DOI:
10.1039/D2SC06425A
(Edge Article)
Chem. Sci., 2023,
14, 4114-4119
Palladium-catalyzed synthesis of 4-sila-4H-benzo[d][1,3]oxazines by intramolecular Hiyama coupling†
Received
22nd November 2022
, Accepted 21st March 2023
First published on 22nd March 2023
Abstract
A palladium-catalyzed synthesis of 4-sila-4H-benzo[d][1,3]oxazines, silicon-switched analogs of biologically relevant 4H-benzo[d][1,3]oxazines, was developed by the intramolecular Hiyama coupling of 3-amido-2-(arylsilyl)aryl triflates. The present reaction is an unusual way of utilizing the Hiyama coupling, enabling the synthesis of value-added organosilanes as the main products. The intramolecular nature of transmetalation with inversion of the stereochemistry at the silicon center was revealed by the mechanistic investigation, and an asymmetric variant of this process was also demonstrated to give silicon-stereogenic 4-sila-4H-benzo[d][1,3]oxazines with relatively high enantioselectivity.
Introduction
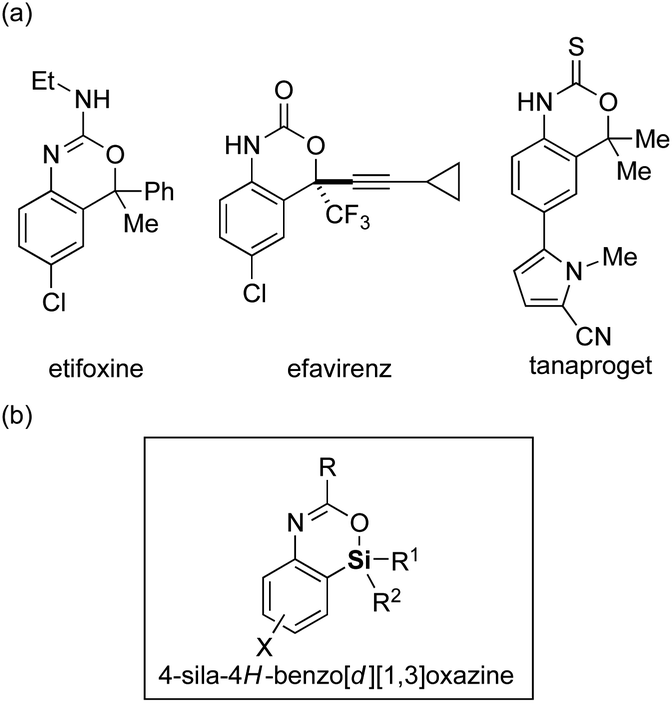
Replacement of a carbon atom of functional organic molecules by a silicon atom (known as “silicon switch”) represents one of the effective ways of improving/modifying their biological activities or physical properties by taking advantage of the difference between carbon and silicon such as in the covalent radius, lipophilicity, and electronegativity, while keeping the similarity as congeners.1 In particular, because nitrogen- and/or oxygen-containing heterocycles often appear as structural motifs for pharmaceuticals and optoelectronic materials, introduction of a silicon atom to these heterocycles would be highly attractive. Although such a strategy had been implemented for the past decades, available synthetic methods and accessible structures of silicon-containing heterocycles are currently still limited.2,3
Among the nitrogen- and oxygen-containing heterocycles, 4H-benzo[d][1,3]oxazines and their derivatives constitute a useful class4 and are often found as core structures of pharmaceuticals such as etifoxine (anxiolytic and anticonvulsant drug),5 efavirenz (antiretroviral medicine),6 and tanaproget (nonsteroidal progestin)7 (Fig. 1a). However, their silicon-switched analogs, sila-4H-benzo[d][1,3]oxazines, have never been reported to date presumably due to the lack of their synthetic methods. To remedy this methodological deficiency, herein we describe a palladium-catalyzed efficient synthesis of 4-sila-4H-benzo[d][1,3]oxazines (Fig. 1b) from 3-amido-2-(arylsilyl)aryl triflates by the use of intramolecular Hiyama coupling, including its asymmetric variant via enantioselective transmetalation.8
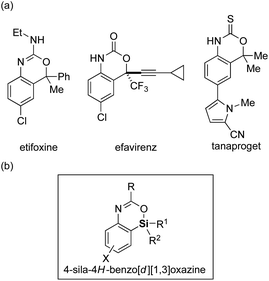 |
| | Fig. 1 (a) Examples of biologically active 4H-benzo[d][1,3]oxazine derivatives and (b) the structure of 4-sila-4H-benzo[d][1,3]oxazine. | |
Results and discussion
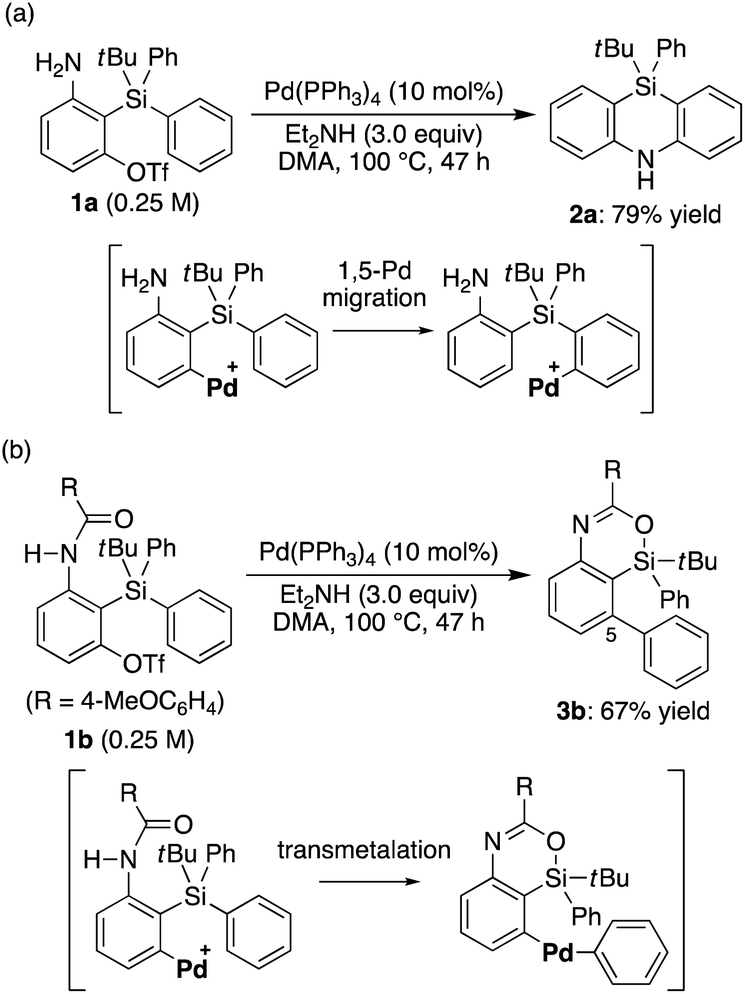
Recently, we reported that the reaction of 3-amino-2-(arylsilyl)aryl triflates such as 1a in the presence of a palladium catalyst selectively gave 5,10-dihydrophenazasilines such as 2avia 1,5-palladium migration followed by an intramolecular C–N bond formation (Scheme 1a).9 In stark contrast, we newly found that, when we employed N-acylated variant 1b as the substrate under the same reaction conditions, 5-phenylated 4-sila-4H-benzo[d][1,3]oxazine 3b was selectively obtained without forming the corresponding 5,10-dihydrophenazasiline (Scheme 1b). This reaction presumably took place through intramolecular transmetalation of a phenyl group from silicon to palladium with the aid of the acyl group on nitrogen instead of 1,5-palladium migration.10 The overall process here can therefore be described as an intramolecular Hiyama coupling reaction, but it is quite different from the typical Hiyama coupling, where a carbon–carbon bond formation between organic (pseudo)halides and organosilanes is the primary process, and the resulting silicon moiety after the reaction is considered as a low-value byproduct and usually treated as waste.11 On the other hand, the present reaction of 1b to give 3b is a rare example in that the Hiyama coupling provides a silicon-containing heterocycle as the value-added main product.12
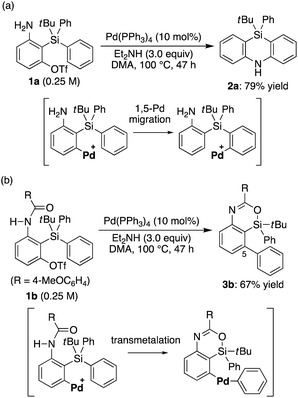 |
| | Scheme 1 (a) Synthesis of 5,10-dihydrophenazasiline from 3-amino-2-(arylsilyl)aryl triflate (previous work) and (b) initial finding for the synthesis of 4-sila-4H-benzo[d][1,3]oxazine from 3-amido-2-(arylsilyl)aryl triflate (this work). | |
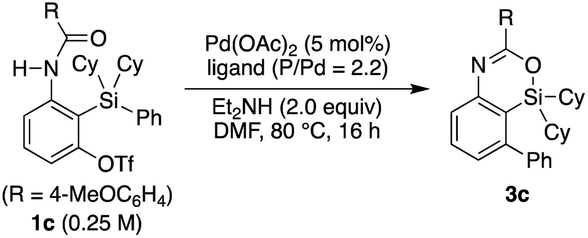
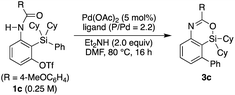
Based on this finding, we initiated our studies for the palladium-catalyzed synthesis of 4-sila-4H-benzo[d][1,3]oxazines by the intramolecular Hiyama coupling of 3-amido-2-(arylsilyl)aryl triflates. As a starting point, we employed 1c as the model substrate and examined the reaction conditions toward 4-sila-4H-benzo[d][1,3]oxazine 3c (Table 1). When the reaction was conducted in the presence of Pd(OAc)2 (5 mol%) and Et2NH (2.0 equiv.) in DMF at 80 °C for 16 h, product 3c was obtained in a moderate yield of 46% (entry 1). The use of PPh3 (11 mol%) as the ligand for palladium did not change the reaction outcome (47% yield; entry 2). In contrast, a significantly higher yield of 3c was achieved by using PCy3 as the ligand (94% yield; entry 3), although the use of bulkier P(tBu)3 resulted in the decrease of the yield (31% yield; entry 4). It was also found that the use of bisphosphine ligands such as dppf and binap could promote this reaction effectively as well (86–93% yield; entries 5 and 6).
Table 1 Palladium-catalyzed reaction of 1c to give 3c
|
|
| Entry |
Ligand |
Conversiona (%) |
Yieldb (%) |
|
Determined by 1H NMR against internal standard (MeNO2).
Isolated yield.
PR3·HBF4/Et2NH was used.
|
| 1 |
None |
50 |
46 |
| 2 |
PPh3 |
58 |
47 |
|
3
|
PCy
3
|
100
|
94
|
| 4c |
P(tBu)3 |
31 |
31 |
| 5 |
dppf |
100 |
93 |
| 6 |
binap |
100 |
86 |
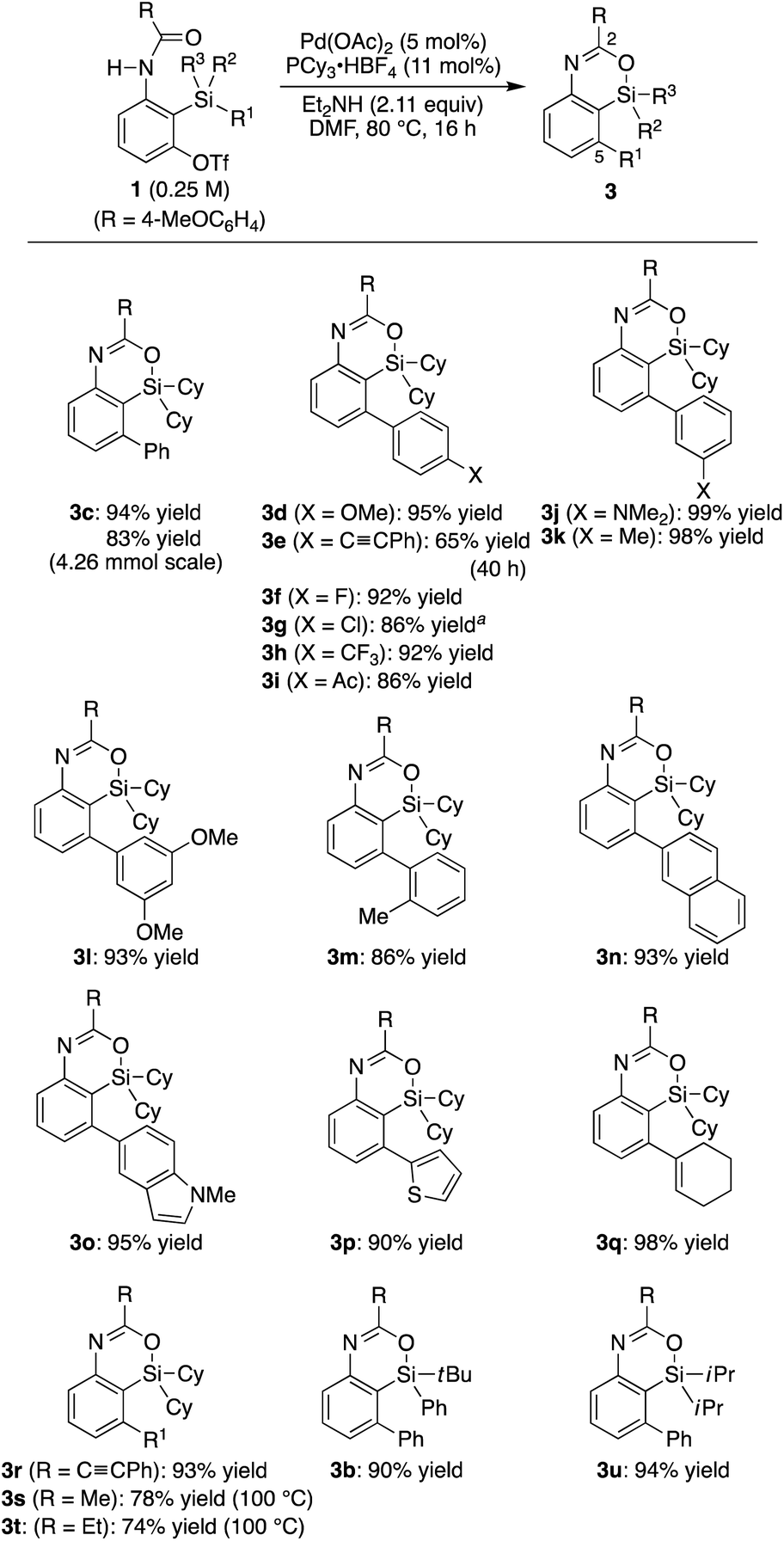
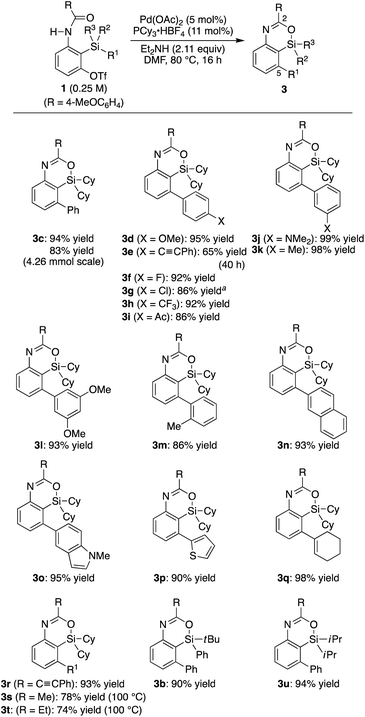
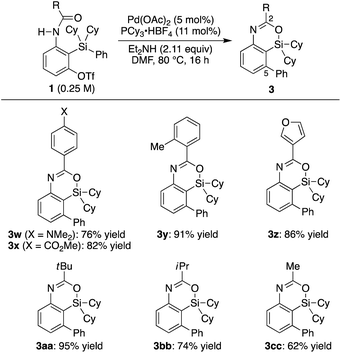
Under the conditions in
Table 1, entry 3, various substituted aryl groups on the silicon atom could be transferred to the 5-position to give corresponding 4-sila-4
H-benzo[
d][1,3]oxazines
3d–m in good to high yields irrespective of the electronic and steric nature of the aryl groups as summarized in
Scheme 2 (65–99% yield). 2-Naphthyl, 5-indolyl, 2-thienyl, 1-cyclohexenyl, and phenylethynyl groups were also applicable as the migrating group from silicon to carbon, giving products
3n–r in similarly high yields (90–98% yield). In addition to these (hetero)aryl, alkenyl, and alkynyl groups, methyl and ethyl groups were successfully transferred as well to give compounds
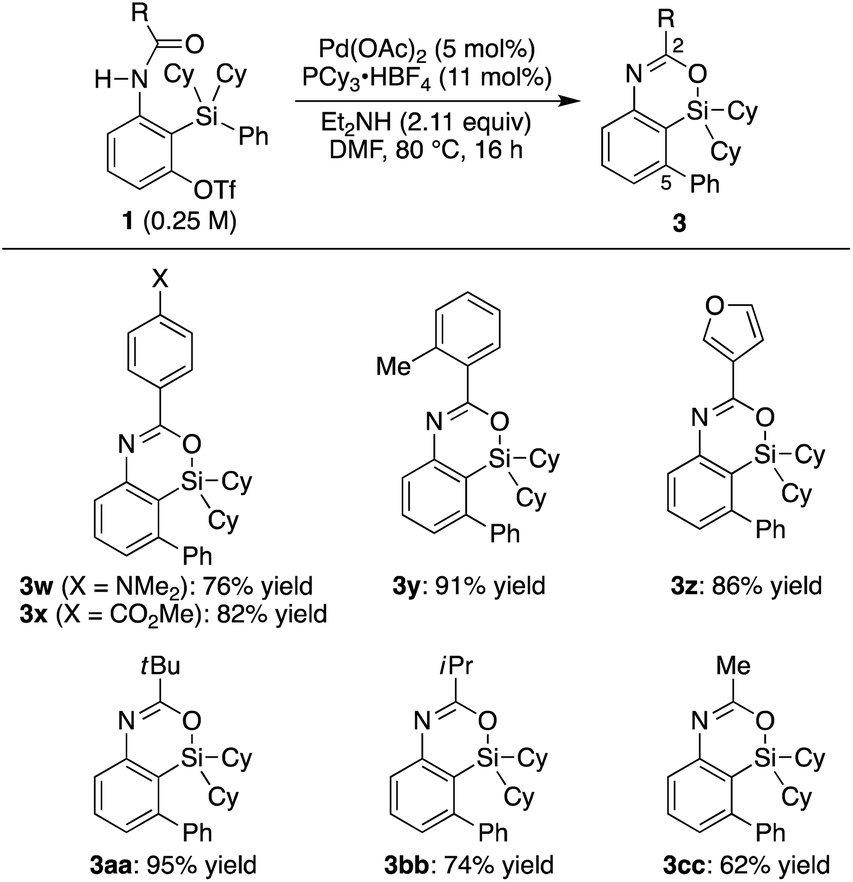
3s–t in 74–78% yield by conducting the reaction at an elevated temperature (100 °C).
13,14 Regarding the ‘spectator’ substituents on the silicon atom, relatively bulky groups were found to be suitable such as
tert-butyl and phenyl (
3b) as well as diisopropyl (
3u) other than dicyclohexyl, but sterically less hindered ones such as dimethyl were not applicable to the present catalysis.
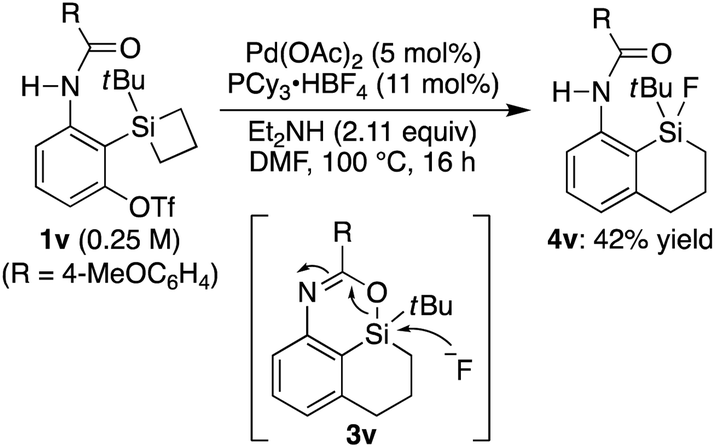
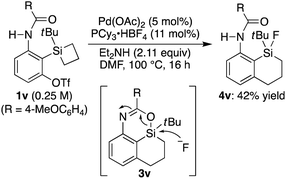
15 On the other hand, substrate
1v having a silacyclobutane as the silicon moiety underwent a ring-expansion reaction with concomitant cleavage of the initially formed silicon–oxygen bond by the fluoride, which is most likely derived from BF
4− of the phosphine ligand salt, to give compound
4v as the major product in 42% yield (
eqn (1)).
12a,16,17 With regard to the substituent of the carbonyl group on nitrogen, various electronically and sterically different (hetero)aryl groups and alkyl groups could be tolerated, and corresponding 2-substituted 4-sila-4
H-benzo[
d][1,3]oxazines
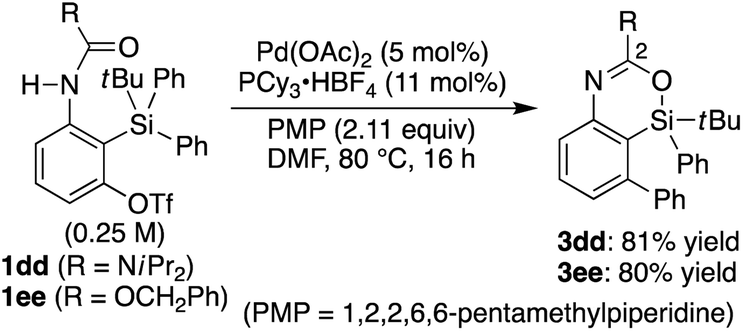
3w–cc were obtained in 62–95% yield (
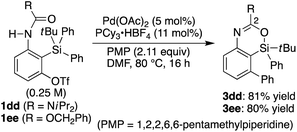
Scheme 3). In addition, diisopropyl urea
1dd and benzyl carbamate
1ee could also be employed in the present catalysis to give 2-heteroatom-substituted 4-sila-4
H-benzo[
d][1,3]oxazines
3dd and
3ee under the slightly modified reaction conditions (
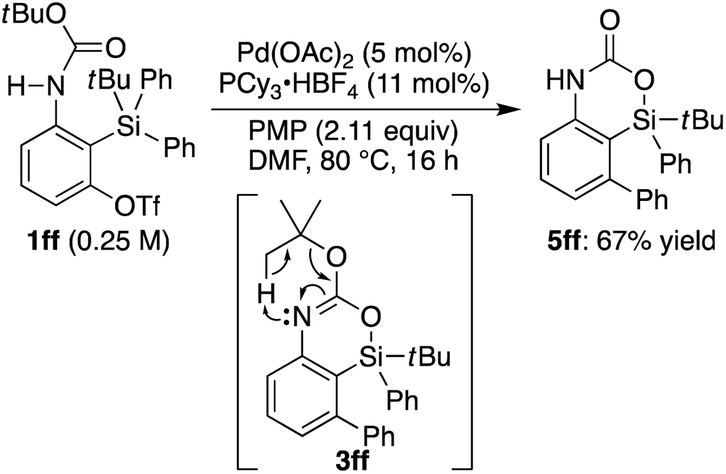
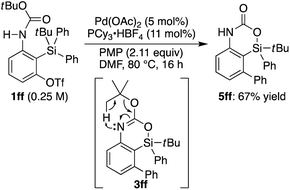
eqn (2)). It is worth noting that
tert-butyl carbamate
1ff gave 4-sila-1,4-dihydro-2
H-benzo[
d][1,3]oxazin-2-one
5ff as the major product, presumably through intramolecular elimination of the
tert-butyl group of initially formed 4-sila-4
H-benzo[
d][1,3]oxazine
3ff under the reaction conditions (
eqn (3)).
| |  | (1) |
| |  | (2) |
| |  | (3) |
 |
| | Scheme 2 Palladium-catalyzed synthesis of 3: scope of R1–R3 on silicon. 0.15 mmol of 1 was used unless otherwise noted. a5.5 mol% of binap was used as the ligand instead of PCy3·HBF4/Et2NH. | |
 |
| | Scheme 3 Palladium-catalyzed synthesis of 3: scope of R on carbonyl. | |
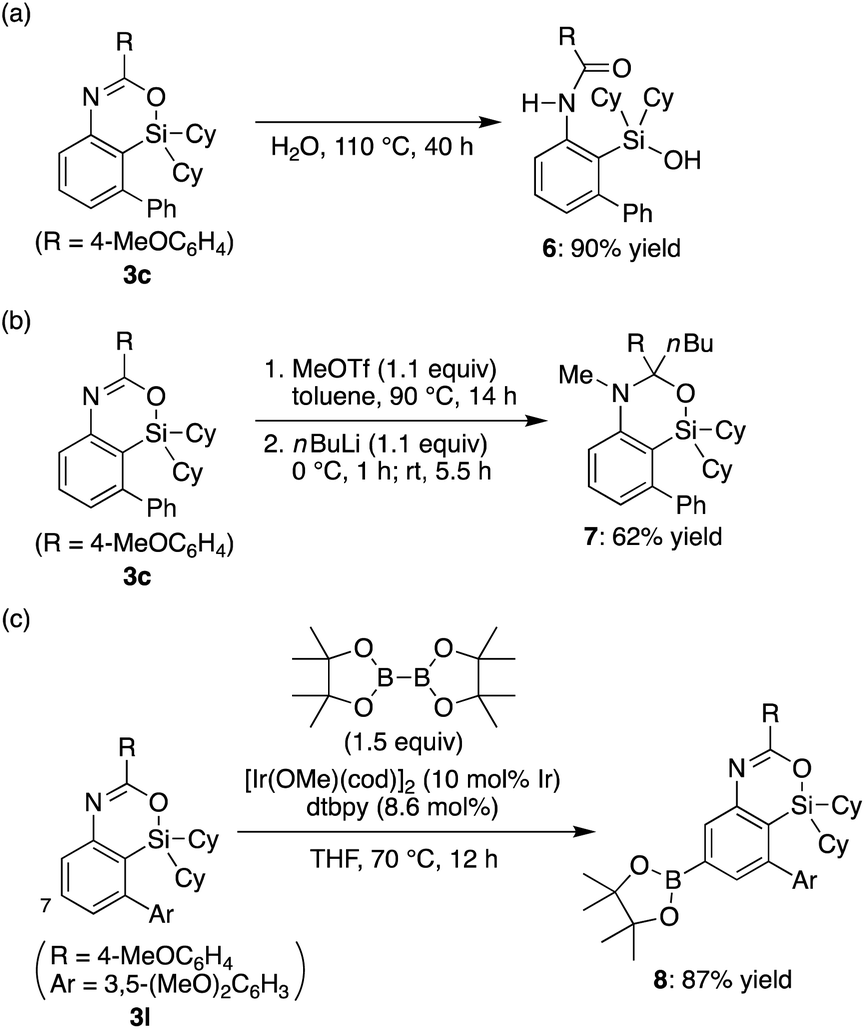
The obtained 4-sila-4H-benzo[d][1,3]oxazines 3 were found to be reasonably stable against H2O under neutral conditions, and complete hydrolytic ring-opening of 3c required a prolonged heating in H2O at 110 °C for 40 h, giving corresponding silanol 6 in 90% yield (Scheme 4a). In addition, in analogy to the derivatization of 4H-benzo[d][1,3]oxazines,18 the C–N double bond of 3c could be dialkylated by sequential treatment with methyl triflate and n-butyllithium to give compound 7 in 62% yield with retaining the silacyclic structure (Scheme 4b). Furthermore, selective C–H borylation at the least hindered 7-position of 3l was achieved under iridium catalysis to give compound 8 in 87% yield (Scheme 4c).19
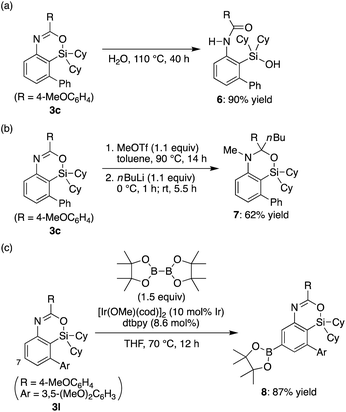 |
| | Scheme 4 Transformation of 4-sila-4H-benzo[d][1,3]oxazines 3. dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridyl. | |
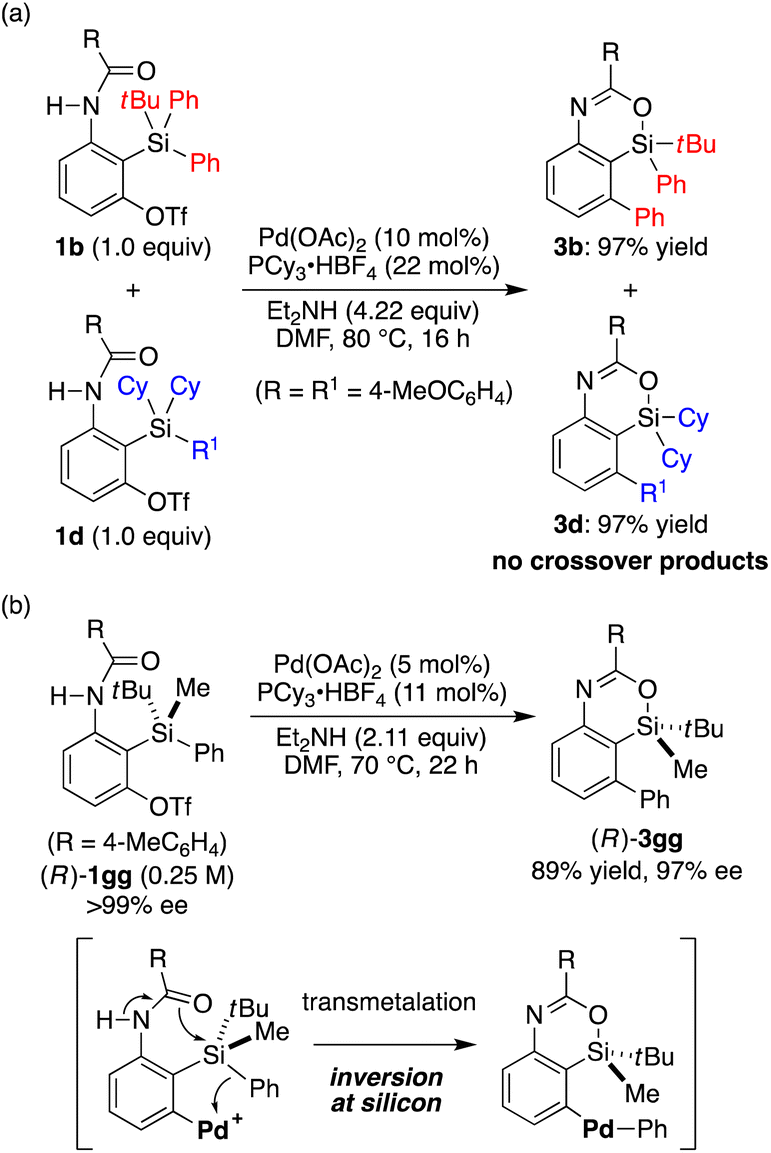
To gain insights into the reaction mechanism of the present catalysis, we conducted a control experiment. When the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of substrates 1b and 1d was subjected to the present reaction conditions, products 3b and 3d were obtained in nearly quantitative yields without forming any crossover products (Scheme 5a). This result indicates that the transmetalation of an aryl group from silicon to palladium takes place intramolecularly once the arylpalladium species is generated by oxidative addition of an aryl triflate to the palladium catalyst.20 We also prepared silicon-stereogenic enantiopure substrate (R)-1gg17,21 and conducted the present reaction to probe the stereochemical outcome at the silicon atom (Scheme 5b). As a result, 4-sila-4H-benzo[d][1,3]oxazine 3gg was obtained in 89% yield with 97% ee (R),17,22 confirming that the transmetalation of the phenyl group from silicon to palladium takes place with inversion of configuration at the silicon stereocenter in a stereospecific manner. This represents the first example of the stereochemical investigation at silicon during transmetalation in the Hiyama coupling reaction as far as we are aware,23,24 although the present result may not be directly generalized due to its intramolecular nature.
:1 mixture of substrates 1b and 1d was subjected to the present reaction conditions, products 3b and 3d were obtained in nearly quantitative yields without forming any crossover products (Scheme 5a). This result indicates that the transmetalation of an aryl group from silicon to palladium takes place intramolecularly once the arylpalladium species is generated by oxidative addition of an aryl triflate to the palladium catalyst.20 We also prepared silicon-stereogenic enantiopure substrate (R)-1gg17,21 and conducted the present reaction to probe the stereochemical outcome at the silicon atom (Scheme 5b). As a result, 4-sila-4H-benzo[d][1,3]oxazine 3gg was obtained in 89% yield with 97% ee (R),17,22 confirming that the transmetalation of the phenyl group from silicon to palladium takes place with inversion of configuration at the silicon stereocenter in a stereospecific manner. This represents the first example of the stereochemical investigation at silicon during transmetalation in the Hiyama coupling reaction as far as we are aware,23,24 although the present result may not be directly generalized due to its intramolecular nature.
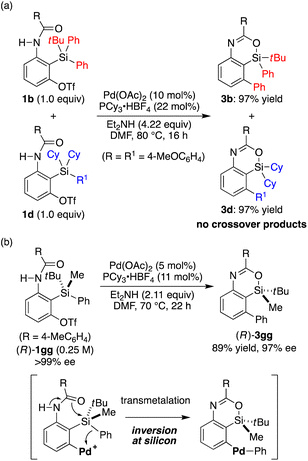 |
| | Scheme 5 Mechanistic insights of the present catalysis. | |
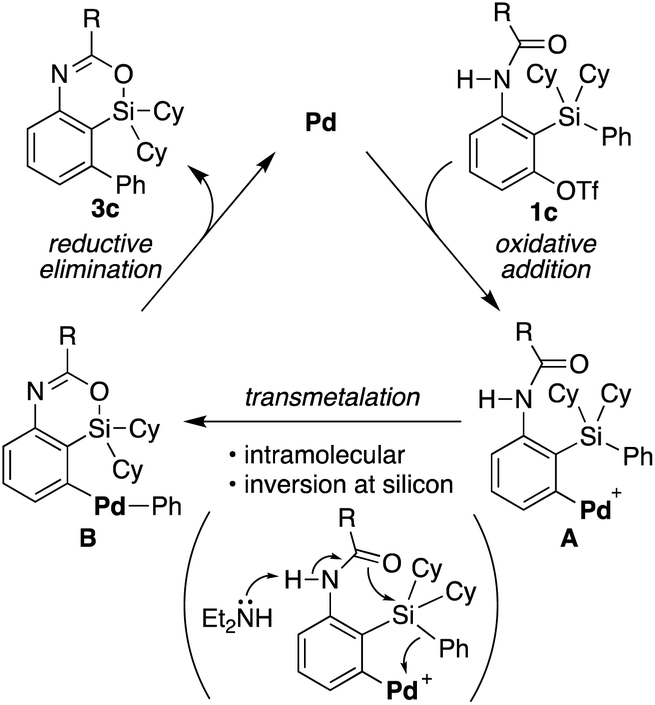
Based on the above consideration, a proposed catalytic cycle for the synthesis of 3c from 1c in the present catalysis is shown in Scheme 6. Oxidative addition of aryl triflate of 1c to palladium(0) gives arylpalladium(II) species A. This then undergoes intramolecular transmetalation via inversion at silicon to give diarylpalladium(II) species B. Subsequent carbon–carbon bond-forming reductive elimination gives 4-sila-4H-benzo[d][1,3]oxazine 3c along with regeneration of palladium(0).
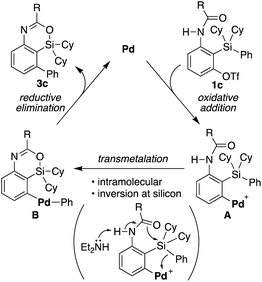 |
| | Scheme 6 A proposed catalytic cycle for the palladium-catalyzed synthesis of 3c from 1c. | |
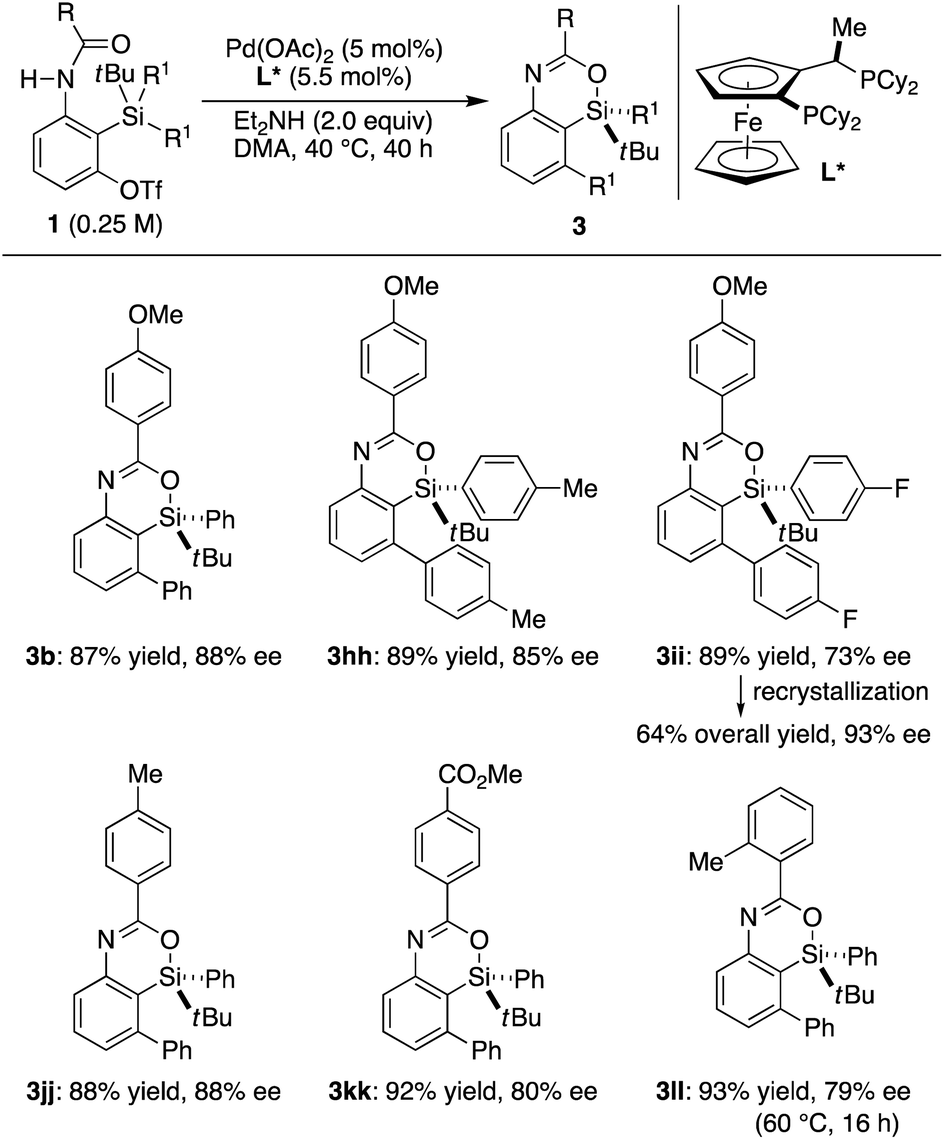
In our preliminary study, we found that the present catalysis could also be applied to enantioselective synthesis of silicon-stereogenic 4-sila-4H-benzo[d][1,3]oxazines by using prochiral substrates in the presence of an appropriate chiral ligand.25 According to our survey, Josiphos-type ligand L* was found to be promising in inducing relatively high enantioselectivity. Thus, the reaction of 1b having two phenyl groups on the silicon atom led to the formation of compound 3b in 87% yield with 88% ee (R) by selective transmetalation of one phenyl group over the other in the presence of a Pd/L* catalyst (Scheme 7).17 Other aryl groups could also be accommodated on the silicon atom for this desymmetrization with moderate to good efficiency (3hh and 3ii). Electronically or sterically different benzoyl groups on nitrogen were tolerated as well to give corresponding silicon-stereogenic 4-sila-4H-benzo[d][1,3]oxazines 3jj–ll with up to 88% ee. Although further improvement on the enantioselectivity is desirable, the ee of 3ii could be upgraded from 73% to 93% by recrystallization from hexane, and the result obtained here is a rare example of asymmetric catalysis involving an enantioselective transmetalation to give silicon-stereogenic compounds.8
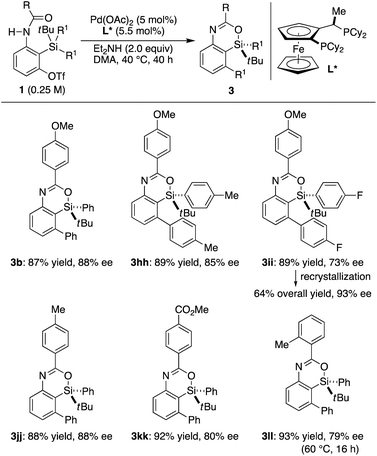 |
| | Scheme 7 Palladium-catalyzed asymmetric synthesis of silicon-stereogenic 3via enantioselective transmetalation. | |
Conclusions
We developed a palladium-catalyzed synthesis of 4-sila-4H-benzo[d][1,3]oxazines, silicon-switched analogs of biologically relevant 4H-benzo[d][1,3]oxazines, by the intramolecular Hiyama coupling of 3-amido-2-(arylsilyl)aryl triflates. The present reaction represents an unusual way of utilizing the Hiyama coupling toward the synthesis of functional organosilanes as the main products and it proceeds through intramolecular transmetalation with inversion of the stereochemistry at the silicon center. An asymmetric variant of this process was also demonstrated by employing prochiral substrates in the presence of a Josiphos-type chiral ligand to give silicon-stereogenic 4-sila-4H-benzo[d][1,3]oxazines with relatively high enantioselectivity. Future studies will be directed toward further expansion of this process for the synthesis of various functional organosilicon compounds.
Data availability
Experimental procedures, characterization data, X-ray crystallographic data, and NMR spectra are available in the ESI.†
Author contributions
R. S. conceptualized the project. D. L. conducted all the experiments and compound characterizations. D. L. and R. S. wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Support has been provided in part by JSPS KAKENHI Grant Number JP20H02741 (Grant-in-Aid for Scientific Research (B); R. S.) and JST, the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2125 (D. L.).
Notes and references
- For reviews:
(a) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779 CrossRef CAS PubMed;
(b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed;
(c) G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551 CrossRef CAS PubMed;
(d) J. Y. Corey, Adv. Organomet. Chem., 2011, 59, 181 CrossRef CAS;
(e) J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714 CrossRef CAS.
- For reviews:
(a) G. C. Nandi, Eur. J. Org. Chem., 2021, 587 CrossRef CAS;
(b) G. Rousseau and L. Blanco, Tetrahedron, 2006, 62, 7951 CrossRef CAS.
- For selected recent examples:
(a) X.-X. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2018, 6, 1023 RSC;
(b) L.-J. Wu, Y. Yang, R.-J. Song, J.-X. Yu, J.-H. Li and D.-L. He, Chem. Commun., 2018, 54, 1367 RSC;
(c) A. Choi and S. C. Miller, Org. Lett., 2018, 20, 4482 CrossRef CAS PubMed;
(d) S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668 CrossRef CAS PubMed;
(e) W.-T. Zhao, Z.-Q. Lu, H. Zheng, X.-S. Xue and D. Zhao, ACS Catal., 2018, 8, 7997 CrossRef CAS;
(f) J. Zhang, S. Park and S. Chang, J. Am. Chem. Soc., 2018, 140, 13209 CrossRef CAS PubMed;
(g) A. Nishii, H. Takikawa and K. Suzuki, Chem. Sci., 2019, 10, 3840 RSC;
(h) L. Noël-Duchesneau, J. Maddaluno and M. Durandetti, ChemCatChem, 2019, 11, 4154 CrossRef;
(i) S.-J. Woo, Y. Kim, Y.-H. Kim, S.-K. Kwon and J.-J. Kim, J. Mater. Chem. C, 2019, 7, 4191 RSC;
(j) H. Fang, K. Xie, S. Kemper and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 8542 CrossRef CAS PubMed;
(k) W. Wang, S. Zhou, L. Li, Y. He, X. Dong, L. Gao, Q. Wang and Z. Song, J. Am. Chem. Soc., 2021, 143, 11141 CrossRef CAS PubMed.
- For reviews:
(a) M. Asif and M. Imran, Int. J. New Chem., 2020, 7, 60 CAS;
(b) D. S. Zinad, A. Mahal, R. K. Mohapatra, A. K. Sarangi and M. R. F. Pratama, Chem. Biol. Drug Des., 2020, 95, 16 CrossRef CAS PubMed;
(c) E. V. Gromachevskaya, F. V. Kvitkovskii, T. P. Kosulina and V. G. Kul’nevich, Chem. Heterocycl. Compd., 2003, 39, 137 CrossRef CAS.
- For reviews:
(a) P. Nuss, F. Ferreri and M. Bourin, Neuropsychiatr. Dis. Treat., 2019, 15, 1781 CrossRef CAS PubMed;
(b) P. Poisbeau, G. Gazzo and L. Calvel, World J. Biol. Psychiatry, 2018, 19, 536 Search PubMed.
- For reviews:
(a) S. J. Bunu, D. Ere and O. Miediegha, Asian J. Pharm. Clin. Res., 2020, 13, 26 CAS;
(b) F. Maggiolo, J. Antimicrob. Chemother., 2009, 64, 910 CrossRef CAS PubMed.
-
(a) A. Fensome, R. Bender, R. Chopra, J. Cohen, M. A. Collins, V. Hudak, K. Malakian, S. Lockhead, A. Olland, K. Svenson, E. A. Terefenko, R. J. Unwalla, J. M. Wilhelm, S. Wolfrom, Y. Zhu, Z. Zhang, P. Zhang, R. C. Winneker and J. Wrobel, J. Med. Chem., 2005, 48, 5092 CrossRef CAS PubMed;
(b) Z. Zhang, A. M. Olland, Y. Zhu, J. Cohen, T. Berrodin, S. Chippari, C. Appavu, S. Li, J. Wilhem, R. Chopra, A. Fensome, P. Zhang, J. Wrobel, R. J. Unwalla, C. R. Lyttle and R. C. Winneker, J. Biol. Chem., 2005, 280, 28468 CrossRef CAS PubMed.
-
(a) R. Shintani, E. E. Maciver, F. Tamakuni and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 16955 CrossRef CAS PubMed; See also:
(b) R. Kumar, Y. Hoshimoto, H. Yabuki, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 11838 CrossRef CAS PubMed.
-
(a) Y. Sato, C. Takagi, R. Shintani and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 9211 CrossRef CAS PubMed;
(b) N. Misawa, T. Tsuda, R. Shintani, K. Yamashita and K. Nozaki, Chem.–Asian J., 2018, 13, 2566 CrossRef CAS PubMed.
- For relevant reviews:
(a) S. Curpanen, G. Poli, J. Oble and A. Perez-Luna, Eur. J. Org. Chem., 2021, 1055 CrossRef CAS;
(b) Y. Nakao and T. Hiyama, J. Synth. Org. Chem., Jpn., 2011, 69, 1221 CrossRef CAS.
- For reviews on the Hiyama coupling:
(a) Y. Minami and T. Hiyama, Chem.–Eur. J., 2019, 25, 391 CrossRef CAS PubMed;
(b) T. Komiyama, Y. Minami and T. Hiyama, Synlett, 2017, 28, 1873 CrossRef CAS;
(c) F. Foubelo, C. Nájera and M. Yus, Chem. Rec., 2016, 16, 2521 CrossRef CAS PubMed;
(d) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982 CrossRef CAS PubMed;
(e) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC;
(f) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC;
(g) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486 CrossRef CAS PubMed.
-
(a) Y. Qin, J.-L. Han, C.-W. Ju and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 8481 CrossRef CAS PubMed;
(b) Y. Qin, L. Li, J.-Y. Liang, K. Li and D. Zhao, Chem. Sci., 2021, 12, 14224 RSC; See also:
(c) Y. Nakao, H. Imanaka, A. K. Sahoo, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 6952 CrossRef CAS PubMed.
-
(a) Y. Hatanaka and T. Hiyama, Tetrahedron Lett., 1988, 29, 97 CrossRef CAS;
(b) Y. Nakao, M. Takeda, T. Matsumoto and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4447 CrossRef CAS PubMed.
- Primary alkyl groups are known to undergo transmetalation preferentially over secondary alkyl groups. The same trend would be expected for methyl group vs. secondary alkyl groups. See ref. 13b.
- 2-(4-Methoxyphenyl)benzo[d]oxazole was obtained as the major product presumably through the aryne formation from 2-(dimethyl(phenyl)silyl)aryl triflate. See the ESI for details.†.
- See the ESI for details.†.
- Deposition numbers 2194996 (for S2), 2194997 (for 3gg·HOTf), 2194998 (for 3b), and 2247904 (for 4v) contain the supplementary crystallographic data for this paper.†.
- S. Rajkumar, M. Tang and X. Yang, Angew. Chem., Int. Ed., 2020, 59, 2333 CrossRef CAS PubMed.
-
(a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed;
(b) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS.
-
(a) C. Mateo, C. Fernández-Rivas, A. M. Echavarren and D. J. Cárdenas, Organometallics, 1997, 16, 1997 CrossRef CAS;
(b) C. Mateo, C. Fernández-Rivas, D. J. Cárdenas and A. M. Echavarren, Organometallics, 1998, 17, 3661 CrossRef CAS.
- The absolute configuration was determined by X-ray crystallographic analysis of the corresponding phenol S2 (precursor of the triflate).
- The absolute configuration was determined by X-ray crystallographic analysis of the corresponding TfOH salt 3gg·HOTf.
- For selected examples of the mechanistic studies on the Hiyama coupling:
(a) A. Gordillo, M. A. Ortuño, C. López-Mardomingo, A. Lledós, G. Ujaque and E. de Jesús, J. Am. Chem. Soc., 2013, 135, 13749 CrossRef CAS PubMed;
(b) C. Amatore, L. Grimaud, G. Le Duc and A. Jutand, Angew. Chem., Int. Ed., 2014, 53, 6982 CrossRef CAS PubMed;
(c) S. A. Tymonko, R. C. Smith, A. Ambrosi, M. H. Ober, H. Wang and S. E. Denmark, J. Am. Chem. Soc., 2015, 137, 6200 CrossRef CAS PubMed;
(d) A. W. E. Stewart, H. Z. Ma, G. K. Weragoda, G. N. Khairallah, A. J. Canty and R. A. J. O'Hair, Organometallics, 2021, 40, 1822 CrossRef CAS;
(e) Q. Wang, H.-M. Jiang, S. Zhuo and L.-P. Xu, Catal. Sci. Technol., 2022, 12, 135 RSC.
- For stereochemical investigations on the carbon stereocenters of organosilicon nucleophiles in the Hiyama coupling reactions:
(a) Y. Hatanaka and T. Hiyama, J. Am. Chem. Soc., 1990, 112, 7793 CrossRef CAS;
(b) Y. Hatanaka, K. Goda and T. Hiyama, Tetrahedron Lett., 1994, 35, 1279 CrossRef CAS;
(c) S. E. Denmark and N. S. Werner, J. Am. Chem. Soc., 2010, 132, 3612 CrossRef CAS PubMed.
- For recent reviews on the catalytic asymmetric synthesis of silicon-stereogenic oragnosilanes:
(a) W. Yuan and C. He, Synthesis, 2022, 54, 1939 CrossRef;
(b) L. Zheng, X.-X. Nie, Y. Wu and P. Wang, Eur. J. Org. Chem., 2021, 6006 CrossRef CAS;
(c) R. Shintani, Synlett, 2018, 29, 388 CrossRef CAS;
(d) Y.-M. Cui, Y. Lin and L.-W. Xu, Coord. Chem. Rev., 2017, 330, 37 CrossRef CAS;
(e) J. O. Bauer and C. Strohmann, Eur. J. Inorg. Chem., 2016, 2868 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab