
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWide-range color-tunable polycyclo-heteraborin multi-resonance emitters containing B–N covalent bonds†
Guoyun
Meng‡
a,
Hengyi
Dai‡
a,
Jianping
Zhou
a,
Tianyu
Huang
a,
Xuan
Zeng
a,
Qi
Wang
a,
Xiang
Wang
a,
Yuewei
Zhang
a,
Tianjiao
Fan
a,
Dezhi
Yang
 c,
Dongge
Ma
c,
Dongdong
Zhang
*a and
Lian
Duan
*ab
c,
Dongge
Ma
c,
Dongdong
Zhang
*a and
Lian
Duan
*ab
aKey Lab of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing, P. R. China. E-mail: ddzhang@mail.tsinghua.edu.cn; duanl@mail.tsinghua.edu.cn
bLaboratory of Flexible Electronics Technology, Tsinghua University, Beijing, 100084, P. R. China
cInstitute of Polymer Optoelectronic Materials and Devices State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, P. R. China
First published on 19th December 2022
Abstract
Boron- and nitrogen (BN)-fused polycyclic aromatic frameworks with amine-directed formation of B–N covalent bonds have the potential to form a new family of facile-synthesis multi-resonance luminophores, which, however, still face imperative challenges in diversifying the molecular design to narrow the emission bandwidth and tune the emission colors. Here, we demonstrate a strategic implementation of B–N bond containing polycyclo-heteraborin multi-resonance emitters with wide-range colors from deep-blue to yellow-green (442–552 nm), small full-width at half-maxima of only 19–28 nm and high photoluminescence efficiencies, by stepwise modifying the basic para B–π–B structures with heteroatoms. The corresponding electroluminescent devices show superior maximum external quantum efficiencies with an exceptional low-efficiency roll-off, retaining 21.0%, 23.6% and 22.1% for the sky-blue, green and yellow-green devices at a high luminance of 5000 cd m−2, respectively.
Introduction
Multiple resonance (MR) fluorophores, whereby the frontier molecular orbital (FMO) distributions are located alternatively on single atoms of a rigid π-conjugation scaffold to minimize the bonding/antibonding characters and suppress vibrational coupling and structural relaxation of the excited states, have shown the ability to provide highly efficient narrowband emissions as emitters in organic light-emitting diodes (OLEDs).1–4 The judicious strategy for developing MR emitters is to incorporate heteroatoms with opposite resonance effects into polycyclic aromatic hydrocarbons (PAHs), with the representative ones being boron (B) and nitrogen (N) atom embedded PAHs (B,N-PAHs) such as DABNA.1 Following the pioneering work by Hatakeyama et al., plenty of works have been devoted to modulating the skeletons of B,N-PAHs to obtain wide-range emission colors that cover the entire visible region and extremely small full-width at half-maxima (FWHMs).3,5–9The most effective strategy to modulate the optoelectronic properties of B,N-PAHs is to enlarge the extension of the π-skeleton, which has been theoretically predicted by Olivier et al.10 For instance, by fusing two DABNA resonating cores through a meta B–π–B motif, the sky-blue v-DABNA realized a leading-edge FWHM of only 14 nm.2 Also, by extending the π-conjugation length of the DABNA skeleton, Yang et al. obtained deep-blue emitters with an extremely small FWHM of 15 nm.11 Yasuda et al. applied this strategy to the carbazole-embedded PAHs by modulating the positions and numbers of B and N atoms, generating the first species of full-color MR emitters.3 Our group reported deep-red/near-infrared (DR/NIR) MR emitters featuring X-shaped B–π–B and N–π–N extended carbazole-containing structures.12 Besides simply extending the MR frameworks, insertion of heteroatoms, including oxygen (O), sulfur (S), and selenium (Se) atoms, to modulate MR properties has also been adopted.13–17 Based on this concept, Wang et al. constructed a pure green emitter with a FWHM of only 19 nm based on a ternary B–N–O fused PAH.13 Despite all those efforts, the molecular design strategies for MR B,N-PAHs still rely on the initial parent triangulene structures with low yields and sophisticated multistep synthesis procedures based on the utilization of dangerous lithium reagents.3,18,19
In contrast to the above B,N-doped MR skeletons, other types of B- and N-doped nanographenes have undergone remarkable developments in their synthetic methodology and thus have rich molecular skeletons.20–23 In particular, isoelectronic B–N covalent bonds have been adopted to replace the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in PAHs, affording novel π-conjugated BN-doped systems with unique optical-electronic properties as well as feasible and efficient synthesis processes. However, those B–N backbones did not show the MR effect with relatively large FWHMs and only a few of them have been adopted as emitters in OLEDs with poor device performance and color purity.24,25 Very recently, our group successfully combined the advantages of MR of B,N-doped skeletons and the feasible synthesis procedures of B–N groups and developed the first example of a MR emitter containing B–N covalent bonds. This was realized by using N atoms to form amine-directed B–N covalent bonds while the other N atoms in the para-position was used to maintain the MR character, thus affording easy-to-access sky-blue MR emitters with a FWHM of 29 nm.26 This work not only provides a new family of MR BN-PAHs with facile synthesis procedures free of lithium reagents and with high yields, but also provides an opportunity to finely manipulate the electronic structures of BN-PAHs through B–N covalent bonds, which may uncover unforeseen optoelectronic properties.27–30 However, at this initial stage, the performance of B–N embedded emitters still lags far behind their classical B,N-PAH counterparts. For instance, the FWHMs of those emitters are still larger than the cutting-edge values of B,N-MR emitters and they still need to demonstrate their color tunability for full-color emission.31,32 Besides, it is still imperative to pursue outstanding design and synthetic methodology to expand the diversity of molecular frameworks for unique molecular architectonics that is full of significant and fascinating pure chemistry charm.
C bond in PAHs, affording novel π-conjugated BN-doped systems with unique optical-electronic properties as well as feasible and efficient synthesis processes. However, those B–N backbones did not show the MR effect with relatively large FWHMs and only a few of them have been adopted as emitters in OLEDs with poor device performance and color purity.24,25 Very recently, our group successfully combined the advantages of MR of B,N-doped skeletons and the feasible synthesis procedures of B–N groups and developed the first example of a MR emitter containing B–N covalent bonds. This was realized by using N atoms to form amine-directed B–N covalent bonds while the other N atoms in the para-position was used to maintain the MR character, thus affording easy-to-access sky-blue MR emitters with a FWHM of 29 nm.26 This work not only provides a new family of MR BN-PAHs with facile synthesis procedures free of lithium reagents and with high yields, but also provides an opportunity to finely manipulate the electronic structures of BN-PAHs through B–N covalent bonds, which may uncover unforeseen optoelectronic properties.27–30 However, at this initial stage, the performance of B–N embedded emitters still lags far behind their classical B,N-PAH counterparts. For instance, the FWHMs of those emitters are still larger than the cutting-edge values of B,N-MR emitters and they still need to demonstrate their color tunability for full-color emission.31,32 Besides, it is still imperative to pursue outstanding design and synthetic methodology to expand the diversity of molecular frameworks for unique molecular architectonics that is full of significant and fascinating pure chemistry charm.
Herein, a parent skeleton, [B-N]N (Scheme 1), was first provided to construct the B–N bond containing MR emitter with a narrower FWHM (19 nm) and blue-shifted emission compared with the known sky-blue emissive analogue BCz–BN based on an aza-triangulene core.4,33 Such a feature of the skeleton is expected to decrease the electron accepting strength of the B atom and afford a large space for color tunability. Encouraged by the modulation strategy of B,N-PAHs, we further demonstrated a strategic implementation of B–N bond containing polycyclo-heteraborin MR emitters featuring para B–π–B skeletons, which were modified by stepwise replacing nitrogen atoms with oxygen to tune the emission colors. Three novel MR emitters were synthesized based on amine-directed twofold electrophilic arene borylation, free of lithium reagents and with high yields. Thus, a wide color range from deep-blue to yellow-green (442–545 nm) was obtained with narrowband pure emissions with FWHMs of 19–28 nm, which were clearly narrower than those of our previously reported B–N bond embedded emitters. The TADF-sensitized devices using p[B-N]O, p[B-N]NO and p[B-N]N as emitters presented ultrahigh brightness of over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2, high maximum external quantum efficiencies (EQEmaxs) of 26.3%, 27.6% and 24.6% and greatly alleviated efficiency roll-off, respectively. Our results presented here greatly diversified the skeletons of MR emitters embedded with B–N covalent bonds, providing not only a large color tunability with cutting-edge narrow bandwidths but also superior device performances.
000 cd m−2, high maximum external quantum efficiencies (EQEmaxs) of 26.3%, 27.6% and 24.6% and greatly alleviated efficiency roll-off, respectively. Our results presented here greatly diversified the skeletons of MR emitters embedded with B–N covalent bonds, providing not only a large color tunability with cutting-edge narrow bandwidths but also superior device performances.
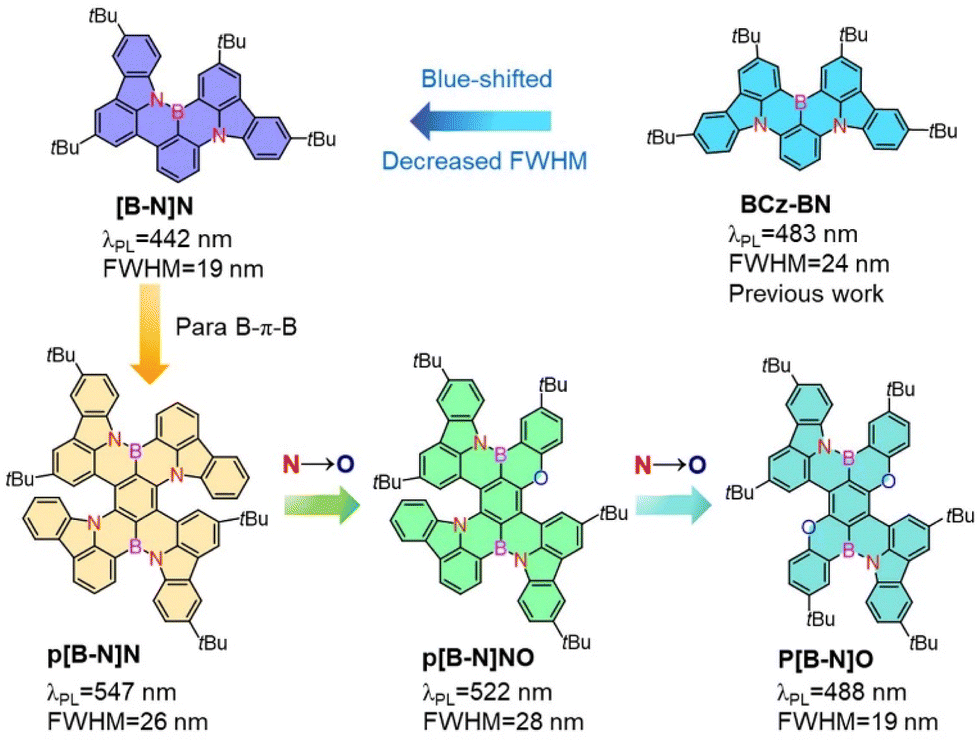 | ||
| Scheme 1 The chemical structures of the previously reported B,N-doped MR emitter, the parent skeleton [B-N]N and the para B–π–B skeletons which were modified by stepwise replacing nitrogen atoms with oxygen to tune the excited-state energies and color purities. | ||
Results and discussion
Molecular design and theoretical calculations
Scheme 1 illustrates the structure of the parent molecular [B-N]N, which combined both the MR effect and B–N bond simultaneously. Such a feature of the skeleton is expected to decrease the electron-accepting strength of the B atom and the intramolecular short-range charge transfer (SRCT) character of the para-distributed B and N architecture, thus leading to a blue-shift in emission color and decreased FWHM compared with the sky-blue emissive analogue BCz–BN. Furthermore, the [B-N]N core can be further extended by a para-position fusing strategy to finely regulate the excited-state energies and color purities. The targeted molecular structures are illustrated in Scheme 1, for which the aim was not only to tune the emission colors and FWHMs but also to expand the library of such MR molecular frameworks. On one hand, we focused on constructing new BN-doped MR emitters by embedding para B–π–B/N–π–N patterns into π-conjugated structures (Scheme 1). Such a molecular configuration could enhance delocalized excited states by increasing the electron donating and accepting strengths, resulting in a red-shifted emission. It is worth noting that the introduction of B–N bonds into this π-conjugated structure at the para-position boron atoms can not only decrease the electron accepting strength of the B atoms but also weaken the intramolecular SRCT character; therefore, a blue-shift in color emission and narrower spectral width can be anticipated compared with the linear B–π–B/N–π–N structures. On the other hand, the structure was further modified by the stepwise substitution of N atoms with weak electron-donating O atoms, enabling great versatility for tailoring the energy levels and skeleton structures (e.g., para N–π–O and O–π–O patterns) and thus further expanding the emission color range. In addition, the incorporation of bulky tert-butyl groups on the backbone structure can effectively increase the intermolecular distance and suppress aggregation-caused concentration quenching.To illustrate the validity of the above design strategy for those compounds, the spatial distributions of the highest occupied and lowest unoccupied molecular orbitals (HOMOs and LUMOs, respectively) were obtained based on density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations at the B3LYP/6-31G(d, p) level (Fig. 1). The HOMO and LUMO distributions of [B-N]N showed almost the same electronic distributions compared with the analogous emitter BCz–BN. Interestingly, the N atoms on the aza-aromatic rings replaced by less electron-rich O atoms forming oxa-aromatic rings still retain the intrinsic resonance patterns, and further stabilize the HOMO levels by restricting the π-conjugation of the HOMO, thus generating an enlarged energy gap (Eg). The Natural Transition Orbital (NTO) analysis performed using multiwfn34 found that the S1 and T1 state of those emitters exhibit a partially intramolecular SRCT excitation character; the hole densities are localized on the N atoms and at the meta-position relative to the B atoms, while the electron densities are localized on the B atoms and at the ortho- and para-positions relative to it, accompanied by a spatial overlap integral (Oh/e) of 0.637–0.691, which is relatively larger than that of the parent molecule BCz–BN (Oh/e of ∼0.582) (Fig. S1–S3†). To gain in-depth insight into the molecular geometrical change and vibrational relaxation occurring during the excitation, as shown in Fig. S4,† the ground (S0) and excited (S1) state geometries of those molecules were superposed and found to possess small root-mean-square displacement (RMSD) values of 0.047 Å, 0.057 Å, 0.135 Å, and 0.091 Å for [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N, respectively. Those extremely small values resulted in weak vibration couplings from the vibrational energy levels of S1 to S0 with small reorganization energies (λ) of 0.17 eV, 0.21 eV, 0.19 eV and 0.19 eV for [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N, respectively. The above results imply that the vibrational coupling and configuration relaxation of excited states can be greatly suppressed to narrow the linewidth. Additionally, all compounds exhibit helical structures between the peripheral B–N bond linked tert-butyl carbazole rings and aza-/oxa-aromatic rings with dihedral angles from 21.4° to 61.1° (Fig. 1). Due to the π-extended delocalization of those compounds, enhanced transition oscillator strengths (f) of 0.35–0.56 were observed and thus highly radiative decay rates can be anticipated.
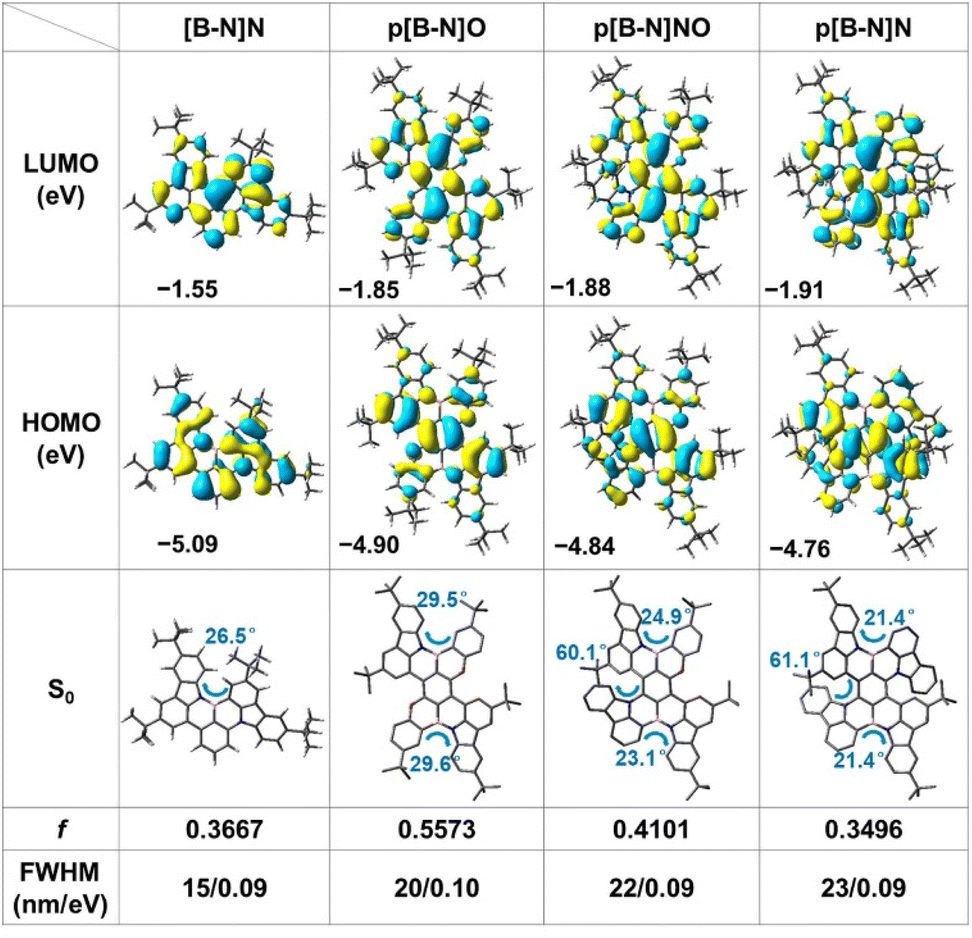 | ||
| Fig. 1 The spatial distributions of HOMO and LUMO, optimized S0 structures, oscillator strengths (f) and FWHMs of the simulated spectra at the B3LYP/6-31G (d, p) level for all compounds. | ||
Based on these optimized S0 and S1 state structures, Franck–Condon spectra for the S1–S0 transitions were further simulated at TD-DFT levels. In particular, not only extremely small FWHMs in the range of 15–23 nm (Fig. S5†), but also a wide peak-wavelength range of 445–564 nm was obtained for those four compounds, validating the effectiveness of the molecular design strategy. It is noted that compared with [B-N]N, relatively broadened spectra were observed for p[B-N]O, p[B-N]NO and p[B-N]N. To gain insight into the bandwidth discrepancy of those compounds, the Huang–Rhys (HR) factors associated with the spectral FWHM were calculated using MOMAP software to quantify the coupling strength between the structural displacement and the vibrational modes.35 The vibration mode analyses display the normal mode in the low-frequency region at 72.95 cm−1, 28.84 cm−1, 11.98 cm−1 and 20.35 cm−1 with the highest HR factors (0.29, 0.39, 2.82 and 1.58) as the ones mainly involved in the spectral progression of [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N, respectively, corresponding to the twisting vibration modes of the tert-butyl groups (Fig. 2a). Obviously, [B-N]N had the smallest HR factor among the four compounds, thus leading to the narrowest FWHM. More interestingly, the traditional BO resonance structure with the B–N bonds (e.g., p[B-N]O) has a smaller HR factor compared with p[B-N]N, and also exhibits emission band narrowing. These results suggest that the small HR values for each vibrational mode are conducive to a narrow FWHM. Meanwhile, the high-frequency vibrational modes (1228.41–1411.47 cm−1) show significant contributions to the reorganization energies for the four molecules, which were dominated by stretching/scissoring vibrations of B–N bonds and the twisted skeleton structures (Fig. S6–S9†). This should be responsible for the presence of weaker vibronic bands at the lower energy of each emitter.
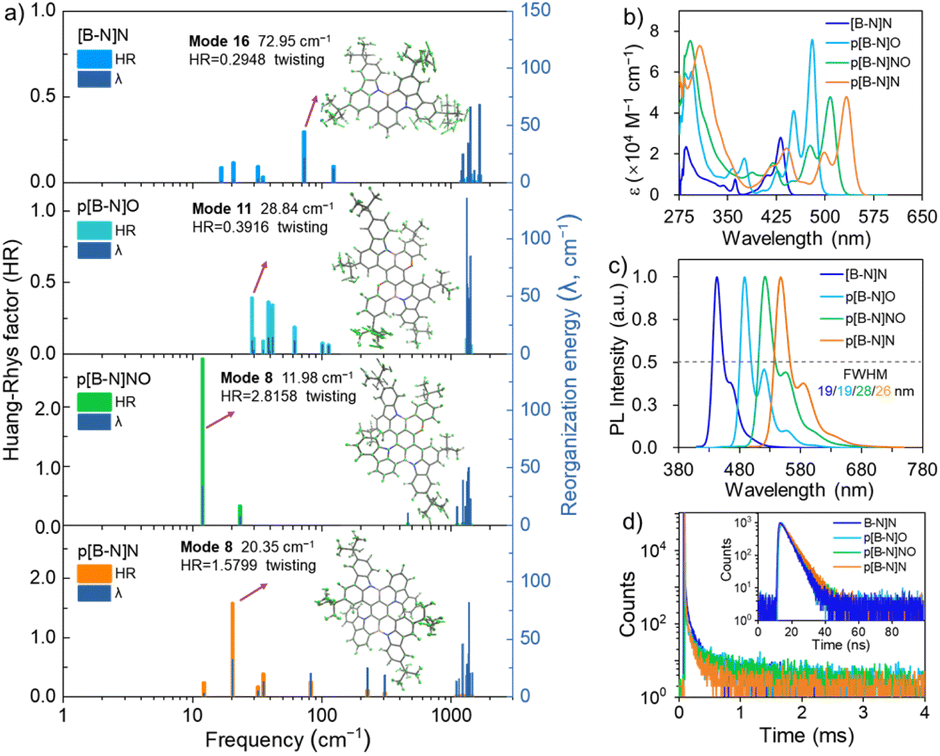 | ||
| Fig. 2 (a) The Huang–Rhys factor (HR) and reorganization energy (λ) as a function of frequency under the representative vibration modes for S1–S0 transition of [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N. (b) The absorption, (c) PL spectra and (d) transient PL decay curves of four emitters in toluene (10−5 M) under inert conditions. | ||
Material synthesis, and thermal and electrochemical properties
The chemical structures and detailed synthetic routes of the designed compounds are presented in Scheme 1 and the ESI.† Aromatic nucleophilic substitution and palladium-catalyzed Suzuki–Miyaura coupling reactions were involved, followed by an amine-directed twofold electrophilic borylation reaction. All the final compounds were purified by temperature-gradient vacuum sublimation and characterized using 1H and 13C NMR spectroscopy and mass spectrometry. Thermogravimetric analysis demonstrated the excellent thermal stabilities of those compounds with all 5% weight-loss temperatures (Tds) exceeding ∼410 °C (Fig. S10†). Besides, the HOMO and LUMO energy levels were estimated using the oxidation and reduction potentials of the cyclic voltammetry (CV) measurements (Fig. S11†), being −5.65 and −2.74 eV for [B-N]N, −5.47 and −2.94 eV for p[B-N]O, −5.34 and −3.01 eV for p[B-N]NO, and −5.29 and −3.06 eV for p[B-N]N, respectively. Notably, p[B-N]N had a shallower HOMO energy level and a deeper LUMO energy level than those of the parent skeleton [B-N]N, thus leading to a reduced HOMO–LUMO energy gap. And the stepwise replacement of N atoms with O gradually deepened the HOMO levels and thus blue-shifted the emissions compared with p[B-N]N.Photophysical characterization
The UV-vis absorption, photoluminescence (PL) spectra and time-resolved transient PL decay of all molecules analyzed in solution are shown in Fig. 2, and they are also summarized in Table 1. These compounds exhibited sharp and intense absorption bands at low energy with large molar extinction coefficients (ε) over 104 M−1 cm−1 (Fig. 2b), originating from the HOMO to LUMO transitions. Interestingly, the stepwise replacement of nitrogen atoms with oxygen on the backbones resulted in a prominent hypsochromic shift of the maximum absorption and emission peaks (λabs/λPL) in the order of p[B-N]N (533/547 nm), p[B-N]NO (508/522 nm) and p[B-N]O (480/488 nm) (Fig. 2c), accompanied with considerably small Stokes shifts for each compound. Compared with the dilute solution, the PL characteristics of all compounds exhibited negligible red-shifted emission peaks (ca. 5–9 nm) in the doped films (1.0 wt% doped in 9-(3-(9H-carbazol-9-yl)phenyl)-9H-3,9′-bicarbazole, mCPBC). These films exhibited emission peaks in the range of 450–552 nm (Fig. S12†) and all had ΦPL values of over ∼80%, which should be mainly ascribed to the large f values that arise from the highly rigid conjugated skeletons. Accordingly, all compounds can retain high purity emissions with narrow FWHM values of ca. 19–30 nm both in solution and thin films. It is of great importance to note that those B–N bond embedded emitters exhibited cutting-edge narrower spectral widths even compared with the classical B,N-doped emitters (e.g., BCz–BN, BNO1 and R-TBN).4,5,12,26 Note that slight shoulder peaks were observed in their PL spectra, which were mainly attributed to the stretching vibrations of the central B–N bonds as well as the significant delocalized π-bonding orbitals caused by the embedded para-positioned acceptor–π–acceptor and donor–π–donor structures, which were consistent with the theoretical calculations and previously reported B,N-MR emitters.2,3,11,13| Compound | λ abs [nm] 298 K | λ PL [nm] 298 K | Stokes shifta [nm] | FWHMb [nm (eV)] | Φ PL [%] 298 K | ΔESTd [eV] | τ PF [ns] | k r [108 s−1] | HOMO/LUMOg [eV] |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in toluene (10−5 M) at room temperature. b Full-width at half-maximum of the PL spectrum given in wavelength and energy. c Absolute PL quantum yield measured in deaerated/aerated toluene solutions. d Singlet-triplet energy gap: ΔEST = ES1 − ET1. Lowest excited singlet (ES1) and triplet (ET1) energies were estimated from peaks of the fluorescence and low-temperature phosphorescence spectra recorded at 298 K and 77 K, respectively. e Emission lifetime of fluorescence. f Rate constant of fluorescence radiative decay, Kr = ΦPF/τPF. g The HOMO and LUMO energies were determined according to cyclic voltammetry: EHOMO/LUMO = −(Eox/Ered + 4.8) eV. | |||||||||
| [B-N]N | 285, 361, 431 | 442 | 11 | 19/0.12 | 84/81 | 0.28 | 4.4 | 1.91 | −5.65/−2.74 |
| [B-N]N-film | — | 450 | — | 25/0.15 | 83 | 0.26 | 4.2 | 1.98 | — |
| p[B-N]O | 294, 375, 451, 480 | 488 | 8 | 19/0.10 | 93/89 | 0.43 | 4.3 | 2.16 | −5.47/−2.94 |
| p[B-N]O-film | — | 497 | — | 23/0.11 | 91 | 0.44 | 4.9 | 1.86 | — |
| p[B-N]NO | 292, 419, 477, 508 | 522 | 14 | 28/0.13 | 90/81 | 0.40 | 4.7 | 1.91 | −5.34/−3.01 |
| p[B-N]NO-film | — | 529 | — | 30/0.13 | 89 | 0.37 | 5.3 | 1.68 | — |
| p[B-N]N | 307, 440, 499, 533 | 547 | 14 | 26/0.11 | 86/80 | 0.36 | 4.0 | 2.15 | −5.29/−3.06 |
| p[B-N]N-film | — | 552 | — | 28/0.11 | 83 | 0.32 | 4.5 | 1.84 | — |
To further evaluate the excited state behaviors of these compounds, the characteristics of the transient PL decay curves were measured, as illustrated in Fig. 2d and S12.† It can be seen that the four compounds exhibited negligible TADF behaviors in degassed solution or doped films, only showing prompt decay lifetimes in the order of nanoseconds. Relatively high ΦPLs in aerated solution were also measured, being 81% for [B-N]N, 89% for p[B-N]O, 81% for p[B-N]NO, and 80% for p[B-N]N, implying that almost no excited triplet states were quenched by oxygen. Based on the fluorescence and phosphorescence spectra, ΔEST values of 0.28–0.43 eV were estimated for the four compounds (Fig. S13 and S14†). These obtained ΔEST values were larger than those of many conventional MR-TADF emitters, and this is also reflected in the relatively larger Oh/es. Notably, the analogous emitter BCz–BN exhibited similar hole and electron distributions but with a relatively smaller Oh/e of ∼0.582 that is responsible for its lower ΔEST of 0.13 eV than [B-N]N (Oh/e of ∼0.637, ΔEST ∼0.28 eV). Additionally, when further expanding its π-conjugated skeletons by a para-position fusing strategy, the total volume of the molecule increases but not as much as the increase of its overlapped volume. The Oh/e values of these emitters are up to 0.665–0.688, further enlarging the ΔESTs. Notably, p[B-N]O showed the highest Oh/e value of 0.688 among the four BN-MR emitters that is responsible for its largest ΔEST, plausibly due to the weak MR effect through B and O atoms with opposite electronic characteristics.15 Remarkably, these results are similar to the previous reports of indolocarbazole-based MR emitters and other reported B,N-emitters that possess MR effects but did not have TADF characters owing to their relatively large ΔESTs.36–41 Based on the ΦPLs and prompt lifetimes, the rate constants of radiative decay (kr) for these compounds were estimated to be as large as 1.68–2.16 × 108 s−1 in solution and doped films (Table 1). Thus, all those emitters showed high krs and narrow-band emissions, rendering them ideal final emitters for OLEDs.
OLED device performances
Given the relatively large ΔESTs of these MR emitters, binary devices based on [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N as emitters exhibited low EQEs of 4.9–6.6% (Fig. S15 and S16†), which could be explained by poor triplet exciton utilization. A routine strategy to address this issue is to adopt a TADF sensitizer to assist in triplet harvesting by adopting a ternary component emitting layer consisting of a wide-energy-gap host, a TADF sensitizer, and a MR emitter.42,43 In this system, triplet exciton generation occurred via the TADF sensitizer under electrical excitation and the triplet excitons upconverted to the S1 state of the TADF sensitizer, and then all singlets transferred into the S1 state of the MR emitters, thus leading to 100% exciton efficiency (Fig. S17†). As a consequence, the influence of the up-conversion process of the MR emitters can be avoided and it is the PL efficiency of those MR emitters that directly determines the device performance. OLED devices were fabricated using the structures of ITO/HATCN (5 nm)/NPB (30 nm)/TCTA (10 nm)/emitting layers (24 nm)/CzPhPy (15 nm)/DPPyA (30 nm)/LiF (0.5 nm)/Al (150 nm), where 1,4,5,8,9,11-hexaazatriphenylene-hexacarbonitrile (HATCN) and LiF were used as the hole- and electron-injection layers; 4,4′-N,N′-bis[N-(1-naphthyl)-N-phenylamino]biphenyl (NPB) and 9,10-bis(6-phenylpyridin-3-yl)anthracene (DPPyA) served as the hole- and electron-transporting layers; tris(4-(9H-carbazol-9-yl)phenyl)amine (TCTA) and 4,6-bis(3-(9H-carbazol-9-yl)phenyl)pyrimidine (CzPhPy) served as the electron- and hole-blocking layers, respectively. In the emitting layer, the material mCPBC was selected as a host due to its wide bandgap and high triplet energy (3.0 eV) that can provide both efficient exciton confinement within the emitting layer and hole transport abilities. The TADF materials, m4TCzPhBN,44 5TCzBN,45 3CTF,46 and DACT-II,47 were successively selected as sensitizers for [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N, respectively, to promote the exciton utilization efficiency, due to their intrinsic TADF features and high reverse intersystem crossing rate constants. They also provided a good spectral overlap between their emission spectra and the absorption/emission spectra of these emitters (Fig. 3). To illustrate an effective energy transfer from TADF sensitizer to MR emitter, the Förster energy transfer radius (R0) was estimated using a previous method.48 Thus, the corresponding R0 values of 2.8, 3.0, 3.2, and 3.3 nm were obtained for [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N, respectively, implying that more efficient energy transfer would occur in these sensitized systems. Furthermore, the films with 30 wt% sensitizer:1 wt% emitter in mCPBC exhibited shorter delayed lifetimes compared with the intrinsic decay curves of the films with 30 wt% sensitizer in mCPBC, agreeing with previously reported sensitized systems and verifying the advantage of the sensitizing process. The detailed device configurations, energy level diagrams and molecular structures of the materials used in the devices are depicted in Fig. 4a and S18, and Table S2.†
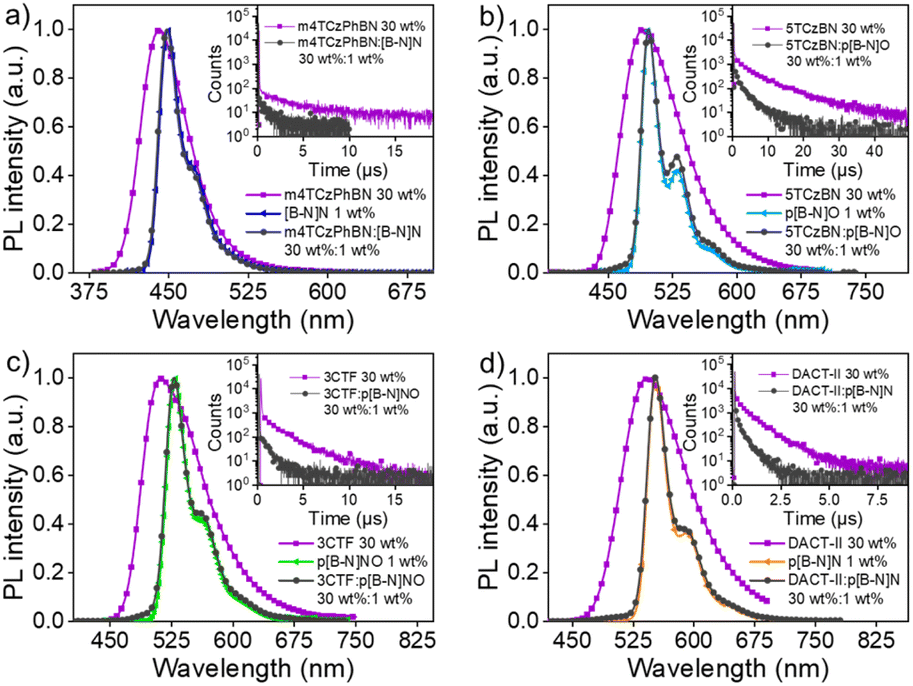 | ||
| Fig. 3 The PL spectra of 1 wt% MR emitter, 30 wt% sensitizer, and 30 wt% sensitizer:1 wt% MR emitter doped in mCPBC. The MR emitter is (a) [B-N]N, (b) p[B-N]O, (c) p[B-N]NO, and (d) p[B-N]N. The insert charts exhibit the transient PL decay curves of the 30 wt% sensitizer and 30 wt% sensitizer:1 wt% MR emitter doped in mCPBC. | ||
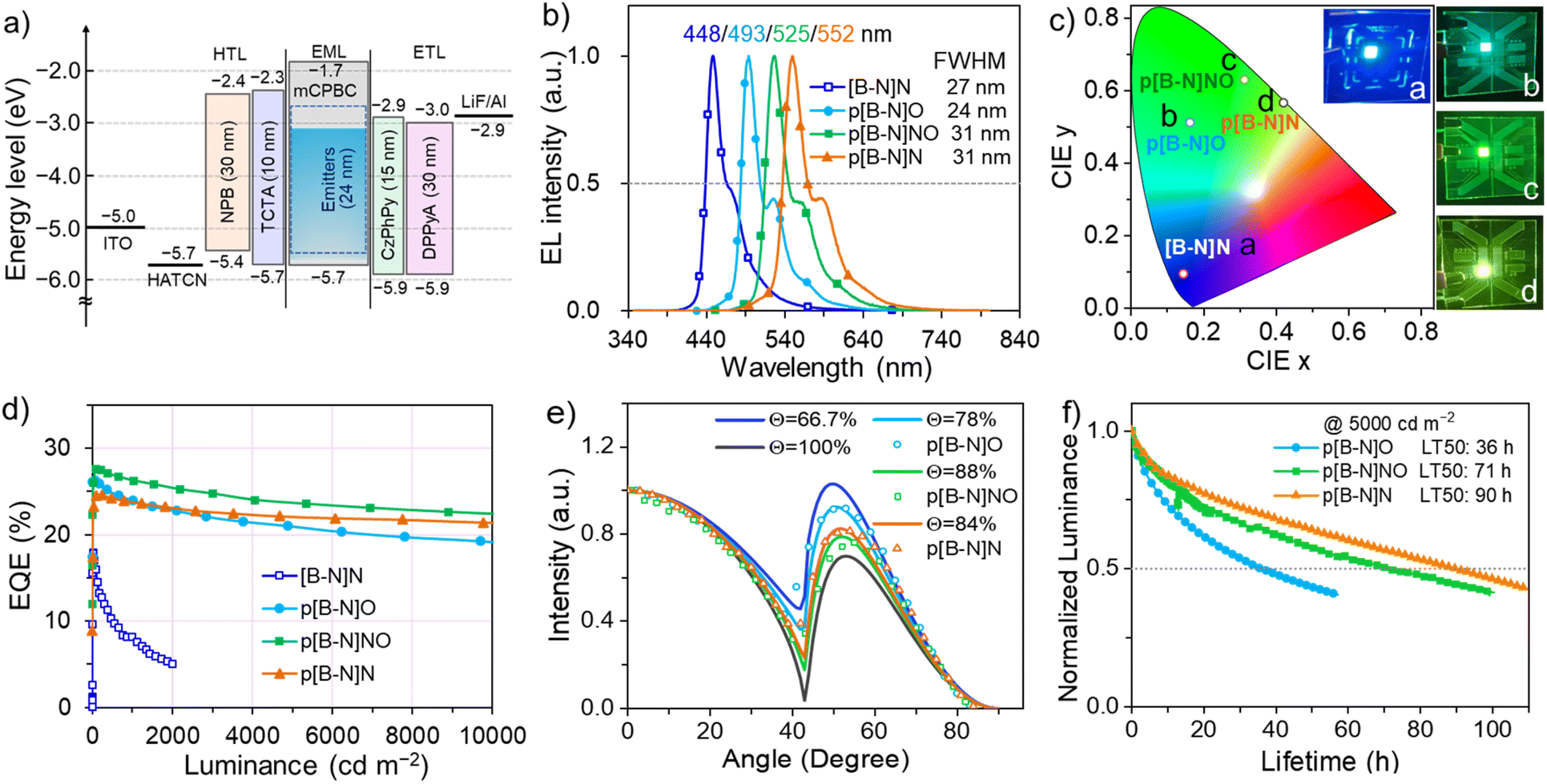 | ||
| Fig. 4 (a) Device configuration with energy-level diagram. (b) The EL spectra of devices were recorded at 1000 cd m−2. (c) The EL color coordinates in the CIE 1931 chromaticity diagram and the photographs show the emission colors of the device. (d) EQE versus current density curves of the emitters-based devices. (e) Angle-dependent PL spectra of three emitters doped into mCPBC. The measured curves are compared to simulated curves with different horizontal dipole ratios Θ// (fully horizontal dipoles: Θ// = 100% and isotropic dipole orientation: Θ// = 66.7%). (f) The EL operational stability of p[B-N]O, p[B-N]NO and p[B-N]N-based devices under a constant current density of 8–10 mA cm−2 (corresponding to 5000 cd m−2). | ||
As depicted in Fig. 4b, the EL characteristics of the devices based on [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N exhibited sharp peaks at 448, 494, 526, and 552 nm, with FWHMs of 27 (0.16), 24 (0.13), 32 (0.14), and 31 (0.13) nm (eV), respectively. These results coincided well with the PL spectra of the doped films. The spectral stability of the devices at elevated operation voltages was also examined and they exhibited entirely identical spectra (Fig. S19†), suggesting that the device emissions solely arose from the emitters. The corresponding CIE 1931 chromaticity coordinates were recorded to be (0.151, 0.079), (0.163, 0.514), (0.306, 0.648), and (0.414, 0.571), representing deep-blue to yellow-green EL spectra, as illustrated in Fig. 4c. Besides, low turn-on voltages in the range of 2.6–3.1 V were observed for the p[B-N]O, p[B-N]NO and p[B-N]N based devices, which also exhibited maximum luminance of over 100000 cd m−2 at ca. 8 V (Table 2).
| Emitter | x wt% | λ EL [nm] | FWHMb [nm (eV)] | V on [V] | L max [cd m−2] | PEmax, 1000, 5000e [lm W−1] | CEmax, 1000, 5000f [cd A−1] | EQEmax, 1000, 5000g [%] | CIEx,ya |
|---|---|---|---|---|---|---|---|---|---|
| a Value recorded at a luminance of around 1000 cd cm−2. b Full width at half maximum of EL given in wavelength and energy. c Turn-on voltage at the luminance of 1 cd m−2. d Maximum luminescence (Lmax). e Maximum PE value at 1000 and 5000 cd cm−2. f Maximum CE, and values recorded at 1000 and 5000 cd cm−2. g Maximum EQE, and values recorded at 1000 and 5000 cd cm−2. | |||||||||
| [B-N]N | 1.0 | 448 | 27.3/0.16 | 3.2 | 2002 | 12.8, 3.4, — | 14.9, 7.1, — | 16.7, 7.6, — | (0.151, 0.079) |
| p[B-N]O | 1.0 | 493 | 24.6/0.13 | 2.6 | 112000 |
73.2, 49.8, 34.4 | 66.8, 60.2, 52.6 | 26.3, 24.0, 21.0 | (0.163, 0.514) |
| p[B-N]NO | 2.0 | 525 | 31.2/0.14 | 2.6 | 106000 |
101.0, 75.9, 53.9 | 102.0, 96.6, 85.8 | 27.6, 26.3, 23.6 | (0.306, 0.648) |
| p[B-N]N | 1.0 | 552 | 31.3/0.13 | 3.1 | 126900 |
71.9, 56.9, 45.1 | 83.0, 79.6, 74.7 | 24.6, 23.6, 22.1 | (0.414, 0.571) |
The EQE, current efficiency (CE) and power efficiency (PE) as a function of luminance for all devices are illustrated in Fig. 4d and the ESI.† The results are summarized in Tables 2 and S4.† Of importance is that the devices based on [B-N]N, p[B-N]O, p[B-N]NO and p[B-N]N displayed excellent EL efficiencies (EQE, CE, and PE) of up to (16.7%, 14.9 cd A−1, 12.8 lm W−1), (26.3%, 66.8 cd A−1, 73.2 lm W−1), (27.6%, 102.0 cd A−1, 101.0 lm W−1), and (24.6%, 83.0 cd A−1, 71.9 lm W−1), respectively. Notably, there is little difference in λEL, FWHM, and EQEmax with increasing dopant concentration from 1 wt% to 3 wt% for p[B-N]O, p[B-N]NO, and p[B-N]N (Fig. S20–S25†), benefiting from the steric structure of the tert-butyl groups on the conjugated skeletons, which effectively suppresses concentration aggregation to achieve a high ΦPL and device performance. What deserves further mentioning is that the p[B-N]O, p[B-N]NO, and p[B-N]N based devices presented low EQE roll-offs, retaining 21.0%, 23.6% and 21.7%, respectively, at a current density of 10 mA cm−2 (ca. 5000 cd m−2). To the best of our knowledge, the efficiency roll-offs of those devices are significantly smaller than those of previously reported high-performance OLEDs based on the classical B,N-MR emitters,11,13 evidencing one of the advantages of this molecular design strategy. The EL decay curves of those devices were recorded to understand the reason for the small efficiency roll-off, as illustrated in Fig. S26.† Interestingly, short exciton lifetimes were observed under electrical excitation, which should help to effectively inhibit the exciton annihilation processes involving triplet–triplet annihilation and triplet-polaron annihilation and so on. As a consequence, small efficiency roll-offs under a high current density can be anticipated.
We further investigated the emitting dipole orientations of these doped films to provide a deeper understanding of the excellent device performances (Fig. 4e). By virtue of the rigid and quasi-planar π-conjugated backbones of p[B-N]O, p[B-N]NO and p[B-N]N, high ratios of horizontal dipole orientations (Θ//s) of 78–88% were obtained, presenting a natural tendency to horizontally align upon evaporation.49 Meanwhile, the transition dipole moments of three emitters were calculated and are illustrated in Fig. S27,† being parallel to the molecular long axes and thus accounting for the high Θ//. Previous work has validated that a high Θ// will greatly enhance the light outcoupling efficiency of devices, which should explain the high efficiency of those three compounds, in addition to their high ΦPLs. Meanwhile, we also evaluated the operational stability of the devices based on p[B-N]O, p[B-N]NO and p[B-N]N, tested at a constant current under ambient conditions with the current density of 8–10 mA cm−2 (ca. 5000 cd m−2). The chart of luminance intensity versus lifetime is displayed in Fig. 4f. The half-lifetimes (LT50s, lifetime to 50% of the initial luminance) of ca. 36, 71, and 90 hours were obtained for the p[B-N]O, p[B-N]NO, and p[B-N]N based devices, respectively. With a degradation acceleration factor (n) of 1.75 obtained from previous reports, the LT50s of all three devices at an initial luminance of 1000 cd m−2 can be extrapolated to 602, 1187, and 1504 hours, respectively. The excellent operation durability of these devices is comparable to or even better than those of previous B,N-doped analogs,13,49–51 indicating the good stability of these B–N doped MR emitters.
Conclusions
In conclusion, a new set of MR emitters containing B–N covalent bonds was developed with a basic para B–π–B skeleton, which was modified by stepwise replacing the nitrogen atoms with oxygen. Such molecular engineering provides a systematic and versatile material design strategy for fine-tuning excited state energies while simultaneously retaining narrow bandwidths and high luminescence efficiencies. Consequently, wide-range emissions (442–545 nm) together with high ΦPLs and small FWHMs of 19–26 nm were obtained. The corresponding OLEDs with sky-blue, green, and yellow-green emissions achieved high maximum EQEs of up to 26.3%, 27.6% and 24.6%, respectively, and exceptionally small efficiency roll-off with high EQEs of 21.0%, 23.6% and 21.7% even at a current density of 10 mA cm−2, greatly outperforming the OLEDs containing classical B,N-PAH analogs. This study provides a family of wide-range color-tunable MR emitters containing B–N covalent bonds, and should stimulate the further development of novel BN-doped emitters by diversifying the material library for future optoelectronic applications.Data availability
All data supporting this study are available in the ESI.†Author contributions
L. D. conceived and supervised the project. D. Z. and L. D. designed the experiments. G. M. synthesized and characterized the emitters, performed OLED fabrication and measurement, and wrote the manuscript. H. D. and J. Z. helped in the synthesis of the emitters. T. H. and Q. W. provided the TADF sensitizers. X. W. and X. Z. helped in theoretical calculations. Y. Z. and T. F. provided suggestions on device fabrication and experiments. D. Y. and D. M. measured the dipole orientations of the emitters. L. D. and D. Z. supervised and reviewed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Guangdong Major Project of Basic and Applied Basic Research (Grant No. 2019B030302009), the National Science Fund of China (Grant No. 22135004, 51903137 and 61890942), and the Guangdong Basic and Applied Basic Research Foundation (2021B1515120041).Notes and references
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678 CrossRef CAS.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912 CrossRef CAS PubMed.
- Y. Liu, X. Xiao, Y. Ran, Z. Bin and J. You, Chem. Sci., 2021, 12, 9408 RSC.
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS.
- S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882 CrossRef CAS PubMed.
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 17442 CrossRef CAS PubMed.
- J. Bian, S. Chen, L. Qiu, R. Tian, Y. Man, Y. Wang, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Adv. Mater., 2022, 34, e2110547 CrossRef PubMed.
- A. Pershin, D. Hall, V. Lemaur, J. C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- X. Lv, J. Miao, M. Liu, Q. Peng, C. Zhong, Y. Hu, X. Cao, H. Wu, Y. Yang, C. Zhou, J. Ma, Y. Zou and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202201588 CAS.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 20498 CrossRef CAS PubMed.
- X. Cai, J. Xue, C. Li, B. Liang, A. Ying, Y. Tan, S. Gong and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202200337 CAS.
- X. F. Luo, H. X. Ni, A. Q. Lv, X. K. Yao, H. L. Ma and Y. X. Zheng, Adv. Opt. Mater., 2022, 2200504 CrossRef CAS.
- I. S. Park, M. Yang, H. Shibata, N. Amanokura and T. Yasuda, Adv. Mater., 2021, e2107951 Search PubMed.
- Y. X. Hu, J. Miao, T. Hua, Z. Huang, Y. Qi, Y. Zou, Y. Qiu, H. Xia, H. Liu, X. Cao and C. Yang, Nat. Photonics, 2022, 16, 803 CrossRef CAS.
- H. Tanaka, S. Oda, G. Ricci, H. Gotoh, K. Tabata, R. Kawasumi, D. Beljonne, Y. Olivier and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 17910 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311 CrossRef CAS PubMed.
- J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156 CrossRef PubMed.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291 CrossRef CAS PubMed.
- Y. Jia, P. Li, K. Liu, C. Li, M. Liu, J. Di, N. Wang, X. Yin, N. Zhang and P. Chen, Chem. Sci., 2022, 13, 11672 RSC.
- Y. Fu, X. Chang, H. Yang, E. Dmitrieva, Y. Gao, J. Ma, L. Huang, J. Liu, H. Lu, Z. Cheng, S. Du, H. J. Gao and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 26115 CrossRef CAS PubMed.
- M. Franceschini, M. Crosta, R. R. Ferreira, D. Poletto, N. Demitri, J. P. Zobel, L. González and D. Bonifazi, J. Am. Chem. Soc., 2022, 144, 21470 CrossRef CAS PubMed.
- S. Nakatsuka, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562 CrossRef CAS PubMed.
- G. Meng, L. Liu, Z. He, D. Hall, X. Wang, T. Peng, X. Yin, P. Chen, D. Beljonne, Y. Olivier, E. Zysman-Colman, N. Wang and S. Wang, Chem. Sci., 2022, 13, 1665 RSC.
- G. Meng, H. Dai, T. Huang, J. Wei, J. Zhou, X. Li, X. Wang, X. Hong, C. Yin, X. Zeng, Y. Zhang, D. Yang, D. Ma, G. Li, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202207293 CAS.
- P. B. Pati, E. Jin, Y. Kim, Y. Kim, J. Mun, S. J. Kim, S. J. Kang, W. Choe, G. Lee, H. J. Shin and Y. S. Park, Angew. Chem., Int. Ed., 2020, 59, 14891 CrossRef CAS PubMed.
- K. Zhao, Z. F. Yao, Z. Y. Wang, J. C. Zeng, L. Ding, M. Xiong, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2022, 144, 3091 CrossRef CAS PubMed.
- L. Jiang, Y. Wang, D. Tan, X. Chen, T. Ma, B. Zhang and D. T. Yang, Chem. Sci., 2022, 13, 5597 RSC.
- A. S. Scholz, J. G. Massoth, M. Bursch, J. M. Mewes, T. Hetzke, B. Wolf, M. Bolte, H. W. Lerner, S. Grimme and M. Wagner, J. Am. Chem. Soc., 2020, 142, 11072 CrossRef CAS PubMed.
- S. Nakatsuka, N. Hatakeyama and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562 CrossRef CAS PubMed.
- X. Wang, F. Zhang, J. Liu, R. Tang, Y. Fu, D. Wu, Q. Xu, X. Zhuang, G. He and X. Feng, Org. Lett., 2013, 15, 5714 CrossRef CAS PubMed.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 1902142 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- Z. Shuai, Chin. J. Chem., 2020, 38, 1223 CrossRef CAS.
- H. L. Lee, W. J. Chung and J. Y. Lee, Small, 2020, 16, 1907569 CrossRef CAS PubMed.
- V. V. Patil, H. L. Lee, I. Kim, K. H. Lee, W. J. Chung, J. Kim, S. Park, H. Choi, W. J. Son, S. O. Jeon and J. Y. Lee, Adv. Sci., 2021, 8, 2101137 CrossRef CAS PubMed.
- G. Meng, D. Zhang, J. Wei, Y. Zhang, T. Huang, Z. Liu, C. Yin, X. Hong, X. Wang, X. Zeng, D. Yang, D. Ma, G. Li and L. Duan, Chem. Sci., 2022, 13, 5622–5630 RSC.
- Y. T. Lee, C. Y. Chan, M. Tanaka, M. Mamada, K. Goushi, X. Tang, Y. Tsuchiya, H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2022, 10, 2200682 CrossRef CAS.
- K. R. Naveen, S. J. Hwang, H. Lee and J. H. Kwon, Adv. Electron. Mater., 2021, 8, 2101114 CrossRef.
- X. Wang, Y. Zhang, H. Dai, G. Li, M. Liu, G. Meng, X. Zeng, T. Huang, L. Wang, Q. Peng, D. Yang, D. Ma, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202206916 CAS.
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050 CrossRef CAS PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Adv. Mater., 2020, 32, 1908355 CrossRef CAS PubMed.
- D. Zhang, M. Cai, Y. Zhang, D. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145 RSC.
- T. Huang, Q. Wang, S. Xiao, D. Zhang, Y. Zhang, C. Yin, D. Yang, D. Ma, Z. Wang and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 23771 CrossRef CAS PubMed.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- R. Braveenth, H. Lee, J. D. Park, K. J. Yang, S. J. Hwang, K. R. Naveen, R. Lampande and J. H. Kwon, Adv. Funct. Mater., 2021, 31, 2105805 CrossRef CAS.
- Y. Xu, Q. Wang, J. Wei, X. Peng, J. Xue, Z. Wang, S. J. Su and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202204652 CAS.
- F. Liu, Z. Cheng, Y. Jiang, L. Gao, H. Liu, H. Liu, Z. Feng, P. Lu and W. Yang, Angew. Chem., Int. Ed., 2022, 61, e202116927 CAS.
- S. Nam, J. W. Kim, H. J. Bae, Y. M. Maruyama, D. Jeong, J. Kim, J. S. Kim, W. J. Son, H. Jeong, J. Lee, S. G. Ihn and H. Choi, Adv. Sci., 2021, 8, e2100586 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06343c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |