
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeometry-dependent valence tautomerism, magnetism, and electrical conductivity in 1D iron–tetraoxolene chains†
Ashlyn A.
Kamin
 a,
Ian P.
Moseley
b,
Jeewhan
Oh
c,
E. J.
Brannan
a,
Paige M.
Gannon
a,
Werner
Kaminsky
a,
Joseph M.
Zadrozny
b and
Dianne J.
Xiao
*a
a,
Ian P.
Moseley
b,
Jeewhan
Oh
c,
E. J.
Brannan
a,
Paige M.
Gannon
a,
Werner
Kaminsky
a,
Joseph M.
Zadrozny
b and
Dianne J.
Xiao
*a
aDepartment of Chemistry, University of Washington, Seattle, Washington 98195, USA. E-mail: djxiao@uw.edu
bDepartment of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA
cDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 27th March 2023
Abstract
Redox-active tetraoxolene ligands such as 1,4-dihydroxybenzoquinone provide access to a diversity of metal–organic architectures, many of which display interesting magnetic behavior and high electrical conductivity. Here, we take a closer look at how structure dictates physical properties in a series of 1D iron–tetraoxolene chains. Using a diphenyl-derivatized tetraoxolene ligand (H2Ph2dhbq), we show that the steric profile of the coordinating solvent controls whether linear or helical chains are exclusively formed. Despite similar ligand environments, only the helical chain displays temperature-dependent valence tautomerism, switching from (FeII)(Ph2dhbq2−) to (FeIII)(Ph2dhbq3˙−) at temperatures below 203 K. The stabilization of ligand radicals leads to exceptionally strong magnetic exchange coupling (J = −230 ± 4 cm−1). Meanwhile, the linear chains are more amenable to oxidative doping, leading to Robin–Day class II/III mixed-valency and an increase in electrical conductivity by nearly three orders of magnitude. While previous studies have focused on the effects of changing metal and ligand identity, this work highlights how altering the metal–ligand connectivity can be a similarly powerful tool for tuning materials properties.
Introduction
Owing to their strong metal chelating behavior and rich redox activity, dioxolene-based ligands (i.e., catechols, semiquinones, and quinones) have served prominent roles across inorganic, bioinorganic, and materials chemistry (Fig. 1).1,2 In the context of new materials design, metal–dioxolene linkages have been recognized as privileged building blocks due to their ability to couple high crystallinity and good chemical stability with intriguing multifunctional materials properties. For example, metal–dioxolene units have been shown to display stimuli-dependent metal-to-ligand electron transfer (“valence tautomerism”), leading to switchable physical properties.3–6 Strong metal–radical coupling and magnetic ordering can be engendered by their capacity to stabilize ligand radicals,6–8 and high electrical conductivity can result from the close energy alignment between metal and ligand orbitals.9–12 Thanks to these desirable properties, metal–dioxolene-based coordination solids and metal–organic frameworks have been investigated for diverse applications, including spintronics,5,6 electrocatalysis,13 and electrochemical energy storage.14,15Extended metal–dioxolene materials are typically accessed by fusing two or more dioxolene units to form a single conjugated polytopic ligand. Well-known examples include 1,4-dihydroxybenzoquinone (H2dhbq),16,17 which contains two fused dioxolene units (also known as tetraoxolene) (Fig. 1), and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP),10 which contains three fused dioxolene units. We have been interested in studying extended 1D metal–tetraoxolene chains with the goal of identifying structure–property relationships that can be translated to higher dimensional 2- and 3D networks. Simple 1D chains are ideally suited for probing how changes to the metal, ligand, and metal–ligand connectivity influence electronic and magnetic coupling across the metal–tetraoxolene linkages. However, a barrier to the systematic synthesis and structural modification of such materials is the complex coordination chemistry of tetraoxolene ligands. For example, while H2dhbq and its substituted derivatives (H2R2dhbq, where R = F, Cl, Br, NO2, etc.) have been shown to bind metals to form 1-, 2-, and 3D networks (Fig. 1),16 how to select for each structure remains poorly understood. In the case of iron, 3D cubic frameworks,11 2D honeycomb sheets,8,18–25 1D linear chains,49,50 and 1D zig-zag chains6,26 have all been reported.
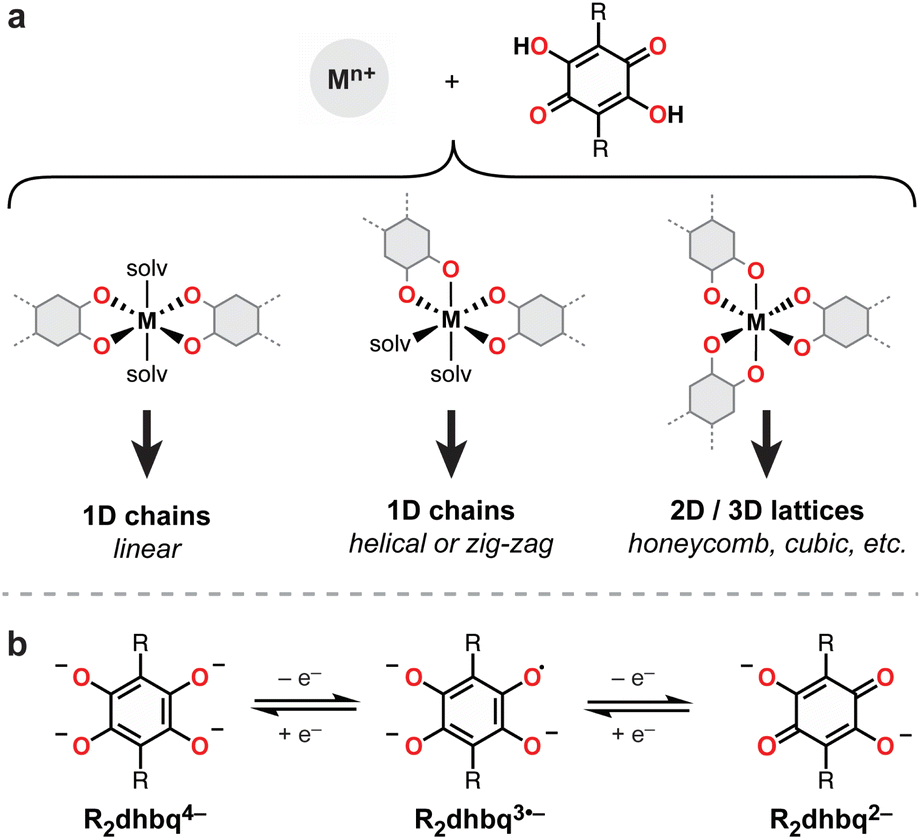 | ||
| Fig. 1 (a) Binding modes for H2R2dhbq ligands and the resulting metal–organic structures. (b) Redox series of R2dhbqn− ligands. The ligand contains a radical in the R2dhbq3˙− state. | ||
Here we show how new linear and helical iron–tetraoxolene chain architectures can be achieved using a more sterically encumbered ligand, H2Ph2dhbq (2,5-dihydroxy-3,6-dipheny-1,4-benzoquinone). Both linear and helical structures can be predictably accessed simply by adjusting the steric bulk of the coordinating solvent. Despite identical bridging ligands, switching the chain structure dramatically alters how charges are distributed across the metal–tetraoxolene linkage. Unlike the linear chain, which remains (FeII)(Ph2dhbq2−) at all observed temperatures, the helical chain undergoes valence tautomerization, adopting a (FeIII)(Ph2dhbq3˙−) configuration at temperatures below 203 K. The stabilization of ligand radicals leads to exceptionally strong antiferromagnetic metal–ligand exchange coupling (J = −230 ± 4 cm−1). Further, we show that the linear chains are more easily oxidatively doped, leading to orders-of-magnitude improvement in electrical conductivity. Together, this work illustrates how changing the metal–ligand connectivity can be a powerful, yet often overlooked, tool to alter materials properties.
Results and discussion
Synthetic control over chain architecture
We hypothesized that functionalizing 1,4-dihydroxybenzoquinone with bulkier substituents would disfavor tris-chelation and limit the formation of 2D and 3D networks. Therefore, we targeted the 3,6-diphenyl substituted derivative, H2Ph2dhbq, also known as polyporic acid (Fig. 2).27 The reduced tetrahydroxy form of this ligand, H4Ph2dhbq, was synthesized in four steps from bromanilic acid with an overall yield of 75%. H4Ph2dhbq was then subjected to aerial oxidation to give the target ligand, H2Ph2dhbq. In addition to simplifying the overall structural landscape, the phenyl substituents in H2Ph2dhbq provide a convenient synthetic handle for future ligand modification. The peripheral phenyl groups can be readily functionalized with electron-donating and withdrawing groups, allowing the steric and electronic properties of the ligand to be tuned. While the coordination chemistry of H2Ph2dhbq has been briefly explored in the past,28–32 this work represents the first fully characterized series of metal–organic chains based on this ligand.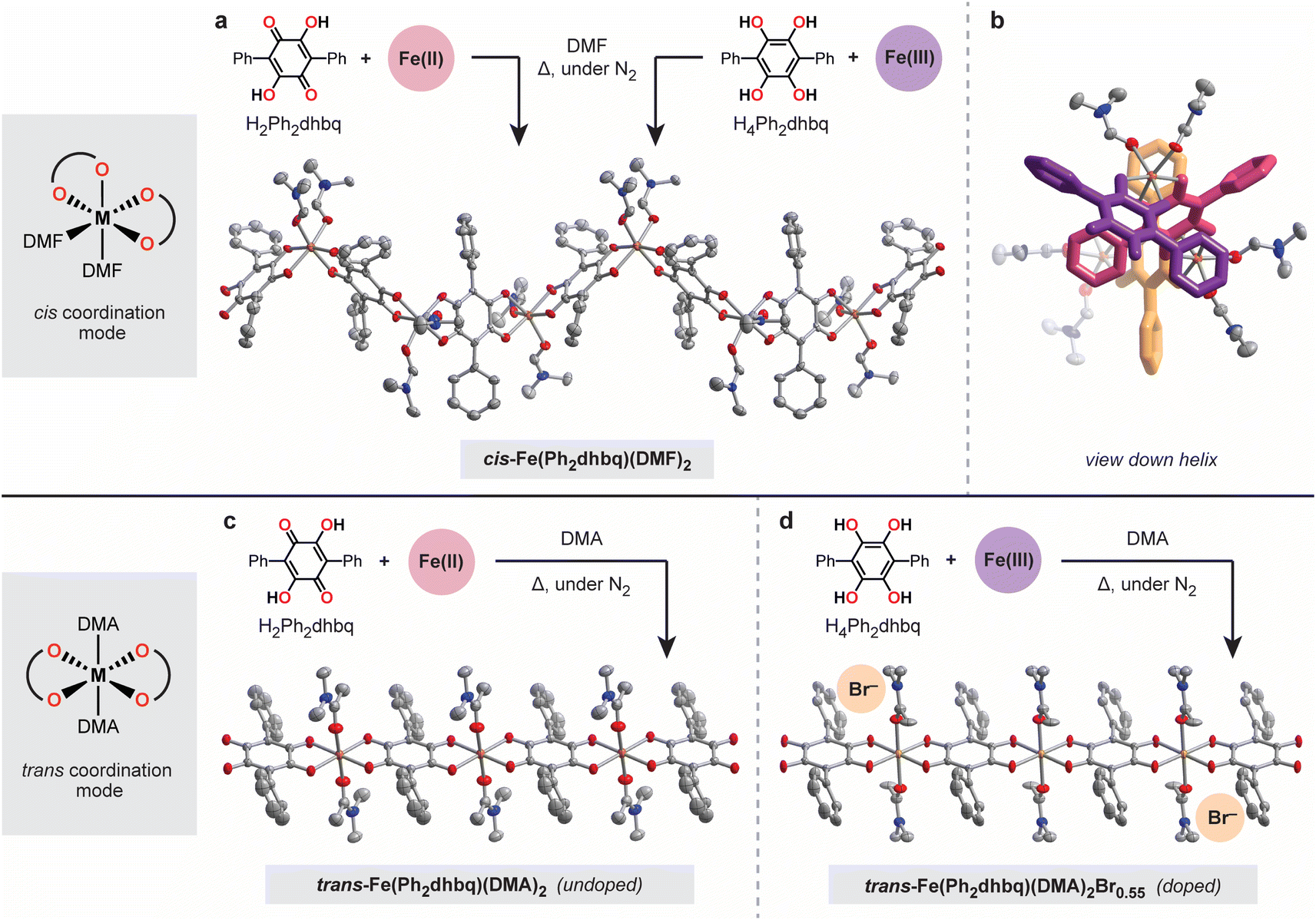 | ||
| Fig. 2 Overview of the synthesis of helical and linear iron–tetraoxolene chains. (a) Routes for the synthesis of the helical chain, cis-Fe(Ph2dhbq)(DMF)2. (b) View down the crystallographic c-axis showing the three-fold helicity of cis-Fe(Ph2dhbq)(DMF)2; terphenyl ligands are highlighted in purple, pink, and orange. (c and d) Routes for the syntheses of the linear chains, trans-Fe(Ph2dhbq)(DMA)2 and trans-Fe(Ph2dhbq)(DMA)2Br0.55, the latter of which is oxidatively doped with FeIII. Thermal ellipsoids are rendered at the 50% probability level. Fe, C, N, and O atoms are represented by orange, grey, blue, and red, respectively. Hydrogen atoms and disorder in both the phenyl groups and coordinated solvent molecules have been omitted for clarity. | ||
The greater steric bulk of the diphenyl substituted ligand provides an opportunity to probe the nuances of 1D chain formation without interference from competing 2D and 3D phases. With H2Ph2dhbq in hand, we were able to carry out a comprehensive survey of different synthesis conditions that led to two important structural findings (Fig. 2). First, we found that the steric bulk of the solvent employed during synthesis dictates the overall chain architecture. Second, we observed that the initial oxidation states of the metal and ligand starting materials greatly impact crystallite size and compositional purity. As we will describe in subsequent sections, these differences in chain architecture lead to significant differences in the physical properties of these chains, including the electrical conductivity and magnetism.
The first surprising structural outcome we observed was that distinct helical and linear chain architectures could be achieved by subtly tuning the steric bulk of the coordinating solvent (Fig. 2). Specifically, heating H2Ph2dhbq and FeCl2 in dimethylformamide (DMF) at 120 °C under an inert atmosphere provides the helical cis-Fe(Ph2dhbq)(DMF)2 chain (Fig. 2a and b). In this structure, the quasi-octahedral iron sites are coordinated by two cis DMF molecules and bridged by bis-bidentate Ph2dhbq ligands to form a helix. To our knowledge, these represent the first chiral 1D chains synthesized from H2dhbq or its substituted derivatives. On the other hand, changing the solvent from DMF to a slightly bulkier dimethylacetamide (DMA) provides trans-Fe(Ph2dhbq)(DMA)2 (Fig. 2c). While the iron sites remain quasi-octahedral in this structure, the bulkier DMA molecules now adopt a trans configuration, leading to linear rather than helical chains.
Given that both FeII and H2Ph2dhbq are redox-active, we next probed how the initial metal and ligand oxidation states impact chain formation. Previous reports have shown that changing the initial ligand oxidation state can lead to improved crystallinity or even entirely new phases.33,34 We hypothesized that similar chain structures could be achieved by combining the fully reduced tetrahydroxy ligand, H4Ph2dhbq, with an FeIII source, as FeIII can bind the ligand as well as serve as an in situ oxidant. Indeed, heating H4Ph2dhbq and FeCl3 in DMF under an inert atmosphere led to the formation of single crystals that are structurally and compositionally identical to those obtained from H2Ph2dhbq/FeII, but with significantly larger crystallite sizes (Fig. 2a). Given the improved crystallinity, all subsequent characterization of cis-Fe(Ph2dhbq)(DMF)2 will focus on samples prepared using H4Ph2dhbq/FeIII.
In contrast to the helical chain, which remains compositionally pure despite changes to the initial metal and ligand oxidation states, the linear chain is more readily over-oxidized. Heating H4Ph2dhbq and FeBr3 at 120 °C in DMA under an inert atmosphere leads to oxidative doping of the 1D chain and incorporation of additional charge-balancing Br− anions. Single crystal X-ray crystallography confirmed the formation of the desired linear chain geometry, and detailed elemental analysis revealed the formula to be trans-Fe(Ph2dhbq)(DMA)2Br0.55 (Fig. 2d). Although the bromine atoms are not observed directly in the crystal structure, it is likely that they reside in the infinite channels along the crystallographic c-axis and their contribution to the diffraction pattern was removed along with that of disordered solvent using SQUEEZE during refinement of the crystal structure.35
Structural characterization and oxidation state assignments
The R2dhbqn− family of ligands has been shown to adopt three different charge states in transition metal complexes, specifically n = 2–4 (Fig. 1b). Structural investigations are particularly helpful for teasing out the distribution of charges, as the bond lengths within the metal–dioxolene chelate ring are highly sensitive to the metal and ligand oxidation states. Specifically, the M–O bond lengths provide insight into the oxidation state of metal, while the C–O bond lengths are reflective of the charges on the ligand.1,16Close analysis of the bond lengths in cis-Fe(Ph2dhbq)(DMF)2 at 100 K and 250 K suggests the chain undergoes valence tautomerism, switching between (FeIII)(Ph2dhbq3˙−) at low temperatures and (FeII)(Ph2dhbq2−) at high temperatures (Fig. 3a). At 100 K, the average Fe–O bond length is 1.991(13) Å, consistent with high-spin FeIII. In addition, the average C–O bond length at this temperature is 1.305(13) Å, which is longer than the ∼1.24–1.28 Å typically observed in first-row transition metal structures containing delocalized R2dhbq2− ligands.16 In contrast, at 250 K, the average Fe–O bond lengths is 2.09(3) Å which is clearly lengthened relative to the Fe–O bond length observed at 100 K. While we were unable to observe a statistically significant change in the average C–O bond length between these temperatures due to lower data quality at 250 K, the average C–O bond length of 1.28(3) Å is in the range expected of R2dhbq2− ligands. Overall, the bond lengths observed at 100 K are more characteristic of FeIII metal centers bound to R2dhbq3˙− radical ligands, while the bond lengths at 250 K resemble previous structures containing FeII metal centers and R2dhbq2− ligands.6,16 While iron-based valence tautomerism is relatively rare,5 similar behavior was recently observed by Harris and coworkers in a structurally related Fe-based chain synthesized from chloranilic acid, [Me4N]2[Fe(Cl2dhbq)Cl2].6 The authors of this earlier work hypothesized that the FeIII state is stabilized by the presence of anionic, π-donating Cl− ligands. In contrast, our system contains only simple DMF ligands, suggesting that the relative arrangement of the R2dhbqn− ligands may be the critical factor, rather than the properties of any additional coordinating ligands.
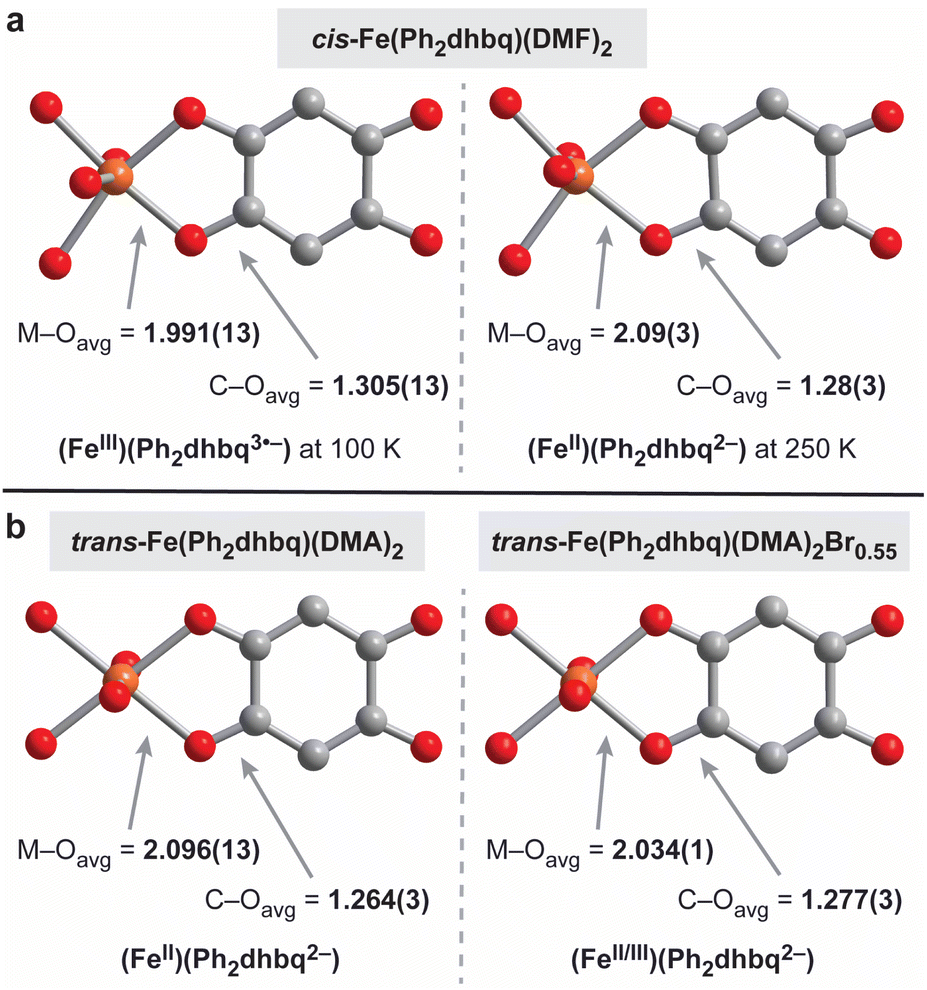 | ||
| Fig. 3 (a) Coordination mode and average bond lengths in Å from the single crystal structures of cis-Fe(Ph2dhbq)(DMF)2 at both 100 and 250 K. (b) Coordination mode and average bond lengths in Å from the single crystal structures of trans-Fe(Ph2dhbq)(DMF)2 (undoped) versus trans-Fe(Ph2dhbq)(DMF)2Br0.55 (oxidatively doped), both at 250 K. | ||
Unlike cis-Fe(Ph2dhbq)(DMF)2, no structural changes indicative of valence tautomerism are observed in trans-Fe(Ph2dhbq)(DMA)2 (Fig. 3b). The Fe–O and C–O bond lengths are consistent with high-spin FeII metal centers and Ph2dhbq2− ligands at all temperatures. Single crystal X-ray diffraction data for trans-Fe(Ph2dhbq)(DMA)2 were collected at 250 K using a synchrotron X-ray source. The Fe–O bond lengths in trans-Fe(Ph2dhbq)(DMA)2 average 2.096(13) Å and C–O bond lengths average 1.264(3) Å.
Finally, the average bond lengths in oxidatively doped trans-Fe(Ph2dhbq)(DMA)2Br0.55 are in between those expected for FeII and FeIII (Fig. 3b).21 The average Fe–O bond length is 2.034(1) Å and the average C–O bond length is 1.277(3) Å at 250 K. Neither of these bond lengths change appreciably (by more than ±0.8%) when the crystal is cooled to 100 K (Table S3†). The C–O bond lengths match the bond lengths observed for cis-Fe(Ph2dhbq)(DMF)2 at 250 K, suggesting that despite the additional positive charge on the iron centers, the ligand is still in the Ph2dhbq2− oxidation state. No significant changes are observed in the νC–O region of the IR spectrum, consistent with metal-centered oxidation (Fig. S6 and S7†).
Mössbauer spectroscopy
To gain further insight into the distribution of charges in our iron–tetraoxolene chains, Mössbauer spectroscopy was used to probe the local chemical environments of the iron centers in cis-Fe(Ph2dhbq)(DMF)2, trans-Fe(Ph2dhbq)(DMA)2, and trans-Fe(Ph2dhbq)(DMA)2Br0.55 (Fig. 4).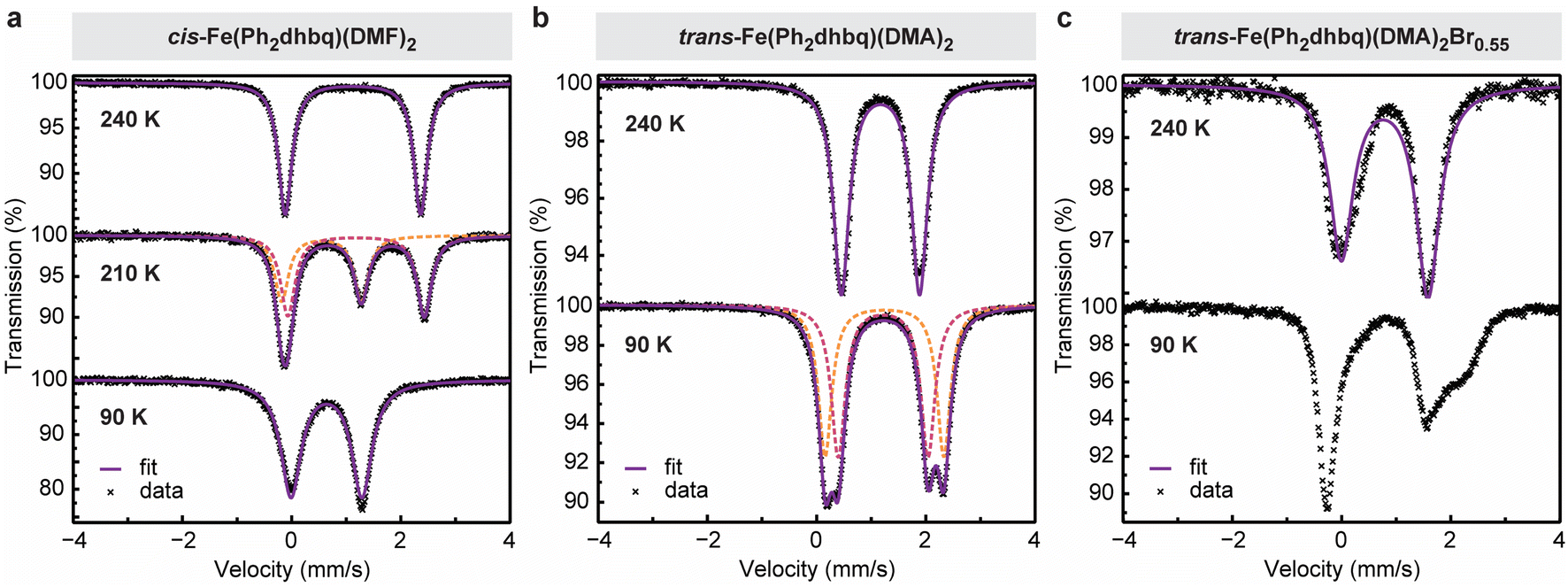 | ||
| Fig. 4 Variable-temperature Mössbauer spectra of (a) cis-Fe(Ph2dhbq)(DMF)2, (b) trans-Fe(Ph2dhbq)(DMA)2, and (c) trans-Fe(Ph2dhbq)(DMA)2Br0.55. Experimental data are represented as individual black crosses, fits for individual doublets within each spectrum are represented by dashed lines, and the overall fit for each spectrum is represented by a solid line. | ||
For cis-Fe(Ph2dhbq)(DMF)2, the Mössbauer spectrum at 240 K shows a single quadrupole doublet with an isomer shift (δ) of 1.12 mm s−1 and quadrupole splitting (|ΔEQ|) of 2.48 mm s−1, which are characteristic of high-spin, octahedral FeII (Fig. 4a). As the sample is cooled to 90 K, the spectrum displays parameters that are more consistent with high-spin, octahedral FeIII (δ = 0.64 mm s−1 and |ΔEQ| = 1.30 mm s−1).6,21 In between these two temperatures, it is possible to resolve three peaks which can be fit by two quadrupole doublets representing a mixture of FeII and FeIII species in roughly equal proportions (Table S5†). Together, these observations are fully consistent with valence tautomerization between (FeII)(Ph2dhbq2−) at high temperatures and (FeIII)(Ph2dhbq3˙−) at low temperatures, as suggested by our structural analysis.
For trans-Fe(Ph2dhbq)(DMA)2, the Mössbauer spectrum does not demonstrate any temperature-dependent change in the iron oxidation state. Instead, the data reveals a single quadrupole doublet with δ = 1.17 mm s−1 and |ΔEQ| = 1.43 mm s−1 at 240 K, which splits into two slightly offset quadrupole doublets at 90 K (δ = 1.22, 1.24 and |ΔEQ| = 1.64, 2.17 mm s−1) (Fig. 4b). All three doublets can unambiguously be assigned to high-spin, octahedral FeII. Again, these assignments agree with the bond lengths observed by single crystal X-ray diffraction.
Unlike the sharp quadrupole doublets observed in the previous two chains, the Mössbauer spectra of oxidatively doped trans-Fe(Ph2dhbq)(DMA)2Br0.55 shows an asymmetric quadrupole doublet at 240 K that broadens as the temperature is lowered (Fig. 4c). The spectrum at 240 K can be fit approximately by a single quadrupole doublet with δ = 0.79 mm s−1 and |ΔEQ| = 1.59 mm s−1. The presence of an averaged quadrupole doublet with an isomer shift that is in between what is typically observed for high-spin FeII and FeIII suggests that the charge is delocalized at this temperature, with electron transfer between the iron centers occurring at a rate that is faster than the Mössbauer time scale (∼10−7 s).21,36,37 This is in agreement with the crystallographically measured average Fe–O bond lengths, which also reside in between typical FeII and FeIII ranges (Fig. 3b). Decreasing the temperature to 90 K (the lowest temperature accessible on our instrument) slows down the rate of charge transfer, leading to increased line broadening.36 Overall, the Mössbauer data obtained for trans-Fe(Ph2dhbq)(DMA)2Br0.55 closely matches the behavior observed in previously reported Robin–Day class II/III mixed-valent iron complexes,36,37 where the extent of electronic coupling lies in an intermediate regime in between fully localized and delocalized.38 The presence of iron-based mixed-valency is further supported by UV-vis-NIR diffuse reflectance spectroscopy. The spectrum for trans-Fe(Ph2dhbq)(DMA)2Br0.55 displays a broad absorption band in the near-IR region with νmax ∼ 4200 cm−1 (Fig. S8†). We tentatively attribute this feature, which is not present in the undoped samples, to intervalence charge transfer (IVCT) between FeII and FeIII centers. The observed band has an asymmetric shape with a sharper cutoff at lower energies, consistent with a class II/III assignment.11,38 Similar IVCT bands in the near-IR region have been observed in other mixed-valence iron complexes and extended materials.36,39
Dc magnetic susceptibility
The structural and spectroscopic analyses described above show that the three chain architectures possess starkly different charge distributions. The magnetic behavior of each chain system was probed using direct-current (dc) magnetic susceptibility measurements to test how these distributions affect the magnetic properties. We discuss cis-Fe(Ph2dhbq)(DMF)2 first. Consistent with the valence tautomerism observed by both X-ray crystallography and Mössbauer spectroscopy, dc magnetic susceptibility measurements of cis-Fe(Ph2dhbq)(DMF)2 show strong temperature-dependent magnetic behavior (Fig. 5). At 300 K, χMT = 3.59 cm3 K mol−1, which is consistent with the expected value for uncoupled high-spin FeII ions (S = 2). As the temperature is decreased, a sigmoidal feature centered at 202.5 K is observed and χMT increases rapidly to 10.4 cm3 K mol−1 at 194 K. This feature is consistent with valence tautomerization between high-spin FeII with Ph2dhbq2− and high-spin FeIII with Ph2dhbq3˙−. A similar feature was observed by Harris et al. at the valence tautomerization transition temperature (T1/2) of the related chain, [Me4N]2[Fe(Cl2dhbq)Cl2].6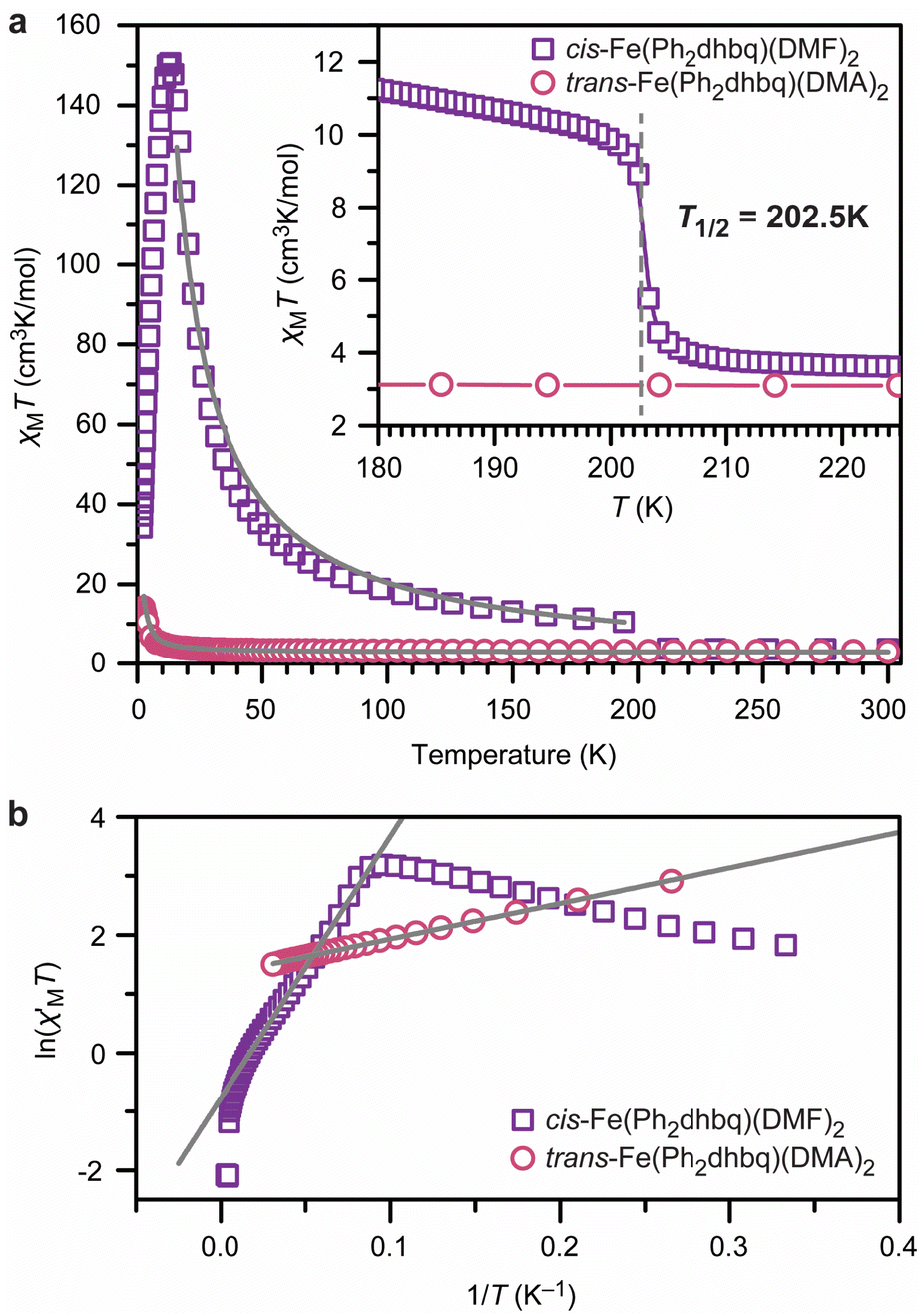 | ||
| Fig. 5 (a) Dc magnetic susceptibility (χMT) measurements for cis-Fe(Ph2dhbq)(DMF)2 (purple squares) and trans-Fe(Ph2dhbq)(DMA)2 (pink circles) under an applied field of 1000 G. The solid grey lines are best fits with Jcis = −230 cm−1 and Jtrans = +0.30 cm−1. Inset: zoom in of the valence tautomerization temperature (T1/2) of the cis chain. (b) Variable-temperature zero-field dc magnetic susceptibility (χM′) of cis-Fe(Ph2dhbq)(DMF)2 and trans-Fe(Ph2dhbq)(DMA)2. The grey lines are linear fits that yield correlation lengths of 12 for the cis chain and 4 for the trans chain. Data were collected at zero dc field with an applied ac field frequency of 1 Hz. See main text and ESI† for fitting details. | ||
The presence of the ligand radical, Ph2dhbq3˙−, at lower temperatures should give rise to strong metal–ligand magnetic coupling. Indeed, below the valence tautomerization transition of cis-Fe(Ph2dhbq)(DMF)2, the value of χMT exceeds the value expected for uncoupled high-spin FeIII (S = 5/2) with Ph2dhbq3˙− radicals (S = 1/2). This discrepancy likely indicates that magnetic coupling between the metal ions and ligand radicals is enabled as soon as valence tautomerization takes place. The best simulations of this data suggest a strong antiferromagnetic metal–radical exchange coupling constant of J = −230 ± 4 cm−1. Due to the differing numbers of unpaired electrons on the metal versus the ligand, the χMT increases with decreasing temperature, reaching a maximum at 13 K (χMTmax = 150 cm3 K mol−1). For strongly magnetically coupled 1D systems, this increase in susceptibility is indicative of an increasing intrachain spin correlation length (2ξ) between domain walls.40 Below 13 K, the magnetic behavior becomes governed by defect sites that limit the chain growth.41 As this chain growth becomes limited, χMT decreases precipitously, reaching 34 cm3 K mol−1 at 1.8 K.
To the best of our knowledge, the magnitude of the exchange coupling constant observed in cis-Fe(Ph2dhbq)(DMF)2 (J = −230 ± 4 cm−1) is the largest reported for any 1D chain, and is almost three times that observed for [Me4N]2[Fe(Cl2dhbq)Cl2] (J = −81 cm−1).6,42 The relative increase in the magnitude of the exchange coupling constant observed in chains constructed from Ph2dhbq versus Cl2dhbq may be attributable to improved orbital overlap between the metal ion and ligand radicals.43 While the average Fe–O bond lengths can be assigned to FeIII in both cases, the Fe–O bond length in cis-Fe(Ph2dhbq)(DMF)2 is 1.991(13) Å, which is shorter than the value of 2.027(2) Å observed in the Cl2dhbq chain,6 which should promote stronger magnetic coupling.
In contrast to the cis chain, dc magnetic susceptibility measurements of trans-Fe(Ph2dhbq)(DMA)2 exhibit no signatures of valence tautomerism. Instead, the susceptibility data reflect the expected behavior for a chain of ferromagnetically-coupled FeII centers (Fig. 5). Best fits of the data suggest a modest ferromagnetic exchange coupling, with J = +0.30 ± 0.05 cm−1. The trans-Fe(Ph2dhbq)(DMA)2Br0.55 chain exhibits similar magnetic behavior to trans-Fe(Ph2dhbq)(DMA)2, neither of which display the valence tautomerization behavior of the cis chain (Fig. S10 and S11†). Based on the Mössbauer and UV-vis-NIR analysis, the dc susceptibility of trans-Fe(Ph2dhbq)(DMA)2Br0.55 was fit assuming contributions from both S = 2 and S = 5/2 ions (see ESI† for further discussion). We note that the simulation of magnetic data in one-dimensional mixed valence systems is far from trivial.44 Nevertheless, best fits using this method yielded an exchange coupling value (J = +0.64 ± 0.01 cm−1) similar in magnitude to trans-Fe(Ph2dhbq)(DMA)2, yet still small relative to cis-Fe(Ph2dhbq)(DMF)2.
Magnetic hysteresis was not observed for any of the chains (Fig. S18–S20†), and charging curves reveal negligible interchain interactions owing to the absence of a low-field inflection in magnetization with increasing field (Fig. S21–S23†).
Ac magnetic susceptibility
Alternating current (ac) magnetic susceptibility measurements of all chains were performed at zero applied magnetic field to test for slow magnetic relaxation of the coupled chains and to provide a second quantification of the magnetic coupling. The out-of-phase ac susceptibility (χM′′) data show slow magnetic relaxation for both cis-Fe(Ph2dhbq)(DMF)2 and trans-Fe(Ph2dhbq)(DMA)2, as evidenced by a peak appearing between frequencies of 1 and 1500 Hz which shifts to higher frequencies with increasing temperature (Fig. S12–S14†). However, the relative intensity of χM′′ to χM′ in both cis-Fe(Ph2dhbq)(DMF)2 and trans-Fe(Ph2dhbq)(DMA)2 suggests that most of the sample is not undergoing slow relaxation. In addition to the weak signal, the broadness and asymmetry of the observed peaks indicate a wide window of accessible relaxation times, likely due to polydispersity in coupled chain lengths.The in-phase ac susceptibility (χM′), measured as a function of temperature at a fixed ac frequency (the ‘zero-field dc susceptibility’), reveals the effective lengths of the chains in these solids (Fig. 5b and S10†) when plotted as ln(χM′T) versus 1/T.40,45 Here, the linear region was fit (see ESI† for details) to yield the domain wall energy (Δζ). These values are considerably larger for the cis chain versus the trans chains, with 37 cm−1 for cis-Fe(Ph2dhbq)(DMF)2 and 6 cm−1 for trans-Fe(Ph2dhbq)(DMA)2. The extension of the chains with increased exchange coupling follows expectations for 1D molecular magnetic materials.40,45 Likewise, trans-Fe(Ph2dhbq)(DMA)2Br0.55 exhibits a larger domain wall energy relative to the undoped chain (12 cm−1), which is expected given the increased exchange coupling.
The correlation length (n) was determined as described in the ESI† and gives the average number of contiguous metal ions coupled via the exchange interaction (J). As expected, for the cis chain we obtain n = 12 while trans-Fe(Ph2dhbq)(DMA)2 yields n = 4 (Table S12†). Interestingly, the doped trans-Fe(Ph2dhbq)(DMA)2Br0.55 yields the same correlation length as the undoped chain (n = 4), despite possessing larger exchange coupling as well as a larger domain wall energy. Overall, the magnetic characterization demonstrates clear differences between each of the three chains.
Electrical conductivity
The introduction of mixed-valency, centered on either the ligand or metal, has been shown to be an effective strategy to improve the electrical conductivity of metal–organic materials.46,47 This literature precedent motivated us to investigate the conductivity differences between cis-Fe(Ph2dhbq)(DMF)2 and trans-Fe(Ph2dhbq)(DMA)2, which both contain FeII at room temperature, and the mixed-valence trans-Fe(Ph2dhbq)(DMA)2Br0.55, where the iron centers have been partially oxidized.Two-probe pressed pellet conductivities for all chains were collected at ambient temperature in air immediately following removal of the samples from the glovebox. The electrical conductivities of cis-Fe(Ph2dhbq)(DMF)2 and trans-Fe(Ph2dhbq)(DMA)2 were found to be similar, with measured values of 2.31 ± 0.13 × 10−8 and 6.0 ± 1.5 × 10−9 S cm−1, respectively. In contrast, the electrical conductivity of mixed-valence trans-Fe(Ph2dhbq)(DMA)2Br0.55 was found to be 2.5 ± 1.3 × 10−6 S cm−1, nearly three orders of magnitude higher. We note that pressed pellet conductivity measurements are strongly affected by grain boundaries, particularly in highly anisotropic systems such as the 1D chains reported here. Nevertheless, the dramatic improvement suggests that the conductivity of these chains may be further improved through controlled oxidative doping. A more comprehensive study on the conductivity of these materials, including single crystal measurements and controlled oxidation and reduction experiments, will be the subject of future work.
Conclusions
The combined synthetic and characterization efforts described here provide a new perspective on the rational design of tetraoxolene-based extended materials. While previous studies have shown that changes to the metal ion and tetraoxolene ligand greatly impact electronic structure and magnetic behavior,17,48 this work shows that dramatic changes in physical properties can be observed even when the metal and bridging ligand identities remain constant. Slight alterations to the chain geometry are sufficient to change the preferred distribution of oxidation states across the metal–tetraoxolene linkages, turning on properties such as valence tautomerism and strong magnetic coupling. Future work will focus on using these 1D chains as building blocks to construct higher dimensionality porous frameworks with unique geometries and, hence, tunable and diverse electronic and magnetic properties.Data availability
Additional experimental data supporting this article are included in the ESI.†Author contributions
AAK and DJX designed the research and wrote the manuscript; AAK and EJB carried out the synthesis, routine characterization, and conductivity measurements; PMG and WK collected and refined the crystallographic data; JO collected and analysed the Mössbauer data; IPM and JMZ collected and analysed the magnetic data. All authors interpreted the results and contributed to reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthetic work described here was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021966 (DJX). The characterization work was partially supported by the Arnold and Mabel Beckman Foundation through a Beckman Young Investigator Award (DJX) and the Research Corporation for Scientific Advancement (JMZ). AAK and IPM are supported by NSF graduate research fellowships. AAK was additionally supported by the state of Washington through a University of Washington (UW) Clean Energy Institute graduate fellowship. EJB was supported by a Mary Gates research scholarship. The X-ray facility of the UW Department of Chemistry is supported by NSF Award Number CHE-0840520. This research utilized resources of the Advanced Light Source (ALS), which is a DOE Office of Science User Facility under Contract Number DE-AC02-05CH11231. We thank Prof. Theodore A. Betley (Harvard University) for facilitating the collection of Mössbauer data, and Drs Simon Teat and Nick Settineri for assisting with the collection of synchrotron single crystal X-ray diffraction data at ALS beamline 12.2.1.Notes and references
- C. G. Pierpont and R. M. Buchanan, Coord. Chem. Rev., 1981, 38, 45–87 CrossRef CAS.
- W. P. Griffith, Transition Met. Chem., 1993, 18, 250–256 CrossRef CAS.
- C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99–125 CrossRef CAS.
- E. Evangelio and D. Ruiz-Molina, Eur. J. Inorg. Chem., 2005, 2005, 2957–2971 CrossRef.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- J. A. DeGayner, K. Wang and T. D. Harris, J. Am. Chem. Soc., 2018, 140, 6550–6553 CrossRef CAS PubMed.
- A. Dei, D. Gatteschi, L. Pardi and U. Russo, Inorg. Chem., 1991, 30, 2589–2594 CrossRef CAS.
- I.-R. Jeon, B. Negru, R. P. Van Duyne and T. D. Harris, J. Am. Chem. Soc., 2015, 137, 15699–15702 CrossRef CAS PubMed.
- J. A. DeGayner, I.-R. Jeon, L. Sun, M. Dincă and T. D. Harris, J. Am. Chem. Soc., 2017, 139, 4175–4184 CrossRef CAS PubMed.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- L. E. Darago, M. L. Aubrey, C. J. Yu, M. I. Gonzalez and J. R. Long, J. Am. Chem. Soc., 2015, 137, 15703–15711 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- E. M. Miner, L. Wang and M. Dincă, Chem. Sci., 2018, 9, 6286–6291 RSC.
- M. E. Ziebel, C. A. Gaggioli, A. B. Turkiewicz, W. Ryu, L. Gagliardi and J. R. Long, J. Am. Chem. Soc., 2020, 142, 2653–2664 CrossRef CAS PubMed.
- J. W. Gittins, C. J. Balhatchet, Y. Chen, C. Liu, D. G. Madden, S. Britto, M. J. Golomb, A. Walsh, D. Fairen-Jimenez, S. E. Dutton and A. C. Forse, J. Mater. Chem. A, 2021, 9, 16006–16015 RSC.
- S. Kitagawa and S. Kawata, Coord. Chem. Rev., 2002, 224, 11–34 CrossRef CAS.
- M. L. Mercuri, F. Congiu, G. Concas and S. A. Sahadevan, Magnetochemistry, 2017, 3, 17 CrossRef.
- S. Benmansour, A. Abhervé, P. Gómez-Claramunt, C. Vallés-García and C. J. Gómez-García, ACS Appl. Mater. Interfaces, 2017, 9, 26210–26218 CrossRef CAS PubMed.
- C. J. Kingsbury, B. F. Abrahams, D. M. D'Alessandro, T. A. Hudson, R. Murase, R. Robson and K. F. White, Cryst. Growth Des., 2017, 17, 1465–1470 CrossRef CAS.
- J. Chen, Y. Sekine, Y. Komatsumaru, S. Hayami and H. Miyasaka, Angew. Chem., Int. Ed., 2018, 57, 12043–12047 CrossRef CAS PubMed.
- S. A. Sahadevan, A. Abhervé, N. Monni, C. Sáenz de Pipaón, J. R. Galán-Mascarós, J. C. Waerenborgh, B. J. C. Vieira, P. Auban-Senzier, S. Pillet, E.-E. Bendeif, P. Alemany, E. Canadell, M. L. Mercuri and N. Avarvari, J. Am. Chem. Soc., 2018, 140, 12611–12621 CrossRef CAS PubMed.
- J. Chen, Y. Sekine, A. Okazawa, H. Sato, W. Kosaka and H. Miyasaka, Chem. Sci., 2020, 11, 3610–3618 RSC.
- R. Murase, C. J. Commons, T. A. Hudson, G. N. L. Jameson, C. D. Ling, K. S. Murray, W. Phonsri, R. Robson, Q. Xia, B. F. Abrahams and D. M. D'Alessandro, Inorg. Chem., 2020, 59, 3619–3630 CrossRef CAS PubMed.
- Y. Sekine, J. Chen, N. Eguchi and H. Miyasaka, Chem. Commun., 2020, 56, 10867–10870 RSC.
- M. P. van Koeverden, B. F. Abrahams, D. M. D'Alessandro, P. W. Doheny, C. Hua, T. A. Hudson, G. N. L. Jameson, K. S. Murray, W. Phonsri, R. Robson and A. L. Sutton, Chem. Mater., 2020, 32, 7551–7563 CrossRef CAS.
- B. F. Abrahams, K. D. Lu, B. Moubaraki, K. S. Murray and R. Robson, J. Chem. Soc., Dalton Trans., 2000, 1793–1797 RSC.
- P. R. Shildneck and R. Adams, J. Am. Chem. Soc., 1931, 53, 2373–2379 CrossRef CAS.
- R. L. Frank, G. R. Clark and J. N. Coker, J. Am. Chem. Soc., 1950, 72, 1827–1829 CrossRef CAS.
- Ch. Robl and A. Weiss, Zeitschrift für anorganische und allgemeine Chemie, 1987, 546, 152–160 CrossRef CAS.
- C. Robl and W. F. Kuhs, J. Solid State Chem., 1988, 74, 21–26 CrossRef CAS.
- K. Heinze, G. Huttner, L. Zsolnai, A. Jacobi and P. Schober, Chem.–Eur. J., 1997, 3, 732–743 CrossRef CAS.
- A. Garci, J.-P. Mbakidi, V. Chaleix, V. Sol, E. Orhan and B. Therrien, Organometallics, 2015, 34, 4138–4146 CrossRef CAS.
- B. F. Abrahams, T. A. Hudson, L. J. McCormick and R. Robson, Cryst. Growth Des., 2011, 11, 2717–2720 CrossRef CAS.
- L. Wang, R. J. Papoular, N. E. Horwitz, J. Xie, A. Sarkar, D. Campisi, N. Zhao, B. Cheng, G. L. Grocke, T. Ma, A. S. Filatov, L. Gagliardi and J. S. Anderson, Angew. Chem., Int. Ed., 2022, 61, 45 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- A. I. Gaudette, I.-R. Jeon, J. S. Anderson, F. Grandjean, G. J. Long and T. D. Harris, J. Am. Chem. Soc., 2015, 137, 12617–12626 CrossRef CAS PubMed.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2686 CrossRef CAS PubMed.
- D. M. D'Alessandro and F. R. Keene, Chem. Rev., 2006, 106, 2270–2298 CrossRef PubMed.
- R. Murase, B. F. Abrahams, D. M. D'Alessandro, C. G. Davies, T. A. Hudson, G. N. L. Jameson, B. Moubaraki, K. S. Murray, R. Robson and A. L. Sutton, Inorg. Chem., 2017, 56, 9025–9035 CrossRef CAS PubMed.
- C. Coulon, H. Miyasaka and R. Clérac, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, Springer, Berlin, Heidelberg, 2006, pp. 163–206 Search PubMed.
- K. S. Pedersen, A. Vindigni, R. Sessoli, C. Coulon and R. Clérac, in Molecular Magnetic Materials, John Wiley & Sons, Ltd, 2017, pp. 131–159 Search PubMed.
- X. Liu, X. Feng, K. R. Meihaus, X. Meng, Y. Zhang, L. Li, J.-L. Liu, K. S. Pedersen, L. Keller, W. Shi, Y.-Q. Zhang, P. Cheng and J. R. Long, Angew. Chem., 2020, 132, 10697–10705 CrossRef.
- C. Hua, J. A. DeGayner and T. D. Harris, Inorg. Chem., 2019, 58, 7044–7053 CrossRef CAS PubMed.
- Z. G. Soos, in Low-Dimensional Cooperative Phenomena: The Possibility of High-Temperature Superconductivity, ed. H. J. Keller, Springer US, Boston, MA, 1975, pp. 45–64 Search PubMed.
- H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420–3437 CrossRef CAS PubMed.
- L. S. Xie, L. Sun, R. Wan, S. S. Park, J. A. DeGayner, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2018, 140, 7411–7414 CrossRef CAS PubMed.
- R. Murase, C. F. Leong and D. M. D'Alessandro, Inorg. Chem., 2017, 56, 14373–14382 CrossRef CAS PubMed.
- M. E. Ziebel, L. E. Darago and J. R. Long, J. Am. Chem. Soc., 2018, 140, 3040–3051 CrossRef CAS PubMed.
- S. Kawata, S. Kitagawa, H. Kumagai, T. Ishiyama, K. Honda, H. Tobita, K. Adachi and M. Katada, Chem. Mater., 1998, 10, 3902–3912 CrossRef CAS.
- K. Kon, K. Uchida, K. Fuku, S. Yamanaka, B. Wu, D. Yamazui, H. Iguchi, H. Kobayashi, Y. Gambe, I. Honma and S. Takaishi, ACS Appl. Mater. Interfaces, 2021, 13, 38188–38193 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2215980–2215984. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06392a |
| This journal is © The Royal Society of Chemistry 2023 |