerized. We note that although the spatial resolution will not allow us to resolve blocks that are smaller than 20 nm, this size-scale begins to mathematically approach the statistical case. To validate these observations, a 50/50 mixture of [(L1)Cu2Br2] and [(L2)Cu2Br2] was analyzed in a similar manner, where the F map is shown to clearly differentiate between the two sets of bundles due to the intensity difference (Fig. S29†). This result suggests that there are no distinct blocks formed. Instead, these data are consistent with MONTs being present as statistical copolymers.
Conclusions
We have synthesized and characterized the first copolymerized MONTs by incorporating a mixture of two di-triazole ligands in the ratios of 0:1, 1:4, 1:1, 4:1, and 1:0 with copper bromide. These MONTs were then characterized by numerous methods in the bulk phase including SCXRD, ssNMR, and PXRD. SCXRD and ssNMR confirm that the composition of the ligands is in line with the ratios employed during the solvothermal reactions. Notably, these techniques allow for the quantification of the copolymerization of the ligands without destroying the MONT. Linker quantification is typically determined in MOFs by acid digestion followed by NMR. SCXRD precludes the case of alternating copolymerization, while high resolution PXRD measurements show that the ligands are mixed within the material. Nanoscale measurements via HAADF-STEM and EDX confirm that the elemental composition is the same as in the bulk phase, which is critical for these anisotropic materials. Furthermore, the possibility of block copolymerization was eliminated through observation of the fluorine EDX mapping. These combined experiments verify that the copolymerization of the ligands is statistical.
Data availability
X-ray structures are deposited in CCDC. Additional data can be found in ESI.†Author contributions
J. A. B. performed ligand synthesis and characterization and bulk scale MONT reactions along with their characterization (NMR, IR, DART-MS, PXRD, SCXRD, gas adsorption, and TGA). N. D. R. performed nanoscale MONT reactions and microscopy measurements (EDX, HAADF-STEM, and SAED). K. G. provided mentorship in microscopy experiments and their data analysis. X. B. C. collected and solved SCXRD measurements of L2 and provided additional assistance on MONT SCXRD analysis. N. C. G. and D. M. J. designed and supervised the project. The manuscript was written by J. A. B., N. C. G., and D. M. J. and with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. A. B. and D. M. J. thank the National Science Foundation (NSF DMR-2207224) and the University of Tennessee for support. N. D. R. and N. C. G. thank the National Science Foundation (NSF DMR-2207269) for support. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205), the MRSEC program (NSF DMR-1720139) at the Materials Research Center, the International Institute for Nanotechnology (IIN), the Keck Foundation, and the State of Illinois, through the IIN.References
- J.-G. Jia and L.-M. Zheng, Coord. Chem. Rev., 2020, 403, 213083 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- J. Bitzer and W. Kleist, Eur. J. Chem., 2018, 25, 1866–1882 CrossRef PubMed.
- F. Dai, H. He and D. Sun, J. Am. Chem. Soc., 2008, 130, 14064–14065 CrossRef CAS PubMed.
- D. K. Unruh, K. Gojdas, A. Libo and T. Z. Forbes, J. Am. Chem. Soc., 2013, 135, 7398–7401 CrossRef CAS PubMed.
- J.-G. Jia, J.-S. Feng, X.-D. Huang, S.-S. Bao and L.-M. Zheng, Chem. Commun., 2019, 55, 2825–2828 RSC.
- Y. Zhou, S. Yao, Y. Ma, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2018, 54, 3006–3009 RSC.
- T.-T. Luo, H.-C. Wu, Y.-C. Jao, S.-M. Huang, T.-W. Tseng, Y.-S. Wen, G.-H. Lee, S.-M. Peng and K.-L. Lu, Angew. Chem., Int. Ed., 2009, 48, 9461–9464 CrossRef CAS PubMed.
- K.-i. Otake, K. Otsubo, K. Sugimoto, A. Fujiwara and H. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 6448–6451 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- H. H.-M. Yeung, W. Li, P. J. Saines, T. K. J. Köster, C. P. Grey and A. K. Cheetham, Angew. Chem., Int. Ed., 2013, 52, 5544–5547 CrossRef CAS PubMed.
- Y.-B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS PubMed.
- T. W. Goh, C. Xiao, R. V. Maligal-Ganesh, X. Li and W. Huang, Chem. Eng. Sci., 2015, 124, 45–51 CrossRef CAS.
- C. Schlüsener, D. N. Jordan, M. Xhinovci, T. J. Matemb Ma Ntep, A. Schmitz, B. Giesen and C. Janiak, Dalton Trans., 2020, 49, 7373–7383 RSC.
- M. Mon, R. Bruno, E. Tiburcio, M. Viciano-Chumillas, L. H. G. Kalinke, J. Ferrando-Soria, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2019, 141, 13601–13609 CrossRef CAS PubMed.
- S. Patial, P. Raizada, V. Hasija, P. Singh, V. K. Thakur and V. H. Nguyen, Mater. Today Energy, 2021, 19, 100589 CrossRef CAS.
- Y. Wang, H. Lv, E. S. Grape, C. A. Gaggioli, A. Tayal, A. Dharanipragada, T. Willhammar, A. K. Inge, X. Zou, B. Liu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 6333–6338 CrossRef CAS PubMed.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- L. Liu, K. Konstas, M. R. Hill and S. G. Telfer, J. Am. Chem. Soc., 2013, 135, 17731–17734 CrossRef CAS PubMed.
- L. C. Applegate and T. Z. Forbes, CrystEngComm, 2020, 22, 3406–3418 RSC.
- L.-L. Chen, Y.-Y. Wu, W.-W. Wu, M.-M. Wang, H.-J. Lun, D.-B. Dang, Y. Bai and Y.-M. Li, Inorg. Chem., 2022, 61, 8629–8633 CrossRef PubMed.
- C. R. Murdock and D. M. Jenkins, J. Am. Chem. Soc., 2014, 136, 10983–10988 CrossRef CAS PubMed.
- K. M. Vailonis, K. Gnanasekaran, X. B. Powers, N. C. Gianneschi and D. M. Jenkins, J. Am. Chem. Soc., 2019, 141, 10177–10182 CrossRef CAS PubMed.
- K. Gnanasekaran, K. M. Vailonis, D. M. Jenkins and N. C. Gianneschi, ACS Nano, 2020, 14, 8735–8743 CrossRef CAS PubMed.
- S. I. Vasylevs'kyy, G. A. Senchyk, A. B. Lysenko, E. B. Rusanov, A. N. Chernega, J. Jezierska, H. Krautscheid, K. V. Domasevitch and A. Ozarowski, Inorg. Chem., 2014, 53, 3642–3654 CrossRef PubMed.
- C. R. Murdock, N. W. McNutt, D. J. Keffer and D. M. Jenkins, J. Am. Chem. Soc., 2014, 136, 671–678 CrossRef CAS PubMed.
- A. Horváth, Synthesis, 1995, 9, 1183–1189 CrossRef.
- R. Voelkel, Angew. Chem., Int. Ed., 1988, 27, 1468–1483 CrossRef.
- H. A. Habib, A. Hoffmann, H. A. Höppe, G. Steinfeld and C. Janiak, Inorg. Chem., 2009, 48, 2166–2180 CrossRef CAS PubMed.
- L. N. Appelhans, M. Kosa, A. V. Radha, P. Simoncic, A. Navrotsky, M. Parrinello and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 15375–15386 CrossRef CAS PubMed.
- T. Panda, S. Horike, K. Hagi, N. Ogiwara, K. Kadota, T. Itakura, M. Tsujimoto and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 2413–2417 CrossRef CAS PubMed.
- K. Metavarayuth, O. Ejegbavwo, G. McCarver, M. L. Myrick, T. M. Makris, K. D. Vogiatzis, S. D. Senanayake, O. M. Manley, A. M. Ebrahim, A. I. Frenkel, S. Hwang, T. Rajeshkumar, J. D. Jimenez, K. Chen, N. B. Shustova and D. A. Chen, J. Phys. Chem. Lett., 2020, 11, 8138–8144 CrossRef CAS PubMed.
- M. C. Wasson, J. Lyu, T. Islamoglu and O. K. Farha, Inorg. Chem., 2019, 58, 1513–1517 CrossRef CAS PubMed.
- Z. Xue, K. Liu, Q. Liu, Y. Li, M. Li, C.-Y. Su, N. Ogiwara, H. Kobayashi, H. Kitagawa, M. Liu and G. Li, Nat. Commun., 2019, 10, 5048 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, including selected NMRs, IRs, X-ray analysis, MS, gas adsorption data, PXRD, TGA, SAED, STEM, and EDX mapping images (PDF). CCDC 2183216–2183220. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06084a |
| This journal is © The Royal Society of Chemistry 2023 |