
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel N(SCF3)(CF3)-amines: synthesis, scalability and stability†
Yi
Yang
a,
Nathalie
Saffon-Merceron
b,
Julien C.
Vantourout
 a and
Anis
Tlili
*a
a and
Anis
Tlili
*a
aInstitute of Chemistry and Biochemistry (ICBMS–UMR CNRS 5246), Univ Lyon, Université Lyon 1, CNRS, CPE-Lyon, INSA, 43 Bd du 11 Novembre 1918, 69622 Villeurbanne, France. E-mail: anis.tlili@univ-lyon1.fr
bInstitut de Chimie de Toulouse (FR 2599), Université de Toulouse, CNRS, 118 Route de Narbonne, 31062 Toulouse, France
First published on 13th March 2023
Abstract
We disclosed herein a straightforward strategy for the synthesis of unprecedented N-((trifluoromethyl)thio), N-(trifluoromethyl) amines using a combination of isothiocyanates with a fluoride source and an electrophilic trifluoromethylthiolation reagent. More interestingly, the scalability of the methodology has been demonstrated and the stability of the new motif has been studied.
Organofluorine chemistry is by far one of the most active fields of research in modern organic chemistry. The unique properties induced by the fluorine atom or fluorinated motifs including high lipophilicity, increased solubility and metabolic stability have witnessed the efforts for extensive development during the last few years.1 Nowadays, fluorinated molecules are extensively used in agrochemicals2 as well as for pharmaceutical3 applications. In this span of time, several efficient procedures for the incorporation of trifluoromethyl and trifluoromethyl chalcogens have been reported in the literature through reagent and/or catalyst design as well as methodology developments.4
Nitrogen is also a predominant atom in life science technologies;5 therefore the association of amines with fluorine-based motifs such as trifluoromethyl groups has been the quest of several research groups. For example, N-trifluoromethyl azoles have demonstrated excellent in vitro aqueous stability which might improve metabolic stability and membrane permeability compared to their N-methyl counterparts.6
In this context, several direct methodologies have been developed to access N-CF3 amines making use of electrophilic trifluoromethylation reagents, namely Umemoto7 and Togni8 reagents or radical trifluoromethyl sources (Scheme 1A).9,10 However, these strategies offer limited scope especially with regard to the amine starting material. A breakthrough was disclosed by the Schoenebeck group in 2017. In their study, the authors demonstrated that a wide range of trifluoromethylamines could be accessed via in situ generation of thiocarbamoyl fluoride using bench stable (Me4N)SCF3 (Scheme 1A).11 Afterwards, several groups designed new strategies to access the key thiocarbamoyl fluoride intermediate.12–15 Finally, the group of Xu recently reported an elegant oxidative approach for the synthesis of trifluoromethyl amine reagents which have been used for the transfer of the N-CF3 moiety.16
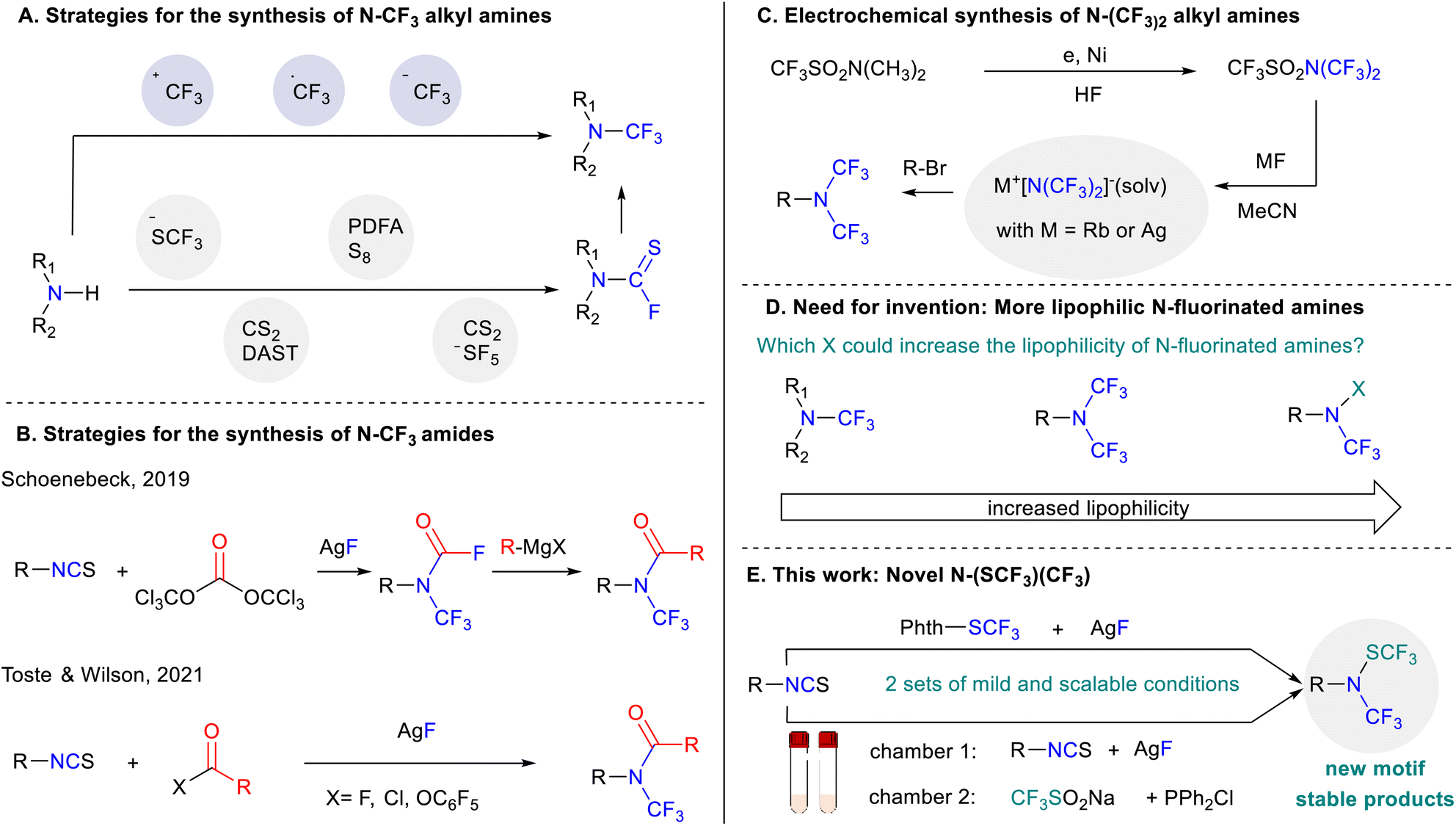 | ||
| Scheme 1 (A–E) State of the art for the synthesis of trifluoromethylated amines and the need for accessing new motifs. | ||
Another way to access trifluoromethylamines relies on the use of isothiocyanates. Indeed, in 1965, Shepard17 initially reported that nucleophilic amines could be formed from isothiocyanates using mercury fluoride. Inspired by this precedent, the group of Schoenebeck18 elegantly demonstrated that silver fluoride could efficiently replace the mercury-based reagent offering a practical and general way to synthesize N-trifluoromethyl carbamoyl fluoride (Scheme 1B). This procedure has been also adapted by the groups of Toste and Wilson19 for the synthesis of N-trifluoromethyl amides (Scheme 1B).
N,N-bis(trifluoromethyl) amines represent another valuable motif due to their enhanced lipophilicity and stability in comparison to aliphatic and aromatic N-trifluoromethylamines.20,21 Despite these interesting properties, access to N-(CF3)2 amines remains very scarce with most promising synthesis employing a combination of N,N-bis(trifluoromethyl)trifluoromethanesulphonamide and a metal-based reagent (Scheme 1C).22
Therefore, there is a clear need for developing new motifs that could be easily and robustly accessible while modulating the properties around the nitrogen atom such as the lipophilicity (Scheme 1D).23 In this context, we report herein an unprecedented, mild and efficient protocol for accessing novel N-((trifluoromethyl)thio)-N-(trifluoromethylamines) starting with isothiocyanates and electrophilic trifluoromethylthiolation reagents (Scheme 1E).
From a reaction design standpoint, inspired by the work of Schoenebeck, we envisioned that a trifluoromethylamino nucleophile intermediate could be first in situ generated by the reaction of isothiocyanate with silver fluoride before subsequently reacting with an electrophilic trifluoromethylthiolating source to furnish the desired compounds.
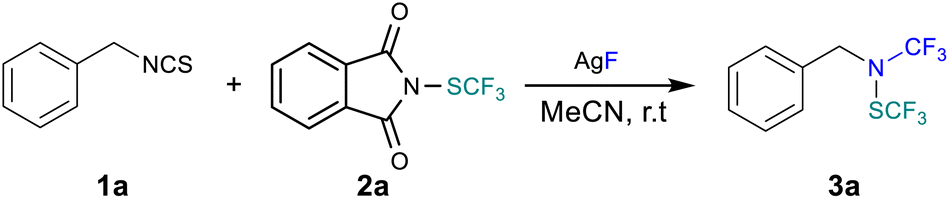
We initiated our study by using benzyl isothiocyanate 1a as a model substrate in the presence of three equivalents of silver fluoride (Table 1). We found that the formed nucleophilic amine was able to react with the electrophilic Munavalli's24N-trifluoromethylthiophthalimide 2a in MeCN at room temperature delivering the desired product 3a in 50% yield (Table 1, entry 1). This encouraging result decided us to further investigate parameters that could enhance the reaction outcome. We first studied the impact of the solvent on the formation of the desired product. THF and 1,4-dioxane as well as DCM were not suitable for the formation of 3a (Table 1, entries 2 & 3). Lower yields were obtained when using DMSO and DMF, 30% and 40% respectively (Table 1, entries 2 and 3). In addition, increasing the temperature was found to be detrimental since only 35% of 3a was obtained at 50 °C (Table 1, entry 7). Then, the impact of the trifluoromethylthiolating reagent was investigated. Surprisingly, the attempt in switching to the more electrophilic Shen's reagent (N-trifluoromethylthiosaccharin)25 resulted in lower efficiency with compound 3a only obtained in 30% yield (Table 1, entry 8). To our delight, adding one equivalent of cesium fluoride allowed for the formation of the desired product in 75% yield (Table 1, entry 9). Finally, potassium fluoride did not improve the overall efficiency of the process (Table 1, entry 10).
|
|
||
|---|---|---|
| Entrya | Deviation from standard conditions | Yieldb (%) |
| a Reactions were performed with 1a (0.2 mmol, 1 equiv.), 2a (0.2 mmol, 1 equiv.), AgF (0.6 mmol, 3 equiv.) and solvent (1 mL) for 16 hours. b Determined by 19F NMR spectroscopy with PhCF3 as an internal standard. Shen's reagent 2b (N-trifluoromethylthiosaccharin). | ||
| 1 | None | 50 |
| 2 | THF or dioxane instead of MeCN | 0 |
| 3 | DCM instead of MeCN | 0 |
| 4 | PhMe instead of MeCN | 5 |
| 5 | DMSO instead of MeCN | 30 |
| 6 | DMF instead of MeCN | 40 |
| 7 | 50 °C instead of rt | 35 |
| 8 | 2b (Shen's reagent) instead of 2a | 30 |
| 9 | With 1 equiv. of CsF | 75 |
| 10 | With 1 equiv. of KF | 40 |
With the best set of conditions in hand, we evaluated the effectiveness of the protocol for different isothiocyanate starting materials. Initial tests were devoted to subjecting benzylic isothiocyanate to our reaction conditions. The desired compounds were obtained from low to very good yields (Scheme 2, products 3a–c). Afterwards, aliphatic isothiocyanates were exposed to the reaction conditions and proved compatible with yields up to 67% (Scheme 2, products 3d–3i). Noteworthily, the melatonin precursor derivative (Scheme 2, compound 3d) was derivatized in a synthetically useful isolated yield of 35%. Several protected tertiary amines were also found to be effective under the reaction conditions (Scheme 2, products 3g–3i). Next, aromatic isothiocyanates were evaluated. Electron rich aryl starting materials were tolerated using our protocol and the desired products were obtained with yields up to 78% (Scheme 2, products 3j–3q). It should be mentioned that ortho, meta or para substituted isothiocyanates are suitable partners.
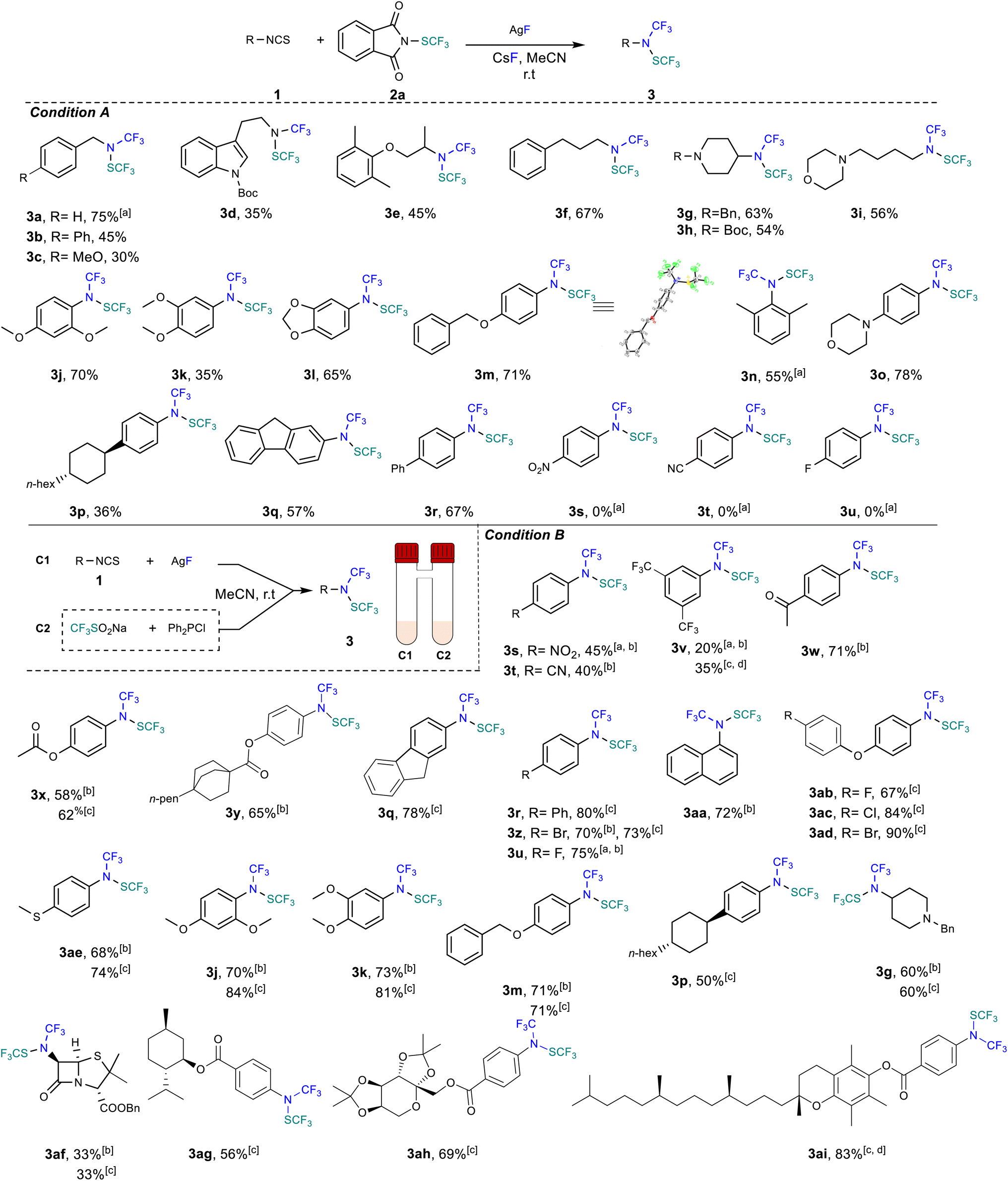 | ||
| Scheme 2 Substrate scope. Condition A: reactions were performed with 1 (0.2 mmol, 1 equiv.), 2a (0.2 mmol, 1 equiv.), AgF (0.6 mmol, 3 equiv.) and CsF (0.2 mmol, 1 equiv.) in MeCN (1 mL) Yields of isolated compounds. Yields of isolated compounds for 16 hours. a Yield determined by 19F NMR spectroscopy with PhCF3 as an internal standard. b Condition B: reactions were performed with chamber 1 (C1): 1 (0.2 mmol, 1 equiv.) and AgF (0.8 mmol, 4 equiv.) in MeCN (1 mL); chamber 2 (C2): CF3SO2Na (0.6 mmol, 3 equiv.) and Ph2PCl (0.6 mmol, 3 equiv.) in MeCN (1 mL) for 16 hours, yields of isolated compounds. c Reactions were performed with chamber 1 (C1): 1 (0.2 mmol, 1 equiv.) and AgF (0.8 mmol, 4 equiv.) in MeCN (1 mL); chamber 2 (C2): CF3SO2Na (0.6 mmol, 3 equiv.) and Ph2PCl (1.2 mmol, 6 equiv.) in MeCN (1 mL) for 16 hours, yields of isolated compounds. d 1.5 mL of ether and 0.5 mL of ACN as a solvent mixture in tube C1. | ||
The para substituted phenylisothiocyanate with a phenyl ring furnishes the desired product in 67% yield (Scheme 2, 3r). Interestingly, the structure of 3m was unambiguously confirmed by X-ray crystallographic analysis.26 Afterwards, we evaluated the influence of electron withdrawing substituents on the starting aryl isothiocyanate derivatives. Unfortunately, the presence of strong electron withdrawing groups including NO2, CN or fluorine was detrimental to the reaction and no product formation was observed (Scheme 2, products 3s–3u) probably due to a decrease of the nucleophilicity of the resulting trifluoromethyl amine anion intermediate. Knowing that Shen's reagent would be too electrophilic (vide supra) and based on the relative trifluoromethylthio cation-donating scale,27 we identified the trifluoromethylthio dimer ((SCF3)2) as the most promising reagent to employ. In addition, the in situ generation of such a reactive species has been well documented in the literature.4a Indeed, the Langlois reagent can react with phosphine derivatives to yield nucleophilic SCF3 (ref. 28) which can easily be oxidized to the desired dimer. Disappointingly, our initial in situ test turned out to be ineffective. Being aware of the incompatibly issues that could arise from mixing together of all the reaction components, we decided to use a two-chamber reactor. In chamber 2, formation of the –SCF3 anion was achieved by reacting chloro diphenylphosphine with the Langlois reagent.
This unstable anion readily collapses to afford the trifluoromethylthio dimer ((SCF3)2) (see ESI for details†).The formation of nucleophilic trifluoromethylamine through fluorinative desulfurization with AgF also yield the Ag2S by-product that could potentially oxidize AgSCF3 to afford the trifluoromethylthio dimer ((SCF3)2). Our hypothesis turned out to be effective when isothiocyanate was mixed with silver fluoride in the first chamber while Langlois's reagent reacted with PPh2Cl in the second chamber. Under these conditions, electron poor arenes including NO2, CN, CF3, acetyl, and ester could be transformed into the desired products with isolated yields up to 80% (Scheme 2, products 3s–3r). Halogen substituted arenes including bromo and fluoro derivatives were also obtained in excellent yields of 70% and 75%, respectively (Scheme 2, products 3z, 3u). Naphthalene derivative 3aa was obtained in 72% yield. Finally, we decided to assess this protocol for electron donating arene derivatives as well as for aliphatic compounds to offer a complementary approach to the original one (condition A) using commercially available reagents. It turns out that these new conditions are also effective with electron donating aryl isocyanate derivatives and the desired products are formed with very good yields up to 90% (Scheme 2, products 3ab, 3j–m). Herein also diarylether derivatives substituted with halogens, including fluoro, chloro and bromo were tolerated and the desired products were obtained in an excellent yield up to 90% (Scheme 2, products 3ab, 3ac & 3ad). Moreover, aliphatic product 3g was also obtained in good yield (60%) while the penicillin core structure was derivatized in a synthetically useful yield of 33% (Scheme 2, product 3af). Finally, the robustness of our strategy was further demonstrated starting with other complex structures. Indeed, using DL-menthol derivatives allowed us to obtain the desired product 3ag in good isolated yield of 56%. Also, using the diacetonefructose derivatives allows the formation of the compound 3ah in very good yield. Finally, vitamin E derivative 3ai was obtained with an excellent yield of 83% when the reaction was performed in a mixture of ether/MeCN to increase the solubility of the starting 1ai.
The robustness of the second protocol was further demonstrated by performing a large-scale experiment. Starting with 10 mmol of isothiocyanate and using a 300 mL two-chamber reactor allowed both desired products 3m and 3r to be synthesized in very good yields of 60% and 59%, respectively (Scheme 3A).
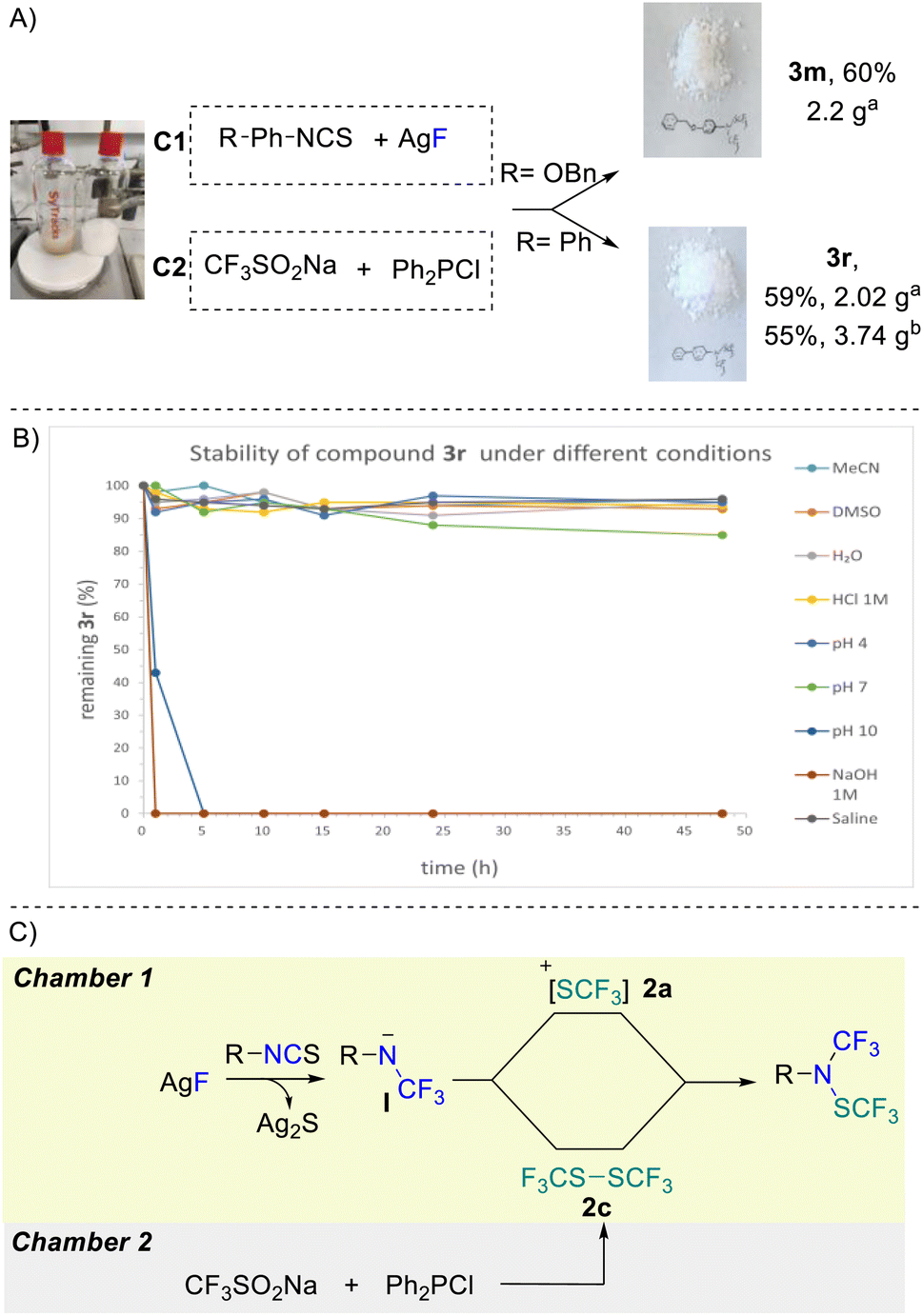 | ||
| Scheme 3 (A) Scale-up experiments: a reactions were performed with chamber 1 (C1): 1 (10 mmol, 1 equiv.) and AgF (40 mmol, 4 equiv.) in MeCN (50 mL); chamber 2 (C2): CF3SO2Na (30 mmol, 3 equiv.) and Ph2PCl (60 mmol, 6 equiv.) in MeCN (50 mL) for 24 hours. b Reactions were performed with chamber 1 (C1): 1 (20 mmol, 1 equiv.) and AgF (80 mmol, 4 equiv.) in MeCN (100 mL); chamber 2 (C2): CF3SO2Na (60 mmol, 3 equiv.) and Ph2PCl (120 mmol, 6 equiv.) in MeCN (100 mL) for 24 hours yields of isolated compounds. (B) Stability of compound 3r in various media. (C) Proposed mechanism. | ||
Unsurprisingly, product 3r turned out to be completely stable in CH3CN and DMSO as well as in water for more than 48 hours (Scheme 3B). This compound also demonstrated high stability under acidic conditions (HCl 1 M and pH 4) and in pH = 7 buffer with more than 90% recovery. Rapid degradation was observed under basic conditions (pH 10 and NaOH (1 M)). Interestingly, very high stability under saline conditions was also observed.
From a mechanistic standpoint, condition A proceeds via the generation of trifluoromethyl amine anion I that subsequently reacts with the electrophilic trifluoromethylthiolating reagent 2a yielding the desired product (Scheme 3C). On the other hand, for condition B, the key to success is the generation of the electrophilic CF3SSCF3 dimer 2c. Dimer 2c finally reacts with nucleophilic amine I intermediate to deliver the desired product (Scheme 3C).
Conclusions
In summary, we reported the discovery of two efficient and complementary protocols for the synthesis of unprecedented N-((trifluoromethyl)thio), N-(trifluoromethyl) amines. While one uses a shelf-stable electrophilic trifluoromethylthiolation reagent, the other employs a two-chamber reactor for the in situ generation of an electrophilic trilfuoromethylthiolating reagent. The desired products have been obtained in moderate to excellent yields. The scalability of the reaction was demonstrated through the preparation of more than 3.7 grams of the desired compounds. Importantly, N(SCF3)CF3 shows high aqueous stability. We assume that this discovery will pave the way to future developments in this exciting field of research.Data availability
Experimental data have been provided in the ESI.†Author contributions
Y. Y. and A. T. conceived the project. J. C. V. and A. T. supervised the work and wrote the manuscript. N. S. M. performed the X-ray analysis. All the co-authors contributed to the editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the CNRS and the Agence Nationale de la Recherche (ANR-JCJC-2020-CDI-DEOX) is gratefully acknowledged. Y. Y. thanks the CSC (China Scholarship Council) for a doctoral fellowship.Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2013 CrossRef; (b) D. O‘Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- (a) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (c) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- (a) H. Ge, H. Liu and Q. Shen, Organofluorine Chem., 2021, 99–172 CAS; (b) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–776 CrossRef CAS PubMed; (c) T. Besset, P. Jubault, X. Pannecoucke and T. Poisson, Org. Chem. Front., 2016, 3, 1004–1010 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) S. Schiesser, H. Chepliaka, J. Kollback, T. Quennesson, W. Czechtizky and R. J. Cox, J. Med. Chem., 2020, 63(21), 13076–13089 CrossRef CAS PubMed; (b) S. Schiesser, R. J Cox and W. Czechtizky, Future Med. Chem., 2021, 13, 941–944 CrossRef CAS PubMed.
- T. Umemoto, K. Adachi and S. Ishihara, J. Org. Chem., 2007, 72, 6905–6917 CrossRef CAS PubMed.
- (a) K. Niedermann, N. Früh, E. Vinogradova, M. S. Wiehn, A. Moreno and A. Togni, Angew. Chem., Int. Ed., 2011, 50, 1059–1063 CrossRef CAS PubMed; (b) K. Niedermann, N. Früh, R. Senn, B. Czarniecki, R. Verel and A. Togni, Angew. Chem., Int. Ed., 2012, 51, 6511–6515 CrossRef CAS PubMed.
- For a general review see: T. Milcent and B. Crousse, C. R. Chimie, 2018, 21, 771–781 CrossRef CAS.
- For radical reactions see: (a) A. van der Werf, M. Hribersek and N. Selander, Org. Lett., 2017, 19, 2374–2377 CrossRef CAS PubMed; (b) T. Cao, P. Retailleau, T. Milcent and B. Crousse, Chem. Commun., 2021, 57, 10351–10354 RSC.
- (a) T. Scattolin, K. Deckers and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 221–224 CrossRef CAS PubMed; (b) for selected example of the valorization of trifluoromethyl carbamoyl fluorides by the same group please see: S. Bouayad-Gervais, T. Scattolin and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 11908–11912 CrossRef CAS PubMed; (c) C. D.-T. Nielsen, F. G. Zivkovic and F. Schoenebeck, J. Am. Chem. Soc., 2021, 143, 13029–13033 CrossRef CAS PubMed; (d) S. Bouayad-Gervais, C. D.-T. Nielsen, A. Turksoy, T. Sperger, K. Deckers and F. Schoenebeck, J. Am. Chem. Soc., 2022, 144, 6100–6106 CrossRef CAS PubMed; (e) A. Turksoy, S. Bouayad-Gervais and F. Schoenebeck, Chem.–Eur. J., 2022, e202201435 CAS.
- J. Yu, J.-H. Lin and J.-C. Xiao, Angew. Chem., Int. Ed., 2017, 56, 16669–16673 CrossRef CAS PubMed.
- (a) K. Onida, L. Vanoye and A. Tlili, Eur. J. Org. Chem., 2019, 6106–6109 CrossRef CAS , please see also:; (b) K. Onida and A. Tlili, Angew. Chem., Int. Ed., 2019, 58 CrossRef CAS PubMed; (c) M. P. Drapeau and A. Tlili, Tetrahedron Lett., 2020, 61, 12545–12548 Search PubMed.
- A. Taponard, T. Jarrosson, L. Khrouz, M. Médebielle, J. Broggi and A. Tlili, Angew. Chem., Int. Ed., 2022, 61, e202204623 CrossRef CAS PubMed.
- (a) S. Liang, J. Wei, L. Jiang, J. Liu, Y. Mumtaz and W. Yi, Chem. Commun., 2019, 55, 8536–8539 RSC; (b) W. Xu, F. Liu, J. Li, M. Li, J. Xie and C. Zhu, J. Org. Chem., 2021, 86, 12443–12451 CrossRef CAS PubMed.
- S. Liu, Y. Huang, J. Wang, F.-L. Qing and X.-H. Xu, J. Am. Chem. Soc., 2022, 144(4), 1962–1970 CrossRef CAS PubMed.
- W. A. Sheppard, J. Am. Chem. Soc., 1965, 87, 4338–4341 CrossRef CAS.
- T. Scattolin, S. Bouayad-Gervais and F. Schoenebeck, Nature, 2019, 573, 102–107 CrossRef CAS PubMed.
- J. Liu, M. F. L. Parker, S. Wang, R. R. Flavell, F. D. Toste and D. M. Wilson, Chem, 2021, 7, 2245–2255 CAS.
- (a) L. M. Yagupol’skii, Aromatic and Heterocyclic Compounds with Fluoro-containing Substituents, Naukova Dumka, Kiev, 1988 Search PubMed; (b) R. N. Haszeldine, A. E. Tipping and R. H. Valentine, J. Fluorine Chem., 1982, 21, 329–334 CrossRef CAS; (c) L. M. Yagupol’skii, S. S. Shavaran, B. M. Klebanov, A. N. Rechitskii and I. I. Maletina, Pharm. Chem. J., 1994, 28, 813–817 CrossRef.
- Halogenoamines have been also used, for a selected example please see: T. W. Hart, R. N. Haszeldine and A. E. Tipping, J. Chem. Soc., Perkin Trans. 1, 1980, 1544–1550 RSC.
- For selected references for the use of N,N-bis(trifluoromethyl)trifluoromethanesulphonamide please see: (a) P. Sartori, N. Ignat’ev and S. Datsenko, J. Fluorine Chem., 1995, 75, 157–161 CrossRef CAS; (b) V. Hiliarius, H. Buchholz, P. Sartori, N. Ignatiev, A. Kucherina and S. Datsenko, WO2000046180, Merck Patent GmbH, 2000 Search PubMed; (c) L. N. Schneider, E.-M. T. Krauel, C. Deutsch, K. Urbahns, T. Bischof, K. A. M. Maibom, J. Landmann, F. Keppner, C. Kerpen, M. Hailmann, L. Zapf, T. Knuplez, R. Bertermann, N. V. Ignat'ev and M. Finze, Chem.–Eur. J., 2021, 27, 10973–10978 CrossRef CAS PubMed.
- (a) A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS; (b) B. Linclau, Z. Wang, G. Compain, V. Paumelle, C. Q. Fontenelle, N. Wells and A. Weymouth-Wilson, Angew. Chem., Int. Ed., 2016, 55, 674–678 CrossRef CAS PubMed.
- (a) S. Munavalli, D. K. Rohrbaugh, D. I. Rossman, F. J. Berg, G. W. Wagner and H. D. Durst, Synth. Commun., 2000, 30, 2847–2854 CrossRef CAS; (b) T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856–12859 CrossRef CAS PubMed.
- C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed, 2014, 53, 9316–9320 ( Angew. Chem. , 2014 , 126 , 9470–9474 ) CrossRef CAS PubMed.
- CCDC-2211357 (4) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures/.
- M. Li, J. Guo, X.-S. Xue and J.-P. Cheng, Org. Lett., 2016, 18, 264–267 CrossRef CAS PubMed.
- Y. Yang, L. Xu, S. Yu, X. Liu, Y. Zhang and D. A. Vicic, Chem.–Eur. J., 2016, 22, 858–863 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2211357. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06542h |
| This journal is © The Royal Society of Chemistry 2023 |