
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic chlorine-induced polyene cyclizations harnessing hypervalent chloroiodane–HFIP assemblies†
Julia
Binder
ab,
Aniruddha
Biswas
 a and
Tanja
Gulder
*ab
a and
Tanja
Gulder
*ab
aInstitute of Chemistry and Mineralogy, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany. E-mail: tanja.gulder@uni-leipzig.de
bDepartment of Chemistry, Technical University Munich, Lichtenbergstrasse 4, 85748 Garching, Germany
First published on 14th March 2023
Abstract
While bromo- and iodocyclizations have recently been successfully implemented, the challenging chlorocyclizations have been scantly investigated. We present a selective and generally applicable concept of chlorination-induced polyene cyclization by utilizing HFIP–chloroiodane networks mimicking terpene cyclases. A manifold of different alkenes was converted with excellent selectivities (up to d.r. >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). The cyclization platform was even extended to several structurally challenging terpenes and terpenoid carbon frameworks.
:5). The cyclization platform was even extended to several structurally challenging terpenes and terpenoid carbon frameworks.
1 Introduction
The power of terpene cyclases cannot be overemphasized. Starting from simple, achiral, linear C5 isoprene-derived precursors 1, nature can generate an entire collection of structurally diverse and highly complex (poly)cyclic carbon frameworks, in a single step.1 Among the utilized cationic pathways, the head-to-tail (HT) cyclization commences with electrophilic activation of the terminal (head) functional unit (cf.Fig. 1a). While the protonation of epoxides and alkenes constitute the most common initiation step, cyclizations triggered by halenium (X+) addition to the terminal olefin (1, Fig. 1a) are likewise possible.2 The latter has already given rise to over 200 terpene scaffolds,3 isolated from natural producers. These have mostly been brominated but can also be chlorinated, with hamiltonin C (9),4 haterumaimide L (10),5 and azamerone6 (11, Fig. 1b) among the chlorinated representatives.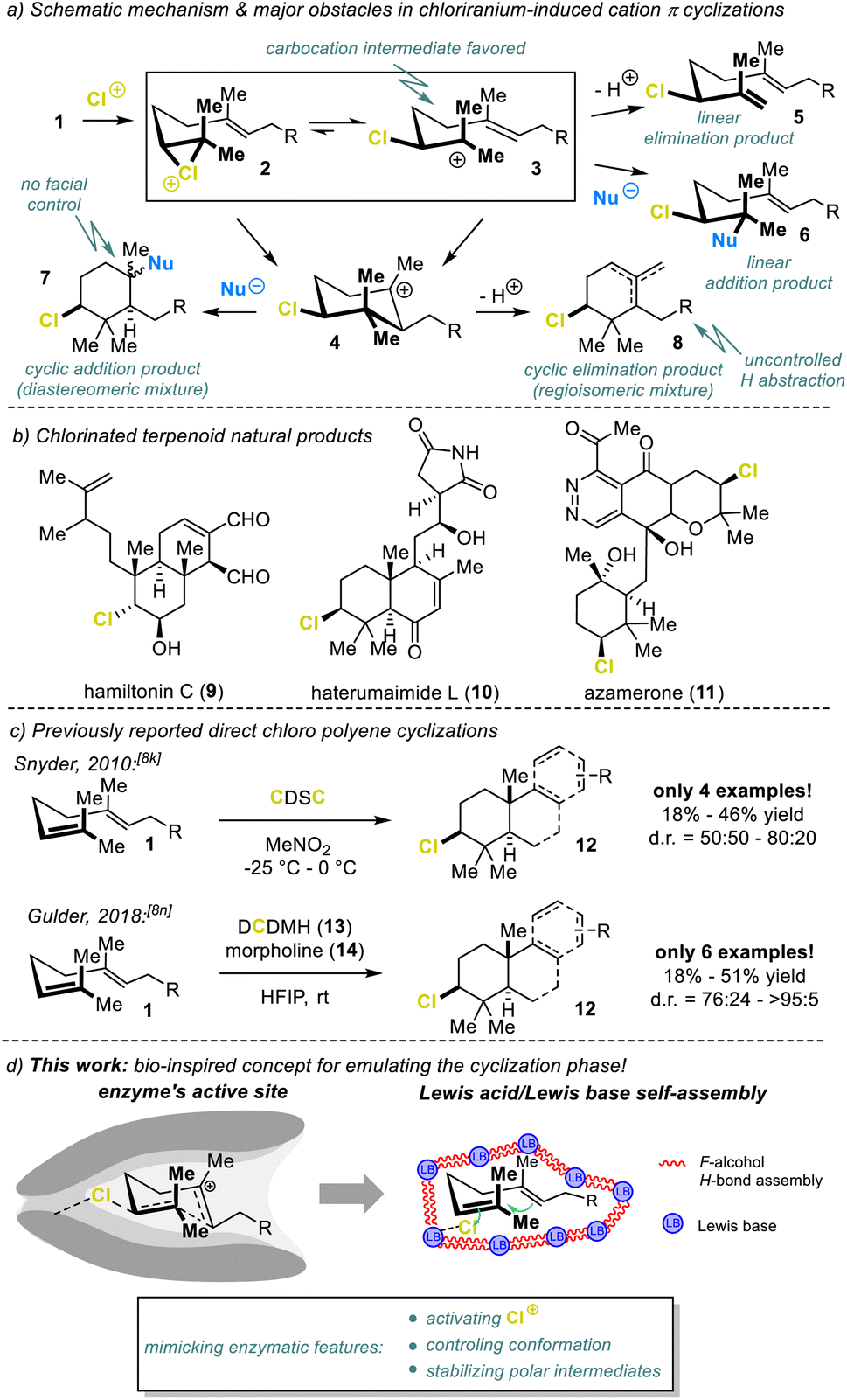 | ||
| Fig. 1 (a) Mechanistic challenges in chlorination-assisted polyene cyclizations, (b) chlorine-containing terpenes, (c) previous direct chlorocyclizations, and (d) concept of this work. | ||
Despite significant progress in developing selective and generally applicable bromo- and iodocyclization methods,2,7–9 chlorocyclizations have barely been touched as they are significantly more challenging. For chlorination-induced cyclizations, the tendency for the closed-ring intermediate 4 to form is generally very low (Fig. 1a). Typically, linear products 5 and 6 result. In addition, the chemical instability of the chlorine addition intermediate 2 constitutes another severe problem. The electronegativity and small radius of the chlorine atom render the linear, carbocationic form 3 preferable over the cyclic haliranium ion 2.10 Consequently, the pendant nucleophile attack proceeds via an SN1 mechanism, resulting in low product stereoselectivity 7.11 In addition, β-H abstraction is the predominant cation 3 trapping process, forming the desired products 7 and 8 in small quantities.
To our knowledge, only two direct chloriranium-triggered terpene cyclization methods are known (Fig. 1c).8k,n,12 In 2010, Snyder et al.8k employed their newly developed chlorodiethylsulfonium hexachloroantimonate (CDSC, not shown) as the chlorinating agent. Unfortunately, most transformations lacked any diastereoselection. Our group recently published a haliranium-mediated polyene cyclization protocol8n using HFIP13 emulating the intrinsic features of terpene cyclases.1,14 This method is in principle applicable to chlorine-assisted ring closures but here we were able to target just a small set of electron-rich alkenes 1 (6 examples) albeit with good diastereomeric excess. Nevertheless, these first examples marked milestones in the yet unresolved problem of chloriranium-triggered polyene cyclizations. Encouraged by our preliminary studies, we have advanced our bioinspired concept and provided a generally applicable, direct chlorocyclization to polyenes, thus addressing the general problems associated with such terpene formations (Fig. 1d).
2 Results and discussion
For our investigations here, we chose geranyl acetate (15) as the model substrate which constitutes a particular challenge for polyene cyclizations in general but has shown promising results in our brominative ring closures.8n Subjecting 15 to the optimized conditions reported earlier by our group8n indeed furnished the desired cyclic alcohol 16, albeit in 9% yield, together with the linear chloropolyene 17 as the main product (52%, Table 1, entry 1). More importantly, however, the formation of 16 occurred with a promising stereoselection (d.r. = 86:14), which was similar to that observed for bromocyclization (d.r. = 82:18). This result clearly shows that our confined HFIP–Cl+ network15 exerts significant stereocontrol, even for chlorine-assisted cation-π cyclizations. Further screening of chloronium reagents, such as N-chloro morpholine (18), Palau'chlor, chloramine-T, or N-chlorosuccinimide, neither enhanced the chemical yield nor the stereoselectivity of 16 (entry 2 and ESI†). Even CDSC introduced by Snyder and co-workers for chlorocyclizations of 15 in nitromethane (entry 12)8k led to a messy conversion of 15 in HFIP with no chlorine-containing product detectable (entry 13 and the ESI†). The formation of 16 could only significantly be improved if carboxylic acid-derived, cyclic hypervalent iodanes were employed as electrophilic chlorination reagents (entries 3, 4–11, and the ESI†). Using chloro-iodane 19, the yield of 16 was doubled to 19%, while the d.r. remained high (84:16, entry 3). Simultaneously, acyclic product 17 significantly decreased to 41%. The ether analog 20 did not react at all (entry 4). HFIP played a decisive role in terpene cyclization, and no other solvent (entries 5–7) or solvent mixtures with HFIP (cf. ESI†) delivered the cyclized products. As a consequence, we tried to further foster electrophilic chlorine transfer by fine-tuning the electronic properties of the chlorobenziodoxolone core structure. Introducing a nitro substituent at C-5 to increase the electrophilicity at the iodine atom proved superior. The outcome of 16 was enhanced to 23% (entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cl+ | Solvent | Additive | Yield 16 (d.r.)b | Yield 17 |
| a 19.6 mg geranyl acetate 15 (0.10 mmol, 1.0 eq.) was added to a mixture of the Cl reagent (0.12 mmol, 1.2 eq.) and additive in 1.00 mL solvent at 0 °C. b determined by 1H-NMR spectroscopy of the crude mixture. c 0.14 mmol and 1.4 eq. d 20 mol% additive. | |||||
| 1 | 13 | HFIP | 14 | 9% (86:14) |
52% |
| 2 | 18 | HFIP | — | 10% (86:14) |
52% |
| 3 | 19 | HFIP | 14 | 19% (84:16) |
41% |
| 4 | 20 | HFIP | 14 | <5% | <5% |
| 5 | 19 | iPrOH | 14 | — | — |
| 6 | 19 | TFE | 14 | <5% | 27% |
| 7 | 19 | MeNO2 | 14 | — | <5% |
| 8 | 21 | HFIP | 14 | 23% (83:17) |
19% |
| 9 | 19 | HFIP | — | 19% (83:17) |
21% |
| 10 | 21 | HFIP | 22 | 30% (83:17) |
14% |
| 11 | 23 | HFIP | — | <5% | n.d. |
| 12 (ref. 8k) | CDSC | MeNO2 | — | 18% (69:31) |
— |
| 13 | CDSC | HFIP | — | n.d. | n.d. |
|
|||||

Surprisingly, omitting the Lewis basic additive 14 significantly reduced the formation of the unwanted linear allyl chloride 17 to 21% but had no beneficial effect on the cyclization event (entry 9). This result contrasts observations made for the bromo- and iodocyclizations, where morpholine (14) was decisive in taming the reactivity and thus following a productive pathway.8n Next, additives more acidic than 14 (pKa < 8) were tested (see the ESI†). The pH optimum for this reaction was achieved by employing saccharin (22, pKa = 1.6, entry 10, and the ESI†). Here, a threefold increase in the yield (30%) of chlorocyclohexane 16 was observed with a d.r. of 83:17. The amount of Brønsted acid 22 could even be reduced to catalytic quantities (20 mol%) without any impact on the reaction outcome (entry 10). The in situ generation of chlorosaccharin (23) and its subsequent reaction with substrate 15 was excluded, as directly adding 23 to the reaction mixture did not give the desired product 16 (entry 11).
After the transformation was established, we turned our attention to the generality of this chlorination-assisted terpene cyclization (Scheme 1). First, different geranyl derivatives were explored. Geranyl ester, imide, and carbamate derivatives were rapidly cyclized (15 min), as was the free acid, to the corresponding cyclohexyl products 24–28 in moderate to excellent yields (13–70%) for chloropolyene cyclizations and with diastereoselectivities of up to >95:5. The reaction proved to be tolerant of several functional groups including the acid-sensitive N-Boc-protecting group as showcased by the lysine ester 25. Extended π-systems, such as farnesyl esters (→26), were likewise converted in good yields (37%) but suffered from a low diastereoselection. The carbocationic intermediate 4 (cf.Fig. 1a), generated during the ring-closure of geraniol, was trapped by the weakly nucleophilic fluoro alcohol. This can be explained by the less stabilizing effect of the alcohol substituent compared to that of the corresponding ester and imide moieties (products 24–27).8n The geraniol-derived intermediate 4 is thus more reactive and already trapped by HFIP during the reaction, while 4 stabilized by a carbonyl adduct reacts with water most likely not before workup. This returned 29% HFIP–ether 30 with perfect diastereoselectivity, which constituted an 11% improvement over our previous method.8n
 | ||
| Scheme 1 Substrate scope for direct chloriranium-induced polyene cyclization. The substrate (0.20 mmol, 1.0 eq.) was stirred with 76.8 mg 21 (0.24 mmol, 1.2 eq.) and 7.33 mg 22 (40.0 μmol, 0.2 eq.) in 2.00 mL HFIP at 0 °C for 15 min. The d.r. was determined by 1H-NMR spectroscopy of the crude reaction mixture. The given d.r. refers to the configuration at the carbons highlighted in grey. | ||
Next, substrates equipped with an aryl terminating nucleophile were subjected to our optimized reaction conditions. All compounds smoothly delivered the corresponding di- or tricyclic products 31–37 with excellent diastereoselectivities (up to >95:5) and chemical yields (30–78%), regardless of whether intramolecular O- or C-nucleophiles were applied. For heptadienes, which are one carbon atom shorter than the homogeranyl substrates, only monocyclization to product 34 occurred. This was possibly due to the thermodynamically unfavorable, strained 6–5 ring system (not shown) built by a complete cyclization sequence. In the presence of two competing nucleophiles (aryl versus ester moiety) the cationic intermediate 4 was trapped predominantly by the carbon nucleophile affording 37. It is noteworthy that in general chlorocyclizations of polyenes suffer from low conversion and low diastereoselectivities, thus rendering the chemical yield obtained here good to excellent for this type of transformation.16
Encouraged by our results, we extended the cyclization platform to structurally more challenging terpene and terpenoid carbon frameworks (Table 2). Interestingly, the addition of the chlorenium species to the trisubstituted C-3 olefin in α-humulene (38) started a selective transannular ring-closing event. This led to the 5-6-4 tricyclic carbon skeleton 39 in 25% yield with perfect diastereoselectivity (entry 1), showing a similar framework as that of protoilludene sesquiterpenes. Methyl jasmonate (40) was likewise converted using our method, yielding 51% of the HFIP–acetal 41 (entry 2). The slightly diminished diastereoselectivity may have originated from a partial equilibration of the Z-alkene moiety to its E conformer, prior to electrophilic addition. This assumption was corroborated by kinetic studies (see the ESI†) revealing a delayed starting point (ca. 30 min) for the formation of the minor C-2 epimer of 41 compared to the major isomer 41. Interestingly, no α-chlorination to the ketone functionality in 40 was detectable, showing the chemoselectivity of the developed methodology. Treating the monoterpene S-α-terpineol (42) with 2.4 equivalents of 21 delivered the dichlorinated eucalyptol 43 (40%, d.r. > 95:5, entry 3). Product 43 was furnished for the first time directly from 42,via a complex chlorine addition–elimination-chlorination-cyclization cascade. Previous reports on attempted halogenations of 42 resulted in the formation of acyclic chlorohydrin products (not shown) among other side products.17
| Entry | Substrate | Product | Yieldb (d.r.) |
|---|---|---|---|
| a Substrate (0.20 mmol, 1.0 eq.) was added to 76.8 mg reagent 21 (0.24 mmol, 1.2 eq.) and 7.33 mg saccharin 22 (40 μmol, 0.2 eq.) in 2.00 mL HFIP at 0 °C. b Isolated yield. c 2.4 eq. reagent 21 was used. The d.r. was determined by 1H-NMR spectroscopy of the crude reaction mixture. The given d.r. refers to the configuration at the carbon(s) highlighted in grey. | |||
| 1 |

|
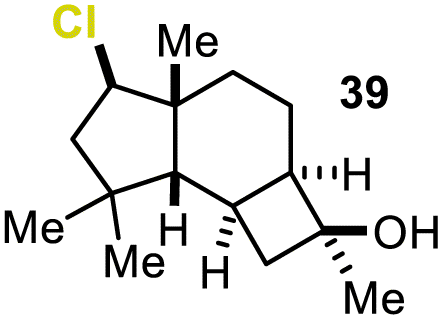
|
25% (>95:5) |
| 2 |

|

|
51% (73:27) |
| 3c |
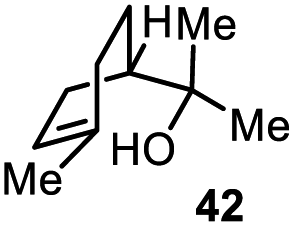
|

|
40% (>95:5) |
To elucidate the interplay of the Cl-iodanes, saccharine (22), and HFIP, NMR experiments were conducted (Fig. 2 and ESI†). To assess the chlorination ability of different chloro λ3-iodanes,18 compared to that of other reagents, we determined their halonium affinity (HalA, see the ESI†), a halogenation reactivity and chemoselectivity parameter.19 A significant influence of the chlorination ability of this reagent type was evident in HFIP. Other commonly used Cl-sources (e.g., 13 and 18) showed only weak interactions (for details see the ESI†). Thus, the iodanes (e.g., 19 and 21) were more activated than the other, more common chlorenium sources resulting in a more productive and selective chlorocyclization. 1H-NMR spectra obtained from the titration of iodane 19 (ref. 20) with HFIP showed a significant downfield shift of the hydroxy-proton signal of the fluoro alcohol (Fig. 2a). Simultaneously, the carbonyl group in 19 was shifted downfield by 4 ppm while a small upfield shift of the adjacent C-6 atom was detected in the corresponding 13C-NMR spectra (Fig. 2b). This strongly implies an attractive non-covalent interaction between the F-alcohol and the Lewis basic carbonyl in 19. This Lewis acid–Lewis base interplay activates 19 for electrophilic chlorine atom transfer and thus, is in line with the observed reactivity. Job's plot analysis (see the ESI†) revealed that this H-bonding interaction already occurs in the 1:1 HFIP–19 complex. As productive chlorocyclizations were only observed in HFIP as solvent, the F-alcohol must have multiple roles in these transformations. Indeed, beyond fine-tuning the chlorination reagent reactivity, the fluorinated alcohol controls the substrate conformation and stabilizes the carbocationic intermediate.21 Addition of the acid-catalyst saccharine (22) to the iodane–HFIP solution shifted the alcohol proton resonance further downfield (3.5 → 3.9 ppm, see the ESI†), while chloro-iodane 19 remained unaffected. Thus, we assume that 22 is integrated in the HFIP–hydrogen-bonding network but without direct interaction with 19. In no experiment the formation of N-chloro saccharine (23) was observed, further excluding 23 as the chlorinating agent (cf.Table 1 and ESI†).
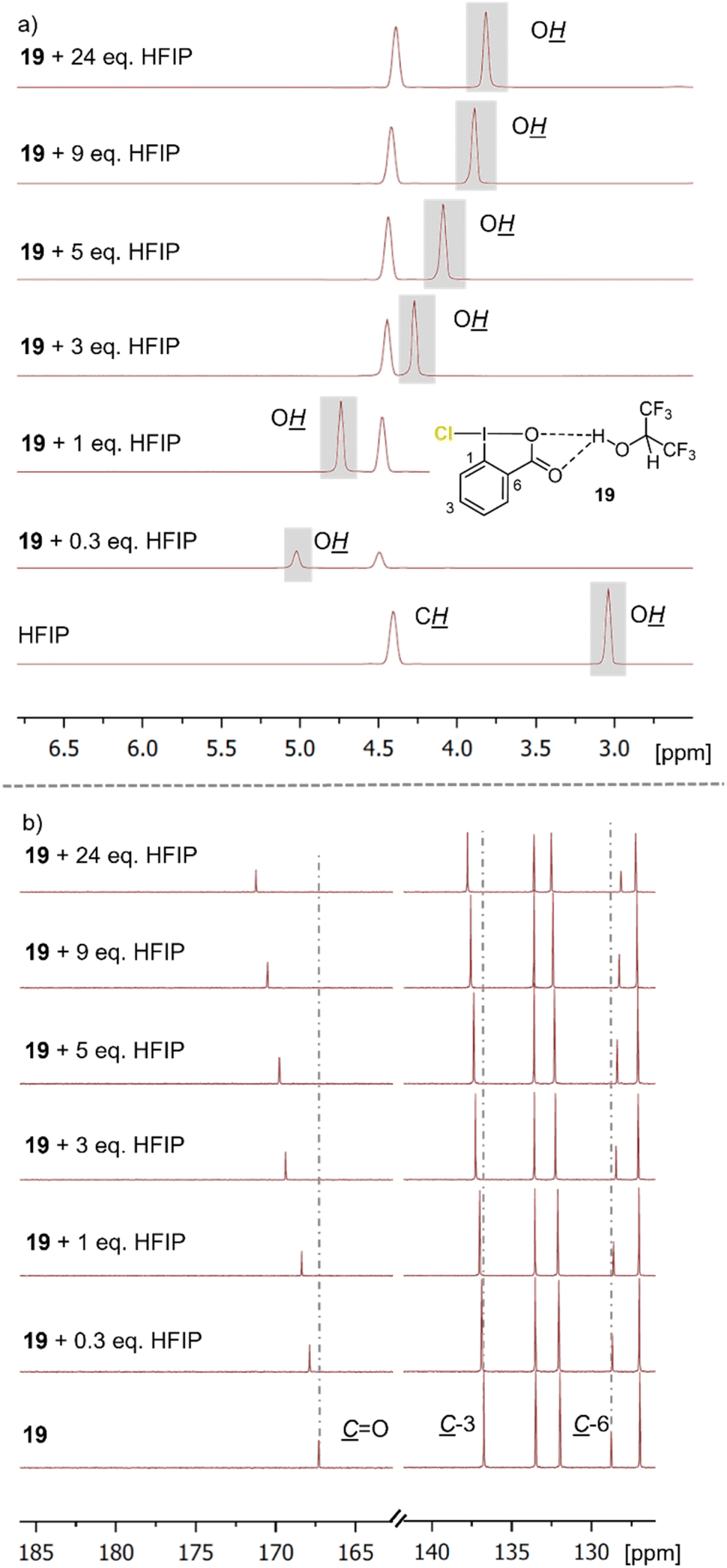 | ||
| Fig. 2 Overlap of the (a) 1H NMR spectra and (b) the 13C NMR spectra of chloro-iodane 19 and different equivalents of HFIP at 25 °C. | ||
3 Conclusion
In summary, we established a mild and straightforward one-step procedure for the challenging chlorination-assisted cation–π-cyclization of linear polyenes 1. Both the Lewis basic chlorenium source and the Lewis acid HFIP, played crucial roles and needed to be finely balanced. Only then did the pair form a heterogeneously microstructured environment imitating the protective and activating enzymatic pockets of terpene cyclases.1,14 The strong H-bonding ability of HFIP15 was utilized to selectively install iodane 21 in this hydrogen-bond network and, consequently, adjust its reactivity. In addition, cationic intermediates such as 2–4 (cf.Fig. 1a) were stabilized inside the H-bonding assembly, and the linear carbon chain of 1 adopted the desired chair-like conformation via solvophobic interactions.15,22 This enabled regio- and diastereoselective transformations (up to d.r. > 95:5) for a broad range of substrates. The general applicability of this method was shown with +20 examples. Differently substituted, electron-rich and -poor alkenes were likewise tolerated. The method was even extended to structurally complex substrates. Another step toward the biomimetic cyclization of polyenes has been reached with the presented method. A direct and more selective access to these powerful molecules is now possible.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.†Author contributions
J. B. conceived the project, designed and carried out the synthetic and mechanistic work, and analyzed the data. A. B. designed, conducted, and analyzed the mechanistic studies. T. G. conceived the project, provided guidance and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr A. M. Arnold for fruitful discussions. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the Emmy-Noether program (GU 1134/3) and – TRR 325 – 444632635.Notes and references
-
(a)
E. M. Davis and R. Croteau, in Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, ed. F. J. Leeper and J. C. Vederas, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000, pp. 53–95 Search PubMed
; (b) D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS PubMed
- R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS PubMed
-
G. W. Gribble, Naturally Occurring Organohalogen Compounds – A Comprehensive Update, Springer-Verlag Vienna, Vienna, 2010 Search PubMed
- J. Pika and D. John Faulkner, Tetrahedron, 1995, 51, 8189–8198 CrossRef CAS
-
(a) J. Uddin, K. Ueda, E. R. O. Siwu, M. Kita and D. Uemura, Bioorg. Med. Chem., 2006, 14, 6954–6961 CrossRef CAS PubMed
-
(a) J. Y. Cho, H. C. Kwon, P. G. Williams, P. R. Jensen and W. Fenical, Org. Lett., 2006, 8, 2471–2474 CrossRef CAS PubMed
-
(a) A. G. M. Barrett, T.-K. Ma and T. Mies, Synthesis, 2019, 51, 67–82 CrossRef CAS
- For selected examples of haliranium-ion-triggered polyene cyclizations see:
(a) E. E. van Tamelen and E. J. Hessler, Chem. Commun., 1966, 411–413 RSC
- For examples of enantioselective, haliranium-ion-triggered polyene cyclizations see:
(a) A. Sakakura, A. Ukai and K. Ishihara, Nature, 2007, 445, 900–903 CrossRef CAS PubMed
-
(a) G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1967, 89, 4744–4752 CrossRef CAS
-
(a) A. J. Cresswell, S. T. C. Eey and S. E. Denmark, Angew. Chem., Int. Ed., 2015, 54, 15642–15682 CrossRef CAS PubMed
- For examples of two-step procedures to yield chlorinated terpenes see reference 6c and S. A. Snyder, D. S. Treitler and A. A. Schall, Tetrahedron, 2010, 66, 4796–4804 CrossRef CAS
- For polyene cyclizations in HFIP not using halogen electrophiles see:
(a) Y. Tian, X. Xu, L. Zhang and J. Qu, Org. Lett., 2016, 18, 268–271 CrossRef CAS PubMed
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed
-
(a) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS
- Reactions leading to the desired chloro cyclization products only in low yields turned out to be sluggish in general giving a plethora of different but undefined products. Exemplarily, the GC traces of the crude mixtures of the reactions to products 29–31, 34, and 36 are given in the ESI† to show the difference between high-yielding and low-yielding transformations.
-
(a) L. Kopperman, R. C. Hallcher, A. Riehl, R. M. Carlson and R. Caple, Tetrahedron, 1976, 32, 1621–1626 CrossRef
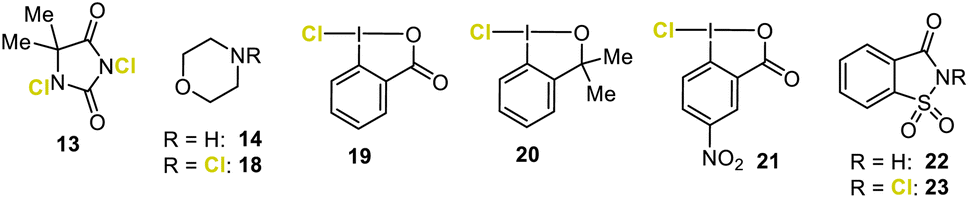
- Hypervalent chloro iodanes 19 and 21 together with other chloro benziodoxolones bearing different substituents at the aromatic portion (for details see the ESI†) were used in this study.
- K. D. Ashtekar, N. S. Marzijarani, A. Jaganathan, D. Holmes, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2014, 136, 13355–13362 CrossRef CAS PubMed
- In further mechanistic studies, iodane 19 replaced the more reactive nitro analog 21 because of the generally low solubility of 21 in CDCl3.
- F.-X. Tian and J. Qu, J. Org. Chem., 2022, 87, 1814–1829 CrossRef CAS PubMed
-
(a) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06664e |
| This journal is © The Royal Society of Chemistry 2023 |