
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe deeper it goes, the brighter it glows: NIR emissive nitro-terrylene diimides with deep LUMOs†
Kundan Singh
Mehra
,
Shivangee
Jha
,
Anila M.
Menon
,
Deepak
Chopra
 and
Jeyaraman
Sankar
*
and
Jeyaraman
Sankar
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, Bhopal Bypass Road, Bhopal, India – 462066. E-mail: sankar@iiserb.ac.in
First published on 30th January 2023
Abstract
Herein, we present the first examples of air-stable, deep-lowest unoccupied molecular orbital (LUMO) polycyclic aromatic molecules with emission in the near-infrared (NIR) region, using nitration as a strategy. Despite the fact that nitroaromatics are non-emissive, the choice of a comparatively electron-rich terrylene core proved to be beneficial for achieving fluorescence in these molecules. The extent of nitration proportionately stabilized the LUMOs. Tetra-nitrated terrylene diimide exhibited a deep-LUMO (≤−4.5 eV) of −5.0 eV vs. Fc/Fc+, the lowest for any larger RDIs. These are also the only examples of emissive nitro-RDIs, with larger quantum yields.
Introduction
Luminescent molecules emitting in the near-IR region (NIR) find wide applications in organic light emitting diodes (OLEDs),1 in organic solar cells,2 and in photobiomodulation (PBM), including for side-effect alleviation after chemoradiotherapy.3 Very few electron-deficient molecules are known to have emission in the NIR.4 This is due to the π-extension or expansion strategy utilized for this purpose. While this method helps in reducing the band gap (Eg), most of the well-known NIR emitters such as porphyrins, phthalocyanines or squaraines, are always used as donors in optoelectronic devices.1,5 Also, the narrow bandgap influences the stability of the molecules on prolonged use.6 Alternatively, electron-deficient rylene diimides (RDIs) have been explored to achieve NIR absorption and emission by the π-extension approach.7 However, this strategy could not sufficiently lower the LUMO levels, to suit their application as efficient acceptors.Open-shell or closed-shell RDIs with electron-withdrawing substituents have classically been exploited as molecules with stabilized LUMOs (Scheme 1).8 In this direction, tetracyanated naphthalene diimide NDI-CN4 has been shown to have a deep-LUMO value of −5.0 eV vs. Fc/Fc+. This is attributed to the smaller π-core connected to multiple electron-withdrawing units.8a Even though this approach helped to achieve low LUMO levels, the resultant molecule lacked any emission. On the other hand, larger RDIs such as perylene diimides (PDIs) and terrylene diimides (TDIs), despite their bathochromically shifted absorption and emission, could not lower the LUMO on simple cyanation (−4.4 eV and −4.49 eV vs. Fc/Fc+, respectively).9 Recently, Li et al. and Tan et al. have reported aggregation induced, emissive RDI-based molecules albeit with LUMO energies at −3.5 and −3.6 eV respectively.10
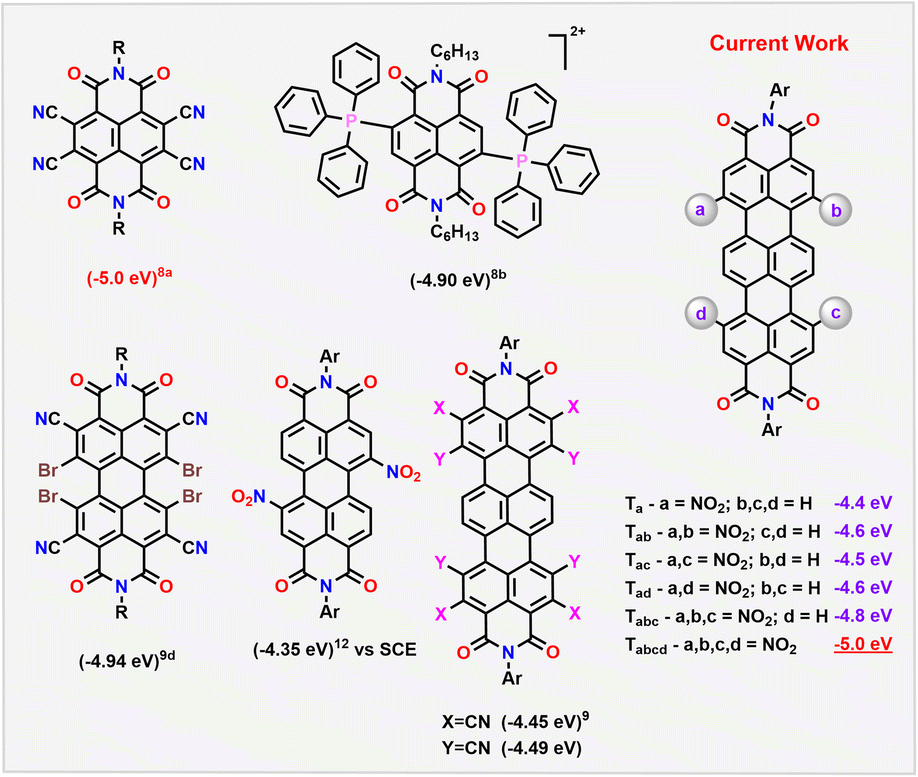 | ||
| Scheme 1 Selected examples of RDIs with their LUMO levels (vs. Fc/Fc+). | ||
Deriving from the positive Hammett constants, the nitro group can be an effective alternative. However, in nitroaromatics, the preferred route of excited state relaxation is non-radiative stemming from efficient intersystem crossing. This non-emissive nature of nitroaromatics makes them less attractive despite their ability to efficiently stabilize the LUMO levels.11 Predictably, dinitro-PDIs with a lower LUMO level of −4.35 eV (vs. Saturated Calomel Electrode (SCE)) exhibited no emission.12
In recent years, there is a renaissance in developing novel luminescent nitroaromatics for modern optoelectronic and biomedical applications.11 One of the exploited routes to achieve emission in these molecules is by the introduction of intramolecular donor–acceptor nature in their excited state. This charge transfer (CT) character can hinder the spin–orbit coupling (SOC), facilitating a reduced overlap between the π-orbital on the aromatic unit (donor) and the n-orbital on the nitro group (acceptor). This results in favourable radiative deactivation. Nitro-PDIs are non-emissive presumably because of the ineffective intramolecular charge transfer (ICT) between electron-deficient PDI and NO2 units.11,12 In this regard, a possible route could be to choose a larger RDI such as TDI.7,13 That (i) will aid in obtaining multiple nitration on the core to achieve deeper LUMOs; (ii) will help to modulate the intramolecular charge transfer in their excited states, rendering them emissive; (iii) will also alleviate any possible steric crowding at the bay positions.
In the current work, we demonstrate that this strategy leads to the formation of a series of nitro derivatives, such as mono, di-, tri- and tetra-nitro TDIs in good yields. With the extent of nitration, their LUMO levels (vs. Fc/Fc+) get stabilized proportionately from Ta (−4.4 eV) to Tabcd (−5.0 eV) (Scheme 1). It is to be highlighted here that Tabcd also emits in the NIR region, thus making it one of the unique electron-deficient molecules with a deep-LUMO level to do so.
Results and discussion
Synthesis
A heterocoupling reaction of perylene monoimide (PMI) with naphthalene monoimide (NMI)7b in the presence of sodium tertiary butoxide and 1,5-diazabicyclo(4.3.0)non-5-ene (DBN) yielded the parent TDI in 36% yield (Scheme 2). Novel nitro-TDI derivatives were obtained by careful addition of concentrated nitric acid to TDI in chloroform under ambient conditions. The extent of nitration could precisely be controlled with reaction duration and equivalence of nitric acid. Unlike the case of PDI, the reaction proceeded without the need for any additive.12 Mono-nitrated TDI Ta was obtained rapidly (85%), within a few minutes of the addition of HNO3 to TDI, along with trace amounts of dinitro-TDIs. The quick formation of Ta reiterates the ease of nitration of TDI. Increasing the equivalents of acid and the reaction duration (100 eq. & 1 h) yielded a complex mixture of three regioisomers of dinitro-TDIs in yields ranging from 20–40%. In contrast to bromination, all three possible regioisomers namely Tac (1,9-) and Tad (1,14-), including Tab (1,6-) could successfully be obtained and characterized.14 Incidentally, this is the first instance in which all three isomers of di-substituted TDIs have been achieved. Through this method, even tri- and tetranitro-TDIs namely Tabc and Tabcd could be obtained in moderate yields (50% and 55% respectively), without the use of any harsh or hazardous conditions. All the molecules have been isolated and purified by column chromatography using CH2Cl2/hexane as eluents and recrystallized from CH2Cl2/MeOH. The complete characterization data are provided in the ESI (Fig. S1–S24†).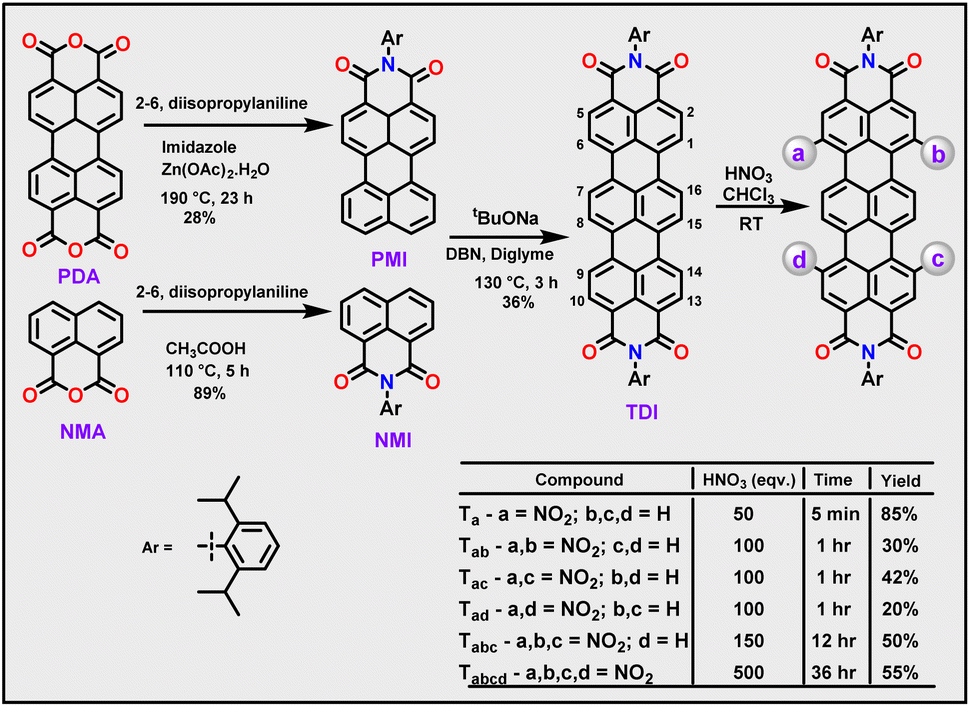 | ||
| Scheme 2 Synthetic scheme for the nitration of terrylene diimides. | ||
SCXRD studies
The structures of the molecules have been unambiguously ascertained by single crystal X-ray diffraction (SCXRD) studies. Despite our best efforts, suitable single crystals could not be obtained for Ta. Except for Ta, all the other nitro-TDIs afforded crystals by a solvent vapour diffusion method. The obtained structures have been given in Fig. 1. The structures of all the molecules show an almost planar TDI core, with the NO2 groups protruding out in varying angles.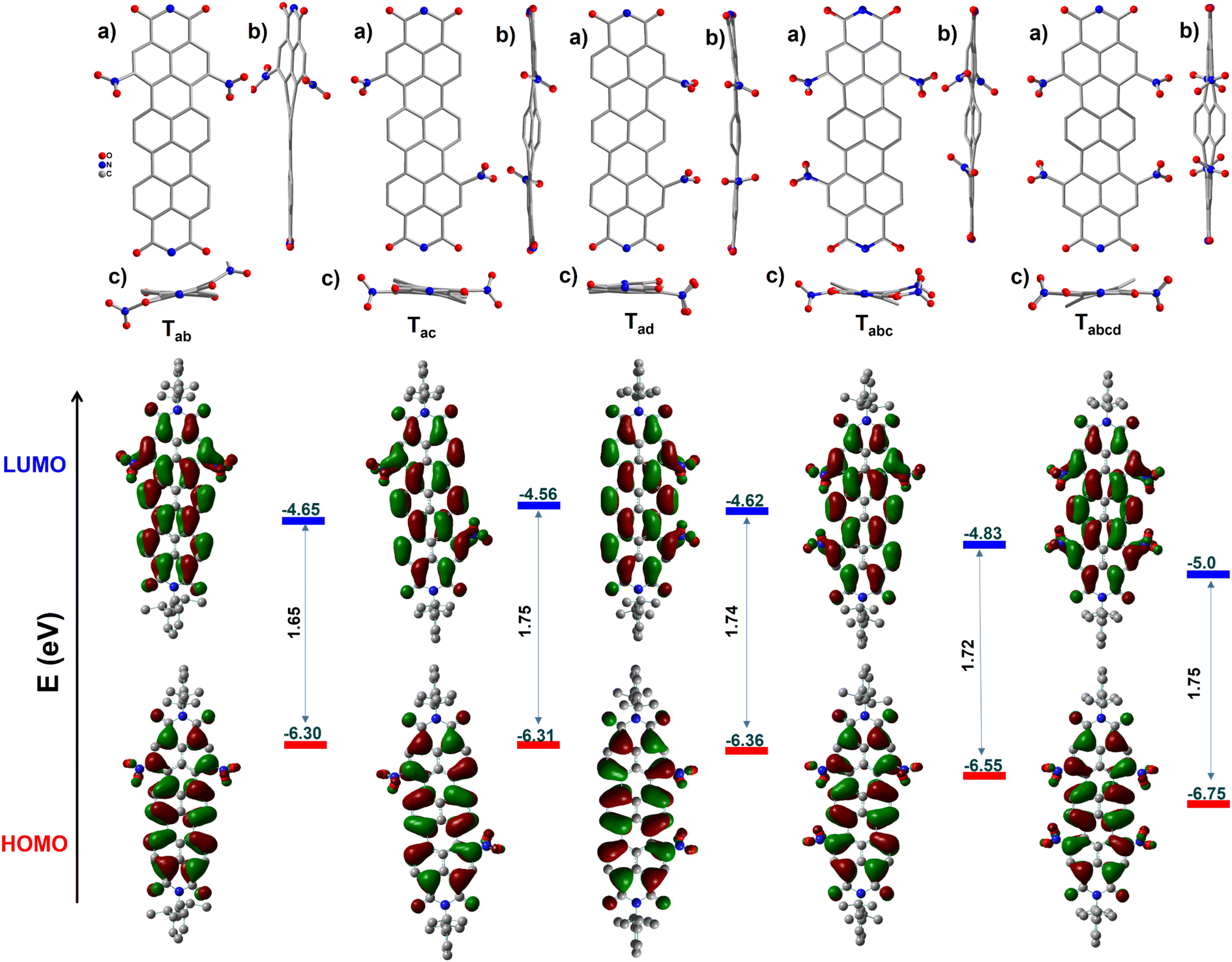 | ||
| Fig. 1 Molecular structures obtained from single crystal X-ray diffraction studies (CCDC 2204224–2204228). (a) Top views, (b and c) side views, and Frontier molecular orbitals (DFT-B3LYP-6-31G**) with experimental energy levels. Hydrogen atoms and lattice solvent molecules have been removed for clarity. | ||
The central naphthalene rings, flanked by two NMI units, twist out of the main plane slightly. This twist was observed to be proportional to the number of NO2 units attached to the bay region and to the positions of the NO2 groups. In the case of Tab, the structure reveals two different conformers in the unit cell; in one of them, the two –NO2 units are placed above the neighbouring naphthalene ring (Tab-cis), maintaining the planarity of the core. In the other conformer, the two groups are placed above and below the neighbouring ring (Tab-trans) as shown in the Fig. 2a inset. This suggests that in solution, this molecule is expected to have at least two conformers contributing to the photophysical properties (vide infra). The solid-state structure of Tad reveals a planar TDI core, with the exception of only two C–H units of the central naphthalene pointing upwards. In Tac, both the naphthalene monoimide units twist symmetrically in opposite directions, with respect to the central naphthalene unit. As expected, Tabc assumes an unsymmetrical structure. One of the NO2 groups in this molecule is positionally disordered between two bay positions. However, the introduction of an additional nitro group to the bay position affords a symmetrically twisted Tabcd with an angle of 19.3° between the mean plane and the central naphthalene ring.
 | ||
| Fig. 2 Solid state packing diagrams for the nitro-TDIs, as obtained from SCXRD studies (a) Tab (inset: Tab-cis & Tab-trans); (b) Tad; (c) Tac; (d) Tabc; (e) Tabcd. | ||
The larger π-surface of these molecules is supposed to facilitate π–π stacking in the solid state. However, a near-perpendicular arrangement of the aryl groups on the imide nitrogens and the NO2 groups at the bay positions prevented strong intermolecular stacking (Fig. 2). Instead, several C–H⋯π contacts and C–H⋯O hydrogen bonds are seen in the packing of these molecules. It is presumed that this arrangement in the solid state is responsible for the ready solubility of these molecules in common organic solvents. It is seen that the lattice solvent molecules, wherever present, bind with the core of the TDI via C–H–π interactions and with the imide oxygens via C–H⋯O hydrogen bonding.
Cyclic voltammetry
In cyclic voltammetric studies (vs. Fc/Fc+), it was observed that the first reversible reduction potentials (Ered11/2) gradually became anodic with the increase in the extent of nitration (Fig. 3a). Ta exhibited Ered11/2 at −0.34 V, which is significantly lower than that observed for TDI (−0.60 V). Strikingly, even the second reduction of Ta is easier (−0.47 V) than the first reduction of TDI (Table 1 & Fig. S36†). Only Ta shows a reversible oxidation wave within the potential range scanned, which is anodic to that of TDI. The dinitro regioisomers exhibited a peculiar trend with the Ered11/2 values of −0.15 V, −0.24 V, and −0.18 V for Tab, Tac and Tad respectively. This underscores the importance of the position of functionalization in fine tuning the electronic energy levels. Tabc and Tabcd can be more easily reduced with Ered11/2 values of 0.03 V and 0.20 V respectively. The stability of these molecules is of paramount importance to utilize them for further applications. To verify the same, Tabcd with the lowest reduction potential had been subjected to a cyclic test at 100 mVs−1 and found to be stable (Fig. 3b) even after 30 cycles. The LUMO values are listed in Fig. 1, Tables 1 and S1.† The trend observed for the values is Ta > Tac > Tad > Tab > Tabc > Tabcd. Tabcd (−5.0 eV) was found to have one of the deepest LUMOs reported for any stable, longer RDI. | ||
| Fig. 3 Cyclic voltammograms of the nitro-TDIs vs. Fc/Fc+ (a); stability of Tabcd after 30 cycles (b), and ESP surfaces for Tabcd, Tabcd•−, Tabcd− (c). | ||
| Comp | λ max,abs [nm] | ε [M−1 cm−1] | E g,opt [eV] | λ max,em [nm] | Stokes shift [cm−1] | ϕ [%] | E red [V] | E ox [V] | LUMOb [eV] | τ av [ns] | k r [s−1] | k nr [s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Quantum yield recorded in hexane (values from paraffin oil in parentheses), using TDI as the reference.15 b LUMO vs. Fc/Fc+. | ||||||||||||
| TDI | 653 | 131![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.81 | 666 | 298 | 90 | −0.60, −0.65 | 1.22 | −4.2 | 3.31 | 2.7 × 108 | 3.02 × 107 |
| Ta | 657 | 76000 |
1.74 | 720 | 1332 | 3(6) | −0.336, −0.47 | 1.4 | −4.4 | 1.49 | 2.0 × 107 | 6.4 × 108 |
| Tab | 669 | 81000 |
1.65 | 696 | 580 | 2(2) | −0.148, −0.46 | — | −4.6 | 1.33 | 1.5 × 107 | 7.3 × 108 |
| Tac | 658 | 82000 |
1.75 | 702 | 952 | 12(14) | −0.24 | — | −4.5 | 1.27 | 9.4 × 108 | 6.9 × 108 |
| Tad | 663 | 75000 |
1.74 | 708 | 980 | 11(14) | −0.18 | — | −4.6 | 1.38 | 7.9 × 107 | 6.4 × 108 |
| Tabc | 666 | 68000 |
1.72 | 732 | 1354 | 2(5) | 0.03, −0.10 | — | −4.8 | 0.98 | 2.0 × 107 | 1.0 × 109 |
| Tabcd | 664 | 82000 |
1.75 | 717 | 1113 | 8((10) | 0.20, 0.07 | — | −5.0 | 1.39 | 5.7 × 107 | 6.5 × 108 |
Absorption and fluorescence studies
The absorption spectra of the nitrated derivatives were broader, with minimal bathochromic shifts, and less intense, compared to those of parent TDI (Table 1 & Fig. 4). This is attributed to the impending deviation of core planarity stemming from steric crowding at the bay positions.7c,14 The di- and tetra-substituted TDIs have marginally higher molar extinction (ε) values. As found in SCXRD studies, these molecules may have other conformers in solution with improved planarity. The absorption onset obtained from the electronic spectra revealed a near identical Eg for all the nitro-TDIs, irrespective of the extent of nitration (Table 1). This is due to the stabilization of both HOMO and LUMO levels evenly.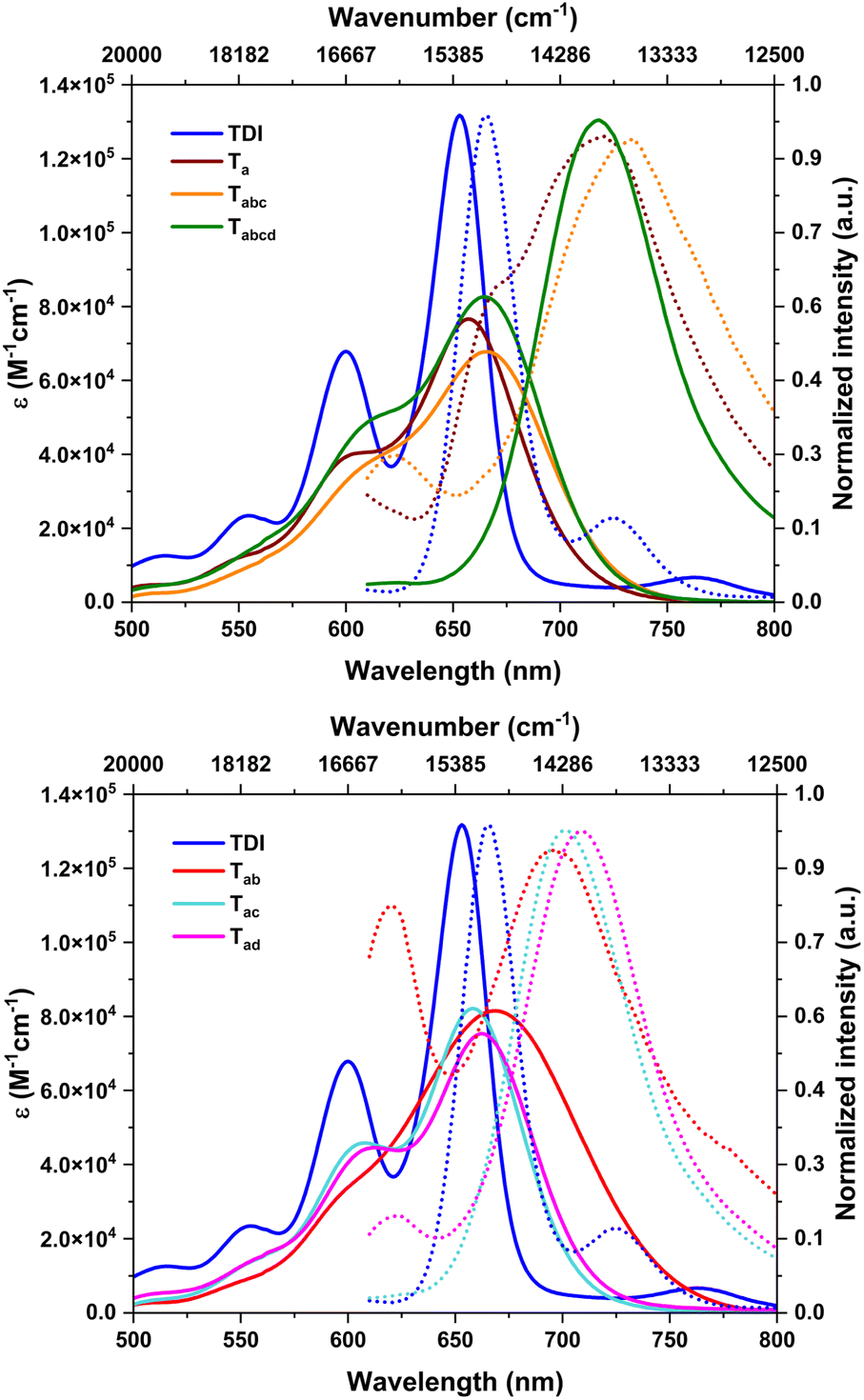 | ||
| Fig. 4 Absorption (solid line) and emission spectra (dotted line) of a nitro-TDI (×10−6 M) solution of CHCl3 (excitation wavelength 600 nm). | ||
As presumed, the nitro derivatives were found to be fluorescent. Even though the absorption profiles were in the visible region, their emission maxima were in the NIR region (>700 nm), owing to larger Stokes shifts (Fig. 4 & Table 1). Nitro-aromatics are often non-emissive in nature. They release the excitation energy effectively via an inter-system crossing (ISC) process. To make them emissive, it is necessary to introduce a charge transfer (CT) character in their excited states, which can be achieved by introduction of a donor-accepter configuration in the molecule. These CT states can hinder ISC and a radiative decay is preferred from the singlet states.11a In the present case, a comparatively electron-rich TDI forms a CT state due to the push–pull configuration with nitro groups. This in turn impedes the inter-system crossing (ISC) and facilitates a radiative channel. The emission of the nitro-TDIs stems from this phenomenon. Our hypothesis was further supported by solvent dependent emission studies. As the CT states are sensitive to solvent polarity, the effect of solvent was investigated.
Nonpolar solvents such as n-hexane could not stabilize the CT state and thus the molecules exhibited emission. However, in polar solvents, the CT states get stabilized effectively and exhibited redshifted emission with reduced intensity (Fig. S25–S30†).11 In the case of the three regio-isomers of dinitro-TDIs, the emission spectra were not identical. This can be attributed to the difference in the electronic nature of the excited states, depending on the position of the nitro groups. As discussed in the X-ray diffraction analysis, Tab, where the two nitro groups are linked to the same naphthalene unit, shows two conformers in the solid state. These conformers might have dynamic interconversion in solution, thus reducing the emission intensity significantly by nonradiative decay, compared to other regio-isomers.
The fluorescence intensity of all the molecules increased significantly at 77 K (Fig. 5a–b & S34). A similar observation was obtained from a viscous paraffin oil solution of the compounds (Fig. S25–S30†). Further, there was no apparent change in emission when recorded under deaerated conditions (Fig. S31†). The emission dependence on solvent viscosity and temperature is partially attributed to the restricted dynamics around the C–N bond. Moreover, under these conditions, the nitro groups may attain co-planarity with the aromatic core and thus enhanced emission was observed. The singlet lifetimes measured were between 1 and 2 ns, much longer than expected for nitroaromatics (sub-ps or sub-ns). The radiative decay rate of ∼108 s−1 further supported the emissive nature.
The electron-deficient nature of Tabcd directs the attention towards probing its electron affinity for possible applications. The absorption spectrum obtained while titrating Tabcd in chloroform against triethyl amine exhibits a new set of bands between 900 nm and 1400 nm, with a decrease in the intensity of the 664 nm band (Fig. 5c and d). The new bands can be attributed to the formation of a dianion, even in the absence of light, reflecting the enhanced electron affinity. The direct formation of a dianion, bypassing the formation of an anion radical, can be originating from the smaller potential gap (130 mV) between the two oxidation states (Fig. 3). Further, the trend observed for the reduction of other nitro-TDIs is linearly correlated with the extent of nitration (Fig. S32†). A measure of the ease of reduction for Tabcd is further emphasized by the formation of a dianion even with a milder base such as DABCO (Fig. S33†). The easier reduction along with their strong absorption in the visible region would make them excellent catalysts.16
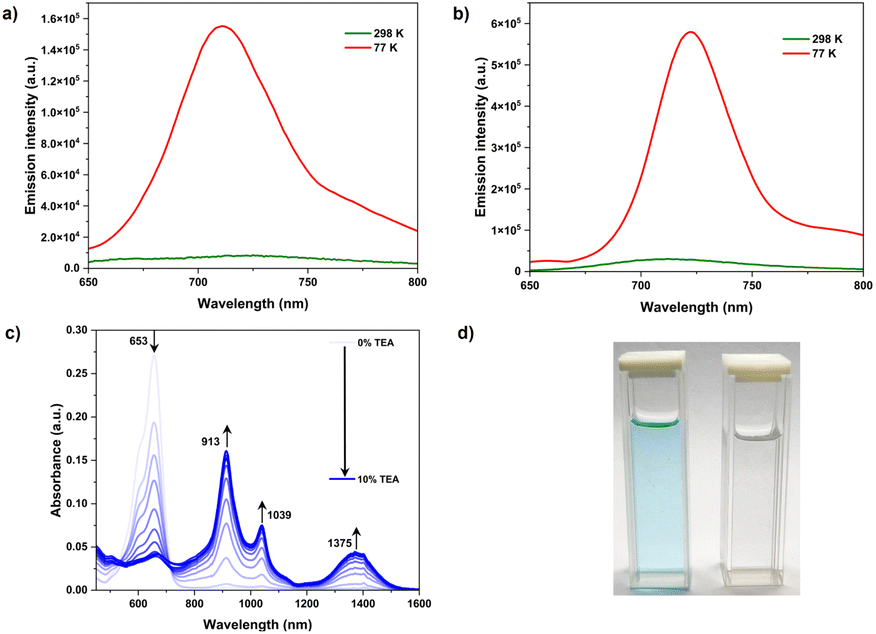 | ||
| Fig. 5 Emission profiles of Ta (a) and Tabcd (b) at 77 K and 298 K; (c) absorption response for Tabcd with the addition of triethylamine in chloroform; (d) photograph of Tabcd before and after TEA addition. | ||
Conclusion
In summary, electron deficient derivatives of TDI with deep-LUMO energy levels have been achieved and are structurally characterized. These molecules demonstrated impressive redox stability and structural rigidity. The tunability of the redox properties of regioisomers highlights the finer flexibility in modulating the electronic nature. Its low lying LUMO enables Tabcd to get reduced even in the presence of bases such as DABCO. Owing to their apt energy levels, all derivatives showed emission in the NIR range. The fluorescence quantum yields are among the best for any electron deficient, NIR emissive molecules. This is attributed to the electronic synergy between the TDI core and the electron withdrawing nitro groups, resulting in longer singlet lifetimes stemming from the modulation of the ICT process. The cryoemission, along with viscosity studies further support the fact that the nonradiative pathways resulting from dynamics can be suppressed to achieve intense emission. Their ultra-low LUMO levels, ambient stability, redox robustness along with emissive nature in the NIR, make nitro-TDIs excellent candidates for modern electronic applications and as catalysts. This work highlights the utility of nitration as an excellent strategy for developing new functional rylene diimides. Further investigations in these directions are currently underway.Data availability
All necessary data have been included in the manuscript and in the ESI.†Author contributions
K. S. M. performed all the synthesis, characterization, and experimental work. K. S. M., S. J. and J. S. analysed the data and wrote the manuscript. A. M. M. and D. C. contributed in SCXRD experiments and structure refinement. J. S. supervised the project. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KSM & SJ thank CSIR, New Delhi and INSPIRE-DST, New Delhi respectively for fellowships. JS thanks DST-SERB-EMR/2016/005768 & DST-SERB-CRG/2020/004632 for funding the UV-vis-NIR spectrophotometer and semi-preparative GPC respectively. The authors thank Dr Ruchika Mishra for useful suggestions and proof-reading of the manuscript. All authors thank IISER Bhopal for infrastructure and funding.Notes and references
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 1807623 CrossRef.
- (a) Z. Y. Wang, Near-Infrared Organic Materials and Emerging Applications, CRC, Boca Raton, 2013 CrossRef; (b) J. Qi, W. Qiao and Z. Y. Wang, Chem. Rec., 2016, 16, 1531–1548 CrossRef CAS PubMed.
- (a) F. Legouté, R. J. Bensadoun, V. Seegers, Y. Pointreau, D. Caron, P. Lang, A. Prévost, L. Martin, U. Schick, B. Morvant, O. Capitain, G. Calais and E. Jadaud, Radiat. Oncol., 2019, 14, 83 CrossRef PubMed; (b) M. A. Naeser, A. S. Marche, M. H. Krengel, M. R. Hamblin and J. A. Knight, Photomed. Laser Surg., 2011, 29(5), 351–358 CrossRef PubMed.
- (a) M. Ito, M. Sakai, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2021, 60, 21853–21859 CrossRef CAS PubMed; (b) X. X. Zhang, H. Qi, Y. L. Liu, S. Q. Yang, P. Li, Y. Qiao, P. Y Zhang, S. H. Wen, H. l. Piao and K. L Han, Chem. Sci., 2020, 11, 11205–11213 RSC.
- (a) A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed; (b) K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynold, Chem. Mater., 2011, 23, 5305–5312 CrossRef CAS; (c) E. J. Peterson, W. Qi, I. N. Stanton, P. Zhang and M. J. Therien, Chem. Sci., 2020, 11, 8095–8104 RSC.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- (a) F. Holtrup, G. Müller, H. Quante, S. de Feyter, F. C. De Schryver and K. Müllen, Chem.–Eur. J., 1997, 3, 219–225 CrossRef CAS PubMed; (b) F. Nolde, J. Qu, C. Kohl, N. G. Pschirer, E. Reuther and K. Müllen, Chem.–Eur. J., 2005, 11, 3959–3967 CrossRef CAS PubMed; (c) K. Peneva, G. Mihov, F. Nolde, S. Rocha, J. i. Hotta, K. Braeckmans, J. Hofkens, H. U. i, A. Herrmann and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 3372–3375 CrossRef CAS PubMed; (d) K. S. Mehra, S. Jha, S. Bhandary, D. Mandal, R. Mishra and J. Sankar, Angew. Chem., Int. Ed., 2022, 61, e202205600 Search PubMed.
- (a) Y. Kumar, S. Kumar, K. Mandal and P. Mukhopadhyay, Angew. Chem., Int. Ed., 2018, 57, 16318–16322 CrossRef CAS PubMed; (b) S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed; (c) S. Kumar, J. Shukla, Y. Kumar and P. Mukhopadhyay, Org. Chem. Front., 2018, 5, 2254–2276 RSC.
- (a) G. Battagliarin, Y. Zhao, C. Li and K. Müllen, Org. Lett., 2011, 13, 3399–3401 CrossRef CAS PubMed; (b) G. Battagliarin, S. R. Puniredd, S. Stappert, W. Zajaczkowski, S. Wang, C. Li, W. Pisula and K. Müllen, Adv. Funct. Mater., 2014, 24, 7530–7537 CrossRef CAS; (c) J. Gao, C. Xiao, W. Jiang and Z. Wang, Org. Lett., 2014, 16, 394–397 CrossRef CAS PubMed; (d) Y. Kumar, S. Kumar, D. Bansal and P. Mukhopadhyay, Org. Lett., 2019, 21, 2185–2188 CrossRef CAS PubMed.
- (a) X. Sun, M.-Y. Liao, X. Yu, Y.-S. Wu, C. Zhong, C.-C. Chueh, Z. Li and Z. Li, Chem. Sci., 2022, 13, 996–1002 RSC; (b) D. Guo, L. Li, X. Zhu, M. Heeney, J. Li, L. Dong, Q. Yu, Z. Gan, X. Gu and L. Tan, Sci. China: Chem., 2020, 63, 1198 CrossRef CAS.
- (a) Y. M. Poronik, B. Sadowski, K. Szychta, F. H. Quina, V. I. Vullev and D. T. Gryko, J. Mater. Chem. C, 2022, 10, 2870–2904 RSC; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS; (c) M. C. Chen, D. G. Chen and P. T. Chou, ChemPlusChem, 2021, 86, 11–27 CrossRef CAS PubMed; (d) W. R. Córdoba, L. G. Arzaluz, F. C. Guzmán and J. Peon, Chem. Commun., 2021, 57, 12218–12235 RSC; (e) K. Skonieczny, I. Papadopoulos, D. Thiel, K. Gutkowski, P. Haines, P. M. McCosker, A. D. Laurent, P. A. Keller, T. Clark, D. Jacquemin, D. M. Guldi and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 16104–16113 CrossRef CAS PubMed.
- (a) K. Y. Chen and T. J. Chow, Tetrahedron Lett., 2010, 51, 5959–5963 CrossRef CAS; (b) H. Y. Tsai, C. W. Chang and K. Y. Chen, Tetrahedron Lett., 2014, 55, 884–888 CrossRef CAS.
- (a) K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef; (b) Z. An, S. A. Odom, R. F. Kelley, C. Huang, X. Zhang, S. Barlow, L. A. Padilha, J. Fu, S. Webster, D. J. Hagan, E. W. Stryland, M. R. Wasielewski and S. R. Marder, J. Phys. Chem. A, 2009, 113, 5585–5593 CrossRef CAS PubMed.
- S. Jha, K. S. Mehra, A. Hasija, D. Chopra, R. Regar and J. Sankar, J. Org. Chem., 2022, 87, 3770–3774 CrossRef CAS PubMed.
- (a) M. Davies, C. Jung, P. Wallis, T. Schnitzler, C. Li, K. Müllen and C. Bräuchle, ChemPhysChem, 2011, 12, 1588–1595 CrossRef CAS PubMed; (b) H. Langhals, A. Walter, E. Rosenbaum and L. B. A. Johansson, Phys. Chem. Chem. Phys., 2011, 13, 11055–11059 RSC.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed; (b) A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 160, 81–86 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2204224–2204228. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06162g |
| This journal is © The Royal Society of Chemistry 2023 |