
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn orthogonally activatable CRISPR-Cas13d nanoprodrug to reverse chemoresistance for enhanced chemo-photodynamic therapy†
Zheng
Liu
 a,
Zhiyuan
Feng
a,
Mohan
Chen
a,
Jiayin
Zhan
a,
Rong
Wu
a,
Yang
Shi
a,
Yunsheng
Xue
b,
Ran
Liu
a,
Jun-Jie
Zhu
a and
Jingjing
Zhang
*a
a,
Zhiyuan
Feng
a,
Mohan
Chen
a,
Jiayin
Zhan
a,
Rong
Wu
a,
Yang
Shi
a,
Yunsheng
Xue
b,
Ran
Liu
a,
Jun-Jie
Zhu
a and
Jingjing
Zhang
*a
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: jing15209791@nju.edu.cn
bJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, School of Pharmacy, Xuzhou Medical University, Xuzhou 221004, China
First published on 20th March 2023
Abstract
Orthogonal therapy that combines CRISPR-based gene editing and prodrug-based chemotherapy is a promising approach to combat multidrug-resistant cancer. However, its potency to precisely regulate different therapeutic modalities in vivo is limited due to the lack of an integrated platform with high spatiotemporal resolution. Taking advantage of CRISPR technology, a Pt(IV)-based prodrug and orthogonal emissive upconversion nanoparticles (UCNPs), we herein rationally designed the first logic-gated CRISPR-Cas13d-based nanoprodrug for orthogonal photomodulation of gene editing and prodrug release for enhanced cancer therapy. The nanoprodrug (URL) was constructed by encapsulating a green light-activatable Pt(IV) prodrug and UV light-activatable Cas13d gene editing tool into UCNPs. We demonstrated that URL maintained excellent orthogonal emission behaviors under 808 and 980 nm excitations, allowing wavelength-selective photoactivation of Cas13d and the prodrug for downregulation of the resistance-related gene and induction of chemo-photodynamic therapy, respectively. Moreover, the photomodulation superiority of URL for overcoming drug resistance was highlighted by integrating it with a Boolean logic gate for programmable modulation of multiple cell behaviors. Importantly, in vivo studies demonstrated that URL can promote Pt(IV) prodrug activation and ROS generation and massively induce on-target drug accumulation by Cas13d-mediated drug resistance attenuation, delivering an ultimate chemo-photodynamic therapeutic performance in efficiently eradicating primary tumors and preventing further liver metastasis. Collectively, our results suggest that URL expands the Cas13d-based genome editing toolbox into prodrug nanomedicine and accelerates the discovery of new orthogonal therapeutic approaches.
Introduction
Despite promising advances in targeted cancer therapy and immunotherapy, chemotherapy remains the mainstay of cancer treatment.1 However, the development of multidrug resistance (MDR) remains a major obstacle to effective cancer chemotherapy due to its mechanistic complexity.1 Thanks to advances in structural biology and cell biology, a series of cancer-overexpressed multidrug transporters have been identified as the major drug efflux regulators contributing to the failure of chemotherapy significantly.2–4 In particular, multidrug resistance associated-protein 1 (MRP1), with the majority of localization at the basolateral membrane, contributes to maintain the cell redox state by transporting the oxidized/reduced forms of glutathione and its drug conjugates; and it is closely associated with a poor clinical outcome (e.g. unfavorable prognosis) in cancers.4 As a result, MRP1 represents one of the “Achilles' Heels” of drug-resistant cancer cells.5 Accumulating evidence has suggested that MRP1-mediated drug resistance can be reversed by downregulating the MRP1 gene with gene silencing methods.6 For example, clustered regularly interspaced short palindromic repeat-associated protein 9 (CRISPR-Cas9) technology has been found to reverse MDR mediated by ATP-binding cassette (ABC) transporters with a significantly higher outcome than other gene editing technologies due to its simple design, flexible target region, higher editing efficiency and multiplexing.7–10 Despite great advances, the majority of CRISPR-Cas9 systems still suffer from some formidable problems, including off-target genomic alterations and genotoxicity, potential immunological risk by Cas9-specific T-cells, and unsatisfactory targeted delivery.8 To address this challenge, CRISPR-Cas13d, a RNA-guided Type VI Cas protein, has been identified for target gene knockdown without altering the genome.11,12 Importantly, compared with the Cas9 protein, Cas13d displayed greatly reduced off-target activity and immune response, minimal targeting constraints, and a remarkably small size suitable for designing an all-in-one delivery system.13 It is therefore our belief that these unique features might be beneficial for overcoming MDR but they have rarely been explored.Also, increasing the drug uptake by prodrug strategies or combination with nanocarriers is also a promising option for overcoming MDR.14–16 In particular, many efforts have been made towards the generation of combinatorial chemo- and gene-therapeutics from various gene regulation tools such as antisense oligonucleotides,17,18 RNAi,19,20 and CRISPR-Cas9,8,9 leading to maximum synergistic activity for reversing chemoresistance. Inspired by these promising achievements, we therefore believe that combining a Cas13d gene editing system with chemotherapy would be an effective approach to treat drug-resistant cancers. However, due to the unique physicochemical properties of Cas13d therapeutics and prodrugs, they often need to be encapsulated in nanocarriers to overcome their weakness in stability, solubility and targeting. More importantly, compared with prodrug chemotherapy, Cas13d-mediated gene knockdown often requires a relatively long time to achieve effective downregulation of the corresponding target protein. In this context, precisely activating Cad13d and prodrugs in a tumor-selective, high spatiotemporal manner is necessary to achieve a synergistic therapeutic effect. One strategy that is appealing involves the use of orthogonal emissive upconversion nanoparticles (UCNPs),21–24 which could remotely manipulate the photoactivation of multiple therapeutics in living cells or even animals on demand with high spatiotemporal precision.25–28 Taking the abovementioned factors into account, developing a nanoprodrug capable of combining targeted delivery, on-demand orthogonal activation, Cas13d gene editing and chemo-photodynamic therapeutic functions into one entity, is highly demanded but challenging.
In this study, we present a conceptual approach for the design of an orthogonally NIR-light activatable CRISPR-Cas13d nanoprodrug by introducing a green light-activatable Pt(IV) prodrug and UV light-activatable Cas13d gene editing tool into the orthogonal emissive core/multi-shell UCNPs. The Pt(IV) prodrug was rationally designed by covalent coupling of an oxaliplatin-based Pt(IV) complex to the Rose Bengal (RB) photosensitizer, whose absorbance spectra possess a maximum overlap only with the green UCL band of UCNPs. Such a design specifically triggers the activation of prodrugs under 980 nm light irradiation, thus rapidly releasing parent drugs through Pt–O bond cleavage and generating fluorescence imaging probes concurrently with reactive oxygen species, such as singlet oxygen (Fig. 1a). On the other hand, a CRISPR/Cas13d gene editing system is constructed by assembling a RNA-guided Cas13d ribonuclease with a MRP1-targeted sgRNA, which is caged by a single-stranded oligonucleotide incorporating three photocleavable (PC) linkers (Fig. 1b). Under 808 nm light irradiation, the UV light emitted by UCNPs could break PC linkers and activate the CRISPR/Cas13d system to cleave intracellular MRP1 mRNA, leading to efficient downregulation of the membrane MRP1 protein and thus, drug resistance attenuation. To provide cancer-targeting delivery as well as improve the in vivo stability of the nanoprodrug, it was encapsulated into a cyclic RGD peptide moiety (cRGD)-functionalized phospholipid, termed as URL (Fig. 1b). We demonstrated that URL maintained excellent orthogonal emission behaviors under 808 and 980 nm excitations, allowing wavelength-selective photoactivation of oxaliplatin prodrug-induced chemo-photodynamic therapy and Cas13d-mediated gene editing. Moreover, in vivo studies demonstrated that such an orthogonally activatable nanoprodrug can promote the activation of the Pt(IV) prodrug and ROS generation and massively induce on-target drug accumulation with the help of Cas13d-mediated drug resistance attenuation, delivering an ultimate chemo-photodynamic therapeutic performance in efficiently eradicating primary tumors and preventing further liver metastasis (Fig. 1c).
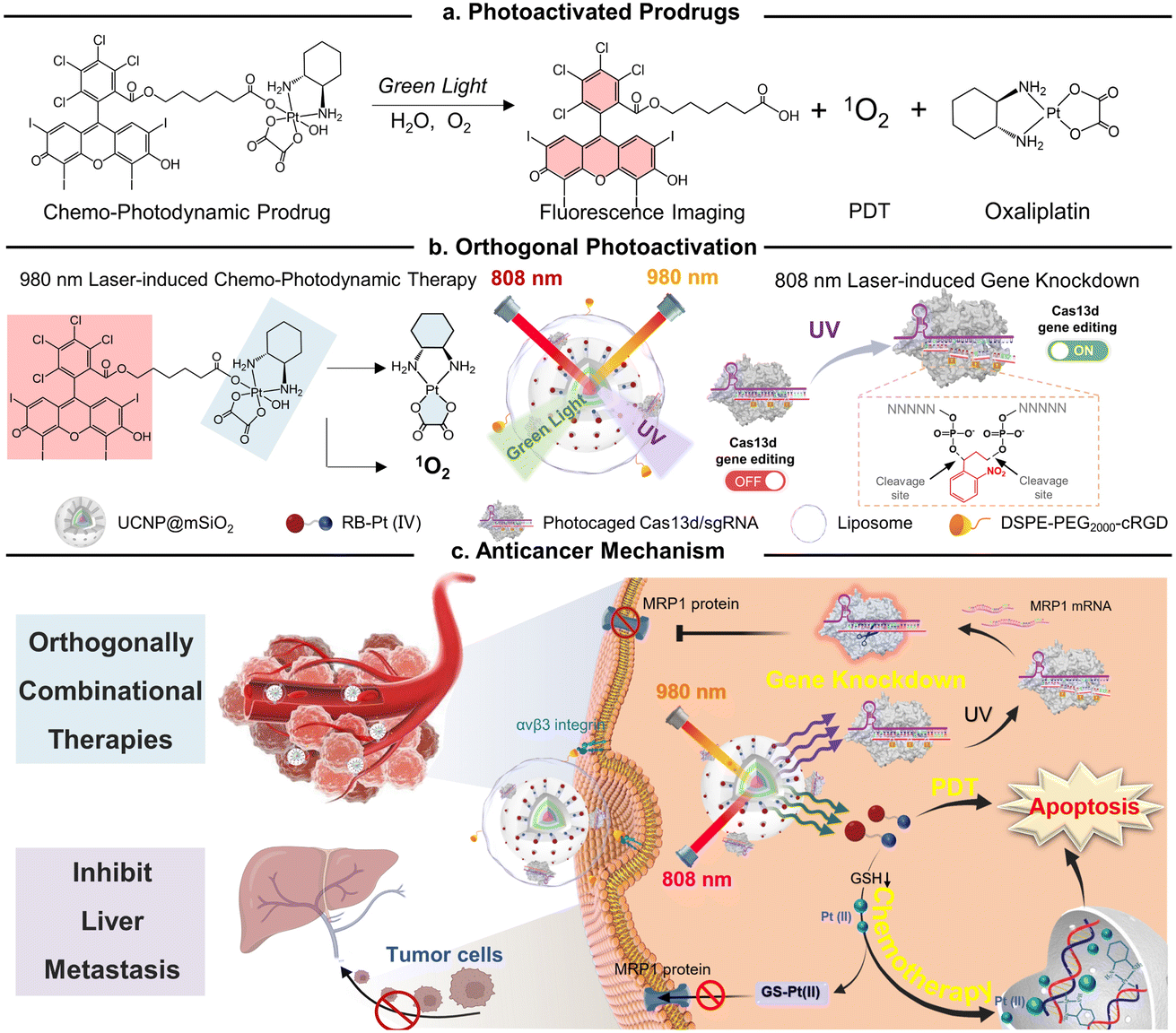 | ||
| Fig. 1 Schematic showing the design of an orthogonally activatable CRISPR-Cas13d nanoprodrug for reversing chemoresistance and enhanced chemo-photodynamic therapy. (a) Chemical structure of the chemo-photodynamic prodrug and its photoactivation mechanism. (b) The orthogonal photoactivation of oxaliplatin prodrug-induced chemo-photodynamic therapy and Cas13d-mediated gene editing. (c) On-demand activation of the nanoprodrug with sequential MRP1 knockdown and efficient glutathione-mediated oxaliplatin(II) release behavior for reversing drug resistance, resulting in enhanced combinational therapy in vivo. | ||
Results and discussion
Design and characterization of a green light activatable oxaliplatin(IV) prodrug
A typical photoactivatable Pt(IV) prodrug is usually composed of a biologically inactive drug derivative and a specific photosensitizer connected by a variable linker, where the photosensitizer could absorb light and transfer its energy to the relevant reaction centers to facilitate Pt(IV) reduction. Through this elaborate design, a series of Pt(IV) prodrugs have been developed and demonstrated their utility in photoactivation for improved therapeutic outcomes.14,29 Despite substantial progress, the majority of these photoactivatable prodrugs suffer from drawbacks such as limited wavelength selectivity. On the other hand, as a proof of concept, we selected here the photocleavable 2-nitrobenzyl linker-based DNA strand to generate the photocaged CRISPR-Cas13d gene editing system, since such a PC linker that is commercially available is already widely used for DNA modification.30–34 Given that the PC linker selectively responds to light in the UV region (λ < 400 nm), it is necessary to develop a Pt(IV) prodrug responding only to light beyond the UV region for the ultimate goal of orthogonal photoactivation. With these considerations in mind, we began by surveying commercially available photosensitizers for designing a wavelength-selective prodrug (Fig. S1†), which is essential for sequential and wavelength-orthogonal photomodulation of multiple photoagents. RB is one of the most studied photosensitizers for medical applications that effectively generates singlet oxygen upon irradiation with green light (λabs,max = 565 nm). In addition, RB has very low absorption within the UV region (300–400 nm), and is extensively used for PDT because of its high photodynamic efficiency. Therefore, we chose to construct a green light activatable prodrug, rosebplatin (RB-Pt), in which RB was conjugated to the axial hydroxido ligand of oxaliplatin.As illustrated in Fig. 2a, RB-Pt was prepared in a few steps through esterification coupling reactions with moderate yields. The structure of RB-Pt was first validated by nuclear magnetic resonance spectroscopy (1H NMR and 13C NMR) (Fig. S2 and S3†). We then evaluated the optical properties of RB-Pt. UV-vis absorption results showed intense UV absorbance in the green spectral region which originated from the characteristics of RB (Fig. 2b), and a red shift (9 nm) in the absorption maximum of RB-Pt compared to that of unconjugated RB, further confirming the successful conjugation of RB with oxaliplatin. In addition, RB-Pt also displayed the characteristic red emission peak of RB (Fig. S4†), which is advantageous in that it enables monitoring of the cell fate upon prodrug uptake or activation using fluorescence readouts. Next, we sought to validate whether RB-Pt could be photoactivated to release oxaliplatin and RB under irradiation of green light using ESI-MS (Fig. 2c). After 5 min of green light irradiation, the ESI-MS spectrum of RB-Pt displays two main peaks at 1086.55 and 1499.61 m/z, belonging to the [RB-HA-H]− and RB-Pt [M − H]− ions, respectively. Such a coexistence after green light irradiation suggested the dissociation of the axial RB ligand and the release of oxaliplatin correspondingly.
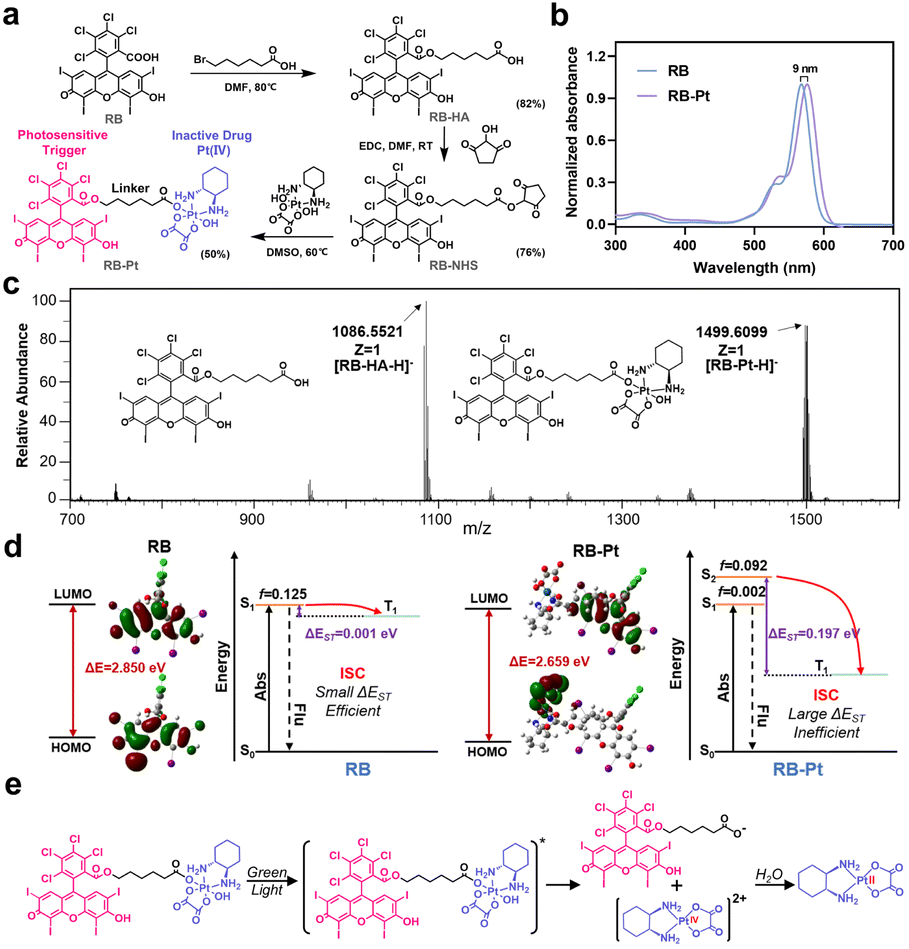 | ||
| Fig. 2 Design and characterization of a green light activatable oxaliplatin(IV) prodrug. (a) Synthetic route to the RB-Pt prodrug. (b) UV-vis absorption spectra of RB and RB-Pt. (c) ESI-MS results of RB-Pt under irradiation of green light. The m/z range is 700–1600. [RB-HA-H]−, calculated 1086.81, found 1086.55; [RB-Pt-H]−, calculated 1500.10, found 1499.61. (d) The leading edge orbital of the ground state (S0) and the highest individually occupied molecular orbital of the lowest excited triplet state (T1) of RB and RB-Pt, and the fitted excited state features of the corresponding theoretically predicted energy gap. (e) Proposed mechanism of photo-induced reduction of RB-Pt. | ||
To gain insight into the fracture mechanism, we performed density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations. We found that covalently conjugating RB to the axial hydroxido ligand of oxaliplatin(IV) results in a dramatically decreased HOMO–LUMO gap (ΔE) from 2.850 eV to 2.659 eV (Fig. 2d), indicating a strong intramolecular charge transfer (ICT) in RB-Pt, consistent with the observed red shift in the UV-vis spectra shown in Fig. 2b. In addition, TDDFT analysis showed that the ΔEST value of RB-Pt is significantly increased to 0.197 eV compared with that of RB (0.001 eV), indicating a potentially inefficient intersystem crossing (ISC) and decreased photosensitizing activity of RB-Pt. The reason for the reduced photoactivity of RB-Pt is that the energy absorbed by RB-Pt is not only used to produce singlet oxygen, but also transferred to the photochemical reaction center of RB-Pt to promote the reduction of Pt(IV). This result is similar to that observed in other Pt(IV) prodrugs, such as coumaplatin14 or phorbiplatin.29 Based on the above observations, together with the recent findings that the Pt(IV) prodrug can be efficiently photoactivated via water oxidation,14 the possible photoreduction mechanism of RB-Pt could be divided into three main steps, including irradiation-induced transition to the excited state, the cleavage of the Pt–O bond of axial ligands, and water-mediated Pt(IV) to oxaliplatin(II) reduction through electron transfer (Fig. 2e).14
Characterization of the CRISPR-Cas13d nanoprodrug in vitro
Despite Pt(IV)-based prodrug research making some progress, drug resistance is still the greatest obstacle for its clinical translation.4 The main reason is that drug resistance pathways are complex processes that involve multiple resistance-associated proteins like MRP1.3,35 In particular, accumulating evidence indicates that regulating MRP1 function is often beneficial to conquer multidrug resistance in many cancer types, but there is still a remaining challenge for precise MRP1 intervention in prodrug-based therapy. Disruption of gene expression has been considered as a powerful approach for the regulation of target protein function, which has been accelerated since the invention of CRISPR technologies. For example, CRISPR-Cas9-based prodrugs have been developed as promising gene editing therapeutics in many diseases.8,9 Inspired by this knowledge, we utilized RspCas13d, a newly identified RNA-guided Cas13d ribonuclease derived from Ruminococcus sp.,36 as our gene-editing tool and designed a specific 20 nt sgRNA targeting MRP1 mRNA (NM_004996.4). Next, to prepare the genome editing assembly, RspCas13d with a molecular weight of ∼110 kDa was first expressed in Escherichia coli, purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography, and analyzed using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. S5a†). Meanwhile, the sgRNA was obtained by in vitro transcription, and the purity and size of the obtained sgRNA was characterized using denaturing urea-PAGE (Fig. S5b†). All the oligonucleotide sequences are listed in Table S1.† Next, to investigate the MRP1-triggered cis-cleavage activity of Cas13d/sgRNA, we conducted an in vitro RNA cleavage assay by introducing a 54 nt linearized MRP1-encoding ssRNA as the substrate for Cas13d/sgRNA. Native PAGE analysis confirmed the MRP1-specific cleavage of Cas13d/sgRNA (Fig. S6†). To achieve on-demand Cas13d/sgRNA activation in response to external light, we further designed a 22-mer single-stranded PC-DNA containing three photocleavable (PC) linkers at positions 6, 11, and 17 (full-length numbering, 5′ to 3′), respectively (Fig. S7†). The PC-DNA is designed so that it can hybridize to the target region of the sgRNA to form a photocaged Cas13d/sgRNA/PC-DNA complex (denoted as RNP), which blocks the cis-cleavage activity of Cas13d/sgRNA. Upon UV light irradiation, the three PC linkers were photolyzed, split into four DNA fragments, and subsequently dehybridized from the RNP because of the lowered melting temperature (Fig. S7†). Thus, photo-activation dramatically shifts the hybridization equilibrium of ssDNA and sgRNA in photocaged RNP to form an active RNP.With the green-light responsive prodrug and UV-light (365 nm) responsive RNP in hand, we further asked whether these two photoactivatable therapeutics can be combined with UCNPs to realize orthogonal photomodulation in the deep tissue-penetrable NIR window. Such features are generally considered to be a requirement for improving the therapeutic index by precisely suppressing the MRP1-mediated drug efflux. To this aim, we first designed and synthesized a core/multi-shell structured UCNP according to a previously reported protocol,21 with some modifications. Specifically, Ho3+/Yb3+ were codoped in the core and first shell layers, respectively, to enable UV emission upon 808 nm excitation. Separation of the lanthanide activator (Tm3+) and sensitizer (Nd3+) in different layers is able to prevent cross-relaxation.23,24 The TEM images confirmed an obvious morphology and size change during the step-by-step synthesis, and the final UCNP displayed a uniform nanorod shape, being about 46.6 ± 2.1 nm in width and 64.1 ± 3.4 nm in length (Fig. S8†). Next, we evaluated the upconversion profile of the core/multi-shell UCNP upon irradiation with NIR light of two different wavelengths. As shown in Fig. 3b, the UCNP displays the characteristic emission peaks of Nd3+/Yb3+/Tm3+ in the UV region (345 and 362 nm) and visible blue region (450 and 477 nm) when excited using an 808 nm laser. However, under 980 nm NIR light irradiation, characteristic Ho3+/Yb3+-dominated visible green (538 nm) and red (646 nm) emissions were observed, while the emission peaks in the UV-blue green region (300–500 nm) disappeared completely. These results demonstrated excellent orthogonal emission behaviors of our core/multi-shell UCNP under 808 and 980 nm excitations, thus allowing wavelength-selective photoactivation of the RB-Pt prodrug and Cas13d editing.
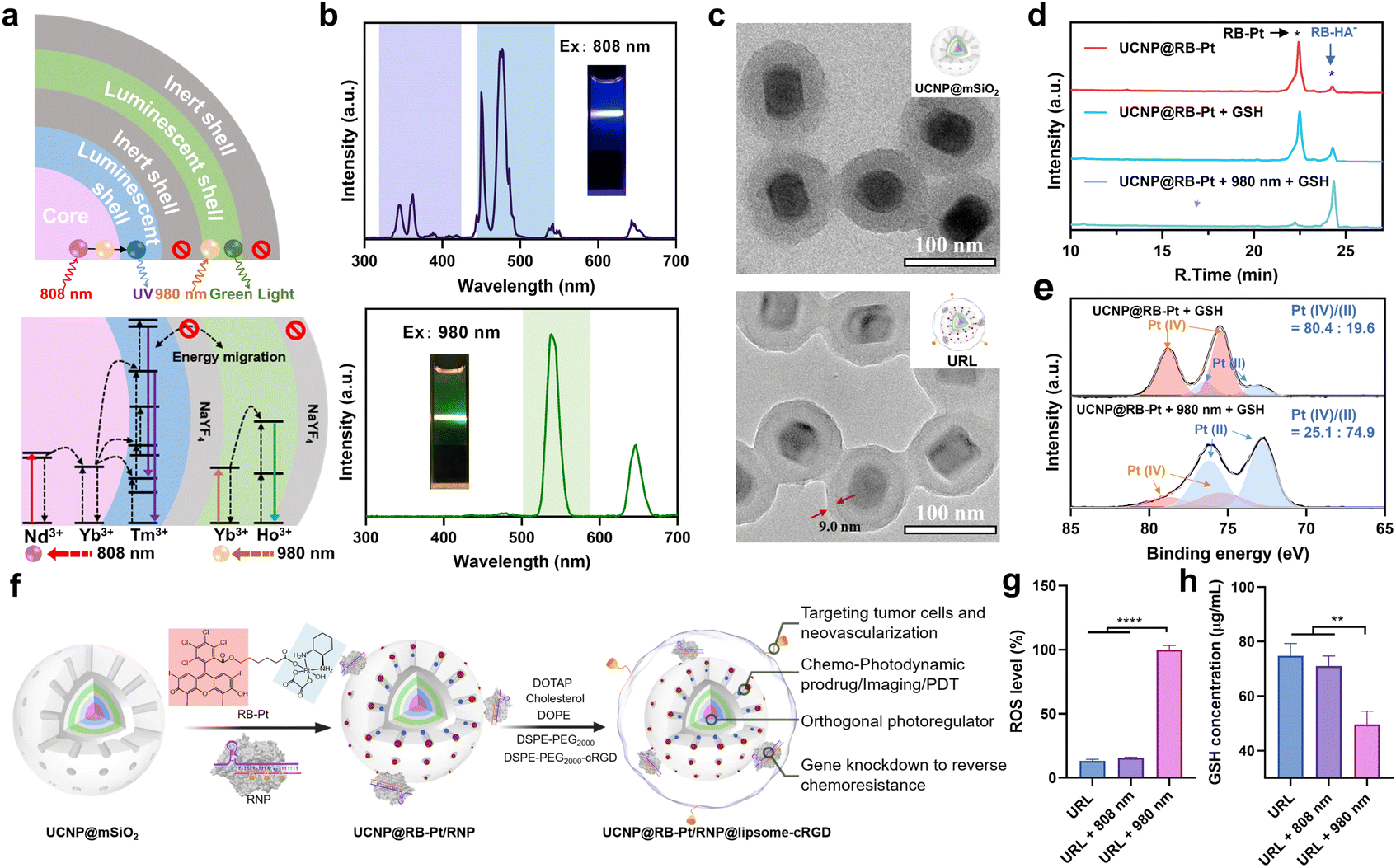 | ||
| Fig. 3 Characterization of the CRISPR-Cas13d nanoprodrug and 980 nm light-controlled prodrug reduction. (a) Schematic illustration of the energy migration pathways of Tm3+ and Ho3+ ions under excitation of an 808 or 980 nm laser. (b) Upconversion luminescence spectra and photograph of URL when irradiated with an 808 or 980 nm laser. (c) TEM images of UCNP@mSiO2 and UCNP@RB-Pt/RNP@liposome (URL). The distance between the red arrows indicates the thickness of the phospholipid layer. (d) HPLC chromatograms of the solution containing UCNP@RB-Pt with different treatments. UCNP@RB-Pt (50 μg mL−1); GSH (10 mM); 980 nm (0.5 W cm−2, 5 min). (e) XPS curves of Pt 4f of UCNP@RB-Pt before and after being irradiated with a 980 nm laser, UCNP@RB-Pt (2 mg mL−1); GSH (10 mM); 980 nm (0.5 W cm−2, 5 min). (f) Schematic illustration of the synthesis of water-soluble URL. (g) ROS level of the solution containing URL NPs with irradiation of an 808 or 980 nm laser. URL (200 μg mL−1); 980/808 nm (0.5 W cm−2, 5 min). (h) Intracellular GSH depletion under irradiation of an 808 or 980 nm laser. URL (100 μg mL−1); 980/808 nm (0.5 W cm−2, 5 min). **P < 0.01, and ****P < 0.0001. | ||
To further endow UCNP with delivery function for the RB-Pt prodrug and gene-editing systems, the UCNP was coated with a mesoporous silica layer (abbreviated as UCNP@mSiO2), followed by (3-aminopropyl)triethoxysilane (APTES) functionalization to yield exposed amine groups. Such a surface modification would allow the efficient loading of anionic Cas13d/sgRNA RNP on the external surface and encapsulation of the RB-Pt prodrug in the mesopores. The success of each step of the construction was evidenced by TEM (Fig. 3c and S9†), HAADF analysis (Fig. S10†), and zeta potential and UV-visible absorption measurements (Fig. S11†).
Encouraged by the promising photophysical properties, we then examined the 980 nm NIR light-triggered activation of the RB-Pt prodrug. Considering the reducing environment of the cell, we conducted a light irradiation test in the presence of GSH (10 mM) to mimic the reducing environment. Briefly, a mixture of UCNP@RB-Pt (50 μg mL−1) and GSH (10 mM) solution was irradiated with 980 nm light for 5 min, followed by 24 h incubation in the dark at 25 °C. Then, DMSO was used to dissolve the product for HPLC analysis. As shown in Fig. 3d, untreated UCNP@RB-Pt exhibited a sharp peak at an elution time of 22.4 min corresponding to RB-Pt; only a very weak peak at an elution time of 24.3 min was observed, which could be assigned to RB-HA− generated by slight reduction of RB-Pt during incubation. In contrast, upon addition of 10 mM of GSH, the peak height for RB-Pt decreased, the peak height for RB-HA− increased simultaneously, and the peak area ratio (PAR) of RB-HA−/RB-Pt increased from 9.5% to 22.7% accordingly. Furthermore, co-treatment with both 980 nm light (5 min) and GSH (10 mM) resulted in a significant decrease of the RB-Pt signal but an obvious increase of the RB-HA− signal. The PAR was calculated to be 1370.6%, which is 60.4 folds higher than that of the GSH-treated prodrug. Consistent with this finding, the Pt-4f XPS analysis further confirmed that 980 nm light irradiation resulted in a substantial increase in the Pt(II)/Pt(IV) ratio from the prodrug relative to the GSH control (Fig. 3e). These observations indicated that 980 nm light irradiation played a vital role in promoting the reduction of the RB-Pt(IV) prodrug to the corresponding Pt(II) form, although a slow, yet inevitable GSH-driven leakage of the prodrug occurred.
Orthogonally regulated prodrug activation and MRP1 knockdown in oxaliplatin-resistant cells
In order to achieve the maximum therapeutic efficiency, the UCNP-based nanocarrier should satisfy two essential prerequisites: (1) sufficient stability to protect chemotherapeutic prodrugs and RNP against preleakage and non-specific degradation during in vivo circulation; (2) active targeting ability to promote tumor accumulation. With these considerations in mind, the UCNP@RB-Pt/RNP was further coated with a lipid layer containing cRGD that can target the αvβ3 integrin overexpressed on most tumor cells, as illustrated in Fig. 3f.37 The obtained UCNP@RB-Pt/RNP@liposome (abbreviated as URL) was verified by TEM (Fig. 3c), zeta potential (Fig. S11a†) and DLS analysis (Fig. S11d†). Given that the production of reactive oxygen species (ROS) plays a major role in PDT by the Pt(IV)-based prodrug, we measured ROS production of URL under NIR irradiation using dichlorofluorescein diacetate (H2DCFDA) as a fluorescent indicator. As shown in Fig. 3g, compared with untreated URL, there was no significant increase in the ROS signal when URL was irradiated by using an 808 nm laser for 5 min. In contrast, 980 nm NIR light notably enhanced ROS production of URL by approximately 7.7 fold. In consistency, CLSM images and flow cytometric results revealed much higher fluorescence signals in the cells treated with URL plus 980 nm light irradiation (5 min) compared with cells exposed to URL only (Fig. S12†). As a control, 808 nm NIR light irradiation of URL led to no obvious generation of intracellular ROS. In the meantime, the intracellular GSH levels were evaluated, and a significant decrease of GSH concentration was observed only in the cells treated with URL plus 980 nm light irradiation (Fig. 3h). Overall, our findings of increased ROS coupled with decreased GSH, together with its wavelength-selective behavior, demonstrated the great potential of our URL in orthogonal UCL-regulated photodynamic therapy.Chemo-photodynamic therapy, which combines anticancer drugs and photosensitizers together into nanocarriers, has emerged as a promising synergistic method with an enhanced therapeutic effect. However, accumulating evidence indicates that drug resistance and toxicity still present obstacles to achieve the full benefits of chemo-photodynamic cancer therapy. The excellent in vitro behaviors of URL motivated us to further investigate whether the chemo-photodynamic therapy triggered by URL upon NIR light irradiation could result in enhanced anticancer activity to overcome drug resistance. As a proof-of-principle experiment, the oxaliplatin-resistant HCT116 colorectal cell line (HCT116/L-OHP) was chosen for an initial investigation. First, the role of the cRGD moiety in guiding URL to αvβ3 integrin-rich HCT116/L-OHP cells was confirmed by a series of experiments, including confocal fluorescence imaging (Fig. S13†), time-dependent internalization study (Fig. S14†), and the tumor sphere model (Fig. S15†). Our MTT assay demonstrated that URL was found to be nontoxic with HCT116/L-OHP cells up to a concentration of 200 μg mL−1 (Fig. S16†), indicating that the nanoprodrug has low toxicity. Upon exposure to 980 nm light for 5 min (0.5 W cm−2), only a slight decrease in cell viability (74.5–95.0%) was detected for all URL concentrations tested. In other words, we did not find the expected inhibition amplitudes to HCT116/L-OHP in the URL plus 980 nm light irradiation group. This result may be attributed to the fact that an HCT116/L-OHP cell displays high-level drug resistance that weakened the efficacy of chemotherapy. To gain insight into the drug resistance mechanism, the expression of the membrane MRP1 protein was first evaluated using western blot (WB) (Fig. S17a†). Compared with the control HCT116 cells, oxaliplatin-resistant HCT116/L-OHP cells exhibited approximately 2-fold higher MRP1 expression (Fig. S17b†). To further corroborate our finding, cellular MRP1 mRNA levels were assessed by qPCR analysis, and a significant upregulation of MRP1 mRNA was observed in HCT116/L-OHP cells (Fig. S17c†). These data suggested that MRP1 may be associated with the above drug resistance. Thus, we speculate that Cas13d-mediated MRP1 mRNA knockdown could contribute to escape from oxaliplatin resistance by repressing MRP1 efflux activity. To confirm our hypothesis, we first evaluated the efficacy of Cas13d-mediated MRP1 knockdown by examining its mRNA and protein levels in HCT116/L-OHP cells post-incubation with URL bearing uncaged RNP for the indicated time points (Fig. S18†). The qPCR and western blot results revealed a significant downregulation of MRP1 expression as early as 6 h, implying high MRP1 knockdown activity of URL bearing uncaged RNP. To assess whether the MRP1 knockdown can be orthogonally regulated by 808 nm NIR light, we next performed the similar qPCR and western blot tests using photocaged URL (Fig. 4a). Compared with cells exposed to URL only, cells treated with URL plus 808 nm light irradiation (5 min) displayed a significant decrease in MRP1 mRNA (Fig. 4b) and protein expression (Fig. 4c and d). As a control, 980 nm NIR light irradiation of URL led to no obvious change of MRP1 at both mRNA and protein levels. Given that the MRP1 protein was mainly located at the cell membrane, the 808 nm light-triggered membrane MRP1 downregulation was further visualized by CLSM. As expected, immunofluorescence staining analysis showed that only in the presence of URL plus 808 nm light did the fluorescence signal decrease significantly (Fig. 4e). Together, these results demonstrate that our URL nanoprodrug could downregulate membrane MRP1 in a wavelength-selective, orthogonal manner, offering great potential to overcome drug resistance for enhanced cancer therapy.
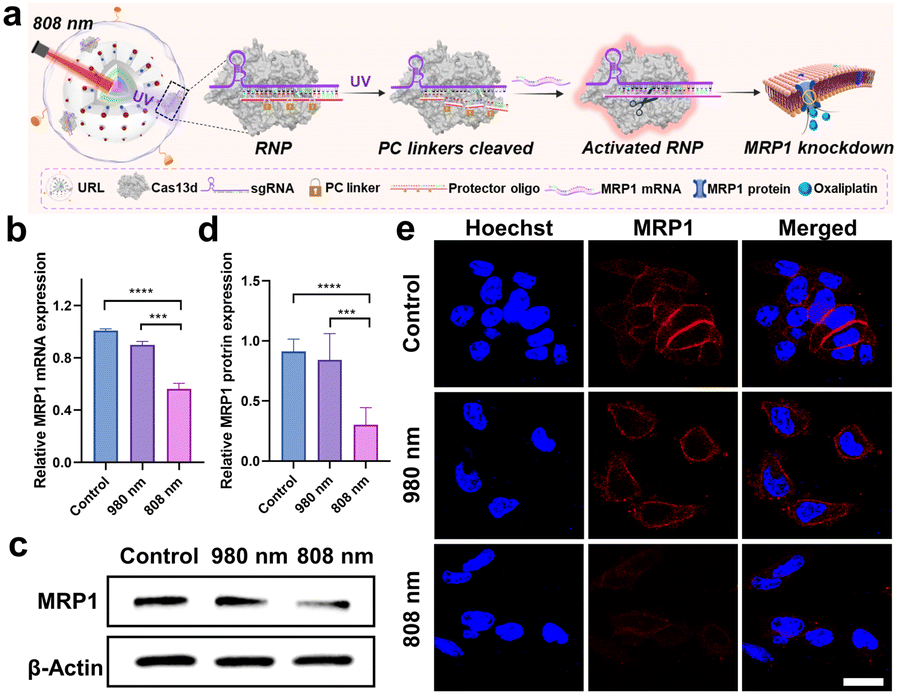 | ||
| Fig. 4 Orthogonal regulation of MRP1 by using the Cas13d nanoprodrug. (a) Schematic illustration of overcoming MRP1-mediated drug resistance upon activation of the nanoprodrug with 808 nm light. (b) Relative MRP1 mRNA expression in HCT116/L-OHP cells post-incubation with URL and harvested at 6 h after 980 or 808 nm treatment. ***P < 0.001 and ****P < 0.0001. (c) Western blot analysis of MRP1 expression in HCT116/L-OHP cells post-incubation with URL and harvested at 12 h after 980 or 808 nm treatment. (d) Quantitative densitometric analysis of MRP1 levels shown in (c). Data are means ± SE (n = 3). (e) CLSM images of HCT116/L-OHP cells with indicated treatments. Scale bar: 20 μm. | ||
Orthogonally regulated cell proliferation, apoptosis, migration, and invasion in vitro
Next, to investigate the orthogonal modulation effect on cell behavior, we conducted a series of experiments under the indicated six sequential logic operations (Group I to VI), where URL, and 980 and 808 nm light were utilized as the three inputs (Fig. 5a). First, a MTT assay was conducted to assess the antiproliferative effect of URL in HCT116/L-OHP cells. As shown in Fig. 5b, cell viability results showed that only a slight antiproliferative effect was observed in Group II (1,0,0) and IV (1,0,1) compared to in the control Group I (0,0,0). In contrast, in the presence of URL plus 980 nm light, cell viability decreased to approximately 62%, indicating a moderate antiproliferative effect in Group III (1,1,0). Moreover, Group V with successively treating URL with 980 and 808 nm NIR light (1,1*,1) showed no obvious change in cell cytotoxicity compared to Group III. These observations indicated that 980 nm light-triggered activation of the RB-Pt prodrug was the major contributor to the antiproliferative effect, whereas only 808 nm light activation of MRP1 knockdown contributed little. Surprisingly, Group VI with successively treating URL with 808 and 980 nm NIR light (1,1,1*) displayed the greatest cytotoxicity in HCT116/L-OHP cells. This was attributed to our orthogonal photomodulation of URL as a second-order logic-gated nanoprodrug, the maximum cytotoxicity of which could be realized only when the first AND-Gated MRP1 knockdown was triggered. Furthermore, we also examined the effect of orthogonally activated nanoprodrugs on apoptosis. Terminal transferase dUTP nick-end labeling (TUNEL) and Annexin V staining were performed to detect apoptotic cells. The CLSM results in Fig. 5c showed that an apparently higher percentage of TUNEL-positive cells in the Group VI was observed than in the other five groups. To further corroborate our finding, Annexin V-FITC/RedNucleus II dual staining-based flow cytometry was conducted (Fig. S19†). Compared to the relatively low levels of apoptosis (<10%) in Group I (0,0,0), II (1,0,0) and IV(1,0,1), Group III (1,1,0) and V (1,1*,1) showed a moderate proportion of early- and late-apoptotic cells (10–20%), while Group VI (1,1,1*) promoted the largest population of HCT116/L-OHP cells to undergo early- and late-stage apoptosis (>20%) (Fig. 5d). These apoptosis profiles were in accordance with the antiproliferation results, confirming the successful orthogonal photoregulation of cell apoptosis by using the URL nanoprodrug. Furthermore, the expression level of cellular apoptosis-related proteins was evaluated by western blot and CLSM analysis. Given that Bax and Bcl-2 function as the pro- and anti-apoptotic proteins, respectively, in mitochondria-mediated apoptosis, an increased ratio of Bax/Bcl-2 indicates the induction of apoptosis. As shown in Fig. 5e and f, Group VI induced a significant decrease in Bcl-2 expression with a concomitant increase in the protein level of Bax, resulting in a 16.2-fold increase in the Bax/Bcl-2 ratio compared to Group I. Moreover, Group VI also displayed the highest protein level of P53, signifying enhanced DNA damage. The CLSM results further confirmed the significantly increased caspase-3 activity in Group VI (Fig. S20†), indicating that the URL nanoprodrug induces apoptosis in a caspase-3-dependent manner. Overall, the antiproliferation and apoptosis findings illustrate a possible mechanism of action of the URL nanoprodrug (Fig. 5g): after uptake by oxaliplatin-resistant cells, 980 nm light irradiation leads to the effective photoreduction of the RB-Pt prodrug to oxaliplatin concurrently with ROS generation; the 808 nm light-induced Cas13d-based editing can inhibit the expression of MRP1 to repress the oxaliplatin drug efflux, thus enhancing the intracellular content of oxaliplatin. The accumulated oxaliplatin could effectively enter the nuclei to induce intense DNA damage and activation of p53-mediated apoptosis, which combines with the ROS-mediated apoptosis effect of PDT. These two pathways cooperatively facilitate apoptosis for an enhanced synergistic antitumor activity. Overall, overexpressed MRP1 promotes cancer cell resistance to chemotherapy, while the URL nanoprodrug is equipped with orthogonally activatable photoagents to downregulate MRP1 expression and subsequently activate the Pt(IV) prodrug for chemotherapy as well as the generation of ROS for PDT, which can potentiate the efficacy of oxaliplatin and reverse oxaliplatin resistance.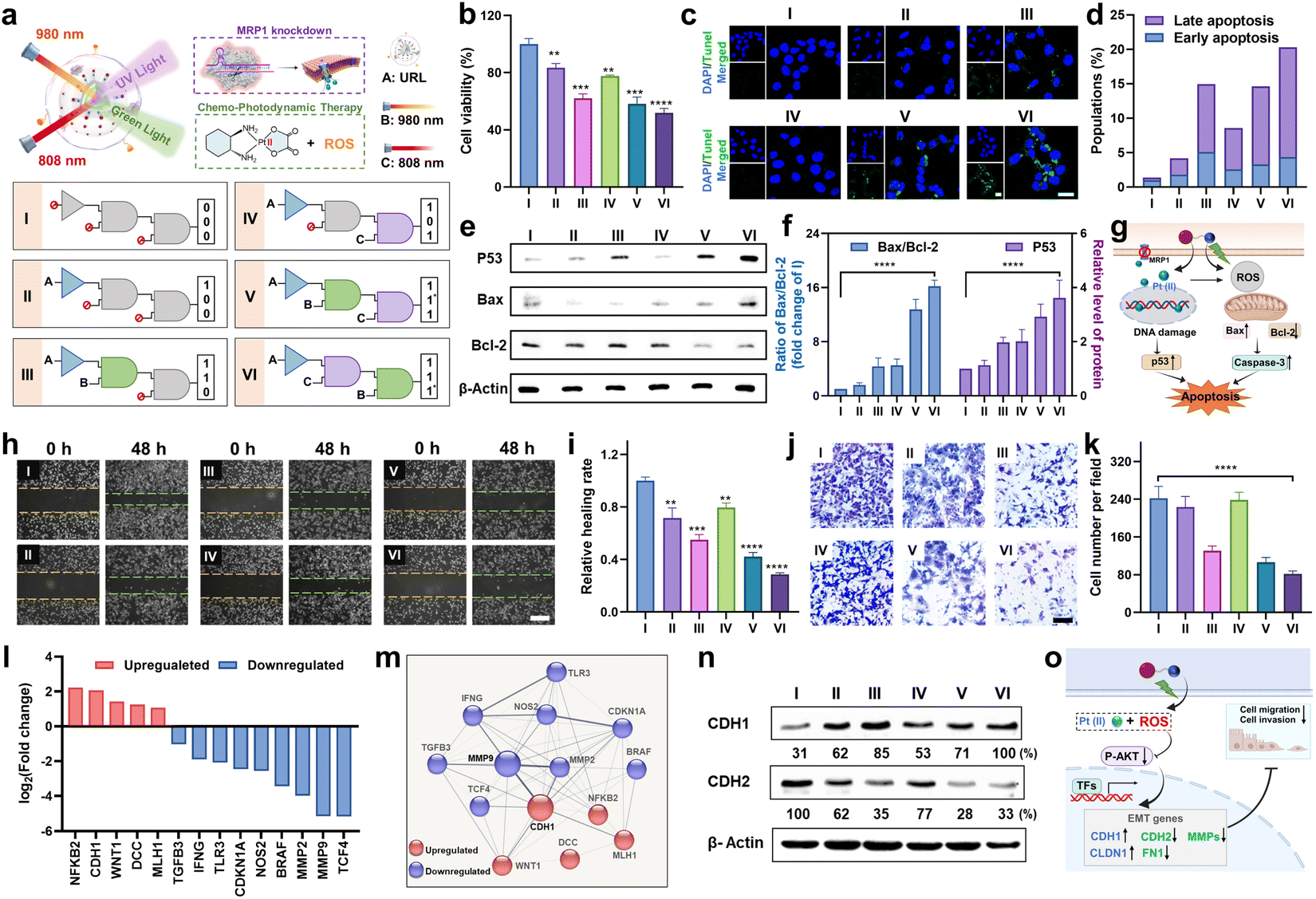 | ||
| Fig. 5 Orthogonally regulated cell migration, invasion, and apoptosis in vitro. (a) Sequential logic response gating system based on multiple signal inputs (three inputs include URL, and 980 and 808 nm irradiation for 5 min). (b) Cell viability of HCT116/L-OHP cells with three-input logic-gated operations. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. (c) Intracellular TUNEL staining of HCT116/L-OHP cells in different groups. DAPI: blue fluorescence; TUNEL: green fluorescence. (d) Quantitative analysis of early and late apoptosis in different groups using flow cytometry. (e) Western blot analysis of the indicated protein expressions in different groups. (f) Quantitative densitometric analysis of p53, Bax, and Bcl-2 levels shown in (e). Data are means ± SE (n = 3). (g) Schematic illustration of enhancement of the chemotherapeutic efficacy of the oxaliplatin prodrug and induction of tumor apoptosis by overcoming the drug resistance. (h) Wound-healing assay of HCT116/L-OHP cells with different treatments. (i) The healing rate with different treatments shown in (h). (j) Matrigel-based transwell assay of HCT116/L-OHP cells with different treatments. (k) The cell number of invaded cells per field with different treatments shown in (j). (l) log2 (fold change) values for fourteen EMT related genes evaluated from the PCR array. (m) STRING network involving MMP9 and CDH1 that were up- or down-regulated in HCT116/L-OHP cells from Group VI. (n) Western blot analysis of CDH1 (E-cadherin) and CDH2 (N-cadherin) expression in different groups. (o) Schematic illustration of the enhancement of the chemotherapeutic efficacy of the oxaliplatin prodrug and inhibition of tumor cell migration and invasion by overcoming the drug resistance. | ||
Extensive research has shown that oxaliplatin-resistant cell lines often exhibit the characteristics of strong migration and invasion, which could be suppressed by reversing drug resistance through combinational therapeutic approaches.38–40 Considering that URL is able to realize the orthogonal photoactivation of the RB-Pt prodrug and MRP1 knockdown, we speculated that this nanoprodrug might hold the potential to inhibit cancer cell metastasis. To validate this hypothesis, we first conducted a scratch wound-healing assay to study the inhibitory effect of URL on tumor cell migration (Fig. 5h). As expected, the cells in the control Group I exhibited potent and aggressive motility. In contrast, compared with the Group I, cells in Group VI migrated more slowly with the lowest in vitro wound healing rate of 28.7% (Fig. 5i), while Group II and IV only displayed a weak migration inhibition effect. Note that moderate cell migration occurred in both Group III and V containing URL plus 980 nm light, suggesting that 980 nm light-triggered oxaliplatin activation and ROS generation may be the major determinant of cell migration inhibition.39 Consistently, transwell migration and Matrigel invasion assays indicated that the migration and invasion abilities of HCT116/L-OHP cells were also significantly suppressed in Group VI (Fig. 5j, P < 0.0001). In addition, quantitative analysis of the number of invasive cells from all tested groups confirmed a similar inhibition effect on cell invasion to that of above migration assays (Fig. 5k). These results strongly supported our hypothesis that the inhibition of metastasis in oxaliplatin-resistant cells could be effectively amplified through orthogonal photoactivation of our URL nanoprodrug.
To gain further insight into the mechanisms by which the URL nanoprodrug inhibits cell metastasis, we examined the differential expression of metastasis-related genes in Group VI compared to in control Group I using a human colorectal cancer metastasis PCR array (Fig. S21†). If more than s 2-fold expression difference was observed between the two groups, the corresponding genes are marked green (downregulation) or red (upregulation), as illustrated in Fig. 5l. A total of 90 genes were quantified, and compared with the control group, nine pro-metastasis genes were significantly downregulated in the URL + 808 + 980 nm-treated group, while five metastasis repressor genes were upregulated concurrently. Moreover, STRING network analysis of the significantly regulated mRNAs revealed that more than 90% of these mRNAs demonstrated strong evidence of a functional relationship with the epithelial–mesenchymal transition (EMT) process, including an upregulated gene cluster centered on CDH1 and a downregulated gene cluster centered on MMP9 (Fig. 5m). Western blot results further confirmed the highest expression of the CDH1 protein (E-cadherin), but the lowest expression of CDH2 (N-cadherin) in Group VI compared to in the other five groups (Fig. 5n), which is a key functional characteristic of the substantial inhibition of the EMT process.41 Taken together, our findings suggest that URL inhibits cell metastasis by regulating EMT-related genes.
Next, we asked whether and how the orthogonal photoactivation of URL contributes to the regulation of EMT-related genes. To answer these questions, four migration-related genes (CDH1, CDH2, CLDN1, and FN1) and three invasion-related matrix metalloproteinase mRNAs (MMP2, MMP3, and MMP9) were chosen for our initial investigation. As shown in Fig. S22,† the qPCR results revealed that Group VI evoked the highest upregulation of CDH1 and CLDN1 mRNAs among all the groups tested, concomitantly with the highest downregulation of the other five mRNAs. Moreover, when compared with control Group I, the corresponding mRNA levels in Group III and V were upregulated or downregulated moderately. These observations were consistent with the logic operation pattern of our URL nanoprodrug in Fig. 5a, confirming the orthogonal regulation of EMT-related genes by URL. On the other hand, considering that the PI3K/AKT signaling 50 pathway is a central regulator in the EMT process, we therefore evaluated the expression levels of phosphorylated AKT (p-AKT) and total AKT in HCT116/L-OHP cells from the above six groups. As shown in Fig. S23,† when compared with control Group I, both Group II and IV showed a similar expression level of p-AKT and a similar p-AKT/total AKT ratio accordingly. In contrast, both p-AKT and the p-AKT/total AKT ratio decreased significantly in Group III, V and VI, while Group VI displayed the lowest values. Despite the differences in logic operation, one thing these three groups have in common is that they all have a 980 nm light irradiation treatment, which triggers ROS generation, which then in turn suppresses the PI3K/AKT signaling pathway. Based on these findings, we proposed a possible mechanism whereby orthogonal inhibition of cell metastasis by our URL nanoprodrug was regulated through the connection of ROS generation, the PI3K/AKT pathway, and the EMT process (Fig. 5o).
Orthogonally reversing chemoresistance for enhanced chemo-photodynamic therapy in vivo
The in vitro behaviors of URL motivated us to further investigate its antitumor efficacy in vivo. A flow chart depicting the major steps in the antitumor studies of URL is illustrated in Fig. 6a. Taking advantage of the luminescence properties of RB-Pt, we initially assessed the tumor-targeting ability and biodistribution profile by intravenously administering URL or cRGD-free URL to HCT116/L-OHP xenografted tumor-bearing nude mice, followed by fluorescence imaging (λex 570/λem 580–640 nm) with an IVIS animal imaging system. The fluorescence signal at the tumor sites of the URL-treated group increased gradually in a time-dependent manner (Fig. S24a†), and the signal intensity peaked at 6 h after URL administration (Fig. S24b†). Compared with cRGD-free URL, URL exhibited enhanced accumulation and a much longer retention time in a tumor microenvironment (Fig. S24b†). Ex vivo imaging was further performed in harvested tumors and major organs at 12 h post-injection (Fig. S24c†). The fluorescence signal in the tumor tissue collected from the URL-treated mice was significantly higher than that from the cRGD-free URL-treated mice, demonstrating a high intratumoral accumulation of URL, consistent with the tumor sphere studies (Fig. S15†). Notably, strong fluorescence signals in the livers and kidneys and negligible fluorescence signals in the hearts, spleen or lungs were observed in all treated groups, which were consistent with previous findings.42–44 However, the cRGD modification resulted in about a 2.5-fold increase of the tumor-to-liver accumulation ratio (Fig. S24d†), confirming the less off-target delivery of URL. Moreover, a considerable colocalization of red fluorescence (URL, λex 570/λem 580–640 nm) with blue fluorescence (DAPI, λex 405/λem 410–460 nm) was observed throughout the tumor section of URL-treated mice (Fig. S24e and f†), indicating that the tumor-targeting capability of URL promotes internalization for potentially enhanced therapy.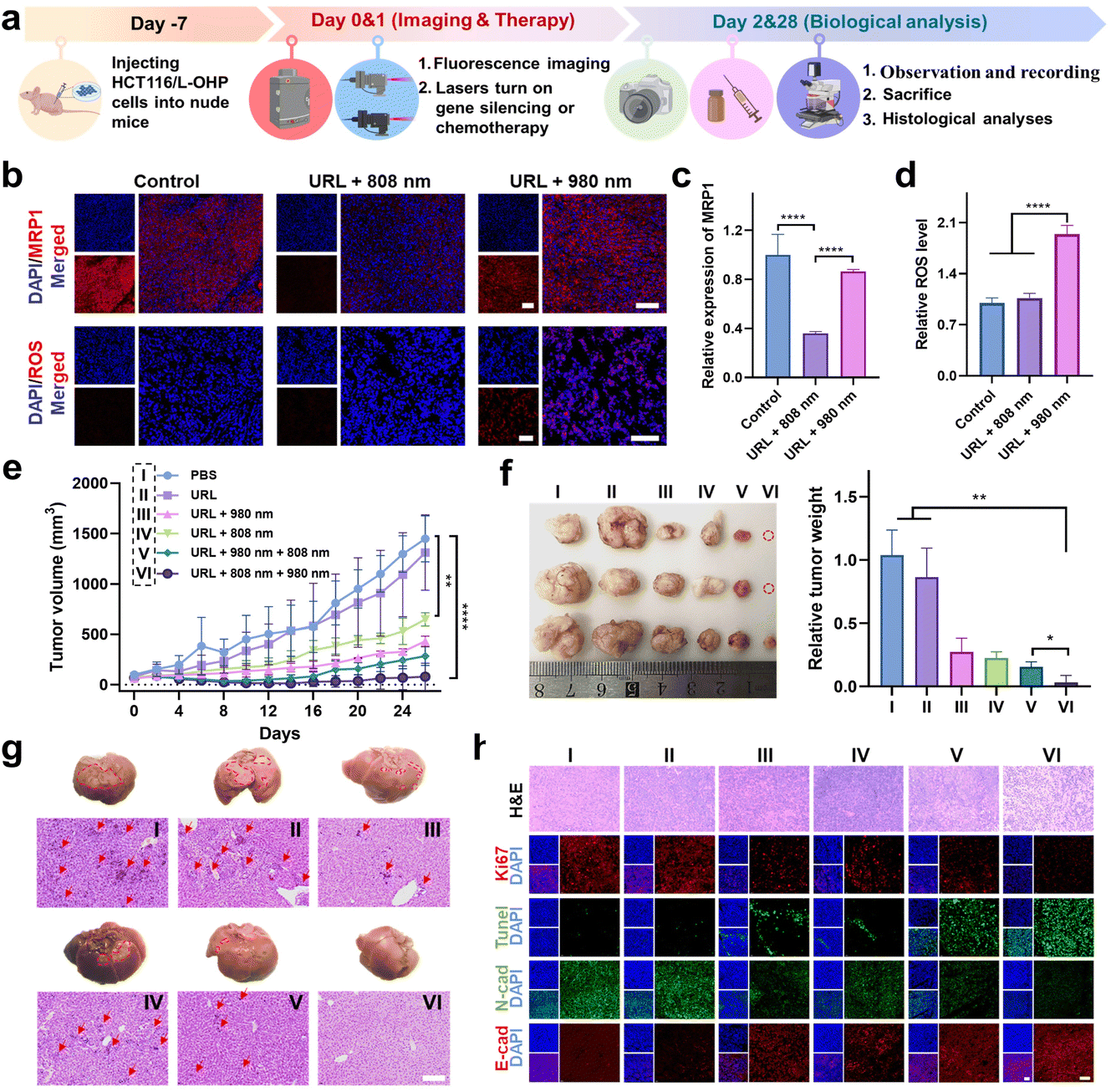 | ||
| Fig. 6 Orthogonally reversing chemoresistance for enhanced chemo-photodynamic therapy in vivo. (a) Scheme of the protocol for establishment of an HCT116/L-OHP tumor model. (b) Histological staining images of MRP1 and ROS levels in tumor slices of mice after the indicated treatments. (c and d) Quantitative histological analysis showing the significantly decreased MRP1 expression (c) and increased ROS levels (d) after NIR light irradiation. (e) Tumor growth curves after treatment. (f) Digital photos and weight of tumor tissues collected from mice at the end of treatment. The red circles indicate that tumors disappeared in the URL + 808 + 980 nm group at the end of treatment. (g) Digital photos and the corresponding H&E staining of livers collected from mice in the different groups at the end of treatment. The red circled area contains the white tissue observed on the liver surface, and the red arrows indicate the tumor focus on the slice. Scale bar: 200 μm. (h) Representative H&E staining, immunofluorescence analysis of proliferation (Ki67, red) and apoptosis (TUNEL, green), and immunofluorescence co-staining for N-cadherin (green) and E-cadherin (red) in tumor sections. Scale bar: 100 μm. **P < 0.01, and ****P < 0.0001. | ||
We then assessed the orthogonal NIR light-mediated regulation of URL in vivo. HCT116/L-OHP tumor-bearing mice were intravenously injected with URL (1 mg kg−1), followed by orthogonal NIR light activation. Histological staining images of the tumor cross-sections showed that 808 nm light irradiation resulted in significantly decreased intratumoral red fluorescence (λex 630/λem 640–695 nm, Fig. 6b), which was 2.8-fold lower than that of the nonirradiated URL group at 12 hours after injection (Fig. 6c). In contrast, 980 nm light irradiation only resulted in slight downregulation of MRP1 (13%) compared to that in the control group (Fig. 6b and c). Furthermore, we used the ROS-sensitive fluorescent dye dihydroethidium (DHE) to detect the ROS generation at 4 hours after URL injection. As shown in Fig. 6b, both the nonirradiated URL and URL plus 808 nm light irradiation group displayed relatively weak intratumoral fluorescence (λex 510/λem 590–620 nm), which may be attributed to the background fluorescence of URL at the excitation wavelength of DHE (510 nm). In contrast, the URL plus 980 nm light irradiation group exhibited a remarkable fluorescence signal with strong colocalization with DAPI throughout the tumor region (Fig. 6b and d), confirming the intense ROS generation during RB-Pt activation only upon 980 nm light irradiation. Taken together, these results demonstrated that Cas13d-based gene editing and the RB-Pt prodrug could be independently activated using dual NIR lights, indicating that the wavelength-orthogonal photomodulation features of URL were also maintained in vivo.
Next, we wondered whether such orthogonal photoactivation of RB-Pt-mediated chemo-photodynamic therapy and Cas13d-based gene editing could act synergistically for inhibiting tumor growth and drug resistance. To this aim, HCT116/L-OHP xenografted tumor-bearing nude mice were randomly divided into six groups, including one PBS control group and five URL-injected groups with the indicated NIR-light irradiation treatments (Fig. 6e). The treatment process was performed via one tail vein injection over 26 days, while the body weight and tumor size were monitored every two days (Fig. 6a). As shown in Fig. 6e, no significant change in the tumor growth rate was observed compared to that in the PBS treatment group (Group I), excluding the effect of nonspecific activation of the URL nanoprodrug in vivo. In contrast, a moderate tumor inhibition effect was observed in mice treated with URL plus 808 nm light (Group IV), with a tumor growth inhibition (TGI) of 47%. Moreover, treatment with URL plus 980 nm light (Group III) or URL plus sequential 980 and 808 nm light (Group V) further improved the tumor inhibition effect, with a TGI of 60.7% and 76.8%, respectively. More importantly, URL plus sequential 808 and 980 nm light irradiation (Group VI) resulted in the highest level of tumor inhibition effect, with the maximum TGI of 92.7%. These findings were consistent with the quantitative results of the final weight of tumors collected from mice after various treatments (Fig. 6f). Collectively, our results demonstrated that the URL nanoprodrug possesses the ability to orthogonally photoactivate gene editing and chemo-photodynamic therapy, leading to enhanced synergistic therapy.
In addition, no significant average weight loss was observed in Group III, V, and VI (<10%) at the end of treatments (Fig. S25†), consistent with the tumor inhibition effect presented in Fig. 6e. Notably, Group I, II, and IV showed an increased weight loss of 18.0%, 17.8%, and 13.0%, respectively. To gain insight into this unusual behavior, mice were sacrificed at the end of treatments, and major organs were collected for H&E staining analysis. No obvious pathological changes were observed in the heart, spleen, lung, and kidney collected from all group mice (Fig. S26†). In contrast, the liver tissues from the mice in Group I, II, and IV exhibited significantly increased hepatic tumor nodules on gross and histologic examination, compared with that of Group III, V, and VI (Fig. 6g). This result indicated a possible connection between liver metastasis and weight loss. Although the latter three groups could all inhibit the formation of hepatic tumor nodules, the statistical results of tumor metastatic foci in average number (Fig. S27a†) and diameter (Fig. S27b†) indicated that, while treatment with URL plus 980 nm light (Group III) or URL plus sequential 980 and 808 nm light (Group V) showed a moderate inhibition effect on liver metastasis, Group VI with treatment of URL plus sequential 808 and 980 nm light displayed the greatest inhibition effect. Given the consistency between liver metastasis inhibition and the antitumor effect, we speculate that the minimum liver metastasis observed in Group VI is believed to be generated by its maximum suppression of the primary tumor.
Finally, we analyzed the tumor pathological changes related to orthogonal URL-based chemo-photodynamic therapy using H&E sections, Ki67 immunofluorescence, and TUNEL staining (Fig. 6h). Compared with PBS-treated Group (I), all URL-treated groups exhibited various degrees of nuclear shrinkage and fragmentation, while Group VI showed obvious nuclear shrinkage and fragmentation, and extensive necrotic areas. In addition, the Ki67 immunofluorescence and TUNEL staining results showed that orthogonal photoactivation of URL (Group VI) displayed the weakest cell proliferation signal but the strongest apoptotic cell signal. Meanwhile, immuno-histochemical analysis indicated the lowest N-cadherin level and highest E-cadherin level in the tumor section obtained from Group VI, confirming the effective inhibition of the EMT process at the tumor site, and thus greatly attenuating the metastasis of colorectal cancer. Overall, the abovementioned results demonstrated that the orthogonal photoactivation of the URL nanoprodrug could trigger efficient Cas13d-mediated MRP1 knockdown to overcome drug resistance, which in turn significantly improved the efficacy of RB-Pt-mediated chemo-photodynamic therapy, ultimately amplifying synergistic primary tumor suppression and metastasis inhibition.
Conclusions
We have demonstrated a dual-NIR-light-mediated nanoprodrug that enables the photoactivation of Cas13d-mediated gene editing and RB-Pt-mediated chemo-photodynamic therapy in an orthogonally controlled manner in living cells and mice. The URL nanoprodrug was established by the combination of a rationally designed, green light-activatable RB-Pt prodrug and a UV-light responsive Cas13d-based scaffold with the orthogonal emissive UCNPs. Both experimental and computational studies confirmed that the RB-Pt(IV) prodrug is able to efficiently generate singlet oxygen concurrently with the release of highly cytotoxic oxaliplatin under 980 nm light irradiation. In addition, western blot and cellular studies demonstrated the efficient downregulation of membrane MRP1 by 808 nm light-activated URL. Moreover, taking inspiration from a Boolean logic gate, we created a set of second-order logic gates by using URL, and 980 and 808 nm light as the inputs, and demonstrated their utility for the programmable modulation of multiple cell behaviors, including proliferation, apoptosis, migration, and invasion. Importantly, the preliminary mechanistic studies suggested a possible inhibition mechanism of cell growth and metastasis, involving ROS generation, PI3K/AKT pathway suppression, and EMT suppression. More importantly, the logic-gated operation of the URL nanoprodrug can be activated on demand in vivo to downregulate MRP1 expression for overcoming drug resistance, which in turn amplifies chemo-photodynamic therapy with robust antitumor efficacy and anti-metastasis ability. Altogether, this proof-of-concept study provides a starting framework for expanding the Cas13d-based genome editing toolbox into prodrug nanomedicine with the ultimate goal of improving therapeutic outcomes.Data availability
A more detailed Experimental section, ESI table S1† including sequences of DNA, and ESI Fig. S1–S27.†Author contributions
Z. Liu and J. Zhang designed the study. Z. Feng helped in protein expression. M. Chen, and Y. Xue assisted in DFT and TDDFT calculations. J. Zhan, R. Wu, Y. Shi, and R. Liu aided with cell culture. J. Zhu helped in supervision. J. Zhang assisted in data analysis, funding acquisition, and final writing. All authors approved the final version.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We greatly acknowledge the financial support from the National Natural Science Foundation of China (no. 22004063 and 22274072), Natural Science Foundation of Jiangsu Province (no. 20200303), and State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM2206). We thank Prof. Hang Xing for very helpful discussions and critical input.References
- N. Vasan, J. Baselga and D. M. Hyman, Nature, 2019, 575, 299–309 CrossRef CAS PubMed.
- R. A. Ward, S. Fawell, N. Floc'h, V. Flemington, D. McKerrecher and P. D. Smith, Chem. Rev., 2021, 121, 3297–3351 CrossRef CAS PubMed.
- S. M. Stefan and M. Wiese, Med. Res. Rev., 2019, 39, 176–264 CrossRef CAS PubMed.
- G. Szakacs, M. D. Hall, M. M. Gottesman, A. Boumendjel, R. Kachadourian, B. J. Day, H. Baubichon-Cortay and A. Di Pietro, Chem. Rev., 2014, 114, 5753–5774 CrossRef CAS PubMed.
- K. M. Hanssen, M. Haber and J. I. Fletcher, Drug Resist. Updat., 2021, 59, 100795 CrossRef CAS PubMed.
- M. Vaghari-Tabari, P. Hassanpour, F. Sadeghsoltani, F. Malakoti, F. Alemi, D. Qujeq, Z. Asemi and B. Yousefi, Cell. Mol. Biol. Lett., 2022, 27, 49 CrossRef CAS PubMed.
- W. Cai, J. Liu, X. Chen, L. Mao and M. Wang, J. Am. Chem. Soc., 2022, 144, 22272–22280 CrossRef CAS PubMed.
- Y. N. Chen and Y. M. Zhang, Adv. Sci., 2018, 5, 1700964 CrossRef PubMed.
- W. Wang, Z. Liang, P. F. Ma, Q. Zhao, M. Y. Dai, J. Zhu, X. Han, H. Xu, Q. Y. Chang and Y. H. Zhen, Bioconjugate Chem., 2021, 32, 73–81 CrossRef CAS PubMed.
- Y. C. Pan, J. J. Yang, X. W. Luan, X. L. Liu, X. Q. Li, J. Yang, T. Huang, L. Sun, Y. Z. Wang, Y. H. Lin and Y. J. Song, Sci. Adv., 2019, 5, eaav7199 CrossRef CAS PubMed.
- H. H. Wessels, A. Mendez-Mancilla, X. Y. Guo, M. Legut, Z. Daniloski and N. E. Sanjana, Nat. Biotechnol., 2020, 38, 722–727 CrossRef CAS PubMed.
- B. B. He, W. B. Peng, J. Huang, H. Zhang, Y. S. Zhou, X. L. Yang, J. Liu, Z. J. Li, C. L. Xu, M. X. Xue, H. Yang and P. Y. Huang, Protein Cell, 2020, 11, 518–524 CrossRef CAS PubMed.
- R. Gupta, A. Ghosh, R. Chakravarti, R. Singh, V. Ravichandiran, S. Swarnakar and D. Ghosh, Front. Cell Dev. Biol., 2022, 10, 866800 CrossRef PubMed.
- Z. Deng, N. Wang, Y. Liu, Z. Xu, Z. Wang, T. C. Lau and G. Zhu, J. Am. Chem. Soc., 2020, 142, 7803–7812 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, Y. Guo, P. Wang, Y. Su, X. Jin, X. Zhu and C. Zhang, Exploration, 2022, 2, 20210172 CrossRef.
- L. J. Zhu, Y. Y. Guo, Q. H. Qian, D. Y. Yan, Y. H. Li, X. Y. Zhu and C. Zhang, Angew. Chem. Int. Ed., 2020, 59, 17944–17950 CrossRef CAS PubMed.
- Q. Mou, Y. Ma, F. Ding, X. Gao, D. Yan, X. Zhu and C. Zhang, J. Am. Chem. Soc., 2019, 141, 6955–6966 CrossRef CAS PubMed.
- Y. X. Chen, R. P. Zhao, L. L. Li and Y. L. Zhao, Angew. Chem. Int. Ed., 2022, 61, 14324–14328 Search PubMed.
- J. C. Barnes, P. M. Bruno, H. V. T. Nguyen, L. Y. Liao, J. Liu, M. T. Hemann and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 12494–12501 CrossRef CAS PubMed.
- H. Xiao, R. Qi, T. Li, S. G. Awuah, Y. Zheng, W. Wei, X. Kang, H. Song, Y. Wang, Y. Yu, M. A. Bird, X. Jing, M. B. Yaffe, M. J. Birrer and P. P. Ghoroghchian, J. Am. Chem. Soc., 2017, 139, 3033–3044 CrossRef CAS PubMed.
- Z. H. Di, B. Liu, J. Zhao, Z. J. Gu, Y. L. Zhao and L. L. Li, Sci. Adv., 2020, 6, eaba9381 CrossRef CAS PubMed.
- X. M. Li, Z. Z. Guo, T. C. Zhao, Y. Lu, L. Zhou, D. Y. Zhao and F. Zhang, Angew. Chem. Int. Ed., 2016, 55, 2464–2469 CrossRef CAS PubMed.
- Z. Zhang and Y. Zhang, Small, 2021, 17, e2004552 CrossRef PubMed.
- Z. Yi, Z. Luo, X. Qin, Q. Chen and X. Liu, Acc. Chem. Res., 2020, 53, 2692–2704 CrossRef CAS PubMed.
- Z. L. Yang, K. Y. Loh, Y. T. Chu, R. P. Feng, N. S. R. Satyavolu, M. Y. Xiong, S. M. N. Huynh, K. Hwang, L. L. Li, H. Xing, X. B. Zhang, Y. R. Chelma, M. Gruebele and Y. Lu, J. Am. Chem. Soc., 2018, 140, 17656–17665 CrossRef CAS PubMed.
- J. Zhao, Z. H. Di and L. L. Li, Angew. Chem. Int. Ed., 2022, 61, e202204277 CAS.
- J. Zhao, J. H. Gao, W. T. Xue, Z. H. Di, H. Xing, Y. Lu and L. L. Li, J. Am. Chem. Soc., 2018, 140, 578–581 CrossRef CAS PubMed.
- Y. L. Shao, B. Liu, Z. H. Di, G. Zhang, L. D. Sun, L. L. Li and C. H. Yan, J. Am. Chem. Soc., 2020, 142, 3939–3946 CrossRef CAS PubMed.
- Z. G. Wang, N. Wang, S. C. Cheng, K. Xu, Z. Q. Deng, S. Chen, Z. F. Xu, K. Xie, M. K. Tse, P. Shi, H. Hirao, C. C. Ko and G. Y. Zhu, Chem, 2019, 5, 3151–3165 CAS.
- Y. X. Chen, X. J. Yan and Y. Ping, ACS Mater. Lett., 2020, 2, 644–653 CrossRef CAS.
- S. R. Wang, L. Wei, J. Q. Wang, H. M. Ji, W. Xiong, J. Liu, P. Yin, T. Tian and X. Zhou, ACS Chem. Biol., 2020, 15, 1455–1463 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, F. Xie, J. Lin and L. Xu, Chem. Sci., 2020, 11, 11478–11484 RSC.
- Y. X. Wu, J. D. Zheng, Q. Zeng, T. Zhang and D. Xing, Nano Res., 2020, 13, 2399–2406 CrossRef CAS.
- E. V. Moroz-Omori, D. Satyapertiwi, M. C. Ramel, H. Hogset, I. K. Sunyovszki, Z. Q. Liu, J. P. Wojciechowski, Y. Y. Zhang, C. L. Grigsby, L. Brito, L. Bugeon, M. J. Dallman and M. M. Stevens, ACS Cent. Sci., 2020, 6, 695–703 CrossRef CAS PubMed.
- S. P. C. Cole, Annu. Rev. Pharmacol., 2014, 54, 95–117 CrossRef CAS PubMed.
- W. X. Yan, S. R. Chong, H. B. Zhang, K. S. Makarova, E. V. Koonin, D. R. Cheng and D. A. Scott, Mol. Cell, 2018, 70, 327–339 CrossRef CAS PubMed.
- J. Gong, H. X. Wang, Y. H. Lao, H. Z. Hu, N. Vatan, J. Guo, T. C. Ho, D. T. Huang, M. Q. Li, D. Shao and K. W. Leong, Adv. Mater., 2020, 32, e2003537 CrossRef PubMed.
- F. Wu, Y. Q. Du, J. N. Yang, B. Y. Shao, Z. S. Mi, Y. F. Yao, Y. Cui, F. He, Y. Q. Zhang and P. P. Yang, ACS Nano, 2022, 16, 3647–3663 CrossRef CAS PubMed.
- C. Y. Lin, S. J. Yang, C. L. Peng and M. J. Shieh, ACS Appl. Mater. Interfaces, 2018, 10, 6096–6106 CrossRef CAS PubMed.
- E. C. Sutton, C. E. McDevitt, J. Y. Prochnau, M. V. Yglesias, A. M. Mroz, M. C. Yang, R. M. Cunningham, C. H. Hendon and V. J. DeRose, J. Am. Chem. Soc., 2019, 141, 18411–18415 CrossRef CAS PubMed.
- T. Shibue and R. A. Weinberg, Nat. Rev. Clin. Oncol., 2017, 14, 611–629 CrossRef PubMed.
- Y. Gu, S. T. Lai, Y. Dong, H. Fu, L. W. Song, T. X. Chen, Y. R. Duan and Z. Zhang, Adv. Sci., 2021, 8, 2002922 CrossRef CAS PubMed.
- Y. Y. Zhao, L. Zhang, Z. X. Chen, B. Y. Zheng, M. R. Ke, X. S. Li and J. D. Huang, J. Am. Chem. Soc., 2021, 143, 13980–13989 CrossRef CAS PubMed.
- Y. Y. Guo, Q. S. Zhang, Q. W. Zhu, J. Gao, X. Y. Zhu, H. J. Yu, Y. H. Li and C. Zhang, Sci. Adv., 2022, 8, eabn2941 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00020f |
| This journal is © The Royal Society of Chemistry 2023 |