
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical energy assisted self-assembling of a porphyrin-substituted benzoic acid in complex environments†
Bingxu
Ma
,
Bowen
Pang
,
Wang
Zeng
*,
Huimin
Fu
,
Yi
Jiang
,
Shenglin
Yao
,
Yida
Yang
,
Kaisheng
Zhu
and
Wei
Zhang
 *
*
South China Advanced Institute for Soft Matter Science and Technology, School of Emergent Soft Matter and Guangdong Provincial Key Laboratory of Functional and Intelligent Hybrid Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: zengwang@scut.edu.cn; weizhang@scut.edu.cn
First published on 28th February 2023
Abstract
Energy input plays a crucial role in the assembling of matter. In our present study, we utilize EDC as a chemical fuel to drive the molecular assembling of POR-COOH. POR-COOH will react with EDC to generate the intermediate POR-COOEDC first, which was well-solvated by solvent molecules. During the subsequent hydrolysis process, EDU and oversaturated POR-COOH molecules at high energy states will be formed and thus allowed the self-assembling of POR-COOH into 2D nanosheets. This chemical energy assisted assembling process could be performed not only under mild conditions with high spatial accuracy but also with high selectivity in complex environments.
Introduction
During the transition of matter from one state to another through assembling, energy input is crucial for activating molecules to overcome the energy barrier during the process (Fig. 1). In general, importing energy into the system and promoting molecules to the required energy level for assembling could be achieved in multiple ways, such as heating, solvation with a good solvent, irradiation with light, excitation under external fields, tuning the pH value, or a combination of them.1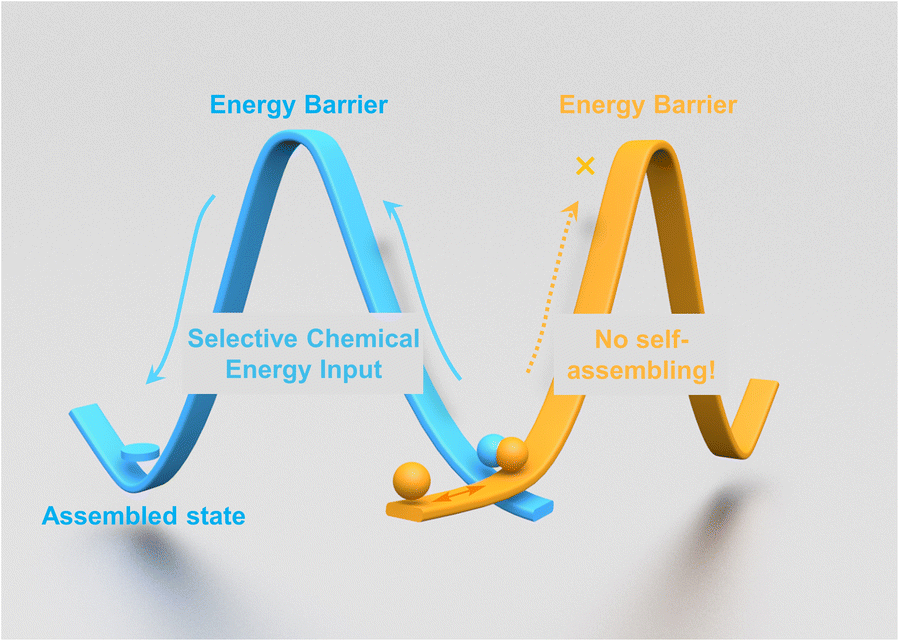 | ||
| Fig. 1 Schematic representation of the chemical energy assisted self-assembling. | ||
In a chemical reaction, the energy will exchange and redistribute among reactants, products and intermediates (if there are any) constantly, thus affording the possibility for the molecules involved in the reaction to assemble. Representative examples have been reported as ‘reaction-triggered self-assemblings’ in the past few decades.2 During these processes, despite the starting matter being consumed and converted into different chemical species, the as-formed products were able to assemble and form ordered supramolecular structures. Here we report that using the energy released during a chemical reaction cycle, we are able to drive molecules to self-assemble and form highly ordered supramolecular structures without changing the chemical structure of molecules at the final stage. More importantly, this chemical energy assisted assembling process could be performed under mild conditions with high spatial accuracy and selectivity.
Results and discussion
We found this chemical energy assisted self-assembling process during the study of the reaction between carbodiimide and carboxylic acid derivates. It is well known that during the reaction between carboxylic acid and carbodiimide (such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, EDC), the corresponding anhydride will be generated as an intermediate first, and then hydrolyze into the original carboxylic acid in the presence of water (Fig. 2a).3 With the continuously supported compound EDC as the chemical fuel, these anhydrides were able to further assemble into functional transient supramolecular structures, as demonstrated by Boekhoven, Hartley and other groups.3b,4 However, in our present study, we noticed that the reaction between EDC and a porphyrin-substituted benzoic acid (POR-COOH, Fig. 2b) did not result in the insoluble anhydride intermediate. For instance, we started from a suspension of amorphously aggregated POR-COOH (AAPOR-COOH, 0.066 mg, 1.0 × 10−4 mmol POR-COOH in 2.0 mL DMSO–H2O (6/4, v/v), Fig. S1 and S2†). When 1.2 mg EDC (6.0 × 10−3 mmol, 60 equiv.) was added into the system, we first found that the brown suspension (Fig. 2c(i)) turned to a transparent pink solution within 10 minutes (Fig. 2c(ii)), and then gradually turned into a turbid suspension (Fig. 2c(iii)) and eventually yielded a red precipitate after 48 h (Fig. 2c(iv)).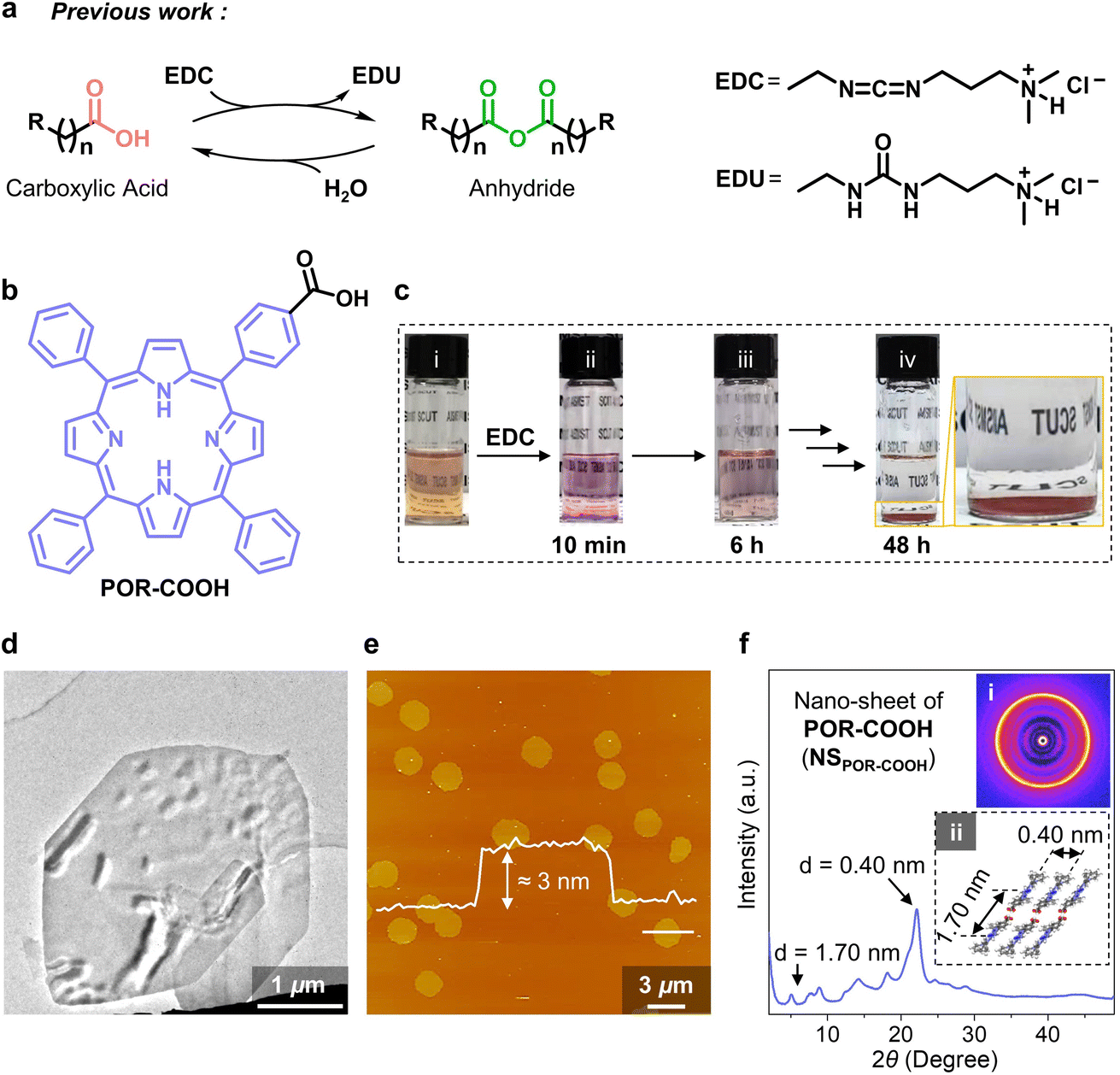 | ||
| Fig. 2 (a) Formation of a transient anhydride intermediate from carboxylic acid derivates driven by EDC. (b) Molecular structure of POR-COOH. (c) Photographs of the reaction mixture of the POR-COOH suspension and EDC at different moments. (d) TEM micrograph of an air-dried dispersion of NSPOR-COOH. (e) Liquid phase AFM micrograph of NSPOR-COOH in the presence of solvent. (f) WAXD profile of NSPOR-COOH and its 2D WAXD pattern (inset i) and proposed packing model (inset ii). | ||
After completely dissolving the red precipitate in DMSO-d6, we found that its 1H NMR spectrum was identical to that of compound POR-COOH (Fig. S3†), which suggested that the red precipitate has an identical chemical composition to starting POR-COOH. In the meantime, the transmission electron microscopy (TEM) images of an air-dried sample allowed for the visualization of nano-sheets with side lengths up to micrometers (Fig. 2d). From scanning electron microscopy (SEM), we found that these nano-sheets stacked together on the substrate during solvent evaporation (Fig. S4†). Fortunately, liquid-phase atomic force microscopy (AFM) allowed us to conduct the measurement without removing the solvent and provided us the opportunity to observe the morphology of the resulting assemblies in situ. As shown in Fig. 2e, the AFM micrograph clearly proved the formation of two-dimensional (2D) nano-sheets with a uniform thickness of 3.0 nm. Considering the reported packing model of porphyrin derivatives,5 we presume that the 2D nano-sheets of POR-COOH (NSPOR-COOH) are constructed from J-aggregated POR-COOH molecules (Fig. 2f and S5†), and the simulated wide-angle X-ray diffraction (WAXD) profile of this proposed stacking model is consistent with the experimental measurement (Fig. S6†). These results indicate that the resulting red precipitate is compound POR-COOH in the form of NSPOR-COOH with a high degree of crystallinity.
Utilizing ultraviolet-visible (UV-vis) absorption and dynamic light scattering (DLS) spectroscopy, we are able to monitor the entire process. The UV-vis absorption spectrum of AAPOR-COOH suspension (0.50 × 10−4 M in DMSO–H2O (6/4, v/v)) showed a maximum absorption peak at 417 nm along with a shoulder peak at 437 nm (Fig. 3a, green curve), characteristic of the J-aggregated POR-COOH.6 After adding EDC (60 equiv.) into the AAPOR-COOH suspension, the shoulder peak at 437 nm disappeared, while the intensity of the peak at 417 nm increased within 10 minutes (Fig. 3a), which suggested that the aggregated POR-COOH was converted into a solvated intermediate.6c After 10 minutes, the peak at 417 nm gradually decreased, with the intensity increase of the shoulder peak at 437 nm, indicating that the solvated intermediate turned into J-aggregated POR-COOH again (Fig. 3b). When we plot the absorption intensity at 417 nm versus time, a maximum absorption was found at 10 minutes after adding EDC (Fig. 3c). From the DLS profiles, we found objects with sizes up to several hundreds of nanometers in the AAPOR-COOH suspension, and they decrease to less than 1 nanometer in 10 minutes after adding EDC (Fig. 3d), followed by gradually increasing to several hundreds of nanometers again in 360 minutes (Fig. 3e). The observed time-dependent size variation pattern (Fig. 3f) matched with time-dependent absorption spectra very well. Later we are able to identify the intermediate utilizing matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass and Fourier transform infrared (FT-IR) spectroscopy. As expected, the MALDI-TOF mass spectra of AAPOR-COOH and NSPOR-COOH displayed a major peak at m/z = 658.20 and 658.50, respectively (Fig. 3g, green and blue curves), which matched with the molecular weight of POR-COOH (calcd m/z = 658.24), since they have identical chemical composition. However, the MALDI-TOF mass spectrum of the soluble intermediate showed the presence of a fraction with a different molecular weight at m/z = 813.38 (Fig. 3g, red curve), and no other chemical composition, such as anhydride, was detected. This value (m/z = 813.38) equals the sum of the molecular weight of POR-COOH and EDC. Considering the reported reaction mechanism between EDC and carboxylic acid derivatives,3b,7 we suspected that an EDC-ester POR-COOEDC was formed as an intermediate first, and then hydrolyzed into POR-COOH, due to its high chemical activity (Fig. 3h). To examine our assumption, we used FT-IR spectroscopy to trace the reaction process. We found that AAPOR-COOH and NSPOR-COOH showed almost identical IR spectra, with the stretch vibration peak of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the carboxylic group at 1690 cm−1 (Fig. 3i, green and blue curves). However, after adding EDC, the intermediate separated from the pink solution displayed two new peaks at 1740 cm−1 and 1650 cm−1 (Fig. 3i, red curve), which could be assigned to the CO bond in ester groups and the CN bond on the side chain, respectively.7a,8 Alternatively, using water-free conditions, we are able to isolate this intermediate POR-COOEDC and identified its chemical structure by 1H NMR spectroscopy (Fig. S7†) clearly.
O bond in the carboxylic group at 1690 cm−1 (Fig. 3i, green and blue curves). However, after adding EDC, the intermediate separated from the pink solution displayed two new peaks at 1740 cm−1 and 1650 cm−1 (Fig. 3i, red curve), which could be assigned to the CO bond in ester groups and the CN bond on the side chain, respectively.7a,8 Alternatively, using water-free conditions, we are able to isolate this intermediate POR-COOEDC and identified its chemical structure by 1H NMR spectroscopy (Fig. S7†) clearly.
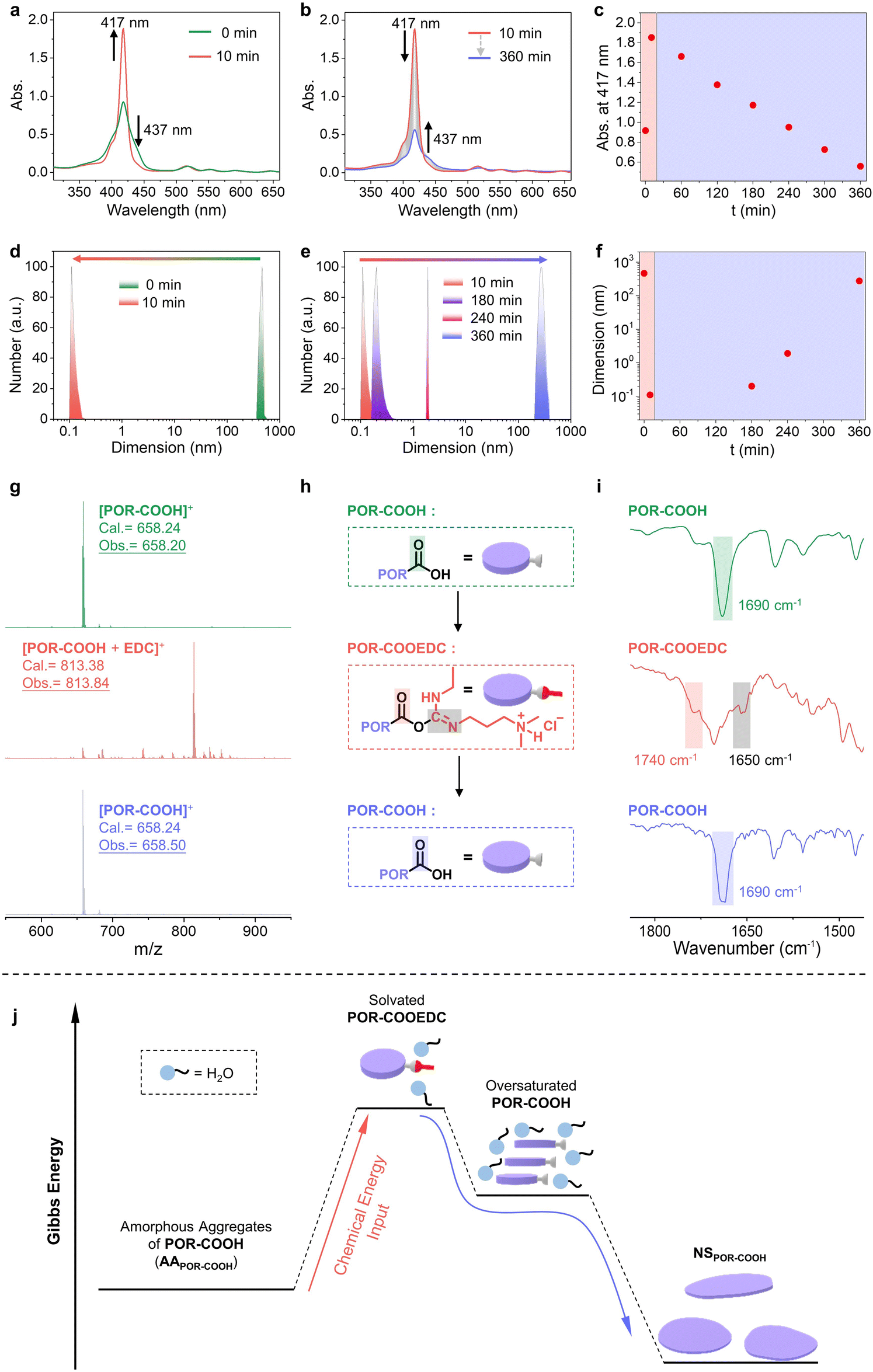 | ||
| Fig. 3 (a and b) Time-dependent absorption spectra of AAPOR-COOH after the addition of EDC. (c) Time-dependent absorption intensity at 417 nm. (d and e) Time-dependent size distribution of AAPOR-COOH monitored by DLS after the addition of EDC. (f) Plot of the particle size versus time, monitored by DLS. (g) MALDI-TOF mass spectra of AAPOR-COOH (green curve), AAPOR-COOH reacts with EDC for several minutes (red curve), and NSPOR-COOH (blue curve). (h) Schematic representation of the reaction cycle between POR-COOH and EDC. (i) FT-IR spectra of AAPOR-COOH (green curve), POR-COOEDC (red curve), and NSPOR-COOH (blue curve). (j) Schematic illustration of the mechanism of the chemical energy assisted self-assembling process in this study. | ||
Taking into account all above results, we are able to conclude the mechanism of this chemical energy assisted assembling process (Fig. 3j). The starting material POR-COOH has low solubility in certain solvents, such as DMSO–H2O (6/4, v/v). Without a proper self-assembling process, POR-COOH tends to aggregate randomly and forms an amorphous aggregated structure (Fig. S1†). Once EDC was added into the system, POR-COOH will be converted into intermediate product POR-COOEDC at the early stage of the reaction. POR-COOEDC has higher solubility and will be solvated by water molecules and form a pink transparent solution. Later, when POR-COOH was re-generated from the hydrolysis of solvated POR-COOEDC, the insoluble POR-COOH molecules were still surrounded by solvent molecules temporarily and thus existed as an oversaturated high energy state, which allowed them to self-assemble into 2D nano-sheets. In this reaction cycle, the net reaction is the high-energy chemical species EDC was converted into the low-energy chemical species EDU, and in the meantime, the chemical energy released during the reaction was transferred to AAPOR-COOH and promoted POR-COOH molecules to a high energy state, followed by self-assembling into 2D nano-sheets.
This chemical energy assisted self-assembling process could be performed under conditions close to solvent-free environments. As shown in Fig. S8†, a thin film of AAPOR-COOH (see the ESI† for fabrication details) shows no birefringence in polarized light microscopy (POM) under crossed polarizers, due to the random alignment of POR-COOH molecules. In contrast, the assembled crystalline NSPOR-COOH shows birefringence due to the highly ordered molecular alignment in the 2D nano-sheet assembly (Fig. S9†). When a thin film composed of AAPOR-COOH was covered by PET stamps (shaped to letters “S”, “C”, “U”, and “T”) soaked with the EDC solution (0.1 M in DMSO–H2O (6/4, v/v)) for 48 h (Fig. 4a), a bright “SCUT” pattern was observed under the crossed polarizers, after removing the chemical energy stamps (Fig. 4b). That means that amorphously aggregated AAPOR-COOH located in the area covered by PET stamps were assembled into NSPOR-COOH with high spatial accuracy. We further confirmed that this chemical energy-assisted assembling process could be repeated many times. Starting from a thin film of AAPOR-COOH as shown in Fig. 4c, we could first promote the molecules on the right side to self-assemble by using a PET stamp soaked with EDC solution (Fig. 4d), erasing the alignment of molecules to amorphous states by heating and rapid cooling (Fig. 4e), and then drive them to assemble into NSPOR-COOH by using the PET stamp again (Fig. 4f).
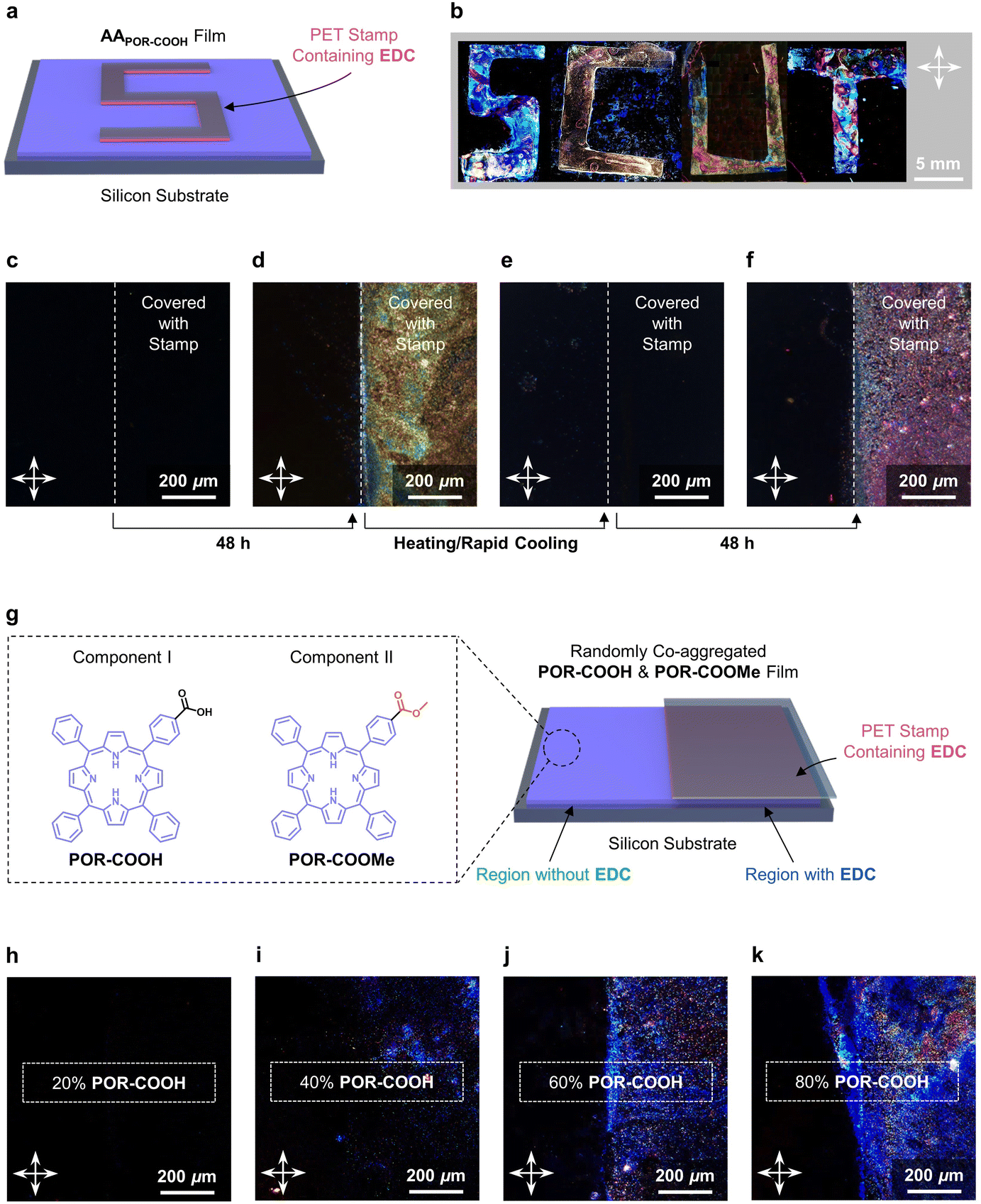 | ||
| Fig. 4 (a) The schematic illustration of the experimental setup for spatial controlled chemical energy-assisted assembling of the AAPOR-COOH film. (b) Polarized light microscopy images of the AAPOR-COOH film covered by PET stamps shaped into “SCUT” letters. (c–f) Polarized light microscopy images of (c) the AAPOR-COOH film, (d) covered with the PET stamp on the right side for 48 h, (e) erasing the alignment of molecules by heating and rapid cooling, and (f) covered with the PET stamp on the right side for 48 h again. (g) Experimental setup of the chemical energy assisted self-assembling process for the spin-coated AAPOR-COOH&POR-COOMe film. (h–k) Polarized light microscopy images of AAPOR-COOH&POR-COOMe films covered with the PET stamp containing EDC for 48 h. Molar contents of POR-COOH in the mixture were (h) 20%, (i) 40%, (j) 60%, and (k) 80%, respectively. The white crossed arrows indicate the direction of the polarizer and the analyzer. | ||
Due to the selectivity of chemical reactions, our chemical energy assisted assembling process could also be performed selectively under complex environments, such as in the presence of porphyrin derivatives with a similar chemical structure (POR-COOMe, Fig. 4g). To demonstrate it, we prepared thin films composed of amorphous aggregates of a mixture of POR-COOH and POR-COOMe (AAPOR-COOH&POR-COOMe, the molar contents of POR-COOH in the mixture were 20%, 40%, 60%, and 80%, respectively) on silicon substrates accordingly (see the ESI† for fabrication details). After covering the right side of these thin films with PET stamps soaked with EDC solution for 48 h, birefringence was detected on the area covered by EDC. More importantly, we found that the intensity of the birefringence was amplified with the increasing amount of POR-COOH in the mixture (Fig. 4h–k). These observations clearly suggested that, despite the structural similarity between POR-COOH and POR-COOMe, only compound POR-COOH was activated by the energy released during this chemical reaction cycle, and assembled into nano-sheets.
Finally, taking advantage of the high selectivity and spatial accuracy of this chemical energy assisted assembling process, we are able to transfer energy to POR-COOH molecules in a mixture selectively and drive POR-COOH molecules to self-assemble in a separated domain. The experiment was carried out on a hydrogel with an inert polyacrylamide framework (Fig. 5a), with the ability to prevent the Brownian motion of the aggregated objects. As shown in Fig. 5b, zone B was always filled with EDC solution (50 mM in 3.0 μL DMSO–H2O (6/4, v/v)). When zone A was loaded with AAPOR-COOMe suspension (0.50 mM in 5.0 μL DMSO–H2O (6/4, v/v)), we found that all POR-COOMe still remained in zone A after 48 h (Fig. 5c), since AAPOR-COOMe was too large to move inside the inert hydrogel framework. In contrast, when zone A was loaded with AAPOR-COOH suspension (0.50 mM in 5.0 μL DMSO–H2O (6/4, v/v)) instead, we found that almost all POR-COOH was moved out from zone A and distributed in the region in-between zones A and B (Fig. 5d), possibly due to the diffusion of the soluble intermediate POR-COOEDC. When amorphous aggregates composed of a POR-COOH (0.50 mM) and POR-COOMe (0.50 mM) mixture were used in zone A, after 24 h, we found that the starting mixture of porphyrin derivatives was separated into two fractions in different domains (Fig. 5e). Combining the results from Fig. 5, we could conclude that the compound POR-COOMe from the mixture remained in zone A (possibly in an amorphously aggregated states), while compound POR-COOH self-assembled into NSPOR-COOH in the presence of EDC fuel and re-located to the region in-between zones A and B. In another word, in the presence of the EDC gradient, despite the structural similarity between POR-COOH and POR-COOMe, POR-COOH molecules were selectively driven into a separated domain during the chemical energy assisted assembling process.
 | ||
| Fig. 5 (a) Experimental setup for chemical energy assisted assembling in a hydrogel with inert frameworks. (b) Schematic drawing of the experimental setup. (c–e) CLSM micrographs of samples when (c) AAPOR-COOMe, (d) AAPOR-COOH, and (e) AAPOR-COOH&POR-COOMe were loaded in zone A, respectively. | ||
Conclusions
In conclusion, we have successfully demonstrated that the energy released during the hydrolysis of EDC to EDU could be utilized to selectively drive a porphyrin-substituted benzoic acid to self-assemble into highly ordered supramolecular structures. Considering that this chemical energy assisted assembling process could be performed not only under mild conditions with high spatial accuracy but also with high selectivity in complex environments, we believe that our strategy will provide a big innovation in both nanofabrication and supramolecular chemistry.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Guangdong Natural Science Foundation (No. 2018B030306039), Science and Technology Projects in Guangzhou (No. 202201010557), China Postdoctoral Science Foundation (No. 2021M701237), 111 Project (No. B18023) and Recruitment Program of Guangdong (No. 2016ZT06C322).References
- (a) D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154 CrossRef; (b) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed; (c) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed; (d) C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543 RSC.
- (a) W. Jin, T. Fukushima, A. Kosaka, M. Niki, N. Ishii and T. Aida, J. Am. Chem. Soc., 2005, 127, 8284 CrossRef CAS PubMed; (b) W. Wan and C. Pan, Macromolecules, 2007, 40, 8897 CrossRef CAS; (c) W. Ji, J. Yan, E. Chen, Z. Li and D. Liang, Macromolecules, 2008, 41, 4914 CrossRef CAS; (d) C. Trinh, M. T. Whited, A. Steiner, C. J. Tassone, M. F. Toney and M. E. Thompson, Chem. Mater., 2012, 24, 2583 CrossRef CAS; (e) J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433 CrossRef CAS PubMed; (f) H. Xia, H. Fu, Y. Zhang, K. C. Shih, Y. Ren, M. Anuganti, M. P. Nieh, J. Cheng and Y. Lin, J. Am. Chem. Soc., 2017, 139, 11106 CrossRef CAS PubMed; (g) S. Panja, A. M. Fuentes-Caparrós, E. R. Cross, L. Cavalcanti and D. J. Adams, Chem. Mater., 2020, 32, 5264 CrossRef CAS PubMed; (h) J. Park, J.-M. Heo, S. Seong, J. Noh and J.-M. Kim, Nat. Commun., 2021, 12, 4207 CrossRef CAS PubMed.
- (a) L. S. Kariyawasam and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 11949 CrossRef CAS PubMed; (b) M. Tena-Solsona, B. Rieβ, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS PubMed.
- (a) M. M. Hossain, J. L. Atkinson and C. S. Hartley, Angew. Chem., Int. Ed., 2020, 59, 13807 CrossRef CAS PubMed; (b) P. S. Schwarz, L. Tebcharani, J. E. Heger, P. Müller-Buschbaum and J. Boekhoven, Chem. Sci., 2021, 12, 9969 RSC; (c) P. S. Schwarz, S. Laha, J. Janssen, T. Huss, J. Boekhoven and C. A. Weber, Chem. Sci., 2021, 12, 7554 RSC; (d) F. Späth, C. Donau, A. M. Bergmann, M. Kränzlein, C. V. Synatschke, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2021, 143, 4782 CrossRef PubMed; (e) J. Heckel, S. Loescher, R. T. Mathers and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 7717 CrossRef PubMed; (f) W. Zeng, C. Fan, X. Xing, H. Cheng, H. Fu, B. Ma, Z. Yang, R. Zhang and W. Zhang, Giant, 2021, 7, 100067 CrossRef CAS; (g) H. Xu, S. Bai, G. Gu, Y. Gao, X. Sun, X. Guo, F. Xuan and Y. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 43825 CrossRef CAS PubMed.
- (a) T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493 CrossRef CAS PubMed; (b) W. Qian, A. González-Campo, A. Pérez-Rodríguez, S. Rodríguez-Hermida, I. Imaz, K. Wurst, D. Maspoch, E. Ruiz, C. Ocal, E. Barrena, D. B. Amabilino and N. Aliaga-Alcalde, Chem.–Eur. J., 2018, 24, 12950 CrossRef CAS PubMed; (c) R. Cao, G. Wang, X. Ren, P. Duan, L. Wang, Y. Li, X. Chen, R. Zhu, Y. Jia and F. Bai, Nano Lett., 2022, 22, 157 CrossRef CAS PubMed.
- (a) S. Okada and H. Segawa, J. Am. Chem. Soc., 2003, 125, 2792 CrossRef CAS PubMed; (b) A. Satake and Y. Kobuke, Org. Biomol. Chem., 2007, 5, 1679 RSC; (c) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed; (d) N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069 CrossRef CAS PubMed; (e) J. Singh, Theory of Excitons, in Excitation Energy Transfer Processes in Condensed Matter. Physics of Solids and Liquids, Springer, Boston, MA 1994, DOI:10.1007/978-1-4899-0996-1_1.
- (a) N. Nakajima and Y. Ikada, Bioconjugate Chem., 1995, 6, 123 CrossRef CAS PubMed; (b) K. A. Totaro, X. Liao, K. Bhattacharya, J. I. Finneman, J. B. Sperry, M. A. Massa, J. Thorn, S. V. Ho and B. L. Pentelute, Bioconjugate Chem., 2016, 27, 994 CrossRef CAS PubMed; (c) M. Tena-Solsona, C. Wanzke, B. Riess, A. R. Bausch and J. Boekhoven, Nat. Commun., 2018, 9, 2044 CrossRef PubMed.
- (a) L. R. Knöpke, N. Nemati, A. Köckritz, A. Brückner and U. Bentrup, ChemCatChem, 2010, 2, 273 CrossRef; (b) L. R. Knöpke, A. Spannenberg, A. Brückner and U. Bentrup, Spectrochim. Acta, Part A, 2012, 95, 18 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06621a |
| This journal is © The Royal Society of Chemistry 2023 |