
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEDA mediated S–N bond coupling of nitroarenes and sodium sulfinate salts†
Juan D.
Lasso‡
 a,
Durbis J.
Castillo-Pazos‡
a,
Malcolm
Sim
a,
Joaquín
Barroso-Flores
b and
Chao-Jun
Li
*a
a,
Durbis J.
Castillo-Pazos‡
a,
Malcolm
Sim
a,
Joaquín
Barroso-Flores
b and
Chao-Jun
Li
*a
aDepartment of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. W., Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bCentro Conjunto de Investigación en Química Sustentable, UAEM-UNAM, Carretera Toluca-Atlacomulco Km 14.5, Unidad San Cayetano, Toluca, Estado de México C. P. 50200, México
First published on 15th December 2022
Abstract
Despite their long-known photochemical properties and their industrial value, the use of nitroarenes as a productive photochemical handle in organic synthesis has remained relatively unexplored. More specifically, the photochemical formation of nitrogen-sulfur bonds from nitroarenes remains to be demonstrated. Herein, we report the design and application of a sulfinate–nitroarene electron donor–acceptor (EDA) complex and its subsequent use in the first light mediated catalyst-free synthesis of N-hydroxy-sulfonamides. The presence of the EDA was assessed spectroscopically and studied via DFT and TD-DFT calculations. A total of 32 examples including both electron withdrawing and electron donating groups were synthesized under our oxygen- and water-tolerant conditions.
Introduction
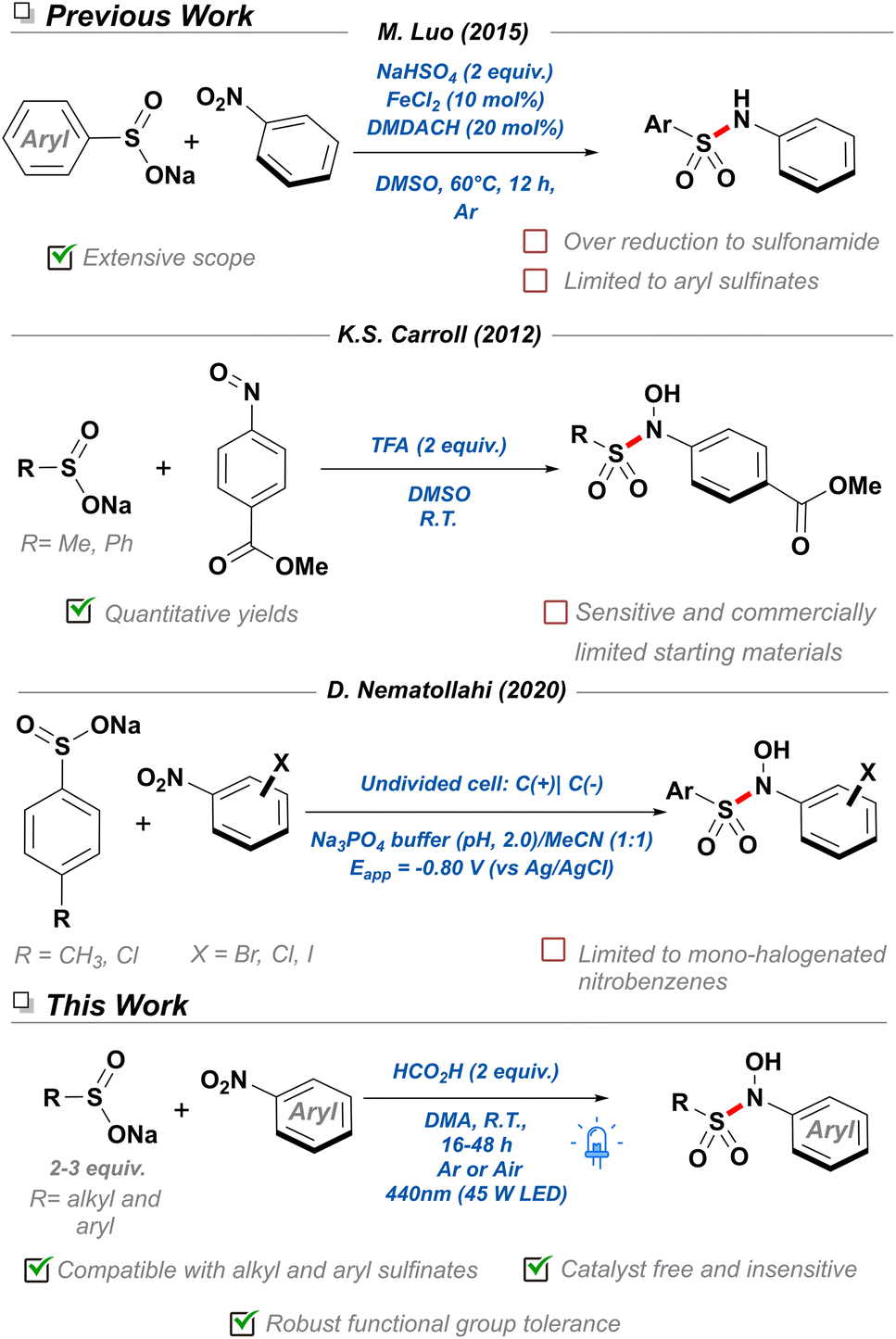
Nitroarenes are indispensable feedstocks for the drugs and materials required in our modern lifestyle. Their production and subsequent reduction to the corresponding aniline derivatives represents one of the fundamental processes of the current chemical industry,1 and as such, innumerable methodologies have been developed for it.2–4 However, their conversion into derived nitrogen-containing chemicals is fraught with issues, since efficient and selective reduction processes typically require the use of super-stoichiometric quantities of iron,5 or scarce transition metals6 and pyrophoric gases.7 Recently, attention has shifted towards the development of novel strategies utilizing the nitro group as a handle for the synthesis of nitrogen-containing building blocks without prior reduction, thereby better conforming to the principles of atom efficiency and green chemistry.8 Additionally, the advent of late-stage functionalization (LSF) reactions has underlined the importance of development of novel reaction methodologies capable of exploiting the innate chemical character of functional groups and co-opting their reactivity for the facile derivatization of biorelevant scaffolds.9,10 Consequently, nitroarenes constitute a unique functionality with dramatically different reactivity profiles to other nitrogenous groups. These advantages include their resistance to oxidation—making them easier to store and use—as well as their highly conjugated nature, where a readily accessible nitro π* orbital allows nitroarenes to undergo photochemical processes.11Thus far, the extensively studied photophysics of nitroarenes —contrary to other light-absorbing functional groups—has typically been relegated to a mechanistic curiosity in physical chemistry, attracting limited interest from synthetic fields.11,12 This is partially due to early works in the area reporting unselective, competing partial photoreductions and photomediated SNAr reactions under strenuous photochemical conditions, often not applicable into either an academic or industrial setting.13,14 Luckily, the recent incorporation of less intense visible light sources as well as potent photocatalysts have aided organic chemists in the selective reduction of a broad range of functional groups.15 However, extensive literature analysis of photochemical reactions of nitroarenes revealed a scarcity of methodologies for the formation of nitrogen-sulfur bonds—well represented in the medicinal chemistry context in the form of sulfonamides.16 More specifically, an interesting subfamily of this class of molecules named N-hydroxy-sulfonamides have only been reported a handful of times, involving the thermo-17 and electrochemical18 nucleophilic addition of sulfinate salts to nitrosoarenes (Scheme 1). Despite the limited toolbox to access N-hydroxy-sulfonamides, these molecules have already found application as organic nitric oxide (NO) sources,19 bioconjugation reagents for selective labeling of cysteine residues,17 NLRP3 inflammasome inhibitors,20 and sulfonylating reagents.21 For this reason, it is crucial to develop new strategies that allow access to this functionality, in order to provide a platform for further discovery of their chemical space, exploration of their reactivity, and uncovering of novel applications. Furthermore, it would be ideal to couple the inherent photochemical properties of nitroarenes with the diversity and availability of sodium sulfinate salts resulting in a more cost-effective methodology that prescinds of super-stoichiometric reducing agents and expensive photocatalysts.22–24 In this work, we report the first visible-light mediated, catalyst-free S–N bond coupling for the synthesis of N-hydroxy-sulfonamide derivatives. Both alkyl and aryl sulfinate salts were amenable to the methodology and tolerant to both oxygen and water. With this methodology we hope to expand the chemical space available to N-hydroxy-sulfonamides and reinvigorate research into this underexplored functional group.
 | ||
| Scheme 1 Comparison of recently reported N–S couplings of sodium sulfinates. | ||
Results and discussion
During the early stages of design for this work, we originally envisioned the use of 10-phenylphenothiazine (PTH) as a photocatalyst to aid in the reduction of 4-chloronitrobenzene—a substrate originally chosen for its reduction potential. Due to the limited solubility of sulfinate salts in most polar aprotic solvents, N,N-dimethylacetamide (DMA) was chosen as a beneficial starting point allowing for the nucleophilic attack on a transient nitrosobenzene intermediate. Such modified reducing conditions worked sufficiently well, resulting in the generation of the desired N-hydroxy-sulfonamide in good yield (Scheme 2, entry 2). Motivated by these results, we performed a short round of control experiments removing the reagents present in solution. Contrary to our initial expectations, control reactions conducted without the presence of PTH were found to perform identically to those including such catalyst (Scheme 2, entry 3). Based on these observations, it was posited that the conversion in the absence of catalyst and the emergence of a yellow color in solution were the result of an alternative mechanism, different to the originally proposed single-electron transfer (SET) between PTH and the nitroarene. Another possible course of action was the formation of an electron donor–acceptor (EDA) complex between the nitroarene and one of the components in our reaction mixture. EDA complexes are defined as the interaction between two molecules—typically unable to absorb light in the visible range on their own— leading to a SET process upon irradiation with light. These compounds are usually evidenced by the formation of a new absorption band, due to population of new molecular orbitals.25 In fact, EDA complexes between nitroarenes and electron donors have been reported in the literature before, yet their inclusion into widespread synthetic methodologies has gone astonishingly unnoticed.26–28 Examples of this phenomenon include phosphine-nitroarenes EDA systems for the synthesis of anilines, as well as C–N couplings of boronic acids with nitroarenes.29,30 Crucially, these new methodologies demonstrate an untapped potential towards the development of green technologies capable of accessing highly sought-after nitrogen-containing functional groups without the use of sensitive hydrides, transition metals, or expensive photocatalysts.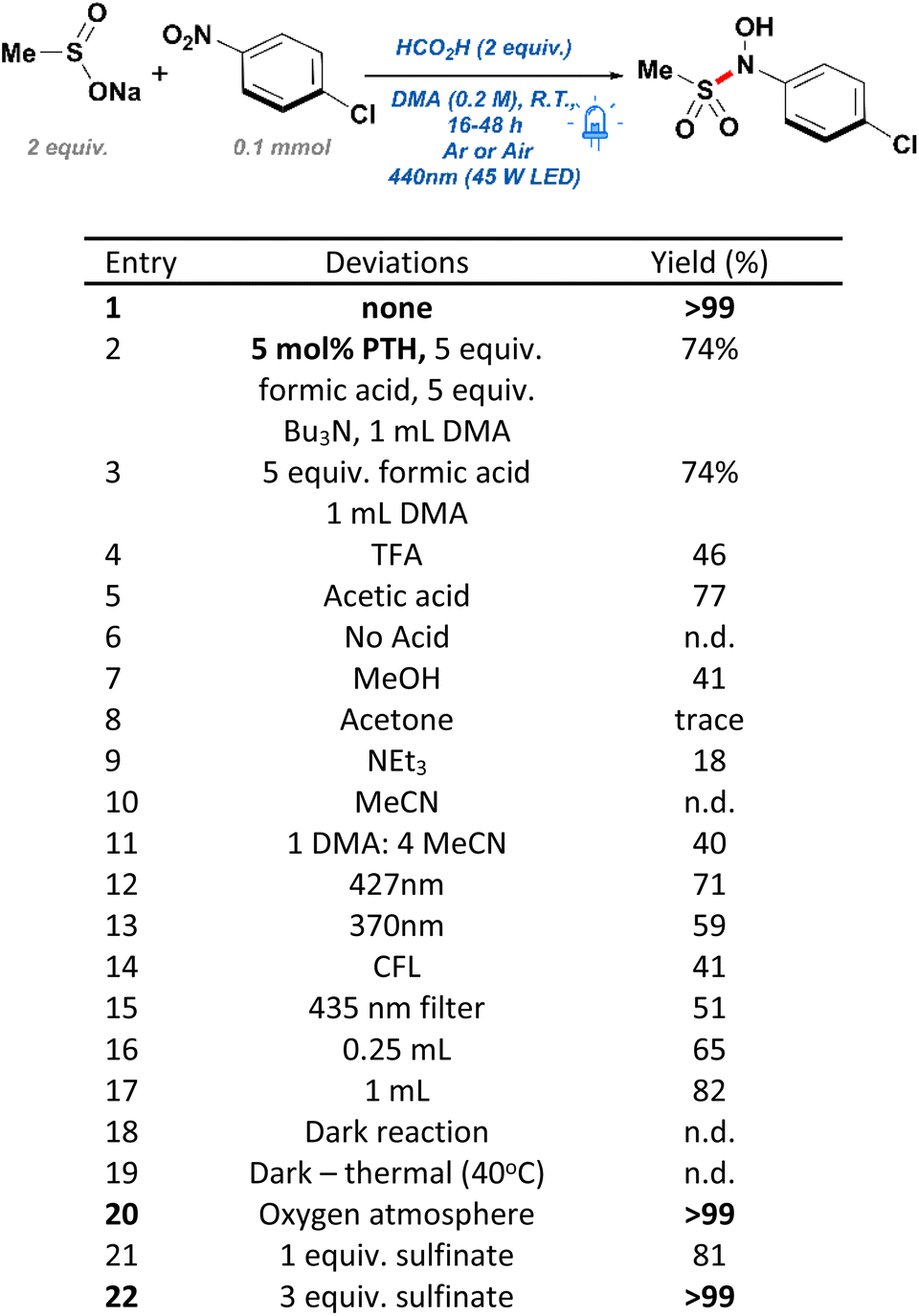 | ||
| Scheme 2 Parameters modified for reaction optimization. | ||
In order to confirm the generation of an EDA complex in our system, we turned to UV-vis spectroscopy to detect the absorption of our components at the concentration of a standard reaction solution. Firstly, p-chloronitrobenzene (p-CNB) dissolved in DMA displayed limited absorbance at 440 nm, which was quickly quenched upon addition of acid. Pleasingly, after adding sodium methane sulfinate, a new absorption band present at 440 nm was formed, explaining the yellow reaction mixture, and confirming the generation of an EDA complex (Scheme 3). Unexpectedly, addition of acid decreased the absorbance of such band without quenching it completely (reaction conditions). Further controls in the absence of acid resulted in poor yields and a low conversion of 49%. Moreover, screening of other acids such as TFA with no product formed, and acetic acid with 77% yield, suggested that acid selection was not solely dependent on pKa and likely was a function of the reducing capabilities of the starting acid (Scheme 2, entries 4–6). Based on these observations, formic acid was deemed the best candidate for our conditions.
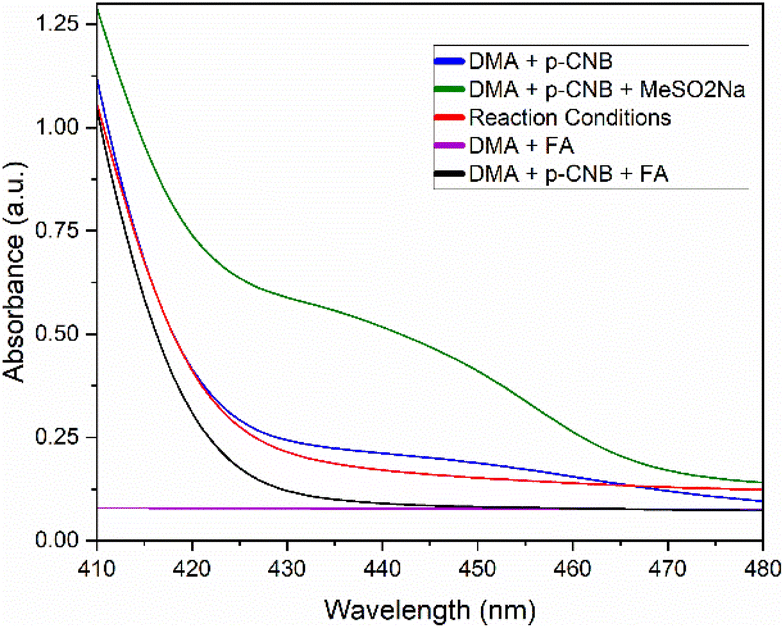 | ||
| Scheme 3 UV-vis spectra of reaction components dissolved in DMA. | ||
Focus then was shifted to the screening of alternative greener solvents, capable of giving similar yields (Scheme 2, entries 7–11). Unfortunately, methanol promoted the undesired reduction of the nitroarene into the corresponding diazene with a moderate conversion towards the desired product. Such over reduction is likely a result of the decomposition of the sulfinate salts in a protic solvent, therefore limiting the capture of the nitrosobenzene intermediate. Consequently, we focused our attention onto utilizing low boiling polar aprotic solvents such as acetone and triethylamine; yet, both solvents were found to provide low conversion and low yields of the desired product. Further attempts with acetonitrile were unsuccessful and resulted in limited conversion of the starting 4-chloronitrobenzene. Attempts to limit quantities of DMA to 20% in acetonitrile were found to enhance the yield of the desired product but resulted in dramatically reduced yields while giving conversions close to 92%. Finally, light sources of 390 nm, 427 nm, and a 25 W commercial compact fluorescent lamp (CFL) were tested for their efficiency at mediating the reaction (Scheme 2, entries 12 and 13). However, despite the capacity of the reaction mixture to absorb well throughout the 200–400 nm range, 427 nm and 390 nm lamps were found to negatively impact the reaction by lowering yields to 71% and 59%, respectively. These reactions also resulted in a slew of undesired byproducts, likely due to the nitroarene's tendency to undergo various photochemical pathways under ultraviolet light. For this reason, it was initially surmised that a low intensity bulb might be capable of performing the transformation more selectively, yet it was discovered that the use of CFL bulbs resulted in a moderate yield of 41% and a low conversion of 59% (Scheme 2, entry 14). Finally, in order to exclude the possibility of ultraviolet light at the edge of the lamp irradiation profile initiating direct reduction by formic acid—as has been reported previously in the literature31,32 —a control reaction with a 435 nm long-pass filter was run to exclude this possibility. Thus, it was found that the reaction proceeded smoothly albeit with a reduced yield of 51% due to the diminished light intensity (Scheme 2, entry 15).
After establishing the optimal reaction conditions, we proceeded to study the scope of the reaction for nitroarenes. Considering the possibility of electron-withdrawing functionalities aiding the initial electron transfer, the transformation was attempted with 4-bromo and 4-iodo nitrobenzene, yielding 67% (Schemes 4 and 3b) and 47% (3c), respectively. While 4-iodobenzene led to lower yields, this was determined to be due to sluggish conversion as opposed to deleterious side reactions such as dehalogenation. Thus, attempts with 3-iodonitrobenzene over 48 hours were able to produce the N-hydroxy-sulfonamide in good yield (3d). Other electron-withdrawing groups were also tested such as 4-nitrobenzonitrile (3f), which provided product in 64% yield. This product generated colourless needles suitable for X-ray diffraction (see ESI Section 2a†). Subsequently, we tested the feasibility of performing the reaction with less electron-deficient substrates: Satisfyingly, nitrobenzene itself was found to be a suitable substrate with a yield of 61% (3e) over 16 hours of irradiation. Weakly electron-donating groups such as a methyl (3j) in the para position were also found to give moderate yields under 24 hours but suffered from slow conversion.
 | ||
| Scheme 4 Exploration of nitroarene scope with optimized N-hydroxyl sulfonamidation conditions. (a) Reaction was conducted under normal ambient atmosphere (b) reaction was allowed to react for 48 hours under visible light illumination. (c) Reaction was conducted with 2-nitrobenzyl chloride and produced di-sulfonylated product. | ||
To determine the probability of conducting difunctionalization, both para- and meta-dinitrobenzene were tested, resulting in only the mono-sulfonylated compound in moderate yield in both cases. It can be inferred that the electron-donating aniline formed after reduction of the first nitro group decreases the probability for a second single electron transfer (SET) leading to the difunctionalized product. Additionally, both dinitro arenes were found to suffer from generation of their corresponding diazo analogues which are unproductive in this methodology, thereby rendering the procedure less effective. SNAr-sensitive substrates such as 4-fluoronitrobenzene (3m) and 2-fluoronitrobenzene (3n) were amenable to the reaction conditions producing 55% and 39% yield, respectively. However, a more SNAr-prone substrate such as 1,3,5-trifluoronitrobenzene was not able to generate any product in detectable amounts and solely resulted in the monosulfonylated SNAr product. In contrast, 4-nitroacetophenone (3o) and 4-nitrobenzaldehyde (3p) showed satisfactory yields of 85% and 61%, respectively, despite their known photo reactivity—pinacol coupling or deleterious side reactions.
During the investigation, it was readily apparent that strong electron-donating groups hindered reduction of the nitro functionality, and thus both 4-nitrophenol and 4-nitroaniline were found to be unaffected by the reaction conditions. Unexpectedly, 4-nitroanisole (3r) was instead found to produce the desired transformation in 42% yield, however the product was observed to be generally unstable and decomposed rapidly over the course of 3 hours. Consequently, it was envisioned that reactivity of phenols could be modulated by the installation of an electron-withdrawing group. Thus, a 3-trifluoromethanesulfonate (3q) analogue was determined to be a suitable substrate producing the desired compound in good yield. Unfortunately, an analogous strategy with 4-nitroaniline proved unfruitful, for both 4 N-acetyl and 4 N-tosyl analogues.
Tolerance of the reaction conditions towards a diverse selection of nitroarenes suggested the potential for the late-stage functionalization of medicinally relevant scaffolds containing a nitro group. While nitroarenes have a limited presence in pharmaceutical compounds, they constitute an important, readily adjustable functionality for the facile synthesis of anilines in drug scaffolds. Therefore, two immediate precursors for biorelevant molecules were assessed. Oxidized analogues of the topical anesthetics benzocaine (3w) and butamben (3x) were both subjected to the reaction conditions giving excellent yields with minimal side products.
Having established the application of a diverse selection of nitroarenes, we shifted our focus to the exploration of the reactivity associated with sulfinate salts. Initially, a small selection of alkyl sulfinates was subjected to the reaction conditions with 4-nitrochlorobenzene. Despite diminished yields, sodium ethane sulfinate (Scheme 5. 4a) and sodium isobutyl sulfinate (4b) produced the desired products in moderate yields. Remarkably, sodium cyclopropyl sulfinate (4c) underwent efficient conversion and delivered an excellent yield for the desired product. On the other hand, sodium cyclohexyl sulfinate (4d) showed a moderate yield of 50% and a tendency towards producing SNAr side products. In contrast to alkyl sulfinates, aryl sulfinates were found to be more sensitive to the reaction conditions, frequently undergoing self-disproportionation and consequently producing sulfonothioates and other sulfur-containing byproducts. As such, these reactions performed best under the presence of 3 equivalents of the corresponding sulfinate salts. While sodium 4-methylphenylsulfinate (4e) gave a yield of 78% under the revised conditions, other electron-deficient sodium aryl sulfinate salts performed at moderate yields—potentially due to reduced propensity to undergo SET or reduced nucleophilicity.
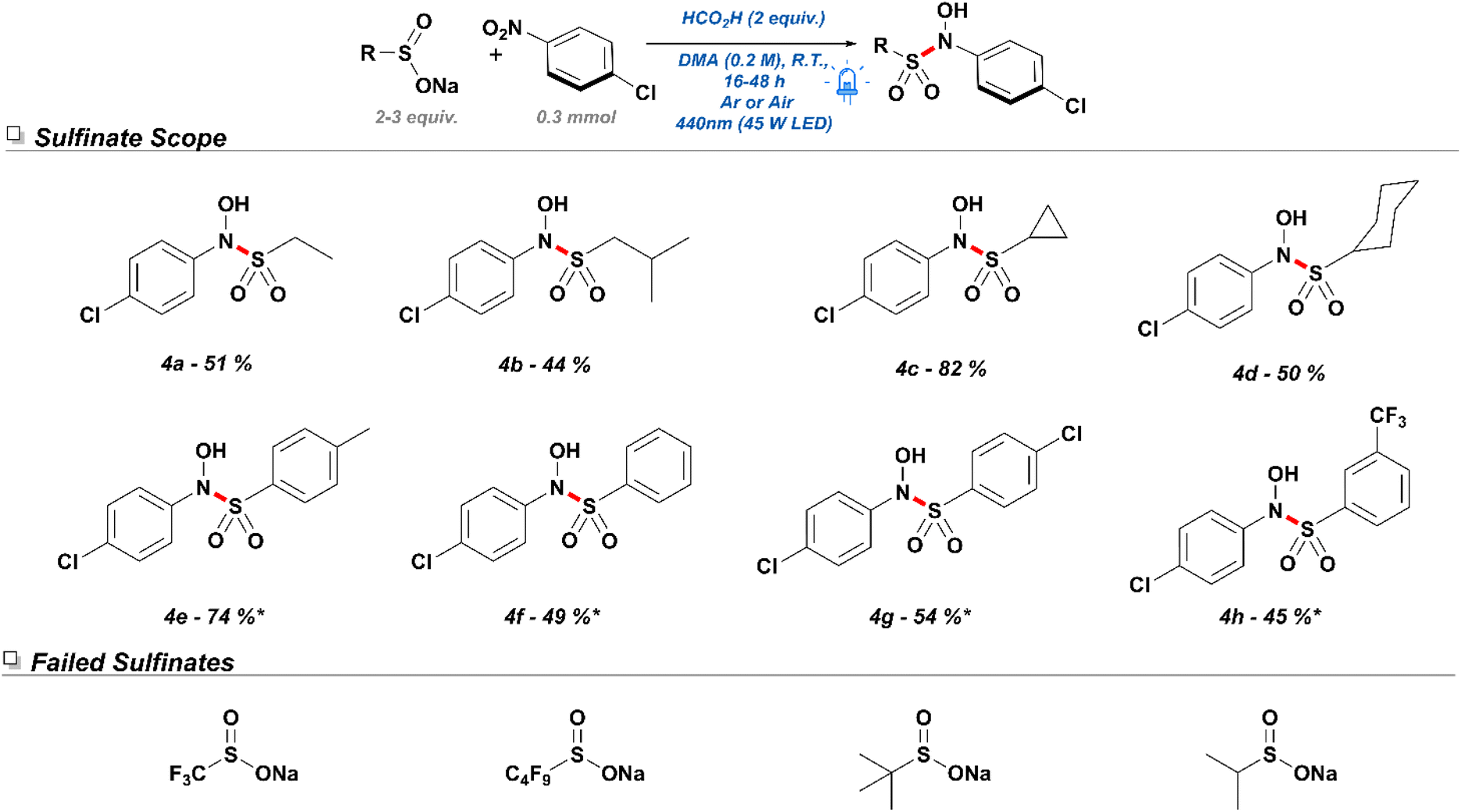 | ||
| Scheme 5 Exploration of sulfinate tolerance in the designed N-hydroxyl sulfonamidation conditions. *Reactions were conducted with 3 equivalents of sodium sulfinate. | ||
Once we understood the overall reactivity of both the nitroarene and sulfinate components in our system, we dedicated our efforts to understand the mechanistic details of this transformation. While the photophysics involved in the reduction of the nitro group are well-established, the exact pathway leading to the formation of the N-hydroxy-sulfonamide remained unknown. The first posed question pertained to the purpose of formic acid in the reaction. While initial studies determined that pKa of the acid played no role in the yield of the reaction, its character as a reducing agent needed to be elucidated. Consequently, the gaseous phase of a standard reaction was analyzed through gas chromatography coupled to a thermal conductivity detector (GC-TCD) to monitor the presence of gaseous byproducts. This experiment exhibited the presence of carbon dioxide, signaling oxidation of the formic acid to CO2, and confirming its role as an active reductant (Section 4, ESI†), thus it can be envisioned the formic acid can function as an auxiliary reductant of the produced nitroarene radical anion and other open shell species formed in solution.
Furthermore, GC-TCD analysis showed the presence of methane gas, likely produced through direct decomposition of the sodium methane sulfinate. Thus, the observation of sulfonothioates as well as methane gas was a strong indicator of SET processes occurring from the sodium sulfinate salts, but also indicated the likely probability that addition of the sulfonyl fragment by either direct SN2 or free radical addition (FRA) as has been previously in the reported in the literature.33 As such, radical quenching studies with TEMPO were conducted with both methane and toluene sulfinates, as it was envisioned that the higher stability of toluene sulfonyl radicals could potentially result in diverging bond forming mechanisms. Interestingly, production of methane N-hydroxy-sulfonamide was slightly impacted to give a lower yield of 77% by NMR indicating a possibility that a majority of the addition occurs through an SN2 addition. Results from the toluene sulfinate further confirmed the presence of a radical component of the reaction sulfinate as yields plummeted to 34% under the presence of TEMPO, despite the large excess of aryl sulfinate. Further studies with a 1,1-diphenyl ethylene again confirmed the generation of sulfonyl radicals as both sodium methane sulfinate and sodium p-toluene sulfinate produced the expected vinyl sulfonyls through radical addition (Scheme 6). Considering all the data available, we therefore propose a mechanism dependent on a radical chain initiated through an EDA complex between a nitroarene and a sodium sulfinate molecule (Scheme 7). The corresponding nitroarene radical anion can be further reduced by DMA through a HAT process (ESI, Section 4†) or formic acid thereby liberating CO2. Thus, through extrusion of a water molecule, a nitrosoarene intermediate can be formed, which can finally be captured through either a direct SN2 with another sulfinate equivalent or through a free radical addition (FRA) from an in situ generated sulfonyl radical.
 | ||
| Scheme 6 Control reactions utilized to validate reaction mechanism. | ||
 | ||
| Scheme 7 Proposed mechanism for the EDA mediated generation of N-hydroxy sulfonamides. | ||
To further cement the plausibility of the envisioned EDA, the envisioned complex was assessed by Density Functional Theory (DFT) and Time Dependent DFT methods. Three different conformations for the nitroarene–sulfinate–sodium complex were explored (Scheme 8, top), their structures were optimized at the ωB97XD/cc-pVTZ level of theory and their excited states were calculated at the same level of theory. The presence of minima on the potential energy surface (PES) was confirmed via vibrational frequency analysis: all reported compounds showed no negative frequencies, indicating that they all correspond to a minimum on the PES. Excitations with the highest value of oscillator strength (f) were selected as the most likely and it was used to calculate their Natural Population Analysis in the excited state. The total natural population for each fragment was compared in the ground and the excited state with the highest value of f, population difference is collected in (Scheme 8, left table). Therein, it is observed that conformations b and c exhibit the highest change in electron population from the sulfinate to the nitroarene (0.467 electrons), indicating that an effective electron transfer occurs in the excited state respect to the ground state in which each species exhibits their expected neutral (nitrobenzene) and negative (methane sulfinate) charges, respectively. These calculations were repeated in the absence of a Na+ cation to assess the participation of the cation in the formation of a suitable template that leads to the EDA complex and their results are collected in (Scheme 8, right table); however no significant population transference was observed. Results from both analyses, suggest that the Na+ cation plays a pivotal role in facilitating the charge transfer.
 | ||
| Scheme 8 Initial proposed conformations for the EDA (top) and their final optimized structures (bottom) at the ωB97XD/cc-pVTZ level of theory. Total natural charges for molecular fragments in the EDA complexes at the ground and excited state (with and without an intermediate Na+ cation). Population differences are ascribed to the transfer of electrons. *Natural charge in the excited state. **Charge transfer was calculated as the charge difference between the excited and ground state for the nitrobenzene molecule. | ||
A third option explored involved the inclusion of an explicit DMA solvent molecule in the nitroarene–sulfinate–sodium complex. The effect of DMA in the formation of the EDA complex could in principle be twofold: it could provide the necessary polarity of the medium to solvate the EDA complex or their components, or it could actively participate as a component of the EDA.34 To assess the intervention of DMA, we calculated the same population analysis scheme on the most probable excited state of such complex, in which a proposed EDA is formed through the addition of an explicit DMA molecule in complex c (Section 5, ESI†). The TD-DFT calculation reveals low intensity excitations, since the largest oscillator strength value found is f = 0.2322 for the 8th excited state. No significant population changes were observed for any of the four components of the EDA (0.025 e nitrobenzene; 0.016 e methane sulfinate; −0.036 e DMA), which leads to the conclusion that DMA provides the necessary polar medium for the formation of the EDA, whose structure should be closer to conformer c, than forming a part of the molecular ensemble constituting the EDA complex.
Conclusions
In this work we describe the formation of a novel EDA complex formed between nitroarenes and sodium sulfinates and demonstrated its applicability to the production of scarcely reported N-hydroxy-sulfonamide analogues. The reaction conditions are mild, tolerant to both oxygen and water, and dramatically increase the available chemical space of this class of molecules. Additionally, TD-DFT calculations suggest the pivotal role of the sodium cation from the sulfinate salt in the charge transfer process.Data availability
The data supporting this study is available within the main text and the associated ESI.†Author contributions
This work was achieved through equal contribution from JDL and DJCP in conceptualization, experimental work, and writing of the manuscript. MS contributed to the reaction optimization and scope phases, while JBF guided and performed all computational studies. All this work was supervised, guided, and revised by CJL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the McGill Chemistry Characterization Facility (MC2) for their contribution to this work, especially Robin Stein on the NMR data, Hatem Titi on SCXRD interpretation, and Nadim Saadé and Alexander Wahba on HRMS. RMS for lending their insight and knowledge. DJCP acknowledges the financial support granted by the Vanier Canada Graduate Scholarship, CONACYT Mexico, and the Richard H. Tomlinson Fellowship. JBF thanks DGTIC—UNAM for the access to their supercomputer ‘Miztli’ and to Citlalit Martínez-Soto for keeping our local computer facilities running.Notes and references
- G. Booth, H. Zollinger, K. McLaren, W. G. Sharples and A. Westwell, Ullmann’s Encyclopedia of Industrial Chemistry, 2012, vol. 11, pp. 676–729 Search PubMed.
- M. Orlandi, F. Tosi, M. Bonsignore and M. Benaglia, Org. Lett., 2015, 17, 3941–3943 CrossRef PubMed.
- L. Zhao, C. Hu, X. Cong, G. Deng, L. L. Liu, M. Luo and X. Zeng, J. Am. Chem. Soc., 2021, 143, 1618–1629 CrossRef PubMed.
- X. Li, R. R. Thakore, B. S. Takale, F. Gallou and B. H. Lipshutz, Org. Lett., 2021, 23, 8114–8118 CrossRef PubMed.
- In Comprehensive Organic Name Reactions and Reagents, ed. Z. Wang, 2010, pp. 284–287, DOI:10.1002/9780470638859.conrr063.
- T. Kahl, K.-W. Schröder, F. R. Lawrence, W. J. Marshall, H. Höke and R. Jäckh, Ullmann’s Encyclopedia of Industrial Chemistry, 2012, vol. 3, pp. 465–478 Search PubMed.
- Q. Zhang, J. Bu, J. Wang, C. Sun, D. Zhao, G. Sheng, X. Xie, M. Sun and L. Yu, ACS Catal., 2020, 10, 10350–10363 CrossRef.
- K. Muto, T. Okita and J. Yamaguchi, ACS Catal., 2020, 10, 9856–9871 CrossRef.
- J. D. Lasso, D. J. Castillo-Pazos and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10955–10982 RSC.
- J. Börgel and T. Ritter, Chem, 2020, 6, 1877–1887 Search PubMed.
- A. Giussani and G. A. Worth, J. Chem. Theory Comput., 2017, 13, 2777–2788 CrossRef CAS PubMed.
- A. Giussani and G. A. Worth, Phys. Chem. Chem. Phys., 2020, 22, 15945–15952 RSC.
- G. G. Wubbels, D. P. Susens and E. B. Coughlin, J. Am. Chem. Soc., 1988, 110, 2538–2542 CrossRef CAS.
- G. G. Wubbels, J. W. Jordan and N. S. Mills, J. Am. Chem. Soc., 1973, 95, 1281–1285 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS.
- M. Lo Conte and K. S. Carroll, Angew. Chem., Int. Ed., 2012, 51, 6502–6505 CrossRef CAS.
- H. Goljani, Z. Tavakkoli, A. Sadatnabi, M. Masoudi-khoram and D. Nematollahi, Sci. Rep., 2020, 10, 17904 CrossRef CAS.
- J. P. Toscano, F. A. Brookfield, A. D. Cohen, S. M. Courtney, L. M. Frost and V. J. Kalish, US Pat., USRE45314, 2014 Search PubMed.
- C. Guo, J. W. Fulp, Y. Jiang, X. Li, J. E. Chojnacki, J. Wu, X.-Y. Wang and S. Zhang, ACS Chem. Neurosci., 2017, 8, 2194–2201 CrossRef CAS PubMed.
- F.-X. Wang, S.-D. Zhou, C. Wang and S.-K. Tian, Org. Biomol. Chem., 2017, 15, 5284–5288 RSC.
- A. Cheruvathoor Poulose, G. Zoppellaro, I. Konidakis, E. Serpetzoglou, E. Stratakis, O. Tomanec, M. Beller, A. Bakandritsos and R. Zbořil, Nat. Nanotechnol., 2022, 17, 485–492 CrossRef CAS.
- G. Xiao, P. Li, Y. Zhao, S. Xu and H. Su, Chem.–Asian J., 2018, 13, 1950–1955 CrossRef CAS.
- R. Rai and D. K. Chand, J. Chem. Sci., 2021, 133, 87 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- M. M. Ayad, Z. Physiol. Chem., 1994, 187, 123–133 CrossRef CAS.
- J. O. Singh, J. D. Anunziata and J. J. Silber, Can. J. Chem., 1985, 63, 903–907 CrossRef CAS.
- M. A. Zon, H. Fernández, L. Sereno and J. J. Silber, Electrochim. Acta, 1987, 32, 71–77 CrossRef CAS.
- K. Manna, T. Ganguly, S. Baitalik and R. Jana, Org. Lett., 2021, 23, 8634–8639 CrossRef CAS.
- Z. Qu, X. Chen, S. Zhong, G.-J. Deng and H. Huang, Org. Lett., 2021, 23, 5349–5353 CrossRef CAS PubMed.
- A. Cors, S. M. Bonesi and R. Erra-Balsells, Tetrahedron Lett., 2008, 49, 1555–1558 CrossRef CAS.
- K. Tsutsumi, F. Uchikawa, K. Sakai and K. Tabata, ACS Catal., 2016, 6, 4394–4398 CrossRef CAS.
- A. van der Werf, M. Hribersek and N. Selander, Org. Lett., 2017, 19, 2374–2377 CrossRef CAS.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
Footnotes |
| † CCDC 2208621. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06087f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |