
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed enantioselective rearrangement of dienyl cyclopropanes†
Qi
Xu‡
,
Chuan-Jun
Lu‡
 ,
Chang-Qiu
Guo
,
Jia
Feng
and
Ren-Rong
Liu
*
,
Chang-Qiu
Guo
,
Jia
Feng
and
Ren-Rong
Liu
*
College of Chemistry and Chemical Engineering, Qingdao University, Qingdao, 266071, P. R. China. E-mail: renrongliu@qdu.edu.cn
First published on 2nd March 2023
Abstract
Vinyl cyclopropanes (VCPs) are among the most useful three-carbon building blocks in organic synthesis. They are commonly used as dienophiles in a range of cycloaddition reactions. However, VCP rearrangement has not received much attention since its discovery in 1959. In particular, the enantioselective rearrangement of VCP is synthetically challenging. Herein, we report the first palladium-catalyzed regio- and enantioselective rearrangement of VCPs (dienyl or trienyl cyclopropanes) for the construction of functionalized cyclopentene units in high yields and with excellent enantioselectivities and 100% atom economy. The utility of the current protocol was highlighted by a gram-scale experiment. Moreover, the methodology provides a platform for accessing synthetically useful molecules containing cyclopentanes or cyclopentenes.
Introduction
Chiral five-membered carbocycles are ubiquitous structural motifs in a myriad of biologically active natural products and pharmaceuticals,1–4 such as laurokamurene A, vibralactone, silphinene and (+)-multifidene (Scheme 1A). In addition, they also serve as intermediates in various total syntheses of unnatural and natural products.5,6 However, in contrast to six-membered carbocycles, which can be readily accessed via asymmetric Diels–Alder reactions, chiral 5-membered all-carbon rings are difficult to synthesize. Thus, the development of enantioselective and flexible synthetic routes to these frameworks is highly warranted.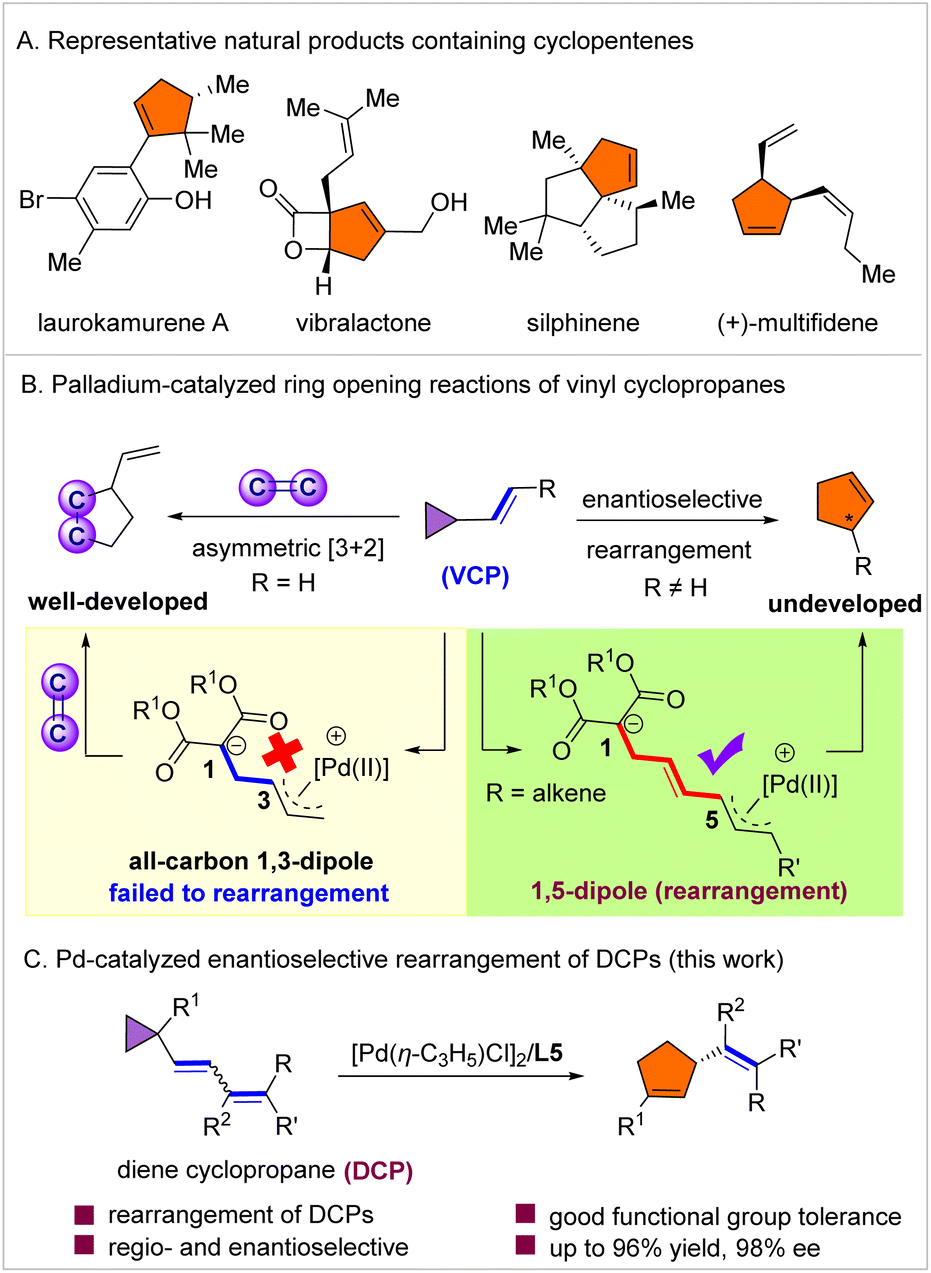 | ||
| Scheme 1 Representative natural products containing cyclopentenes and asymmetric reactions of vinyl cyclopropanes. | ||
Vinyl cyclopropanes are among the most useful three-carbon building blocks in organic synthesis.7–10 They are well known to generate a dipole, which allows for their application in a range of cycloaddition reactions with unsaturated compounds.11–16 Among these, the palladium-catalyzed asymmetric [3 + 2] annulation of VCPs with activated alkenes is by far one of the most studied transformations, providing a powerful approach to optically active cyclopentane derivatives (Scheme 1B).17 In 2011, Trost and co-workers achieved the first palladium-catalyzed enantioselective synthesis of chiral cyclopentanes via [3 + 2] annulation of vinyl cyclopropanes.18 Since then, significant advancements have been made in this field using activated alkenes.19–28 On the other hand, VCPs are also known to undergo rearrangement to afford cyclopentenes.29 However, compared with cycloaddition, VCP rearrangement has been largely overlooked since its discovery in 1959 due to the necessity for harsh conditions (normally proceeds at 300–500 °C).30–35 Only a handful of studies have been reported regarding VCP rearrangement under mild conditions catalyzed by transition metals such as Rh,36–39 Pd,40–42 Ni,43–45 and Cu,46–48 that proceed via coordination of the metal catalyst to the vinyl substituent of VCP. Nevertheless, the direct enantioselective rearrangement of VCP has remained a formidable challenge and the development of strategies for achieving this objective is highly desirable. Inspired by the recently reported palladium-catalyzed asymmetric functionalization of dienes,49–54 we herein report the first palladium-catalyzed enantioselective isomerization of dienyl cyclopropanes (activated VCPs) under mild conditions (Scheme 1C) for the formation of chiral cyclopentene derivatives, which are otherwise challenging to synthesize.
Results and discussion
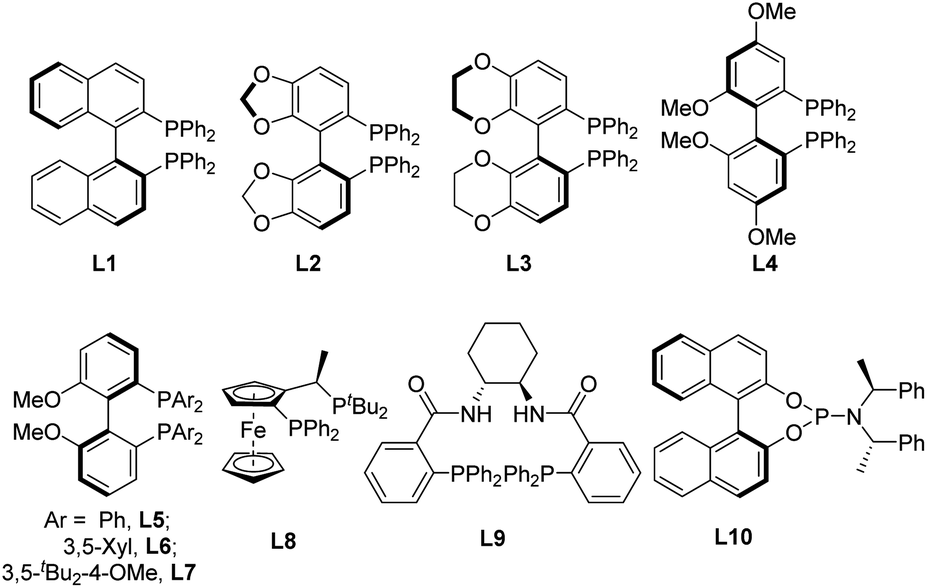
We initiated our investigation using 1a as a model substrate. Substrate 1a was obtained as a Z/E mixture in high yield via the Wittig reaction (see ESI† for details). Selective rearrangement of dienyl cyclopropane 1a was initially attempted using Pd(OAc)2 (5 mol%), (S)-BINAP (L1) (7.5 mol%) in toluene at 80 °C, affording 5-membered cyclopentene derivative 2a in 75% yield and with 83% ee as a single E-isomer (Table 1, entry 1). The use of Pd(dba)2 instead of Pd(OAc)2 delivered comparable results (entry 2), while a slightly higher yield was observed by employing [Pd(η-C3H5)Cl]2 as the catalyst (entry 3). After screening several other solvents, including THF, dioxane and DMF (entries 4–6), it was found that toluene remained the best choice. Various phosphine ligands were then screened to improve the ee of 2a (entries 7–15). In general, palladium complexes with electron-rich phosphine ligands afforded good results for this rearrangement. The use of (S)-SEGPHOS (L2) as the ligand improved the enantioselectivity significantly to 94% (entry 7). Comparable results (94% yield, 94% ee) were obtained when the reaction was performed with (R)-MeO-BIPHEP (L5) as the ligand (entry 10). (S)-SYNPHOS (L3) and (S)-GARPHOS (L4) displayed comparable activities, both affording product 2a in 87% yield and with 88% and 90% ee, respectively (entries 8–9). Interestingly, the use of highly electron-donating (R)-DTBM-BIPHEP (L7) or (R,S)-JOSIPHOS (L8), sharply reduced the yields and enantioselectivities (entries 12–13). The use of palladium/Trost ligand (L9) or palladium/Feringa ligand (L10) complexes failed to give any desired rearrangement product (entries 14–15). The enantioselectivity could not be further enhanced by lowering the reaction temperature to 60 °C (entry 16). Although the rearrangement proceeded smoothly at room temperature, the yield was moderate (entry 17). Increasing the concentration of 1a to 0.2 M decreased the yield of 2a to 87% without affecting the enantioselectivity (entry 18). Halving the catalyst loading did not significantly affect the results, with the enantioselectivity remaining unchanged and the yield was slightly lower (entry 19).|
|
||||||
|---|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.1 mmol, E/Z mixtures), [Pd] (5.0 mol%), and ligand (12 mol%) in toluene (1.0 mL) at 80 °C for 3 h. b Isolated yield. c Determined by chiral HPLC. d Reaction performed at 60 °C. e Reaction performed at 25 °C for 20 h. f Reaction performed with 0.2 M concentration of 1a. g [Pd(η-C3H5)Cl]2 (2.5 mol%), ligand (6 mol%), reaction time: 10 h. | ||||||
| Entry | L* | [Pd] | Solvent | Yieldb (%) | eec (%) | |
| 1 | L1 | Pd(OAc)2 | Toluene | 75 | 83 | |
| 2 | L1 | Pd(dba)2 | Toluene | 77 | 83 | |
| 3 | L1 | [Pd(η-C3H5)Cl]2 | Toluene | 85 | 83 | |
| 4 | L1 | [Pd(η-C3H5)Cl]2 | THF | 80 | 82 | |
| 5 | L1 | [Pd(η-C3H5)Cl]2 | Dioxane | 84 | 80 | |
| 6 | L1 | [Pd(η-C3H5)Cl]2 | DMF | 82 | 62 | |
| 7 | L2 | [Pd(η-C3H5)Cl]2 | Toluene | 91 | 94 | |
| 8 | L3 | [Pd(η-C3H5)Cl]2 | Toluene | 87 | 88 | |
| 9 | L4 | [Pd(η-C3H5)Cl]2 | Toluene | 87 | 90 | |
| 10 | L5 | [Pd(η-C3H5)Cl]2 | Toluene | 94 | 94 | |
| 11 | L6 | [Pd(η-C3H5)Cl]2 | Toluene | 89 | 90 | |
| 12 | L7 | [Pd(η-C3H5)Cl]2 | Toluene | 50 | 42 | |
| 13 | L8 | [Pd(η-C3H5)Cl]2 | Toluene | 20 | 40 | |
| 14 | L9 | [Pd(η-C3H5)Cl]2 | Toluene | <5% | — | |
| 15 | L10 | [Pd(η-C3H5)Cl]2 | Toluene | <5% | — | |
| 16d | L5 | [Pd(η-C3H5)Cl]2 | Toluene | 75 | 94 | |
| 17e | L5 | [Pd(η-C3H5)Cl]2 | Toluene | 45 | 95 | |
| 18f | L5 | [Pd(η-C3H5)Cl]2 | Toluene | 87 | 93 | |
| 19g | L5 | [Pd(η-C3H5)Cl]2 | Toluene | 83 | 94 | |
|
||||||
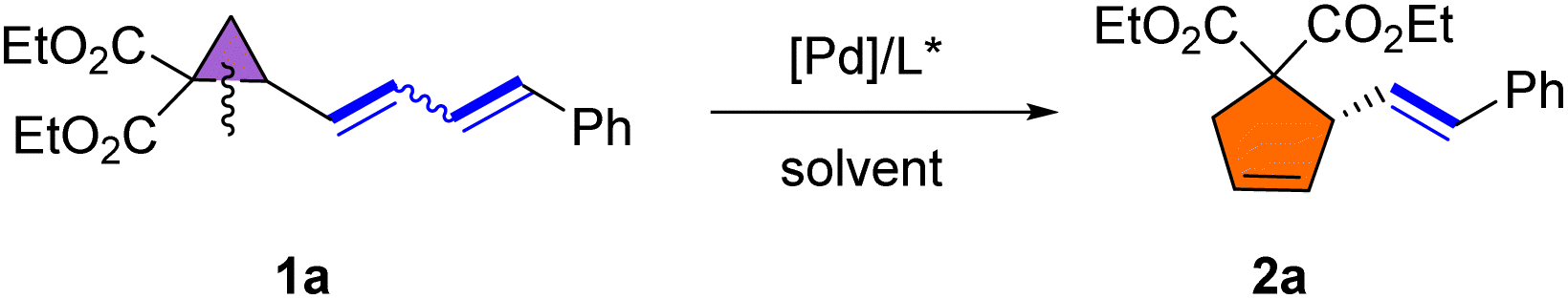
After establishing the optimal reaction conditions, we explored the scope of the rearrangement reaction. As shown in Scheme 2, the dienylsubstituent R was first examined. Most of the tested para- and meta-substituted aromatic dienyl cyclopropanes (1a–1m) underwent the rearrangement smoothly to afford vinylcyclopentene adducts 2a–2m in 85–96% yield and with 90–98% ee. It is noteworthy that in all cases, the E/Z mixtures of dienyl cyclopropanes 1 afforded pure E-isomers. The absolute configuration was confirmed based on single crystal X-ray analysis of 2d (CCDC 2183663). Substrates bearing halogens, including fluorine (2e, 2m), chlorine (2f, 2l) and bromine (2g, 2k), were rearranged smoothly in good yields and with high ees. 3,4-Disubstituted aromatic dienes were also well tolerated (2q–2u). It is worth noting that ortho-substituted aromatic dienes also rearranged to give good yields and high enantioselectivities (2n–2p). The inclusion of 2-naphthyl (1v), thienyl (1w) and furyl (1x) functionalities on the diene was successful, affording products 2v–2x in 80–88% yield and 93–95% ee. Importantly, we found that ester-substituted dienes 1y–1z rearranged satisfactorily into cyclopentene 2y and 2z. Moreover, 1,2-disubstituted dienyl cyclopropane 1aa was also applicable in this rearrangement reaction to afford 2aa in high yield and with good enantioselectivity (93% ee). However, 1,1-disubstituted dienyl cyclopropane 1ad failed to undergo the rearrangement. Furthermore, the terminal dienyl cyclopropane 1ab afforded 2ab in 90% yield with a moderate ee, which may be caused by the reduced steric hindrance of terminal olefin during rearrangement. The cyclopropane substituents (R1, E) were examined next. Dienyl cyclopropane 1ac, bearing geminal methyl and dienyl groups, reacted favorably under the reaction conditions to afford 2ac in 90% yield and with 86% ee. In addition, changing the substituents on the ester functional groups had little effect on this rearrangement reaction (2ae). Other electron-withdrawing substituents, such as CN or SO2Ph substituted vinyl cyclopropanes were also suitable for this rearrangement reaction to afford 2af and 2ag in high yields with good enantioselectivities. Interestingly, rearrangement of acetyl substituted cyclopropane under the standard conditions afforded the corresponding Cloke–Wilson type vinyl-dihydrofuran 3 in 57% yield with 29% ee, while 2ah was achieved in 17% yield with 87% ee.
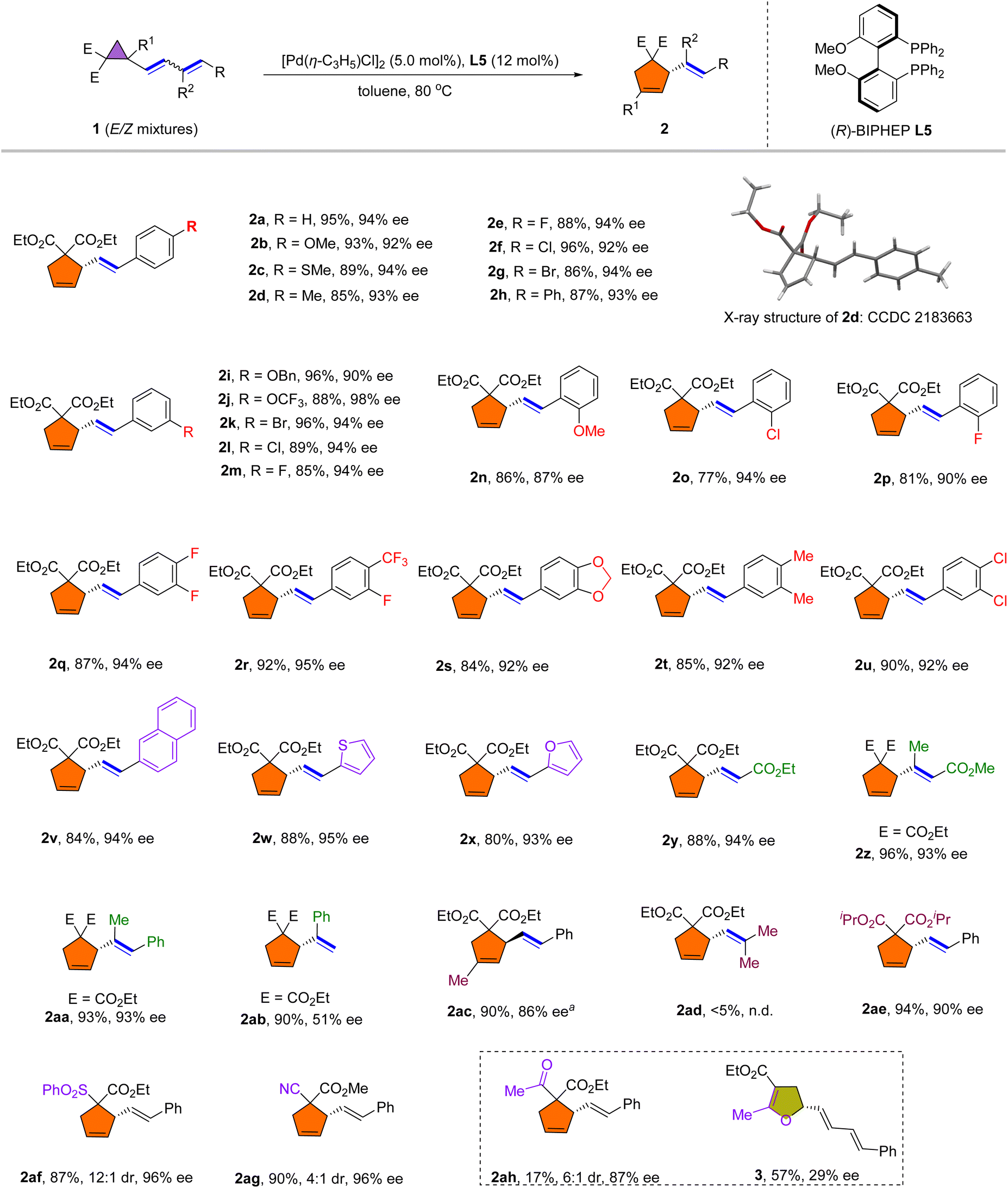 | ||
| Scheme 2 Scope of dienyl cyclopropanes, reaction conditions: 1 (0.1 mmol, E/Z mixtures), [Pd(η-C3H5)Cl]2 (5.0 mol%), L5 (12 mol%), in toluene (1.0 mL) at 80 °C for 3–5 h. aL2 was used as the ligand instead of L5. | ||
To further verify the universality of the reaction, we examined the rearrangement of triene cyclopropane substrates (Scheme 3). To our delight, the rearrangement occurred smoothly to afford 5a and 5b with 98% ee and 86% ee, respectively. The regiochemistry is intriguing in that a five-membered-ring product was again observed, even in the case of triene rearrangement, while seven-membered-ring formation did not occur.
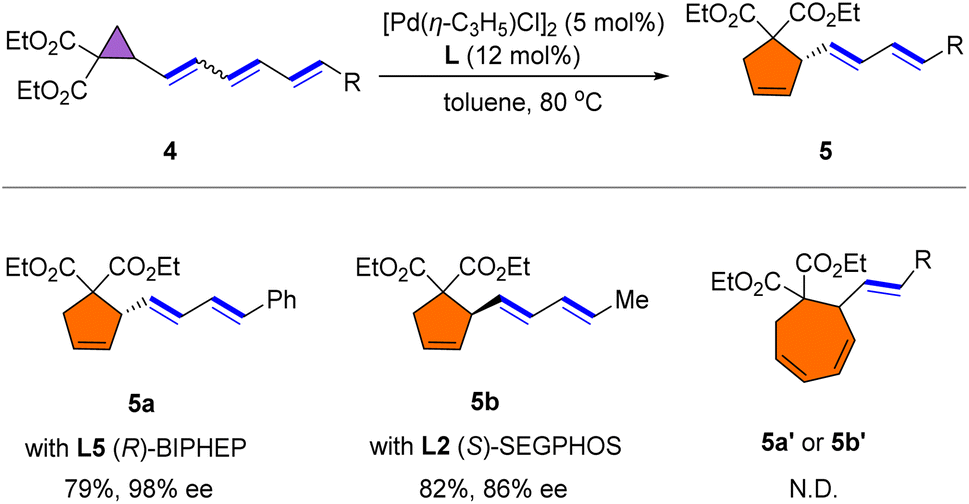 | ||
| Scheme 3 Reaction conditions: 3 (0.1 mmol), [Pd(η-C3H5)Cl]2 (5.0 mol%), L (12 mol%), in toluene (1.0 mL) at 80 °C for 3 h. | ||
Polysubstituted cyclopentane units are widely distributed in pharmaceuticals and biologically active compounds; thus, we envisaged the synthesis of polysubstituted cyclopentane using the developed rearrangement. As shown in Scheme 4, 2-formylcyclopropane 8 was synthesized in 85% yield with 94% ee and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr via a one-pot organocatalytic domino Michael/α-alkylation using bromomalonate 6 and crotonaldehyde 7.55 The subsequent Wittig reaction afforded dienyl cyclopropane substrate 9 in 77% yield. Under the standard conditions, polysubstituted cyclopentene 10 was formed in high yield and with excellent diastereoselectivity. The configuration of 10 was assigned to be cis via H–H NOESY analysis (see the ESI†).
:1 dr via a one-pot organocatalytic domino Michael/α-alkylation using bromomalonate 6 and crotonaldehyde 7.55 The subsequent Wittig reaction afforded dienyl cyclopropane substrate 9 in 77% yield. Under the standard conditions, polysubstituted cyclopentene 10 was formed in high yield and with excellent diastereoselectivity. The configuration of 10 was assigned to be cis via H–H NOESY analysis (see the ESI†).
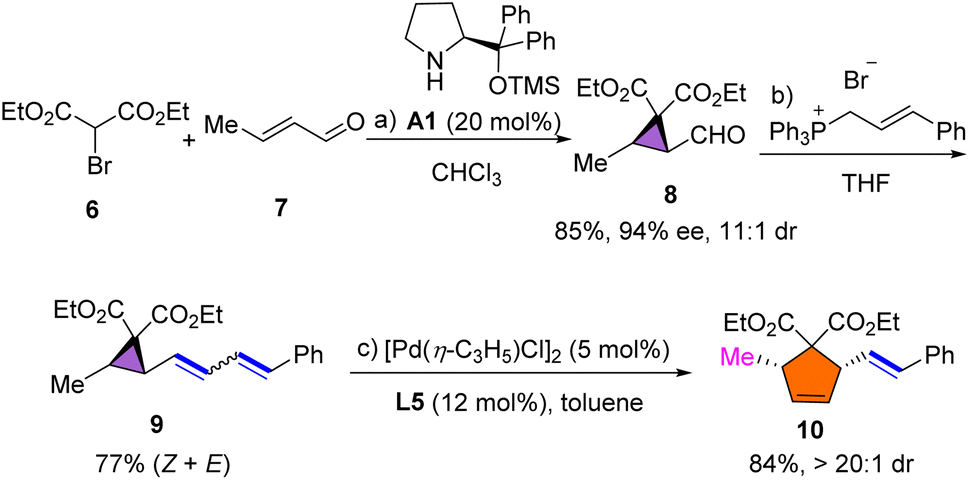 | ||
| Scheme 4 Reaction conditions: a6 (0.5 mmol), 7 (0.6 mmol), A1 (20 mol%), Et3N (0.5 mmol), CHCl3 (2.0 mL), 85% yield. b8 (0.33 mmol), cinnamyltriphenylphosphonium (0.3 mmol), n-BuLi (0.33 mmol), in THF (2.0 mL) at 0 °C for 5 h, 77% yield. c9 (0.1 mmol), [Pd(η-C3H5)Cl]2 (5.0 mol%), L5 (12 mol%), in toluene (1.0 mL) at 100 °C for 24 h, 84% yield, > 20:1 dr. | ||
A gram-scale reaction of 1a was performed by decreasing the loading of [Pd(η-C3H5)Cl]2 and L5, delivering 2a in 83% yield and with comparable enantioselectivity to that of the small-scale reaction (Scheme 5A). The synthetic importance of this dienyl cyclopropane-vinylcyclopentene rearrangement was further highlighted by several transformations of representative compound 2a (Scheme 5B). Reduction of the alkene groups of 2a proceeded smoothly under Pd/C-catalyzed hydrogenation to provide 11 in 90% yield and with 93% ee. Compound 2a was selectively converted to functionalized cyclopentene 12 through epoxidation using m-CPBA in moderate yield. Reduction of the ester group using LiAlH4 proceeded smoothly to afford diol 13 in 70% yield. Subsequently, 2a was decarboxylated employing the Krapcho reaction to deliver 14 in 82% yield.
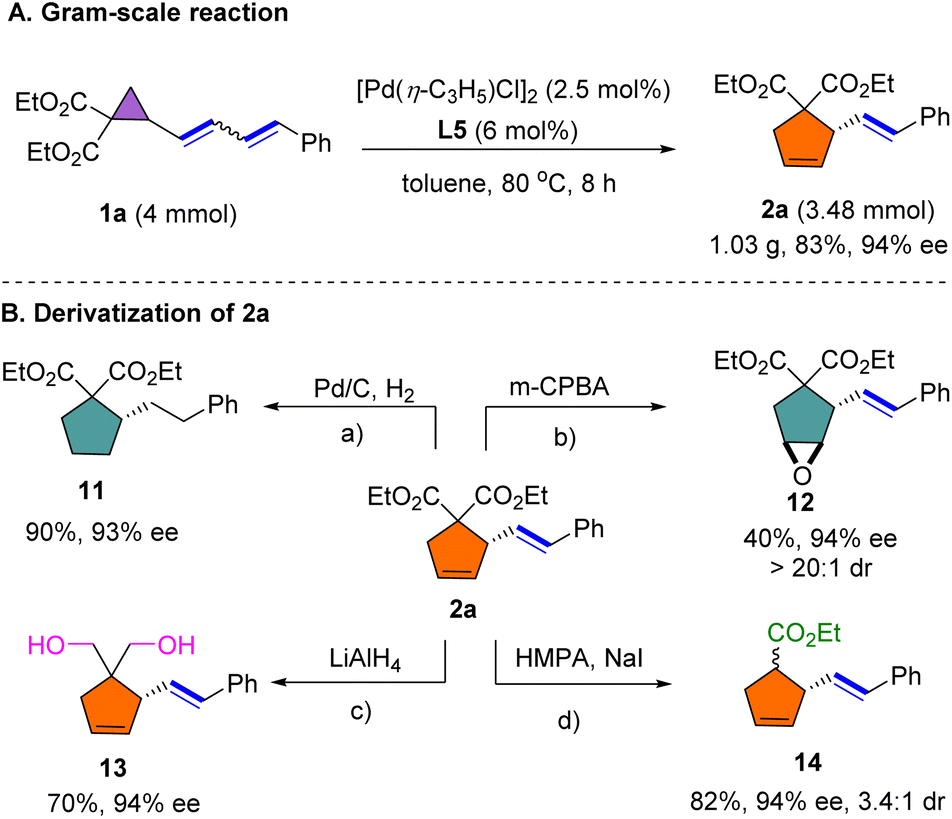 | ||
| Scheme 5 Gram-scale reaction and derivatization of 2a, reaction conditions: a2a (0.1 mmol), Pd/C (20 mol%), H2 (balloon), MeOH (1.0 mL), at RT for 12 h, 90% yield. b2a (0.1 mmol), m-CPBA (0.2 mmol), NaHCO3 (0.12 mmol), in DCM (1.0 mL) at 0 °C for 10 min, then RT for 24 h, 40% yield. c2a (0.1 mmol), LiAlH4 (0.28 mmol), in THF (1.0 mL) at 0 °C for 12 h, 70% yield. d2a (0.1 mmol), NaI (0.14 mmol), in HMPA (0.2 mL) at 110 °C for 24 h, 82% yield, 3.4:1 dr. | ||
To better understand the mechanism of the rearrangement, several control experiments were conducted. As shown in Scheme 6A, pure E-1a and Z-1a were subjected to the standard conditions used in Scheme 2, providing the product E-2a in 92% and with 94% ee, and in 88% and with 94% ee, respectively (eqn (1) and (2)), suggesting that isomerization occurred during the rearrangement of dienyl cyclopropane. However, when vinyl cyclopropane 15 was reacted under the standard conditions, the VCP decomposed and rearrangement product 16 was not detected. A plausible mechanism for the rearrangement is shown in Scheme 6B. Initially, coordination of the double bond of 1a to the palladium complex forms intermediate A. Subsequent oxidative addition of cyclopropane leads to the formation of the syn,syn-η3-allyl palladium complex B, which affords the anti,syn-η3-allyl intermediate C. Dynamic equilibration of C into syn,syn-η3-allyl complex E through π–σ–π (C–D–E) isomerization triggers the formation of cyclopentene product 2via a Re attack on the Pd-π-allyl moiety. However, we couldn't exclude another reaction pathway that proceed via initial coordination of palladium to the distal alkene, followed by the formation of the η-allyl complex accompanied by alkene migration and cyclopropane ring opening to give D.
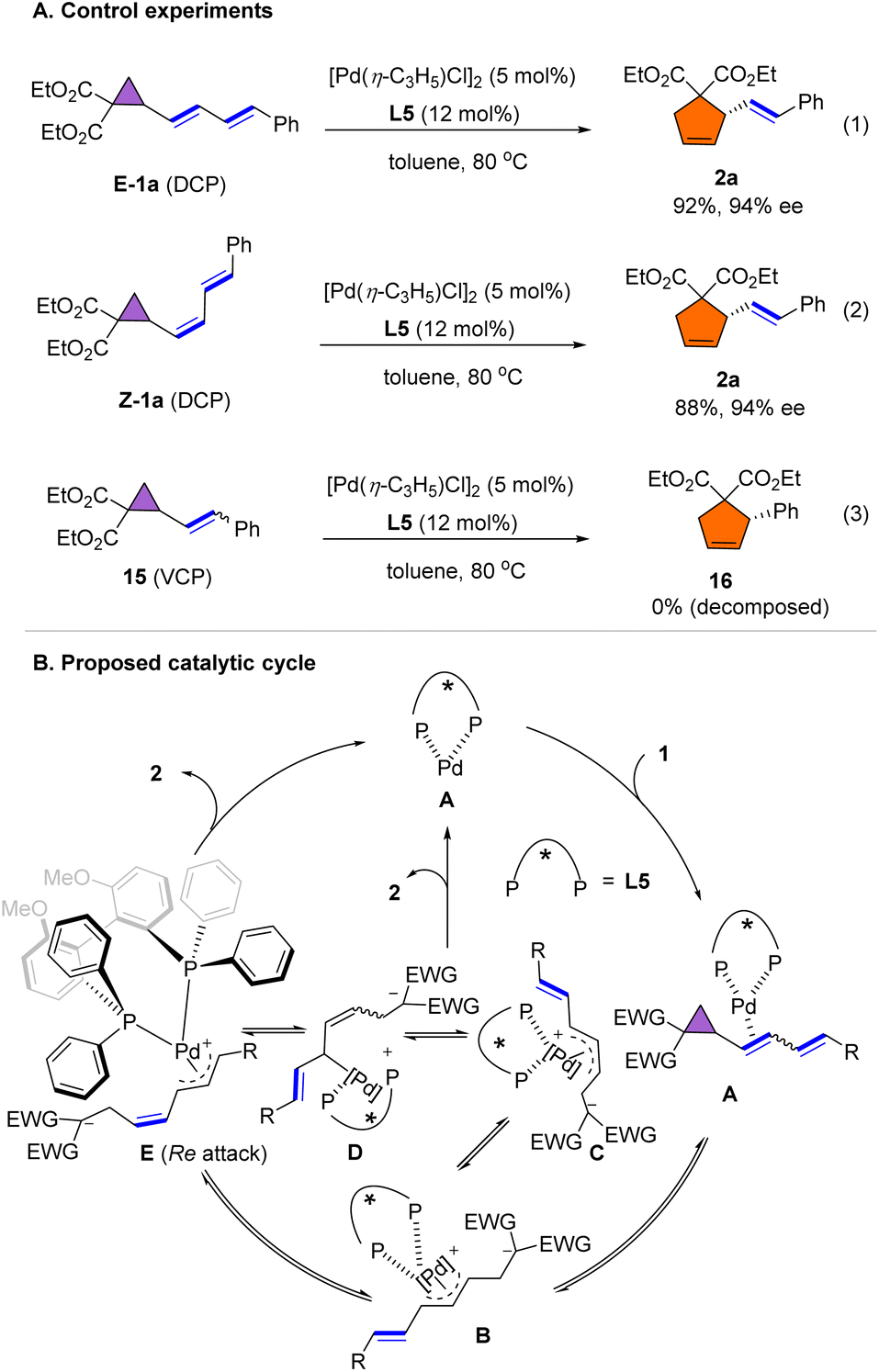 | ||
| Scheme 6 Control experiments and proposed catalytic cycle. | ||
Conclusions
In conclusion, we have developed a mild and efficient palladium-catalyzed enantioselective rearrangement of dienyl or trienyl cyclopropanes. A broad range of functionalized vinylcyclopentene derivatives were conveniently constructed in high yields and with excellent regioselectivities and enantioselectivities. The utility of the current protocol was highlighted by a successful gram-scale experiment. Furthermore, the developed method provides a platform for constructing synthetically useful molecules containing cyclopentanes or cyclopentenes. To the best of our knowledge, the reported palladium-catalyzed enantioselective ring expansion is unprecedented.Data availability
Experimental data associated with this article can be found in the ESI.†Author contributions
R. R. L. conceived, designed, and originated this project. Q. X., C. J. L., C. Q. G and J. F. performed the experiments, obtained all spectroscopic data, and analysed the results. R. R. L. and C. J. L. co-wrote the manuscript. All authors analysed the data, discussed the results and contributed to the relevant discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the generous support from the Taishan Scholar Youth Expert Program in Shandong Province (tsqn201909096), National Natural Science Foundation of China (21901236), and the startup fund from Qingdao University.Notes and references
- G. Mehta and A. Srikrishna, Chem. Rev., 1997, 97, 671–720 CrossRef CAS.
- S.-C. Mao and Y.-W. Guo, J. Nat. Prod., 2006, 69, 1209–1211 CrossRef CAS PubMed.
- D.-Z. Liu, F. Wang, T.-G. Liao, J.-G. Tang, W. Steglich, H.-J. Zhu and J.-K. Liu, Org. Lett., 2006, 8, 5749–5752 CrossRef CAS.
- J. Lebreton, V. Alphand and R. Furstoss, Tetrahedron Lett., 1996, 37, 1011–1014 CrossRef CAS.
- H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS.
- S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744–5893 CrossRef CAS PubMed.
- L. Jiao and Z.-X. Yu, J. Org. Chem., 2013, 78, 6842–6848 CrossRef CAS PubMed.
- T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed.
- M. Meazza, H. Guo and R. Rios, Org. Biomol. Chem., 2017, 15, 2479–2490 RSC.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed.
- I. Shimizu, Y. Ohashi and J. Tsuji, Tetrahedron Lett., 1985, 26, 3825–3828 CrossRef CAS.
- X. B. Huang, X. J. Li, T. T. Li, B. Chen, W. D. Chu, L. He and Q. Z. Liu, Org. Lett., 2019, 21, 1713–1716 CrossRef CAS PubMed.
- A. P. Dieskau, M. S. Holzwarth and B. Plietker, J. Am. Chem. Soc., 2012, 134, 5048–5051 CrossRef CAS PubMed.
- A. K. Turek, M. H. Sak and S. J. Miller, J. Am. Chem. Soc., 2021, 143, 16173–16183 CrossRef CAS PubMed.
- M. Zhu, X.-L. Huang, S. Sun, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2021, 143, 13441–13449 CrossRef CAS.
- M.-M. Zhang, B.-L. Qu, B. Shi, W.-J. Xiao and L.-Q. Lu, Chem. Soc. Rev., 2022, 51, 4146–4174 RSC.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed.
- B. M. Trost and P. J. Morris, Angew. Chem., Int. Ed., 2011, 50, 6167–6170 CrossRef CAS.
- B. M. Trost, P. J. Morris and S. J. Sprague, J. Am. Chem. Soc., 2012, 134, 17823–17831 CrossRef CAS PubMed.
- M. Meazza and R. Rios, Chem. – Eur. J., 2016, 22, 9923–9928 CrossRef CAS PubMed.
- C. Ma, Y. Huang and Y. Zhao, ACS Catal., 2016, 6, 6408–6412 CrossRef CAS.
- W. P. Ding, G. P. Zhang, Y. J. Jiang, J. Du, X. Y. Liu, D. Chen, C. H. Ding, Q. H. Deng and X. L. Hou, Org. Lett., 2019, 21, 6805–6810 CrossRef CAS PubMed.
- B. M. Trost, W.-J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710–6717 CrossRef CAS.
- Q. Cheng, J.-H. Xie, Y.-C. Weng and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS.
- B. M. Trost and Z. Zuo, Angew. Chem., Int. Ed., 2021, 60, 5806–5810 CrossRef CAS PubMed.
- M.-M. Li, Q. Xiong, B.-L. Qu, Y.-Q. Xiao, Y. Lan, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 17429–17434 CrossRef CAS.
- M. Faltracco, K. N. A. van de Vrande, M. Dijkstra, J. M. Saya, T. A. Hamlin and E. Ruijter, Angew. Chem., Int. Ed., 2021, 60, 14410–14414 CrossRef CAS PubMed.
- M.-M. Li, Z.-X. Zhou, Y.-J. Li, M.-Y. Cao, X.-P. Liu, H.-H. Lu, L. Rao, L.-Q. Lu, A. M. Beauchemin and W.-J. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202212444 Search PubMed.
- For a review regarding vinyl cyclopropane rearrangement see: T. Hudlicky and J. W. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864–4876 CrossRef CAS.
- N. P. Neureiter, J. Org. Chem., 1959, 24, 2044–2046 CrossRef CAS.
- E. Vogel, Angew. Chem., 1960, 72, 4–26 CrossRef CAS.
- C. G. Overberger and A. E. Borchert, J. Am. Chem. Soc., 1960, 82, 1007–1008 CrossRef CAS.
- C. G. Overberger and A. E. Borchert, J. Am. Chem. Soc., 1960, 82, 4896–4899 CrossRef CAS.
- E. J. Corey and R. H. Wollenberg, J. Org. Chem., 1975, 40, 2265–2266 CrossRef CAS.
- B. M. Trost and M. J. Bogdanowicz, J. Am. Chem. Soc., 1973, 95, 5311–5321 CrossRef CAS.
- V. Aris, J. M. Brown, J. A. Conneely, B. T. Golding and D. H. Williamson, J. Chem. Soc., Perkin Trans., 1975, 2, 4–10 RSC.
- N. W. Alcock, J. M. Brown, J. A. Conneely and D. H. Williamson, J. Chem. Soc., Perkin Trans., 1979, 2, 962–971 RSC.
- T. Hudlicky, F. J. Koszyk, T. M. Kutchan and J. P. Sheth, J. Org. Chem., 1980, 45, 5020–5027 CrossRef CAS.
- M. Hayashi, T. Ohmatsu, Y.-P. Meng and K. Saigo, Angew. Chem., Int. Ed., 1998, 37, 837–839 CrossRef CAS.
- Y. Morizawa, K. Oshima and H. Nozaki, Isr. J. Chem., 1984, 24, 149–152 CrossRef CAS.
- K. Fugami, Y. Morizawa, K. Oshima and H. Nozaki, Tetrahedron Lett., 1985, 26, 857–860 CrossRef CAS.
- R. W. Coscia and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 2496–2498 CrossRef CAS.
- I. Ryu, K. Ikura, Y. Tamura, J. Maenaka, A. Ogawa and N. Sonoda, Synlett, 1994, 941–2279 CrossRef CAS.
- R. K. Bowman and J. S. Johnson, Org. Lett., 2006, 8, 573–576 CrossRef CAS PubMed.
- G. Zuo and J. Louie, Angew. Chem., Int. Ed., 2004, 43, 2277–2279 CrossRef CAS PubMed.
- L. A. Batory, C. E. McInnis and J. T. Njardarson, J. Am. Chem. Soc., 2006, 128, 16054–16055 CrossRef CAS PubMed.
- E. Rogers, H. Araki, L. A. Batory, C. E. McInnis and J. T. Njardarson, J. Am. Chem. Soc., 2007, 129, 2768–2769 CrossRef CAS PubMed.
- T. J. L. Mustard, D. J. Mack, J. T. Njardarson and P. H.-Y. Cheong, J. Am. Chem. Soc., 2013, 135, 1471–1475 CrossRef CAS PubMed.
- X. Wu and L.-Z. Gong, Synthesis, 2019, 51, 122–134 CrossRef CAS.
- N. J. Adamson, E. Hull and S. J. Malcolmson, J. Am. Chem. Soc., 2017, 139, 7180–7183 CrossRef CAS PubMed.
- N. J. Adamson, K. C. E. Wilbur and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 2761–2764 CrossRef CAS PubMed.
- S.-Z. Nie, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 16450–16454 CrossRef CAS.
- H. Wang, R. Zhang, Q. Zhang and W. Zi, J. Am. Chem. Soc., 2021, 143, 10948–10962 CrossRef CAS.
- M.-M. Li, L. Cheng, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2021, 60, 2948–2951 CrossRef CAS PubMed.
- I. Ibrahem, G.-L. Zhao, R. Rios, J. Vesely, H. Sundén, P. Dziedzic and A. Córdova, Chem. – Eur. J., 2008, 14, 7867–7879 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data for all the new compounds, chiral HPLC spectra for the products. CCDC 2183663. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06548g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |